

Abstract
Hyperhomocysteinemia (HHcy) is a significant and independent risk factor for cardiovascular diseases. Endothelial dysfunction (ED) is the earliest indicator of atherosclerosis and vascular diseases. We and others have shown that HHcy induced ED in human and in animal models of HHcy induced by either high-methionine load or genetic deficiency. Six mechanisms have been suggested explaining HHcy-induced ED. These include 1) nitric oxide inhibition, 2) prostanoids regulation, 3) endothelium-derived hyperpolarizing factors suppression, 4) angiotensin II receptor-1 activation, 5) endothelin-1 induction, and 6) oxidative stress. The goal of this review is to elaborate these mechanisms and to discuss biological and molecular events related to HHcy-induced ED.
Keywords: Hyperhomocysteinemia, endothelial dysfunction, vascular relaxation/vascular contractile responses
Hyperhomocysteinemia and Cardiovascular Diseases
In healthy individuals, plasma homocysteine (Hcy) level is between 5-10 μmol/L. Elevation of plasma Hcy levels creates a condition called hyperhomocysteinemia (HHcy), which is characterized into three ranges; moderate (16-30 μmol/L), intermediate (31 - 100 μmol/L), and severe (>100 μmol/L) HHcy [1]. In the general population, modulate elevations in plasma Hcy and may be due to inherited enzyme variants and/or a relative deficiency of folate, vitamin B6, which are required for the normal metabolism of Hcy. Two strongest determinants of plasma Hcy concentration are folate status and renal function.
McCully suggested that HHcy are causally related to cardiovascular diseases (CVD) in 1969 [2]. However, this theory was neglected for a long time, probably due to the low prevalence of inborn errors of metabolism that were accompanied by severe HHcy. In 1976, HHcy gained a world-wide interest, when Wilcken and Wilcken observed an abnormal rise in plasma Hcy after oral methionine (Met) loading in patients with coronary artery disease [3]. Since then, abundant retrospective and prospective observational studies have shown a relation between plasma total homocysteine (tHcy) level and coronary artery disease, peripheral artery disease, stroke, or venous thrombosis [4, 5]. In a metaanalysis by Boushey et al., an increase of 5 μmol/L in plasma Hcy enhanced the risk of CVD by 1.6-to 1.8-fold [4]. Another recent meta-analysis by den Heijer et al., also shown that an 25% elevation (about 3 μmol/L) tHcy was associated with about a 10% higher risk of cardiovascular events and a 20% higher risk of stroke after adjustment for other known risk factors [5].
Homocysteine Metabolism
The single source of Hcy in humans is the demethylation of essential amino acid, Met, via two intermediate compounds, S-adenosylmethionine (SAM), a major donor of cellular methylation, and S-adenosylhomocysteine (SAH), a potent inhibitor of biological transmethylation (Fig. 1). SAM is the methyl donor in over 115 different cellular methyltransferase reactions, including those of DNA, RNA, proteins and lipids [6]. With the transfer of the methyl group, SAM is converted into SAH. Hcy can utilize adenosine, a normal constituent of all body fluids to forma SAH, a potent inhibitor of cellular methylation, thereby causing cellular hypomethylation. The intracellular SAM/SAH ratio has been used as a predictor of cellular methylation capacity. SHA is further converted into Hcy, a nonproteinogenic amino acid containing a free sulfhydryl group, and adenosine by the SAH hydrolase, which is widely distributed in mammalian tissues. Hcy can be further metabolized via two alternative pathways: it may be irreversibly degraded through the transsulfuration pathway or remethylated to Met via remethylation pathway. Hcy is transsulfurated to cystathionine and cystaine cystathionine β-synthase (CBS), vitamin B6 dependently in the liver and kidneys. Hcy can be also remethylated to Met by folate dependent and non-dependent pathways. When the cellular capacity to metabolize Hcy is exceed, this amino acid will be exported to the extracellular compartment to achieve equilibrium concentration in the circulation.
Fig. (1). The pathways of homocysteine metabolism.
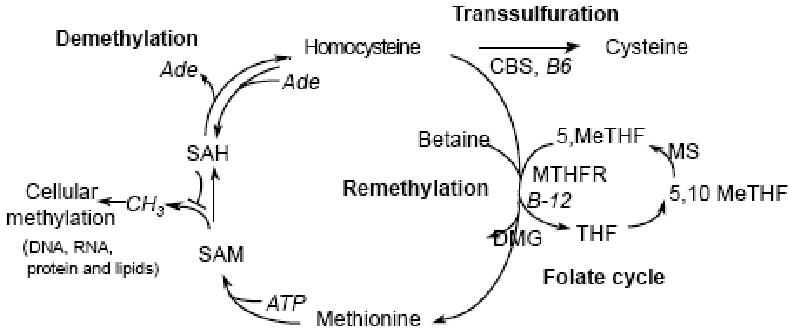
Ade, Adenosine; CBS, cystathionine-b-synthase; DMG, dimethyglycine; MeTHF, methylenetetrahydrofolate; MS, methionine synthase; MTHFR, methylenetetrahydrofolate reductase; SAH,S-adenosylhomocysteine; SAM, S-adenosylmethionine; THF, tetrahydrofolate.
Hcy is present in different forms. About 70-80% is protein-bound. Non-protein-bound Hcy is found in “mixed disulfide” (i.e. the dimmer of Hcy and cysteine), homocystine (the oxidized disulfide of Hcy) and reduced Hcy. Reduced Hcy forms only about 1% of the tHcy content. Laboratories usually measure tHcy concentration, which is the sum of all Hcy fractions [7].
Endothelial Injury as the Primary Mechanism for Hcy-Related CVD
During the last two decades, biomedical research has been actively exploring the role of Hcy in atherosclerosis and the underlying mechanisms [8-10]. Among the proposed mechanisms, we believe that endothelial injury is the primary mechanism for Hcy-related CVD. Our laboratory were the first to report: 1) Hcy, at clinically relevant concentrations, arrests cell growth and suppresses cyclin A transcription in Endothelial cells (ECs) via hypomethylation-related mechanism [8, 11]. These findings, which cannot be mimicked by cysteine, led us to propose that hypomethylation is the major biochemical mechanism in Hcy vascular disease. 2) HHcy accelerates atherosclerosis in mice [12] and inhibits high density lipoprotein biosynthesis in human and mice [13]. 3) Severe HHcy impairs reendothelialization and increases neointimal formation in mice [14]. 4) Hcy demethylates cyclin A promoter and inhibits DNA methyltransferases 1 activity in ECs [15]. 5) Hcy transport is differentially regulated in vascular cells and is predominantly via a lysosomal dependent alanine-serine-cysteine transport system [16]. 6) Hcy inhibits cyclin A transcription and cell growth by inhibiting DNA methylation through suppression of DNA methyltransferase 1 in ECs [17]. It is possible that HHcy induces endothelial injury and ED leading to the development of atherosclerosis.
Homocysteine and Endothelial Dysfunction
The endothelium is a single layer of cells lining all blood vessels. It plays an important role in many physiological functions, including the control of blood cell trafficking, vasomotor tone, vessel permeability, and hemostatic balance. ECs produce a wide variety of substances in response to various physical and chemical stimuli, including vasodilator substances (i.e. nitric oxide, NO; prostacyclin, PGI2; and endothelium-derived hyperpolarizing factor, EDHF), and vasoconstrictor substances (i.e. endothelin-1, ET-1; angiotensin II, Ang II; thromboxane A2, TXA2 or free radicals) [18].
ED is defined as an impairment of endothelium-dependent relaxation of blood vessels. It is the earliest indicator of the development of CVD and precedes the appearance of atherosclerotic plaque and the frank symptoms of peripheral vascular disease. Chronic exposure to physical and chemical stimuli can lead to ED. Impaired endothelial vasomotor function was observed in human and experimental models of HHcy in minipigs, monkey, rats and mice either with high-Met diet or the deletion of the CBS gene [19, 20]. Further, endothelium-dependent vascular relaxation response to acetylcholine (ACh) and bradykinin was also impaired in the pig common carotid arteries pretreated with Hcy (50 and 100 μmol/L) for 24 h, rabbit thoracic aorta rings incubated with Hcy (0.1, 1, and 10 mmol/L) for 3 h [21]. Superfusion of Hcy (1 mmol/L) on the cerebral cortex impaired cerebrovascular vasodilatory responses induced by ACh in rabbits [22]. Six mechanisms have been suggested explaining HHcy-induced ED. These include 1) NO inhibition, 2) prostanoids regulation, 3) EDHF inhibition, 4) angiotensin II receptor 1 (AT1) activation, 5) endothelin-1 (ET-1) induction, and 6) oxidative stress. We summarized these mechanisms in Fig. 2 and discussed the biological and molecular events related to HHcy-induced ED below.
Fig. (2). Potential mechanisms of HHcy-induced endothelial dysfunction.
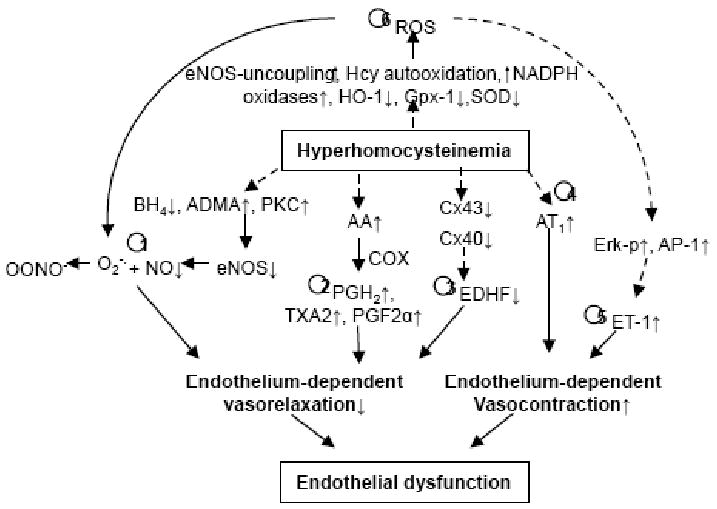
Six mechanisms have been suggested explaining HHcy-induced ED; 1) nitric oxide inhibition, 2) prostacyclin regulation, 3) endothelium-derived hyperpolarizing factors suppression, 4) angiotensin II receptor-1 activation, 5) endothelin-1 induction, and 6) oxidative stress. AA, aracidonic acid; AP-1, activator protein-1; AT1, angiotensin II receptor 1; eNOS, endothelial NO synthase; ADMA, asymmetric dimethylarginie, an eNOS inhibitor; BH4, tetrahydrobiopterin; COX, cyclooxygenase; Cx, connexins; EDHF, endothelium-derived hyperpolarizing factor; Erk-p, phosphorylated extracellular signal-regulated protein kinase, ET-1, endothelin-1; Gpx-1, glutathione peroxidase, an antioxidant enzyme; HO-1, heme oxygenase-1; O2-•, superoxide anion; OONO-, peroxynitrite anion; BH4, tetrahydrobiopterin, a cofactor of eNOS; NADPH, a cofactor of eNOS; NO, nitric oxide; PKC, protein kinase C; PGH2, protaglandin H2; PGF2α, prostaglandin F2α; ROS, reactive oxygen species; SOD, superoxide dismutase; TXA2, thromboxane A2,
1) NO Inhibition
Multiple studies suggested that HHcy induces ED via NO inhibition. NO is a potent vasodilator that activates soluble guanylyl cyclase in vascular muscle, resulting in accumulation of cyclic guanosine monophosphate (cGMP) and relaxation. It was reported that bioavailability of endothelium-derived nitric oxide was reduced in the aorta, mesenteric arterioles, cerebral arterioles and arterials from gracilis muscle of mice with mild HHcy [20]. We found that endothelium-dependent vascular relaxation response to ACh was impaired in the aortic and cremaster microvascular arterials from CBS-/- mice. Preinucabtion with NG-nitro-L-arginine methyl ester (L-NAME), an endothelial nitric oxide synthase (eNOS) inhibitor, abolished vascular relaxation response to ACh, indicating that decreased NO bioavailability was involved in HHcy-induced ED (Fig. 3) [19]. A suppressive effect of Hcy on endogenous bioavailable NO is also supported by the previous finding that incubation of arterial strips with Hcy significantly attenuated the endothelium-dependent vascular relaxation response to calcium ionophore A23187 [23]. NO bioavailability could be diminished by the reaction of NO with superoxide anion (O2-.) or by the reaction of thiol groups in the Hcy molecule with NO. Exposure of ECs to Hcy led to the formation of S-nitrosohomocysteine, decreasing the bioactivity of NO [24].
Fig. (3). Severe HHcy Impaired endothelial dependent arterial relaxation via eNOS inhibition.
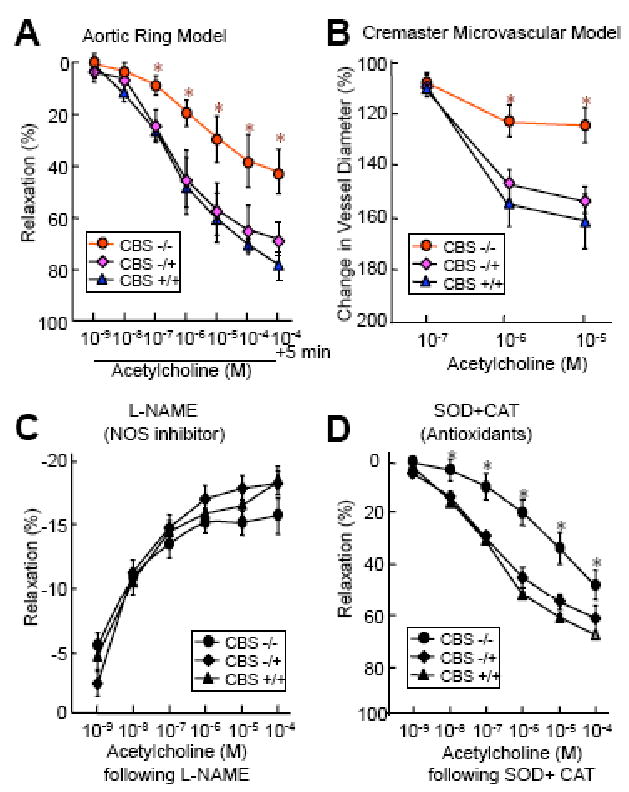
Mouse vascular reactivity was examined using two ex-vivo functional models, aortic ring and intravital videomicroscopy of the cremaster. Mouse thoracic aortas were harvested from CBS KO mice at the age of 10 weeks. A. Ex vivo aortic vasomotor response. Vessel rings were precontracted and then contracted with 10-6 M phenylephrine. Dose-responsive relaxation was measured in response to cumulative addition of acetylcholine (10-9-10-4M) in 1 min interval. B. Cremaster microvascular vasomotor response. Mouse cremaster micrevasculature was pinned radially, visualized through a upright microscope and continually superfused with Krebs buffer. The inner lumen diameter of the arteriole was measured before and after superfusion with acetylcholine (10-7-10-5M) in a 3 min interval. C & D. Vasorelaxation to acetylcholine following NG-nitro-L-arginine methyl ester (L-NAME) and superoxide dismutase (SOD) + catalase (CAT). Aortic rings were precontracted with KCl, treated with 30 mM L-NAME for 20 min or 200 U/ml SOD plus 100 U/ml CAT for 2 min, and contracted with phenylephrine. Dose-response relaxation was measured in response to cumulative addition of acetylcholine at 1 min intervals. n=9, * P≤0.01 verses CBS-/+ or CBS+/+ mice with identical treatment.
Effect of HHcy on eNOS expression on vasculature was controversy by either upregulate [25] or downregulate [19]. We have found that HHcy inhibited eNOS activity in human aortic ECs via PKC and threonine 495 phosphorylation pathways [19]. Recent evidence suggests that asymmetric dimethylarginine (ADMA), an endogenous inhibitor eNOS, may mediate HHcy-induced ED [20]. The supporting evidence for the ADMA hypothesis is that Hcy reduced the activity of dimethylaminohyrolase (DDAH), the enzyme that degrades ADMA, in cultured endothelial cell line ECV304 [26]. However, we did not find an elevated plasma ADMA levels in CBS-/- mice, a genetic model of severe HHcy, by using HPLC analysis [19]. In addition, tetrahydrobiopterin (BH4), a cofactor of eNOS, was recently reported to attenuate Hcy-mediated impairment of endothelium-dependent relaxation in rats aortic rings [27]. ED in the mesenteric artery from pregnant CBS+/- mice was restored by preincubation with sepiaptern, a precursor of BH4 [28]. Very recently, It was reported that 5-MTHF has beneficial effects on endothelial function and vascular superoxide production in human atherosclerosis, by preventing peroxynitrite-mediated BH4 oxidation and improving eNOS coupling [29]. Interestingly, we have found that neither argine (1 mmol/L) nor BH4 (10 μmol/L) or sepiapterin (10 μmol/L) could prevent the inhibitory effect of Hcy on eNOS activity in cultured human and mouse aortic ECs [19].
2) Prostanoids Regulation
Prostanoids including prostaglandins and thromboxanes are generated from aracidonic acid by cyclooxygenase (COX). Prostacyclin (PGI2) is the major vasodilatory prostanoid produced in ECs and is released in response to shear stress, hypoxia, or to substances that stimulate NO formation. PGI2 relaxes vascular smooth muscle by activation of adenylate cyclase (AC), and thus leads to increased production of cyclic adenosine monophosphate (cAMP). In addition to the vasodilatory prostanoids, ECs also produce vasoconstrictive prostanoids such as prostaglandin F2α (PGF2α), prostaglandin H2 (PGH2), or thromboxane A2 (TXA2). Contractile prostaglandins induced vascular contraction by inhibiting activity of AC, and thus lead to decreased generation of cAMP Previous studies found that a high Hcy concentration increased TXA2 and 8-iso-PGF2α (a metabolite of PGF2α) biosynthesis [30-32]. Preincubation with a non-specific COX inhibitor indomethacin or PGH2/TXA2 receptor antagonist SQ 29,548 restored ED in the skeletal muscle arterioles from Wistar rats fed with high-Met (1 g/kg) diet for 4 weeks [31]. In the arterioles of HHcy rats, increased flow-induced constriction was abolished by indomethacin or SQ 29,548 or the TXA2 synthase inhibitor CGS 13,080 [32]. PGI2 generation seems not significantly affected by HHcy [33]. Whether contractile prostanoids induction in HHcy is the result of COX activation and/or due to increased release of arachidonic acid remains unclear. In fact, recent evidence that Hcy induced arachidonic acid release and formation of its end product TXB2 in platelet may provide partial explanation [34].
Taken together, HHcy induces ED, probably partially, via aracidonic acid-prostanoids pathway. The induction of TXA2, PGH2 and PGF2α, but not PGI2, seems contributing to HHcy-induced ED. Further studies exploring the role and mechanisms of HHcy-induced imbalance of vasodilatory and vasocontractive prostanoids should provide important insights.
3) Endothelium-Derived Hyperpolarizing Factors (EDHF) Inhibition
Accumulative evidence is mounting that EDHF is a major determinant of vascular tone, especially in small resistance vessels [18]. The nature of EDHF is still not entirely elucidated. The endothelium-mediated relaxation that remains resistant to NOS and COX inhibition is thought to be mediated by EDHF. Current evidence suggests that EDHF-mediated responses are initiated by activation of endothelial K+ channels with resultant hyperpolarization of ECs. This endothelial hyperpolarization spreads to the underlying smooth muscle layer through myoendothelial gap junctions, or the efflux K+ from the ECs elicit hyperpolarization of the adjacent smooth muscle cells. Epoxyeicosatrienoic acid likely has a regulatory role in this pathway [35]. EDHF may reflect a purely electrical coupling process between cells and involve myoendothelial gap junctions that are constituted, potentially, from the five connexins (Cx 37, 40, 43, 44 and 45) found in vascular tissues [36-38]. Several risk factors for atherothrombotic CVD have been reported to affect the EDHF pathway, including hypertension [39], diabetes [40], hypercholesterolemia [39]. Role of Hcy on EDHF still remains unclear. EDHF-mediated renal vasodilatory response was impaired in the Wistar rats during acute and chronic HHcy [41]. Furthermore, HHcy-induced impairment of EDHF was suggested to be related to redistribution of Cx43 [42], and SAH related down-regulation of Cx44 in ECs [38]. In addition, oxidative stress, implicated as a pathological mechanism in HHcy, has also been shown to disrupt gap junctional communication [43]. Understanding the mechanisms of HHcy-induced EDHF impairment could be beneficial for the prevention and/or therapy of microangiopathy in HHcy.
Endothelium-independent dilation induced by sodium nitroprusside (SNP), a nitric oxide donor, however, has been reported to be reduced in HHcy by some authors [44] or not reduced by others [45]. The differences on endothelium-independent relaxation response to SNP may be due to difference on tHcy levels, duration of HHcy, species, and type of vessels, and synergetic effects of other factors such as hyperglycemia and dyslipidemia.
4) Angiotensin II Receptor-1 Activation
Angiotensin II (Ang II), an octapeptide hormone, is the active component of the renin-angiotensin system. Ang II is a multifunctional peptide that has numerous actions on vascular smooth muscle, such as the modulation of vasomotor tonus. Accordingly, Ang II plays a fundamental role in controlling the functional and structural integrity of the arterial wall and may be important in physiological processes, regulating blood pressure and pathological mechanisms underlying vascular disease. Therefore, understanding whether Ang II is involved in HHcy-induced ED may provide a fundamental insight in the treatment of HHcy-related vascular diseases. However little is known about the effect of this elevation upon the vascular reactivity to Ang II. Recent study found that AT1 activated in mouse aortic ECs treated with Hcy (100 μmol/L) for 48 h [46]. Furthermore, HHcy-induced activation of AT1 receptor involves matrix metalloproteinase-9 and collagen type-1 modulation using extracellular signal-regulated kinase-1/2 and signal transducer and activator of transcription 3 signaling cascades [46]. Valsartan, a AT1 receptor blocker, attenuated pathological ventricular hypertrophy induced by HHcy in rats [47]. Vasoconstraction to Ang II was increased in the carotid artery from Wistar rats fed with Met (0.1, 1 and 2g/kg body weight) in the drinking water for 8 and 16 weeks [48]. Preincubation with indomethacin, normalized the contractile response to Ang II. This study suggested that HHcy-induced vasocontractile response to Ang II appears to be related to the release of vasoconstrictor prostanoids [48].
5) Endothelin-1 Induction
Endothlin-1 (ET-1) is a 21-residue peptide synthesized and secreted by EC. It is classically defined as a potent vasoconstrictor and mitogen for VSMCs. ET-1 often acts as a paracrine hormone, binding to transmembrane ETA receptor expressed on VSMC. However, ET-1 also elicits a vasodilatory response mediated by transmembrane ETB receptors on vascular ECs. Because regulation of vascular tone is one of the most specific endothelial functions so far described, ET-1 is used as a powerful marker to study endothelial function.
Several studies showed that HHcy activate or induce ET-1. Preincubation with Hcy (500 μmol/L) for 24 h increased the secretion and mRNA levels of ET-1 in human umbilical vein ECs [49]. It is suggested that Hcy induces ERK phosphorylation via reactive oxygen species (ROS) accumulation, and activates transcriptional factor AP-1, which in turn elicits additional expression and secretion of ET-1 [49, 50]. Clinically, plasma Hcy level showed significantly positive correlation with plasma ET-1 level in the patients with disturbed glucose metabolism [51]. Hcy-induced ET-1 release is dependent on hyperglycaemia and reactive oxygen species (ROS) production in bovine aortic ECs [52]. Recently, Duan et al., reported that phytoestrogen alpha-zearalanol ablated Hcy-elicited ET-1 secretion/mRNA induction and ROS accumulation [49]. In contrast, other studies shown that pathophysiological concentrations of Hcy reduced both ET-1 production and preproET-1 mRNA levels in cultured EC potentially through oxidative products [53, 54]. Because of the contradictory observations stated, the role of ET-1 in HHcy-induced ED is uncertain and need to be further evaluated.
6) Role of Free Radical
Oxidation has been suggested as a primary biochemical mechanism responsible for HHcy-related pathogenesis. Experiments using animal models with genetically or diet-induced HHcy [55] or Hcy treated cultured ECs [56] showed an increased accumulation of ROS. Five mechanisms have been proposed for Hcy-induced oxidative stress. These include: 1) inhibition of the activity of cellular antioxidant enzymes such as cellular glutathione peroxidase or heme oxygenase -1, 2) Hcy autooxidation, 3) nitric oxide synthase (NOS)-dependent generation of superoxide anion (O-2) via uncoupling of eNOS, 4) disruption of extracellular superoxide dismutase from endothelial surfaces, and 5) activation of NADPH oxidases.
ROS and oxidant stress promote the formation of nitrotyrosine, an indictor of the NO and superoxide radical reaction, resulting in the formation of the strong oxidant peroxynitrite. Peroxynitrite, besides other effects, leads to tyrosine nitration. The letter event may alter protein function, and therefore, induce cellular dysfunction. Pretreatment with catechin, a flavonoid antioxidant that reduces ROS levels, decreased Hcy-dependent formation of nitrotyrosine [57]. Peroxynitrite can also directly damage the electron-transport chain in mitochondria and thereby induce mitochondrial and consequently ED [58]. Apocynin, a NADPH oxidase inhibitor, not only attenuated aortic superoxide and peroxynitrite to control levels but also restored endothelium-dependent relaxation in the aortas of HHcy rats [59]. Tempol, a cell permeable mimetic of SOD, improved endothelial function in the rat femoral arteries preincubation with D,L-Hcy (100 μM) for 4 h [60]. Tempol also improved ED in the absence of Hcy after the initial incubation period [60]. Decreased NO bioavailability by reaction of NO and superoxide is likely a major mechanism on oxidative stress-induced ED. Superoxide also plays an important role in impairment of EDHF by oxidation of myoendothelial gap junction proteins [43].
7) Vascular Contractile Response to Phenylephrine
Phenylephrine (PE) induces smooth muscles cell contraction by stimulating α1-adrenoreceptors, which causes a rapid calcium release from scarcoplasmic reticulum and prevents calcium reuptake. PE also depolarizes the arterial smooth muscle cells and consequently activates voltage-operated Ca2+ channels in the plasma membrane of the smooth muscle, leading to an increased influx of Ca2+. The sustained contractions result from calcium influx through the receptor-operated calcium channels. Vascular contractile response to PE is either increased [61, 62] or decreased in the presence of HHcy [63]. Preincubation with Hcy (10 mmol/L) for 3 h, increased vascular contractile response to PE in pulmonary arteries of guinea-pig [62]. In addition, long-term high Met load (1g/kg body weight) for 4 weeks increased vasoreactivity to PE in the rat skeletal muscle arteriolars [64]. Furthermore, increased contractile response to PE by HHcy was significantly inhibited by the concomitant incubation with either SOD plus catalase, Tiron or PJ34 (an inhibitor of polyADP-ribose polymerase) [62, 64], suggesting the involvement of oxidative stress. Increased contractile response to PE can be also inhibited by preincubation with L-NAME, suggesting HHcy reduced activity of NO in arterioles may contribute to the microvascular impairment described in HHcy [64]. Very recently, Andrade et al., demonstrated that the enhanced PE-induced vascular contraction in carotid artery due to HHcy is endothelium-dependent and involves a loss of the inhibitory effect of relaxant α1D-adrenoceptors by reducing NO biodisponibility [61].
Enhanced vasoreactivity to PE in HHcy may be via endothelium-dependent pathway, and may be related to oxidative stress, decreased NO bioavailability, loss of the inhibitory effect of relaxant α1D- adrenoceptors by reducing NO biodisponibility.
Hcy-Lowering Therapy and ED
Several studies have described an improvement in endothelial function following administration of folic acid (5 or 10 mg/day), but not with low dose of folic acid (400 μg/day) in human [65, 66]. More recently, It was reported that folic acid supplementation at high dose (0.071 μg/g/day, equivalent to a 5 mg/70 kg/day human dose) but not low dose (0.0057 μg/g/day, equivalent to a 400 μg/70kg/day human dose) for two weeks restored ED in the mesenteric artery from CBS (+/-) mice [67]. This suggests that the dosage of folic acid may be critical for improvement of ED in HHcy. Improvement of endothelial function does not consistently correlate with total Hcy lowering [66]. However, some studies found a significant positive correlation between tHcy reduction and improvement in endothelial function [68]. The efficacy of folic acid supplementation on improving ED and prevent Hcy-related cardiovascular events remains an open questions and requires further evaluation.
Conclusion
In summary, extensive experimental evidence, both in vitro and in vivo, indicates that HHcy impairs endothelial function. As summarized in Fig 2, HHcy changes vascular tone by regulating endothelium-dependent vasodilator and constrictor substances, including decreasing NO bioavailability, increasing contractile prostanoids as well as interfering myoendothelial communication. Although oxidative stress and eNOS uncoupling have been suggested to play a critical role in HHcy-induced ED, in our study, preincubation with antioxidant SOD plus catalase did not restored ED in the aortic arterial from CBS-/- mice (Fig. 3) [19]. Further, adenoviral transduced SOD or GPX-1 overexpression did not restore eNOS activity suppressed by Hcy in cultured mouse ECs [19]. Similarly, results of randomized controlled trials of antioxidant vitamin supplements in large numbers of participants has been ambiguous or contradictory [69]. This discrepancy remains to be resolved in future experimental and clinical studies. Studies on the mechanisms of HHcy-induced impairment of microvasculature myoendothelial gap junction, such as Cx43 and Cx44, should be an interesting area for future research. Further, the effect of HHcy on ET-1 secretion/generation may be relevant for CVD in diabetes. Finally, the beneficial effects on the endothelial function by supplementation of high dose folic acid (5-10 mg/day) may provide potential therapeutic advantage for HHcy-related cardiovascular diseases.
Abbreviation
- AC
Adenylate cyclase
- ACh
Acetylcholine
- ADMA
Asymmetric dimethylarginine
- Ang II
Angiotensin II
- AT1
Angiotensin II receptor-1
- BH4
Tetrahydrobiopterin
- cAMP
Cyclic adenosine monophosphate
- CBS
Cystathionine β-systhase
- COX
Cyclooxygenase
- CVD
Cardiovascular disease
- Cx
Connexin
- DDAH
Dimethylaminohyrolase
- ECs
Endothelial cells
- ED
Endothelial dysfunction
- EDHF
Endothelium-derived hyperpolarizing factor
- eNOS
Endothelial nitric oxide synthase
- ET-1
Endothelin-1
- ETA
Endothelin-1 receptor A
- Hcy
Homocysteine
- HHcy
Homocystinemia
- Met
Methionine
- MS
Methionine synthase
- MTHER
5,10-methylenetetrahydrofolate reducatase
- NO
Nitric oxide
- PGH2
Prostaglandin H2
- PGI2
Prostacyclin
- PGF2α
Prostaglandin F2α
- SNP
Sodium nitruprusside
- SOD
Superoxide dismutase
- ROS
Reactive oxygen species
- tHcy
Plasma total homocysteine
- TXA2
Thromboxane A2
References
- 1.Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol. 2004;10(12):1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56(1):111–128. [PMC free article] [PubMed] [Google Scholar]
- 3.Wilcken DE, Wilcken B. The pathogenesis of coronary artery disease. A possible role for methionine metabolism. J Clin Invest. 1976;57(4):1079–1082. doi: 10.1172/JCI108350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. Jama. 1995;274(13):1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- 5.Collaboration HLT. Dose-dependent effects of folic acid on blood concentrations of homocysteine: a meta-analysis of the randomized trials. Am J Clin Nutr. 2005;82(4):806–812. doi: 10.1093/ajcn/82.4.806. [DOI] [PubMed] [Google Scholar]
- 6.Scott JM, Weir DG, Molloy A, McPartlin J, Daly L, Kirke P. Folic acid metabolism and mechanisms of neural tube defects. Ciba Found Symp. 1994;181:180–187. doi: 10.1002/9780470514559.ch11. discussion 187-191. [DOI] [PubMed] [Google Scholar]
- 7.Mudd SH, Finkelstein JD, Refsum H, et al. Homocysteine and its disulfide derivatives: a suggested consensus terminology. Arterioscler Thromb Vasc Biol. 2000;20(7):1704–1706. doi: 10.1161/01.atv.20.7.1704. [DOI] [PubMed] [Google Scholar]
- 8.Lee ME, Wang H. Homocysteine and hypomethylation. A novel link to vascular disease. Trends Cardiovasc Med. 1999;9(1-2):49–54. doi: 10.1016/s1050-1738(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Tan H, Yang F. Mechanisms in homocysteine-induced vascular disease. Drug Discovery Today (Disease mechanisms) 2005;2(1):25–31. [Google Scholar]
- 10.Yang F, Tan HM, Wang H. Hyperhomocysteinemia and atherosclerosis. Sheng Li Xue Bao. 2005;57(2):103–114. [PubMed] [Google Scholar]
- 11.Wang H, Yoshizumi M, Lai K, et al. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J Biol Chem. 1997;272(40):25380–25385. doi: 10.1074/jbc.272.40.25380. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Jiang X, Yang F, et al. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood. 2003;101(10):3901–3907. doi: 10.1182/blood-2002-08-2606. [DOI] [PubMed] [Google Scholar]
- 13.Liao D, Tan H, Hui R, et al. Hyperhomocysteinemia decreases circulating high-density lipoprotein by inhibiting apolipoprotein A-I Protein synthesis and enhancing HDL cholesterol clearance. Circ Res. 2006;99(6):598–606. doi: 10.1161/01.RES.0000242559.42077.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan H, Jiang X, Yang F, et al. Hyperhomocysteinemia inhibits post-injury reendothelialization in mice. Cardiovasc Res. 2006;69(1):253–262. doi: 10.1016/j.cardiores.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jamaluddin MS, Chen I, Yang F, et al. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood. 2007;110 doi: 10.1182/blood-2007-06-096701. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X, Yang F, Brailoiu E, et al. Differential regulation of homocysteine transport in vascular endothelial and smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/ATVBAHA.107.148544. Revision submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jamaluddin MD, Chen I, Yang F, et al. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood. 2007;110(10):3648–3655. doi: 10.1182/blood-2007-06-096701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aird WC. Endothelium as an organ system. Crit Care Med. 2004;32(5 Suppl):S271–279. doi: 10.1097/01.ccm.0000129669.21649.40. [DOI] [PubMed] [Google Scholar]
- 19.Jiang X, Yang F, Tan H, et al. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler Thromb Vasc Biol. 2005;25(12):2515–2521. doi: 10.1161/01.ATV.0000189559.87328.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dayal S, Lentz SR. ADMA and hyperhomocysteinemia. Vasc Med. 2005;10(Suppl 1):S27–33. doi: 10.1191/1358863x05vm599oa. [DOI] [PubMed] [Google Scholar]
- 21.Lang D, Kredan MB, Moat SJ, et al. Homocysteine-induced inhibition of endothelium-dependent relaxation in rabbit aorta: role for superoxide anions. Arterioscler Thromb Vasc Biol. 2000;20(2):422–427. doi: 10.1161/01.atv.20.2.422. [DOI] [PubMed] [Google Scholar]
- 22.Zhang F, Slungaard A, Vercellotti GM, Iadecola C. Superoxide-dependent cerebrovascular effects of homocysteine. Am J Physiol. 1998;274(6 Pt 2):R1704–1711. doi: 10.1152/ajpregu.1998.274.6.R1704. [DOI] [PubMed] [Google Scholar]
- 23.Okatani Y, Wakatsuki A, Reiter RJ. Protective effect of melatonin against homocysteine-induced vasoconstriction of human umbilical artery. Biochem Biophys Res Commun. 2000;277(2):470–475. doi: 10.1006/bbrc.2000.3687. [DOI] [PubMed] [Google Scholar]
- 24.Stamler JS, Osborne JA, Jaraki O, et al. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J Clin Invest. 1993;91(1):308–318. doi: 10.1172/JCI116187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Upchurch GR, Jr, Welch GN, Fabian AJ, Pigazzi A, Keaney JF, Jr, Loscalzo J. Stimulation of endothelial nitric oxide production by homocyst(e)ine. Atherosclerosis. 1997;132(2):177–185. doi: 10.1016/s0021-9150(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 26.Stuhlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation. 2001;104(21):2569–2575. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- 27.Dhillon B, Badiwala MV, Maitland A, Rao V, Li SH, Verma S. Tetrahydrobiopterin attenuates homocysteine induced endothelial dysfunction. Mol Cell Biochem. 2003;247(1-2):223–227. doi: 10.1023/a:1024146501743. [DOI] [PubMed] [Google Scholar]
- 28.Powers RW, Gandley RE, Lykins DL, Roberts JM. Moderate hyperhomocysteinemia decreases endothelial-dependent vasorelaxation in pregnant but not nonpregnant mice. Hypertension. 2004;44(3):327–333. doi: 10.1161/01.HYP.0000137414.12119.f6. [DOI] [PubMed] [Google Scholar]
- 29.Antoniades C, Shirodaria C, Warrick N, et al. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114(11):1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Dudman NP, Wilcken DE. Effects of homocysteine and related compounds on prostacyclin production by cultured human vascular endothelial cells. Thromb Haemost. 1993;70(6):1047–1052. [PubMed] [Google Scholar]
- 31.Ungvari Z, Sarkadi-Nagy E, Bagi Z, Szollar L, Koller A. Simultaneously increased TxA(2) activity in isolated arterioles and platelets of rats with hyperhomocysteinemia. Arterioscler Thromb Vasc Biol. 2000;20(5):1203–1208. doi: 10.1161/01.atv.20.5.1203. [DOI] [PubMed] [Google Scholar]
- 32.Bagi Z, Ungvari Z, Szollar L, Koller A. Flow-induced constriction in arterioles of hyperhomocysteinemic rats is due to impaired nitric oxide and enhanced thromboxane A(2) mediation. Arterioscler Thromb Vasc Biol. 2001;21(2):233–237. doi: 10.1161/01.atv.21.2.233. [DOI] [PubMed] [Google Scholar]
- 33.Davi G, Di Minno G, Coppola A, et al. Oxidative stress and platelet activation in homozygous homocystinuria. Circulation. 2001;104(10):1124–1128. doi: 10.1161/hc3501.095287. [DOI] [PubMed] [Google Scholar]
- 34.Signorello MG, Pascale R, Leoncini G. Effect of homocysteine on arachidonic acid release in human platelets. Eur J Clin Invest. 2002;32(4):279–284. doi: 10.1046/j.1365-2362.2002.00971.x. [DOI] [PubMed] [Google Scholar]
- 35.Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23(8):374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 36.McGuire JJ, Ding H, Triggle CR. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factor(s) Can J Physiol Pharmacol. 2001;79(6):443–470. [PubMed] [Google Scholar]
- 37.Sandow SL, Bramich NJ, Bandi HP, Rummery NM, Hill CE. Structure, function, and endothelium-derived hyperpolarizing factor in the caudal artery of the SHR and WKY rat. Arterioscler Thromb Vasc Biol. 2003;23(5):822–828. doi: 10.1161/01.ATV.0000067425.06019.D7. [DOI] [PubMed] [Google Scholar]
- 38.Heil SG, De Vriese AS, Kluijtmans LA, Mortier S, Den Heijer M, Blom HJ. The role of hyperhomocysteinemia in nitric oxide (NO) and endothelium-derived hyperpolarizing factor (EDHF)-mediated vasodilatation. Cell Mol Biol (Noisy-le-grand) 2004;50(8):911–916. [PubMed] [Google Scholar]
- 39.Eizawa H, Yui Y, Inoue R, et al. Lysophosphatidylcholine inhibits endothelium-dependent hyperpolarization and N omega-nitro-L-arginine/indomethacin-resistant endothelium-dependent relaxation in the porcine coronary artery. Circulation. 1995;92(12):3520–3526. doi: 10.1161/01.cir.92.12.3520. [DOI] [PubMed] [Google Scholar]
- 40.Cheng ZJ, Vaskonen T, Tikkanen I, et al. Endothelial dysfunction and salt-sensitive hypertension in spontaneously diabetic Goto-Kakizaki rats. Hypertension. 2001;37(2 Part 2):433–439. doi: 10.1161/01.hyp.37.2.433. [DOI] [PubMed] [Google Scholar]
- 41.De Vriese AS, Blom HJ, Heil SG, et al. Endothelium-derived hyperpolarizing factor-mediated renal vasodilatory response is impaired during acute and chronic hyperhomocysteinemia. Circulation. 2004;109(19):2331–2336. doi: 10.1161/01.CIR.0000129138.08493.4D. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Brodsky S, Kumari S, et al. Paradoxical overexpression and translocation of connexin43 in homocysteine-treated endothelial cells. Am J Physiol Heart Circ Physiol. 2002;282(6):H2124–2133. doi: 10.1152/ajpheart.01028.2001. [DOI] [PubMed] [Google Scholar]
- 43.Upham BL, Kang KS, Cho HY, Trosko JE. Hydrogen peroxide inhibits gap junctional intercellular communication in glutathione sufficient but not glutathione deficient cells. Carcinogenesis. 1997;18(1):37–42. doi: 10.1093/carcin/18.1.37. [DOI] [PubMed] [Google Scholar]
- 44.Lentz SR, Sobey CG, Piegors DJ, et al. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. J Clin Invest. 1996;98(1):24–29. doi: 10.1172/JCI118771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lentz SR, Piegors DJ, Malinow MR, Heistad DD. Supplementation of atherogenic diet with B vitamins does not prevent atherosclerosis or vascular dysfunction in monkeys. Circulation. 2001;103(7):1006–1011. doi: 10.1161/01.cir.103.7.1006. [DOI] [PubMed] [Google Scholar]
- 46.Sen U, Herrmann M, Herrmann W, Tyagi SC. Synergism between AT1 receptor and hyperhomocysteinemia during vascular remodeling. Clin Chem Lab Med. 2007;45(12):1771–1776. doi: 10.1515/CCLM.2007.354. [DOI] [PubMed] [Google Scholar]
- 47.Kassab S, Garadah T, Abu-Hijleh M, et al. The angiotensin type 1 receptor antagonist valsartan attenuates pathological ventricular hypertrophy induced by hyperhomocysteinemia in rats. J Renin Angiotensin Aldosterone Syst. 2006;7(4):206–211. doi: 10.3317/jraas.2006.039. [DOI] [PubMed] [Google Scholar]
- 48.Bonaventura D, Tirapelli CR, Haddad R, Hoehr NF, Eberlin MN, de Oliveira AM. Chronic methionine load-induced hyperhomocysteinemia enhances rat carotid responsiveness for angiotensin II. Pharmacology. 2004;70(2):91–99. doi: 10.1159/000074673. [DOI] [PubMed] [Google Scholar]
- 49.Duan J, Xu H, Dai S, et al. Phytoestrogen alpha-zearalanol inhibits homocysteine-induced endothelin-1 expression and oxidative stress in human umbilical vein endothelial cells. Atherosclerosis. 2008;197(2):549–555. doi: 10.1016/j.atherosclerosis.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 50.Cheng CM, Hong HJ, Liu JC, et al. Crucial role of extracellular signal-regulated kinase pathway in reactive oxygen species-mediated endothelin-1 gene expression induced by endothelin-1 in rat cardiac fibroblasts. Mol Pharmacol. 2003;63(5):1002–1011. doi: 10.1124/mol.63.5.1002. [DOI] [PubMed] [Google Scholar]
- 51.Gottsater A, Anwaar I, Eriksson KF, Mattiasson I, Lindgarde F. Homocysteine is related to neopterin and endothelin-1 in plasma of subjects with disturbed glucose metabolism and reference subjects. Angiology. 2000;51(6):489–497. doi: 10.1177/000331970005100606. [DOI] [PubMed] [Google Scholar]
- 52.Sethi AS, Lees DM, Douthwaite JA, Dawnay AB, Corder R. Homocysteine-induced endothelin-1 release is dependent on hyperglycaemia and reactive oxygen species production in bovine aortic endothelial cells. J Vasc Res. 2006;43(2):175–183. doi: 10.1159/000090947. [DOI] [PubMed] [Google Scholar]
- 53.Demuth K, Atger V, Borderie D, et al. Homocysteine decreases endothelin-1 production by cultured human endothelial cells. Eur J Biochem. 1999;263(2):367–376. doi: 10.1046/j.1432-1327.1999.00496.x. [DOI] [PubMed] [Google Scholar]
- 54.Drunat S, Moatti N, Paul JL, Cogny A, Benoit MO, Demuth K. Homocysteine-induced decrease in endothelin-1 production is initiated at the extracellular level and involves oxidative products. Eur J Biochem. 2001;268(20):5287–5294. doi: 10.1046/j.0014-2956.2001.02460.x. [DOI] [PubMed] [Google Scholar]
- 55.Robert K, Nehme J, Bourdon E, et al. Cystathionine beta synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterology. 2005;128(5):1405–1415. doi: 10.1053/j.gastro.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 56.Postea O, Krotz F, Henger A, Keller C, Weiss N. Stereospecific and redox-sensitive increase in monocyte adhesion to endothelial cells by homocysteine. Arterioscler Thromb Vasc Biol. 2006;26(3):508–513. doi: 10.1161/01.ATV.0000201039.21705.dc. [DOI] [PubMed] [Google Scholar]
- 57.Perez-de-Arce K, Foncea R, Leighton F. Reactive oxygen species mediates homocysteine-induced mitochondrial biogenesis in human endothelial cells: modulation by antioxidants. Biochem Biophys Res Commun. 2005;338(2):1103–1109. doi: 10.1016/j.bbrc.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 58.Ramachandran A, Levonen AL, Brookes PS, et al. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Radic Biol Med. 2002;33(11):1465–1474. doi: 10.1016/s0891-5849(02)01142-5. [DOI] [PubMed] [Google Scholar]
- 59.Edirimanne VE, Woo CW, Siow YL, Pierce GN, Xie JY, O K. Homocysteine stimulates NADPH oxidase-mediated superoxide production leading to endothelial dysfunction in rats. Can J Physiol Pharmacol. 2007;85(12):1236–1247. doi: 10.1139/Y07-112. [DOI] [PubMed] [Google Scholar]
- 60.Huck JH. Achieving high-quality comprehensive dentistry with i.v. sedation. Dent Today. 1999;18(9):114–115. [PubMed] [Google Scholar]
- 61.de Andrade CR, Fukada SY, Olivon VC, et al. Alpha1D-adrenoceptor-induced relaxation on rat carotid artery is impaired during the endothelial dysfunction evoked in the early stages of hyperhomocysteinemia. Eur J Pharmacol. 2006;543(1-3):83–91. doi: 10.1016/j.ejphar.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 62.Tasatargil A, Sadan G, Karasu E. Homocysteine-induced changes in vascular reactivity of guinea-pig pulmonary arteries: role of the oxidative stress and poly (ADP-ribose) polymerase activation. Pulm Pharmacol Ther. 2007;20(3):265–272. doi: 10.1016/j.pupt.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 63.Wang S, Wright G, Harrah J, et al. Short-term exposure to homocysteine depresses rat aortic contractility by an endothelium-dependent mechanism. Can J Physiol Pharmacol. 2000;78(6):500–506. [PubMed] [Google Scholar]
- 64.Ungvari Z, Pacher P, Rischak K, Szollar L, Koller A. Dysfunction of nitric oxide mediation in isolated rat arterioles with methionine diet-induced hyperhomocysteinemia. Arterioscler Thromb Vasc Biol. 1999;19(8):1899–1904. doi: 10.1161/01.atv.19.8.1899. [DOI] [PubMed] [Google Scholar]
- 65.Chambers JC, Ueland PM, Obeid OA, Wrigley J, Refsum H, Kooner JS. Improved vascular endothelial function after oral B vitamins: An effect mediated through reduced concentrations of free plasma homocysteine. Circulation. 2000;102(20):2479–2483. doi: 10.1161/01.cir.102.20.2479. [DOI] [PubMed] [Google Scholar]
- 66.Doshi SN, McDowell IF, Moat SJ, et al. Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation. 2002;105(1):22–26. doi: 10.1161/hc0102.101388. [DOI] [PubMed] [Google Scholar]
- 67.Clarke ZL, Moat SJ, Miller AL, Randall MD, Lewis MJ, Lang D. Differential effects of low and high dose folic acid on endothelial dysfunction in a murine model of mild hyperhomocysteinaemia. Eur J Pharmacol. 2006;551(1-3):92–97. doi: 10.1016/j.ejphar.2006.08.085. [DOI] [PubMed] [Google Scholar]
- 68.Title LM, Cummings PM, Giddens K, Genest JJ, Jr, Nassar BA. Effect of folic acid and antioxidant vitamins on endothelial dysfunction in patients with coronary artery disease. J Am Coll Cardiol. 2000;36(3):758–765. doi: 10.1016/s0735-1097(00)00809-3. [DOI] [PubMed] [Google Scholar]
- 69.Robinson I, de Serna DG, Gutierrez A, Schade DS. Vitamin E in humans: an explanation of clinical trial failure. Endocr Pract. 2006;12(5):576–582. doi: 10.4158/EP.12.5.576. [DOI] [PubMed] [Google Scholar]