

Abstract
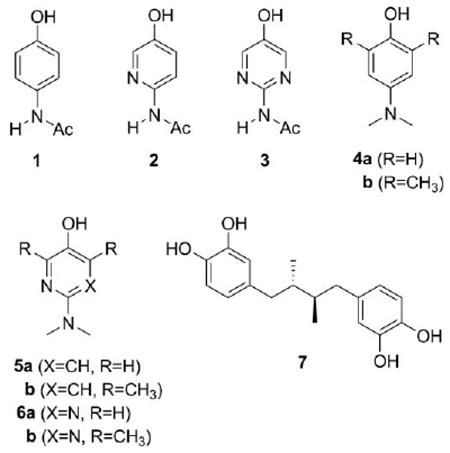
Herein we report an investigation of the efficacy of pyridine and pyrimidine analogs of acetaminophen (ApAP) as peroxyl radical-trapping antioxidants and inhibitors of enzyme-catalyzed lipid peroxidation by cyclooxygenases (COX) and lipoxygenases (LOX). In inhibited autoxidations we find that ApAP, the common analgesic and antipyretic agent, is a very good antioxidant with a rate constant for reaction with peroxyl radicals (kinh = 5 × 105 M−1 s−1) that is higher than many widely-used phenolic antioxidants, such as the ubiquitous butylated hydroxytoluene (BHT). This reactivity is reduced substantially upon incorporation of nitrogen into the phenolic ring, owing to an increase in the O–H bond dissociation enthalpy of pyridinols and pyrimidinols with respect to phenols. Incorporation of nitrogen into the phenolic ring of ApAP was also found to decrease its efficacy as an inhibitor of prostaglandin biosynthesis by ovine COX-1 (oCOX-1). This is explained on the basis of an increase in its oxidation potential and its reduced reactivity as a reducing co-substrate of the peroxidase protoporphyrin. In contrast, the efficacy of ApAP as an inhibitor of lipid hydroperoxide biosynthesis by soybean LOX-1 (sLOX-1) increased upon incorporation of nitrogen into the ring, suggesting a different mechanism of inhibition dependent on the acidity of the phenolic O–H which may involve chelation of the catalytic non-heme iron atom. The greater stability of the 3-pyridinols and 5-pyrimidinols to air oxidation as compared to phenols allowed us to evaluate some electron-rich pyridinols and pyrimidinols as inhibitors of oCOX-1 and sLOX-1. While the pyridinols had the best combination of activities as antioxidants and inhibitors of oCOX-1 and sLOX-1, they were found to be more toxic than ApAP in preliminary assays in human hepatocellular carcinoma (HepG2) cell culture. The pyrimidinols, however, were up to 17-fold more reactive to peroxyl radicals and up to 25-fold better inhibitors of prostaglandin biosynthesis than ApAP, with similar cytotoxicities to HepG2 cells at high levels of exposure.
Introduction
The oxidation of the ubiquitous lipid arachidonic acid (AA) leads to a wide variety of bioactive lipid mediators.1,2 These reactions can occur spontaneously, leading to a myriad of oxidation products, or they can be catalyzed by enzymes, which Nature has evolved to afford specific lipid oxidation products. The non-enzymatic products have been implicated in all major degenerative diseases,3 prompting the extensive study of lipid-soluble radical-trapping antioxidants (e.g. Vitamin E) as therapeutics and/or chemopreventive agents.4 Of the enzymatic products, the prostaglandins and leukotrienes are arguably the most important classes of the enzymatic products, and are collectively known as eicosanoids, along with their downstream products the prostacyclins and thromboxanes. Eicosanoids are involved in the regulation of a whole host of physiological processes including inflammation and immunity, and the rate-limiting steps in their biosynthesis are catalyzed by cyclooxygenases (COXs)5 and lipoxygenases (LOXs),6 respectively.
COXs are bifunctional enzymes that convert AA to prostaglandin H2 (PGH2), the primary prostaglandin, in a two-step sequence (Fig. 1A). In the first step, the enzymes utilize an active site tyrosyl radical to catalyze the bis-dioxygenation of AA to the endoperoxy hydroperoxide prostaglandin G (PGG2) in the so-called cyclooxygenase active site, while in the second step PGG2 is reduced to the endoperoxy alcohol PGH2 in the protoporphyrin-containing peroxidase active site.5 Since the biosynthesis of PGH2 is at the head of the cascade of reactions responsible for pain, fever and inflammation, COX inhibitors are among the most important drug targets. For example, non-steroidal anti-inflammatory drugs (NSAIDs) such as indomethacin, ibuprofen and aspirin are widely used as first-line antipyretic, analgesic and in many cases, anti-inflammatory drugs.
Fig. 1.
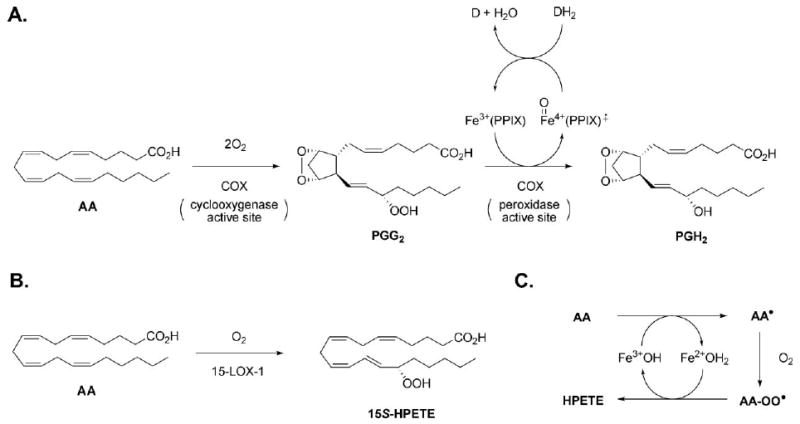
A. Two-step conversion of arachidonic acid (AA) to prostaglandin H2 (PGH2) by the cyclooxygenase and peroxidase active sites of cyclooxygenase (COX). PPIX: Protoporphyrin IX. DH2 (or DH): reduced co-substrate. D (or D•): oxidized co-substrate. B. The oxygenation of AA to a primary hydroperoxyeicosatetraenoic acid (HPETE) as exemplified by 15-LOX-1. C. General mechanism of LOX catalysis, where the amino acid sidechain ligands on the non-heme iron are omitted for clarity.
Since the discovery of the second isoform of COX, whose production is inducible in response to a variety of stimuli, massive efforts have been directed toward the development of inhibitors selective for COX-2 over the constitutively-expressed isoform, COX-1, which regulates many normal cellular processes. Recently reported side effects of selective inhibitors of COX-2, however, lend renewed impetus to the search for new COX inhibitors. Most inhibitors, whether selective or not, bind to the cyclooxygenase active site non-covalently (e.g. indomethacin, ibuprophen), while others covalently modify an active site serine hydroxyl (e.g. aspirin), in order to prevent substrate binding.
Acetaminophen, hereafter referred to as ApAP (1, commonly referred to as either paracetamol or Tylenol®), is a phenolic compound that is believed to inhibit COX in a very different way.7 ApAP is a reducing co-substrate of the peroxidase (DH2 in Fig. 1A), and it is therefore suggested to inhibit COX activity by maintaining the peroxidase active site in a reduced form, thereby preventing formation of the catalytic tyrosyl radical in the cyclooxygenase active site to mediate conversion of AA to PGG2. Although it is one of the most widely used drugs, ApAP overdose is the leading cause for calls to Poison Control Centers (>100,000/year), and has been implicated in nearly 50% of all acute liver failure in the United States.8 Unfortunately, since Nelson and co-workers9 demonstrated that the analgesic potency of three ApAP analogs paralleled their hepatotoxic potential, and both activities parallel the oxidation potentials of the compounds, there appeared to be no room for improvement.
In contrast with COXs, the primary products of LOX-catalyzed oxygenation of AA are not bicyclic endoperoxy hydroperoxides, but acyclic hydroperoxy eicosatetraenoic acids (HPETEs, Fig. 1B),6 with each of 5 mammalian isoforms responsible for oxygenation at a different carbon atom along AA, and each product, a different physiological function.10 To do so, LOXs utilize a single active site which employs a hydroxyl ligated non-heme iron to catalyze the conversion of AA to the HPETEs (Fig. 1C). As for the COXs, LOXs are important drug targets as they are implicated in a variety of diseases, including asthma (5-LOX),1 skin disease (12R-LOX),11 cancer cell proliferation (12S-LOX)12 and heart and neurodegenerative diseases (the 15-LOXs).13,14 Inhibition of LOX catalysis by phenols is epitomized by the catechol nordihydroguaiaretic acid (NDGA, 7), which has been shown to reduce the enzyme from its hydroxide-bound ferric (active) form to its water-bound ferrous (inactive) form.15 Despite the extensive work on NDGA-inhibition of LOXs, structure–activity studies on both phenols and catechols have not revealed a correlation between their potencies and their oxidation potentials, presumably because of sterics.16 This has hampered further development of lipoxygenase inhibitors based on this mechanism, beyond those largely based on the catechol headgroup of NDGA, which are problematic since they are also considered toxic.17
Recently, we have demonstrated that the oxidative stability of electron-rich phenolic compounds can be greatly improved by incorporating nitrogen in the aromatic ring.18 Nitrogen, being more electronegative than carbon, reduces the rate at which these compounds react directly with oxygen and produce superoxide, rendering them useless as antioxidants and usually highly cytotoxic. In doing so, we have designed and developed the most effective phenolic-like peroxyl radical-trapping antioxidants ever reported. For example, even simple, easily accessible compounds such as 5b and 6b react 5-fold and 2-fold faster with lipid peroxyl radicals than α-tocopherol, the most potent form of Vitamin E and the gold standard against which other radical-trapping antioxidants are compared.18c Given the longstanding interest in the abilities of phenolic antioxidants to serve as inhibitors of COX- and LOX-catalyzed lipid peroxidation,19,20 and the widespread use of ApAP as a COX inhibitor despite the high dosages required and its associated hepatotoxicity, we were curious to see how these new classes of phenolic antioxidants fared as inhibitors of enzyme-catalyzed lipid peroxidation. The results of our investigations are presented here.
In addition to simply considering a few of the 3-pyridinol (5) and 5-pyrimidinol (6) antioxidants that we have studied to date,18 we have also synthesized and examined the reactivities of pyridine and pyrimidine isosteres of ApAP (2 and 3). We surmised that the capability to modulate the redox and acid–base properties of ApAP in an isosteric manner would be useful in helping understand the mechanism of action of ApAP. Herein we describe the synthesis, relevant acid–base and redox properties, and the study of pyridine and pyrimidine analogs of ApAP as inhibitors of COX and LOX-catalyzed lipid peroxidation.
Results and discussion
I. Some predicted properties of acetaminophen analogs
Prior to carrying out the chemical synthesis of the pyridine and pyrimidine analogs of ApAP, we sought some assurance that the compounds would have the expected physicochemical properties. In previous work, we used model chemistries based on the B3LYP gradient-corrected density functional21 to predict the O–H bond dissociation enthalpies (BDEs)22 and adiabatic oxidation (ionization) potentials (IPs)23 of phenols, pyridinols and pyrimidinols.18c We carried out the same calculations on ApAP and its analogs as part of this investigation. The results are given in Table 1.
Table 1.
Calculated gas-phase O–H Bond Dissociation Enthalpies (BDEs), Ionization Potentials (IPs) and acidities of acetaminophen and some of its analogs
| O–H BDEa (kcal/mol) | IPb (kcal/mol) | Acidityc (kcal/mol) | |
|---|---|---|---|
| 1 | 82.5 | 176.2 | 342.7 |
| 2 | 83.2 | 184.7 | 337.6 |
| 3 | 83.7 | 193.1 | 331.9 |
| 4a | 76.0 | 157.7 | 347.2 |
| 5a | 77.0 | 164.6 | 345.6 |
| 6a | 78.3 | 174.6 | 340.8 |
| 4b | 72.3 | 152.3 | 346.1 |
| 5b | 73.5 | 157.7 | 345.2 |
| 6b | 74.1 | 167.0 | 341.4 |
O–H BDEs of 1, 2 and 3 were calculated using the B3LYP density functional as in ref. 22. The values for 4–6 were taken from ref. 18c.
IPs of 1, 2 and 3 were calculated using the B3LYP density functional as in ref. 23. The values for 4–6 were taken from ref. 18c.
Acidities were calculated using the B3LYP functional as in ref. 22.
The calculated O–H BDEs increase in the expected way18 on going from ApAP (1) to its 3-pyridinol analog (2) and then 5-pyrimidinol analog (3), i.e. 1 (82.5 kcal/mol) < 2 (83.2 kcal/mol) < 3 (83.7 kcal/mol), as do the calculated IPs, i.e. 1 (176.2 kcal/mol) < 2 (184.7 kcal/mol) < 3 (193.1 kcal/mol), owing to the greater electronegativity of the nitrogen atom(s) in the ring as opposed to carbon. For the same reason, the acidity of 3 (331.9 kcal/mol) is higher than that of 1 (342.7 kcal/mol), with that of 2 (337.6 kcal/mol) intermediate to the two. Thus, while the O–H bond gets marginally stronger, the oxidation potential greatly increases, and the O–H becomes much more acidic.
The increased oxidation potentials of the pyridines and pyrimidines with respect to their phenyl counterparts prompted us to consider also analogs wherein the acetamido group is replaced with the much more electron-donating N,N-dimethylamino group. While the phenols 4a and 4b are generally considered to be too unstable under ambient conditions to be useful, in our previous work we have found that the analogous pyridinols 5a and 5b and pyrimidinols 6a and 6b are in fact quite stable.18 These compounds also have very low O–H BDEs, hence expanding the structure–activity space that can be covered in these studies. Indeed, the O–H BDEs of 5a and 6a are predicted to be 5–6 kcal/mol lower than in 2 and 3, and those for 5b and 6b, 3–4 kcal/mol lower still, while the oxidation potentials are calculated to be significantly higher than that of phenols 4a and 4b, respectively.
II. Synthesis of acetaminophen analogs
The pyridine (2) and pyrimidine (3) analogs of 1 were prepared from 5-iodo-2-aminopyridine (8) and 5-bromo-2-aminopyrimidine (9), respectively, by a benzyloxylation/hydrogenolysis sequence as shown in Scheme 1.18g Since the Cu-catalyzed benzyloxylation is not effective when a free amine is present at the 2-position of the pyridine or pyrimidine rings as in 8 and 9, the amine group was first protected as the dimethylpyrrole to give 10 and 11, followed by coupling to give 12 and 13 and subsequent removal of the protecting group to yield 14 and 15.18g Acetylation of the free amine was then carried out to give 16 and 17, followed by catalytic hydrogenation to remove the benzyl group and yield 2 and 3.
Scheme 1.
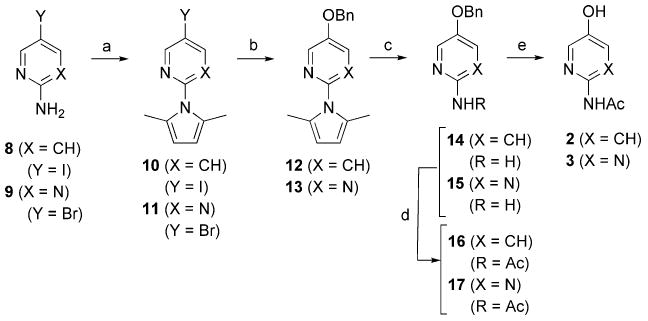
Reagents and conditions: (a) 2,5-hexanedione, pTsOH, toluene, Dean–Stark, 2 h, 82% (for 10), 91% (for 11); (b) CuI, Cs2CO3, BnOH, 90 °C, overnight, 86% (for 12), 82% (for 13); (c) NH2OH·HCl, NEt3, EtOH/H2O (2:1), reflux, 20 h, 81% (for 14), 76% (for 15); (d) CH3C(O)Cl, cat. DMAP, NEt3, CH2Cl2, rt, overnight, 81% (for 16), 69% (for 17); (e) Pd/C, H2, MeOH, rt, 3 h, 95% (for 2), 94% (for 3).
The syntheses of the pyridinols 5a and 5b as well as the pyrimidinols 6a and 6b have been reported elsewhere.18g The preparation of 5a and 6a again utilized 8 and 9 as the starting materials, whereas 5b and 6b utilized the analogous 4,6-dimethylated precursors. Briefly, these starting materials were first halogenated, then reductively alkylated with formaldehyde, and then submitted to the benzyloxylation/hydrogenation sequence.
III. Relevant solution acid–base and redox properties
With the ApAP analogs in hand, we sought some experimental (condensed-phase) confirmation of their predicted (gas-phase) physicochemical properties. Spectrophotometric titrations revealed the expected increase in acidity along the phenol, pyridinol and pyrimidinol series, with the pKa of 2 and 3 determined to be 8.28 and 5.89, respectively, relative to 9.78 for 1 (Fig. 2).
Fig. 2.
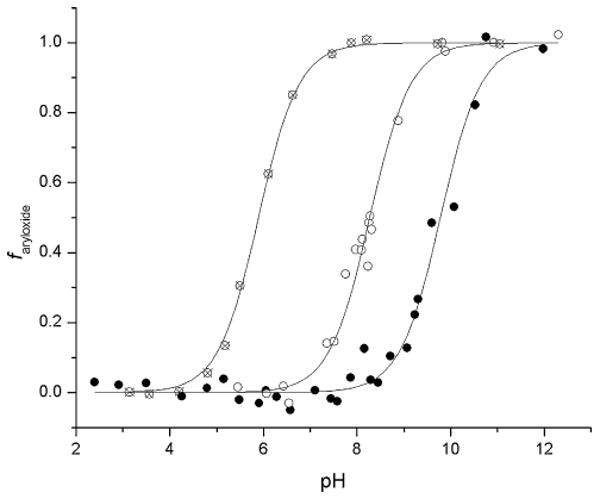
Spectrophotometeric determination of the pKa values of the pyridine (○, 2) and pyrimidine (⊗, 3) analogs of acetaminophen (●, 1). The fraction of aryloxide was obtained as a function of pH from the absorbance at λmax (1: 257 nm, 2: 253 nm, 3: 248 nm).
The two derivatives bearing N,N-dimethylamino groups in place of the acetamido groups in 2 and 3 had elevated pKa values of 9.27 (5a) and 8.18 (6a), respectively, owing to the greater electron-donating nature of this substituent, but still lower than that in 1. The ring-methylated derivatives were further elevated, with pKa values determined to be 10.14 (5b) and 9.00 (6b), respectively. These data are presented in Table 2 alongside other relevant physical properties (vide infra).
Table 2.
Experimental pKa values determined by spectrophotometry,a anodic peak potentials (Epa) determined at the pKa by differential pulse voltammetry,b and rate constants for reactions with peroxyl radicals (kinh) determined by inhibited autoxidation of styrenec
| pKa | Epa @ pKa (V) | kinh (M−1 s−1) | |
|---|---|---|---|
| 1 | 9.78 | 0.32 | 0.5 × 106 |
| 2 | 8.28 | 0.48 | 0.4 × 105 |
| 3 | 5.89 | 1.02 | 1.0 × 104 |
| 5a | 9.27 | 0.12 | 4.8 × 106 d |
| 5b | 10.14 | −0.03 | 1.6 × 107 e |
| 6a | 8.18 | 0.31 | 1.1 × 106 d |
| 6b | 9.00 | 0.17 | 8.6 × 106 e |
Determined at 298 K in 10 mM potassium phosphate containing 200 mM KCl by plotting the fraction of aryloxide present as a function of pH as in Fig. 1 and fitting to the sigmoidal function f aryloxide = 10pH−pKa/(1 + 10pH−pKa).
Determined at 298 K versus Ag/AgCl in 10 mM potassium phosphate containing 200 mM KCl at a scan rate of 10 mV/s.
Determined at 303 K in chlorobenzene and initiated by AMVN as in ref. 37.
Values of 3.7 × 106 (5a) and 1.4 × 106 (6a) were determined by the peroxyl radical clock approach.18c
Carried out at 323 K. Values of 11.6 × 106 (5b) and 6.5 × 106 (6b) were determined at 310 K by the peroxyl radical clock approach.18g
To assess the reducing power of these derivatives we turned to electrochemical techniques. Cyclic voltammograms of solutions of all of the compounds we studied were irreversible even at very high scan rates, which precluded the determination of any clean thermodynamic data. Nevertheless, trends were clear from the anodic (oxidation) peak potentials (Epa values), which we determined as a function of pH by differential pulse voltammetry (DPV). Indeed, the pyridine 2 and pyrimidine 3 are more difficult to oxidize than 1, consistent with the greater electronegativity of the ring nitrogen atoms. The experimental trends track the predicted behaviour nicely, reproducing the order in the calculated IP: 3 > 2 > 1 > 6a > 6b ∼ 5a > 5b (see Fig. 3). Plots of the Epa values as a function of pH reveal decreasing potentials with increasing pH until the pKa is reached, at which point the Epa attains a constant value, as the oxidation is no longer dependent on an unfavourable pre-equilibirum. This is expected, since the negatively-charged aryloxide ions are better electron donors than the neutral aryl alcohol substrates. Slight sigmoidal behavior of 2, 5a and 5b is observed, presumably due to protonation of the ring nitrogen at pH 5–6. This makes the Epa values of 5a < 6b at pH < 6 and 5a > 6b at pH > 6.
Fig. 3.
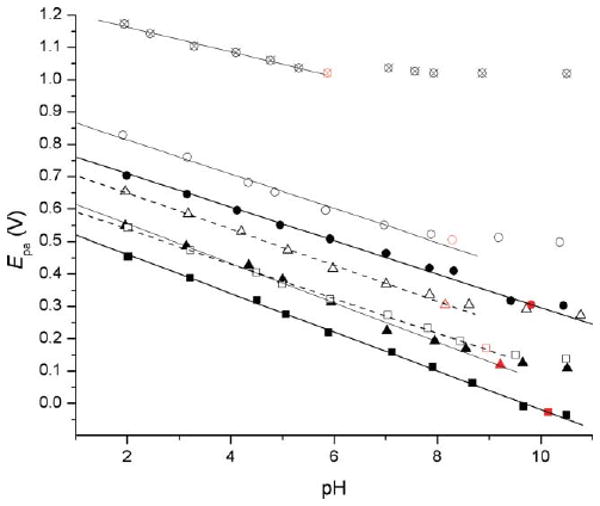
Oxidation (anodic) peak potential of acetaminophen (●, 1) and its analogs as a function of pH (○, 2; ⊗, 3; ▲, 5a; ■, 5b; △, 6a; □, 6b). The peak potential remains essentially constant beyond the pKa.
While we have determined the O–H BDEs of 5b and 6b in previous work using the radical equilibration EPR technique,18a–d those for 1, 2, 3, 5a and 6a are expected to be experimentally inaccessible owing to the short lifetimes of the aryloxyl radicals derived therefrom. Since the rate constants for the reactions of phenols, pyridinols and pyrimidinols with peroxyl radicals (known as inhibition rate constants, kinh) are well known to correlate quantitatively with the strength of the O–H bond being broken in the H-atom transfer reaction,24 these data were determined by inhibited autoxidation of styrene27 and are included alongside the pKa and Epa values in Table 2. Values for 5b and 6b are from some of our previous works,18a–d whereas the values for 1, 2, 3, 5a and 6a were determined here. Again, the experimental trends track the predicted behaviour nicely, reproducing the order in the calculated O–H BDE: 3 > 2 > 1 > 6a > 5a ∼ 6b > 6b.
To the best of our knowledge, this is the first available rate constant for the reaction of ApAP (1) with peroxyl radicals, despite many reports of its radical-trapping antioxidant activity in the literature.25 Its rather large inhibition rate constant of kinh = 5 × 105 M−1 s−1, while smaller than that for α-tocopherol (kinh = 3.2 × 106 M−1 s−1) and the more reactive pyridinols (5a and 5b) and pyrimidinols (6a and 6b), is almost 4-fold larger than that of one of the most widely used phenolic antioxidant, butylated hydroxytoluene (BHT, kinh = 1.4 × 105 M−1 s−1).27b This nicely accounts for the observations that ApAP protects lipids, including lipoproteins, from radical-mediated oxidation.25 The substantially smaller inhibition rate constants for 2 and 3—12.5-fold and 50-fold lower than for 1, respectively—are noteworthy because in previous investigations we have found that equivalently substituted phenols, pyridinols and pyrimidinols have inhibition rate constants that differ by only a few fold.18a–d Since 2 (pKa ∼ 8.3) and 3 (pKa ∼ 5.9) are much more acidic than the pyridinols and pyrimidinols we have studied previously (pKa ∼ 10, similar to phenols), it is likely that the lower rate constants are due to much stronger H-bonding to the solvent for 2 and 3 than for 1.26
IV. Inhibition of COX-1 activity
The efficacy with which the ApAP analogs inhibit prostaglandin biosynthesis was assayed using hematin-reconstituted ovine COX-1 and 14C-labeled AA. The results are shown in Fig. 4. The pyridine (2) and pyrimidine (3) analogs of 1 were determined to be ca. 1.6- and 3-fold less effective inhibitors than 1, respectively. This is consistent with both changes in the calculated O–H BDEs and IPs as well as the experimental Epa and kinh values. The same holds true for the more electron-rich N,N-dimethylated analogs 5a and 6a, which were found to be ca. 12-fold more effective at inhibiting COX-1 activity than ApAP. While ring methylation resulted in a negligible improvement to the activity of the pyridine analog, the pyrimidine 6b was found to be twice as effective as 6a, and therefore 25-fold more effective than ApAP. An increase in efficacy of both ring methylated analogs would be consistent with their lower calculated O–H BDEs and IPs and experimental Epa values. We suspect that the instability of the pyridinol 5b to oxygen under the experimental conditions leads to a lower observed efficacy than would otherwise be documented.
Fig. 4.
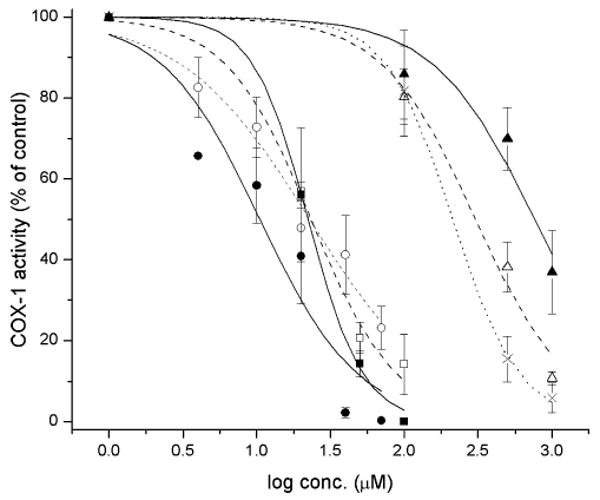
oCOX-1 inhibition by acetaminophen and its pyridine and pyrimidine analogs. oCOX-1 activity is expressed relative to the control where no inhibitor was added (specific activity of 200 mol AA per mol enzyme per min). Each data point represents the mean ± S.E.M of four to six values. IC50 values (μM): 1 (×), 250; 2 (△), 399; 3 (▲), 761; 5a (□) 22.0; 5b (■) 20.1; 6a (○) 18.4; 6b (●) 10.0.
V. Activity as reducing substrates of COX-1 peroxidase
An interesting feature of ApAP and other phenolic compounds as inhibitors of COX-catalyzed conversion of AA to PGH2 is that they are activators of cyclooxygenase activity at low concentrations.30 This has been ascribed to their ability to provide reducing equivalents to the peroxidase active site, thus serving as a substrate along with PGG2 for the formation of PGH2. This is only observed at low concentration, since at higher concentrations both the cyclooxygenase and peroxidase are maintained in the reduced state, thereby inhibiting conversion of AA to PGG2 (vide supra). The efficacy with which reducing agents can serve as substrates for the COX peroxidase can be quantified by monitoring the reduction of a model hydroperoxide, 5-phenyl-4-pentenyl hydroperoxide (PPHP), to its corresponding alcohol, 5-phenyl-4-pentenyl alcohol (PPA).28 We measured the rate of reduction of PPHP to PPA by oCOX-1 in the presence of 1-3, 5 and 6 to assess how the analogs compare to ApAP as reducing co-substrates of the COX peroxidase, and if so, if the trends in their ability to do so were the same as the trends in their abilities to inhibit cyclooxygenase activity.29 The results are shown in Fig. 5.
Fig. 5.
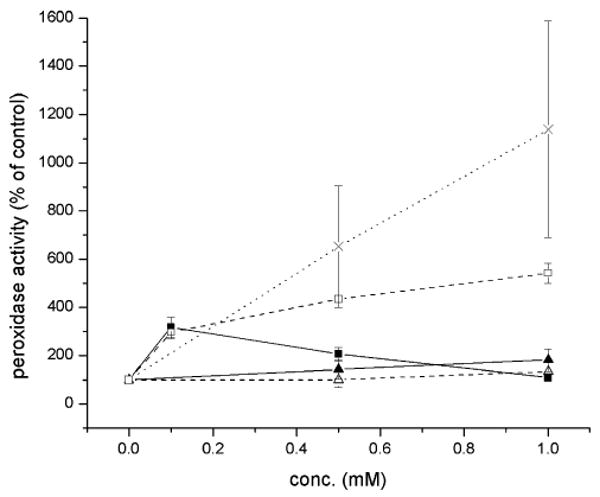
Effect of acetaminophen (1, ×) and its analogs (2, ▲; 3, △; 5a, ■; 6a, □) on the peroxidase activity of oCOX-1 expressed as percent formation of PPA against control where no inhibitor was added. Data points represent the mean ± S.E.M of four values.
When we assayed the effect of 1 and its pyridine (2) and pyrimidine (3) analogs on the peroxidase activity of oCOX-1 (Fig. 5), we found that 1 was most effective, increasing peroxidase activity 11-fold at a concentration of 1 mM, whereas 2 and 3 increased peroxidase activity only slightly at the same concentration, respectively. These trends correlate, at least qualitatively, with the inhibition of cyclooxygenase activity by 1, 2 and 3. The stronger COX inhibitors, 5a and 6a, showed more interesting profiles (also Fig. 5). While they both afforded greater peroxidase activation at low concentration (i.e. at 100 μM) than 1-3, consistent with their trends in inhibiting cyclooxygenase activity (i.e. 5a > 6a), their activities drop off substantially relative to 1 beyond that. In fact, 5a abolishes peroxidase activity above 100 μM. We surmise that the greater oxidative lability of 5a may lend to further reactions leading to inactivation of the peroxidase.
For example, two-electron oxidation of 5a would lead to an excellent alkylating agent, which could covalently modify any nucleophilic active site residues (Scheme 2). It should be noted that the COX peroxidase is irreversibly inactivated by the red wine polyphenol resveratrol through an oxidative mechanism,31 but since it is a m-hydroquinone it cannot form a good Michael acceptor like 5a, and is therefore likely to react via a different mechanism. This should be investigated further, both for its potential role in inhibiting oCOX-1, as well as it potential for the inhibition of other peroxidases.
Scheme 2.
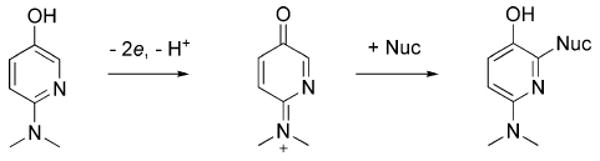
Possible mechanism of irreversible inactivation of the COX peroxidase by the electron-rich pyridine analogs (e.g. 5a) of ApAP.
VI. pH-Dependence of COX-1 inhibition
In order to shed further light on the mechanism of action of ApAP and its analogs, we studied the pH-dependence of the inhibition of oCOX-1. Since the cyclooxygenase and peroxidase active sites are redox-linked, both electron transfer (ET) from the inhibitor to the protoporphyrin radical cation of the peroxidase active site and H-atom transfer (HAT) from the inhibitor to the tyrosyl radical of the cyclooxygenase active site can contribute to the inhibition of cyclooxygenase activity. Since the three isosteres 1, 2 and 3 have distinct pKa values (9.78, 8.28 and 5.89, respectively), the preference for ET or HAT can be inferred from the relative efficacies of these compounds as inhibitors as a function of pH. When the pH > pKa the phenoxide forms of the inhibitors predominate at equilibrium, favouring an ET mechanism, whereas when pH < pKa, the phenolic forms of the inhibitors predominate at equilibrium, favouring an HAT mechanism.
We found that we could reproducibly assay the activity of oCOX-1 between pH 6.6 and 8.5, and therefore settled on this range for our experiments. In the absence of inhibitor, oCOX-1 activity differed by ≤5% throughout this pH range in two separate buffer systems (Tris·HCl and sodium phosphate). The efficacy of ApAP (1) as an inhibitor of oCOX-1 did not change significantly as a function of pH across the tested range (Fig. 6A). This can be attributed to the fact that its protonation state does not change throughout this pH range (pKa = 9.78). In contrast, inhibition of oCOX-1 activity by 2 and 3 was markedly improved at higher pH (Fig. 6B and 6C, respectively), where the pyridinoxide and pyrimidinoxide anions will have higher concentrations at equilibrium, respectively (pKa = 8.28 and 5.89). These data, taken together with the PPHP reduction data, strongly suggest that these analogs inhibit COX activity by reducing the peroxidase protoporphyrin radical cation by ET rather than the tyrosyl radical by HAT. By analogy, these results infer that ApAP reduces the COX protoporphyrin radical cation mainly by ET.
Fig. 6.
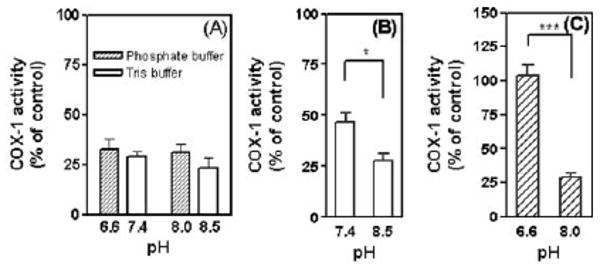
pH-Dependent inhibition of COX-1 activity by 1 (panel A) and its analogs (2, panel B and 3, panel C). Significance was tested by student t-test. * p < 0.05, *** p < 0.001. No significance was found in panel (A).
VII. Inhibition of lipoxygenase activity
The efficacy with which the ApAP analogs inhibit HPETE biosynthesis was assayed using soybean lipoxygenase-1 (sLOX-1). The soybean enzyme was chosen in place of one of the isoforms of mammalian LOX due to its greater accessibility and stability. sLOX-1 activity was assayed with its natural substrate, linoleic acid, under typical conditions with varying amounts of ApAP or the analogs and the results are shown in Fig. 7. Consistent with earlier work,32 we found that ApAP does not inhibit sLOX-1 up to 0.25 mM under these assay conditions. However, to our surprise given the foregoing results with oCOX-1, the isosteric analogs 2 and 3 inhibited sLOX-1 activity. While their potency was not spectacular, with effective IC50 values of 1624 and 564 μM, respectively, the fact that the observed trend in inhibitory activities is markedly different than in our oCOX-1 experiments is very interesting, i.e., the order of efficacy in the inhibition of oCOX-1 is 1 > 2 > 3, whereas for sLOX-1 it is 3 > 2 > 1. Thus, while the inhibition of COX-1 by 1–3 follows the redox chemistry of these analogs, the inhibition of sLOX-1 by 1–3 follows the acid–base chemistry. This suggests a mechanism other than simple reduction of sLOX-1 from its (active) ferric form to its (inactive) ferrous form known for most phenolic (especially catecholic) compounds.
Fig. 7.
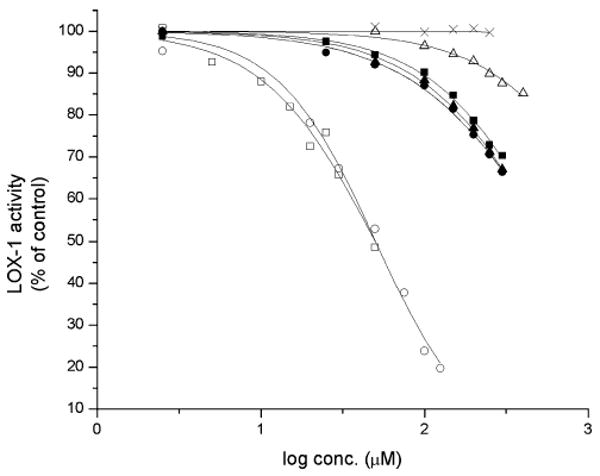
sLOX-1 inhibition by acetaminophen and its pyridine and pyrimidine analogs. sLOX-1 activity is expressed relative to the control where no inhibitor was added. Each data point represents the mean ± S.E.M of four to six values. IC50 values (μM): 1 (×), n/a; 2 (△), 1620; 3 (▲), 564; 5a (□) 49.9; 5b (○) 50.2; 6a (■) 602; 6b (●) 593.
Nelson33 has shown that acidic catechols inhibit sLOX-1 not by reduction, but by bidentate complexation of the iron atom. Our results can be rationalized on a similar basis. Thus, the highly acidic nature of 2 and 3 should facilitate complexation of the iron atom via a ring nitrogen atom and the carbonyl oxygen of the acetamido group (Scheme 3). Clearly, this should be investigated further to substantiate this mechanism, as well as to establish whether this interaction can be exploited in the design of new inhibitors.
Scheme 3.
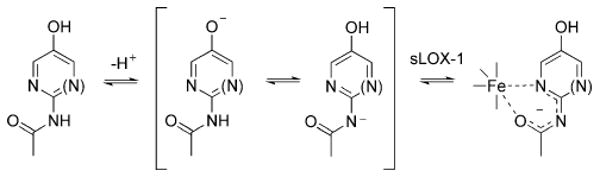
Possible mechanism of inhibition of sLOX-1 by the pyridine and pyrimidine analogs of acetaminophen.
VIII. Cytotoxicity of acetaminophen analogs
Given the encouraging activities of the pyridinols 5a and 5b and pyrimidinols 6a and 6b as inhibitors of lipid peroxidation,18 and now prostaglandin and HETE biosynthesis, we sought to compare their cytotoxicities to that of ApAP, for which much toxicological data exists. The results of preliminary investigations are reported here.
The measurement of intracellular ATP levels is a convenient means to determine cell viability, since these levels will be affected by changes in membrane integrity, mitochondrial function and metabolic activity brought about by cytotoxic events. The cytotoxicities of the analogs were determined by measuring the intracellular ATP concentration in human hepatocellular carcinoma (HepG2) cells using a luciferin/luciferase luminescence assay after a 24 h exposure of the cells to either 1, 5a, 5b, 6a or 6b.34 The results are shown in Fig. 8.
Fig. 8.
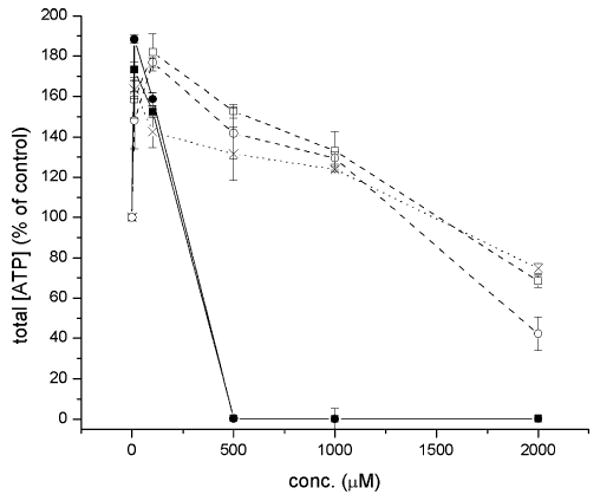
Cytotoxicity of acetaminophen (×, 1) and 3-pyridinol (■, 5a; ●, 5b) and 5-pyrimidinol (□, 6a; ○, 6b) antioxidants to human hepatocellular carcinoma (HepG2) cells, as determined from the intracellular ATP levels using the luciferin–luciferase reaction.
Each of the compounds produced an increase in intracellular ATP levels at low levels of exposure. The levels reached ca. 1.75- and 1.9-fold control levels at 10 μM 5a and 5b and ca. 1.8-fold control levels for 100 μM 6a and 6b. The levels were comparatively lower for ApAP at these concentrations. This effect is likely due to the antioxidant properties of the compounds, as the trend tracks that activity (vide supra). The intracellular ATP concentration then decreased in a dose-dependent manner with increasing concentration of 1 or the analogs. While the pyridinols 5a and 5b were relatively toxic to cells, eliminating ATP production by 500 μM exposure, the pyrimidinols 6a and 6b displayed profiles similar to that of acetaminophen, maintaining the intracellular ATP concentration at or above the control beyond 1 mM. At the highest exposure level we assayed (2 mM), the total intracellular ATP concentrations are decreased by 24.8 ± 1.0% (1), 31.3 ± 1.0% (6a) and 54.7 + 0.8% (6b) when compared to control conditions where no compound was added.
Conclusions
Following the first determination of some of the physicochemical properties of 3-pyridinol and 5-pyrimidinol peroxyl radical-trapping antioxidants in aqueous solution, we have shown that these compounds also inhibit enzyme-catalyzed lipid peroxidation by ovine COX-1 and soybean LOX-1. While incorporation of nitrogen into the phenolic ring of acetaminophen was found to decrease its antioxidant activity and its efficacy as an inhibitor of oCOX-1 because of an increase in its O–H bond dissociation enthalpy and oxidation potential, respectively, its efficacy as an inhibitor of sLOX-1 increased, revealing a different mechanism of inhibition dependent on the acidity of the phenolic O–H.
The more electron-rich 3-pyridinols 5a and 5b were better inhibitors of oCOX-1 and sLOX-1 activity (as well as uncatalyzed lipid peroxidation) than the analogous 5-pyrimidinols 6a and 6b, but were found to be toxic in preliminary viability assays in HepG2 cell culture. The pyrimidinols, however, were comparable inhibitors of lipid peroxidation to α-tocopherol, the most potent form of Vitamin E and Nature's premier phenolic antioxidant, and 12- and 25-fold better inhibitors of prostaglandin biosynthesis than acetaminophen, with similar cytotoxicities to HepG2 cells at high levels of exposure.
Experimental details
General
Unless noted otherwise, all starting materials – including acetaminophen (1) – were obtained from commercial suppliers and used without further purification. Air- and moisture-sensitive reactions were performed under a nitrogen atmosphere. Column chromatography was performed using silica gel 60 (32–63 μm) with the indicated solvents. 1H-NMR (300 or 400 MHz) and 13C-NMR (75 or 100 MHz) spectra were obtained in the given solvents, chemical shifts are reported in ppm relative to TMS and coupling constants are provided in Hz. GC-MS spectra were obtained with a Hewlett-Packard 5890 series II gas chromatograph and 5971 mass selective detector. High-resolution mass spectroscopy was measured using the electrospray technique. Ovine COX-1 was purified from ram seminal vesicles35 and soybean LOX-1 was purified from a commercial preparation as described by Galpin et al.36 Human hepatocellular carcinoma (HepG2) cells were obtained from American Type Tissue Collection (ATCC, Manassas, VA, USA) and cultured in the complete medium containing 10% FBS, 0.1 M non-essential amino acids solution, 100 U/mL penicillin–streptomycin, 1 M sodium pyruvate, and 2 M glutamine in DMEM, with 5% CO2 and 95% air at 37 °C.
Synthesis of acetaminophen analogs
To a solution of either 2-amino-5-benzyloxypyridine (14)18g or 2-amino-5-benzyloxypyrimidine (15)18g (0.152 g, 0.756 mmol) in dry dichloromethane (5 mL) was added DMAP (0.2 equiv.) and triethylamine (0.21 mL, 1.512 mmol), followed by the dropwise addition of acetyl chloride (0.107 mL, 1.512 mmol). The reaction mixture was stirred overnight, quenched by addition of brine, and extracted with ethyl acetate. The combined organic extracts were dried over Na2SO4, the solvent removed under reduced pressure, and the residue purified by silica gel column chromatography to yield 16 in 81% yield; 1H NMR (400 MHz, CDCl3): δ 2.19 (s, 3H), 5.10 (s, 2H), 7.33–7.44 (m, 6H), 8.02 (d, J = 2.8 Hz, 1H), 8.15 (d, J = 9.2 Hz, 1H), 8.26 (bs, 1H); 13C NMR (100 MHz, CDCl3): δ 24.6, 70.9, 114.5, 124.7, 127.5, 128.3, 128.7, 135.2, 136.2, 145.3, 151.9, 168.2; LRMS (ES+): 242 (M+, 7%), 109 (M+ − 133, 11%), 91 (M+ − 151, 100%), 81 (M+ − 161, 18%); HRMS (ES+) calculated (M+) 242.1055, observed 242.1047; and 17 in 69% yield; 1H NMR (400 MHz, CDCl3): δ 2.30 (s, 3H), 5.22 (s, 2H), 7.43–7.46 (m, 5H), 8.55 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 26.2, 71.4, 127.7, 128.9, 129.0, 134.7, 146.0, 152.1, 152.2, 172.3; LRMS (ES+): 243 (M+, 2%), 91 (M+ − 152, 100%); HRMS (ES+) calculated (M) 243.1008, observed 243.1002. Next, a solution of either 16 or 17 (0.175 mmol) in 5 mL of MeOH was treated with 10% Pd/C (0.01 g), and the resulting black suspension was stirred at rt under H2 atmosphere (1 atm) overnight. The catalyst was removed by filtration through a pad of celite, the filtrate was concentrated under reduced pressure, and the residue was subjected to flash chromatography on silica gel to yield 2 in 95% yield; 1H-NMR (DMSO-d6, 300 MHz) δ 1.96 (s, 3H), 7.09 (dd, 1H, J = 9.0, 3.0Hz), 7.75 (d, 1H, J = 3.0Hz), 7.81 (d, 1H, J = 8.7Hz), 9.65 (bs, 1H), 10.1(s, 1H); 13C-NMR (DMSO-d6, 75 MHz) δ 24.0, 114.6, 124.6, 135.2, 144.7, 150.6, 168.6; LRMS (ES+): 152 (M+, 87%), 110 (M+ − 42, 100%), 82 (M+ − 70, 99%); HRMS (ES+) calculated (M) 152.0586, observed 152.0590; and 3 in 94% yield; 1H-NMR (DMSO-d6, 300 MHz) δ 2.08 (s, 3H), 8.13 (s, 2H), 10.20 (bs, 2H), 10.28 (s, 1H); 13C-NMR (DMSO-d6, 75MHz) δ 24.3, 145.3, 148.4, 150.7, 168.7; LRMS (ES+): 153 (M+, 18%), 138 (M+ − 15, 5%), 111 (M+ − 42, 100%), 83 (M+ − 70, 7%); HRMS (ES+) calculated (M) 153.0538, observed 153.0538.
Spectrophotometric determination of pKa values
A solution of 10 mM potassium phosphate and 200 mM KCl was adjusted to the desired pH using 1 M KOH or 1 M HCl in the range of pH 2–14. The buffer solution (2 mL) was transferred to a cuvette and a blank UV spectrum was recorded on an Agilent 8453 diode array spectrophotometer. To this solution, a concentrated solution of inhibitor in absolute ethanol (25mM, 4 μL) was added and the UV spectrum was recorded and the pH remeasured. The fraction of inhibitor in the aryloxide form, f aryloxide, was computed using eqn (1), where A is the absorbance at λmax, and was plotted as a function of pH to obtain the pKa by fitting the f aryloxide data to the equation in Table 2, footnote a.
| (1) |
Electrochemical studies
Differential pulse voltammetry was performed using an electrochemical analyzer from CH Instruments. The experiments were performed in a glass cell with three electrodes. The working electrode was glassy carbon and was polished using 0.05 μm alumina powder between each trial. The reference electrode was Ag/AgCl and the counter electrode was a platinum wire. The buffer solution, 10 mM potassium phosphate and 3 M KCl as a supporting electrolyte, was adjusted to the desired pH using 1 M KOH or 1 M HCl. To 2 mL of buffer, a solution of inhibitor in absolute ethanol (25mM, 10 μL) was added and the pH measured using a pH meter. Argon gas was bubbled through the solution for one minute to remove any dissolved oxygen. The potential was then recorded using the DPV technique. The DPV peak potentials were referenced to the Ag/AgCl electrode used in the experiments. In buffered solutions of pH 7.0 (25mM NaH2PO4/Na2HPO4), the reversible Fe(CN)63−/4− couple was +233 mV versus the Ag/AgCl electrode. The pulse width was 0.05 s and the pulse period was 0.2 s. All experiments were done with a sensitivity of 10−6 A/V.
Inhibited autoxidations
Autoxidation experiments were performed in a two-channel oxygen-uptake apparatus, based on a Validyne DP 15 differential pressure transducer, already described elsewhere.27c The entire apparatus was immersed in a thermostatted bath to ensure a constant temperature within ±0.1 °C. In a typical experiment, an air-saturated chlorobenzene solution of styrene containing the antioxidant (from 1 × 10−6 to 5 × 10−4 M) was equilibrated with the reference solution containing only an excess of α-tocopherol (from 1 × 10−3 to 1 × 10−2 M) in the same solvent at 30 °C. After equilibration, a concentrated chlorobenzene solution of AMVN (1–5 × 10−3 M) or AIBN (1–5 × 10−2 M) was injected into both the reference and sample flasks, and the oxygen consumption in the sample was measured, after calibration of the apparatus, from the differential pressure recorded with time between the two channels. This instrumental setting allowed us to have the N2 production and the oxygen consumption derived from the azo-initiator decomposition already corrected from the measured reaction rates. Initiation rates, Ri, were determined from the length of the inhibited period τ in preliminary experiments using α-tocopherol as a reference antioxidant: Ri = 2[α-tocopherol]/τ. For compounds 5–6, which gave a neat inhibited period, kinh was obtained from the slope of the oxygen consumption trace,27b while for compounds 1–3, which gave a retarding of the oxygen consumption plot, kinh values were obtained by repeating the autoxidation in the absence or presence of variable amounts of the antioxidant and treating the results according to the method developed by Darley-Usmar and coworkers.37
Inhibition of oCOX-1
Ovine COX-1 was preincubated on ice for 30 min with 2 equivalents of hematin in Tris·HCl buffer (pH 8.0) containing 500 μM phenol.30 This solution was then warmed for 2 min at 37 °C in the presence of the inhibitors of designated concentration. [14C]Arachidonic acid (20 μL) (4.8 nCi, 0.5 μM final concentration) in Tris·HCl buffer (pH 8.0) was preincubated at 37 °C for 30 s. The reaction was initiated by adding equivalent activities of COX-1 (5.4 nM final concentration) to a total volume of 200 μL. The reaction was then terminated after 8 s by the addition of 400 μL of ice-cold diethyl ether–methanol–4.0 M citric acid (30:4:1) containing 8 μg of t-butylated hydroxyanisole (BHA) as antioxidant and 8 μg of unlabeled arachidonic acid as a carrier. The organic layer was loaded on a silica plate and eluted with EtOAc–isooctane–water–AcOH (45:25:50:1). Thin layer chromatography plates were analyzed for radioactivity by a Bioscan AR-2000 imaging scanner. Graphical analysis was performed using Win-scan software (Bioscan). PGHS activity was expressed as percentage of the control where no inhibitor was added.
pH-Dependent inhibition of COX-1
Identical experimental conditions to the above assay were used except that no inhibitor was added and each of the following buffers was used: Tris·HCl buffer for pH 8.5, 8.0 and 7.4 and sodium phosphate buffer for pH 8.0, 7.4 and 6.6. All buffers contained 500 μM phenol.30 Identical experimental conditions to the assay for the oxidation of arachidonic acid at different pHs were used in the presence of the inhibitors: 0.5 mM final concentration of 1; 1.0 mM final concentration of 2 and 3. COX-1 activity at each pH and each buffer system was expressed as percentage of the arachidonic acid oxidation against the corresponding control where no inhibitor was added.
Effect of inhibitors on the reduction of PPHP to PPA
COX-1 was preincubated on ice for 30 min with 2 equivalents of hematin in Tris·HCl buffer (pH 8.0). This solution was then warmed for 5 min at 37 °C in the presence or absence of inhibitors. PPHP (20 μL) (10 μM final concentration) in Tris·HCl buffer (pH 8.0) was preincubated at 37 °C for 2 min. The reaction was initiated by adding COX-1 (5.4 nM final concentration) to a total volume of 200 μL. The reaction was terminated after 8 s by the addition of 200 μL of ice-cold diethyl ether-methanol–4 M citric acid (30:4:1). The organic layer was then evaporated under nitrogen to near-dryness and reconstituted with 65% (v/v) methanol–water (final volume 500 μL) for HPLC analysis.
Inhibition of sLOX-1
The effects of added inhibitors on the initial rates of conversion (less than 10%) of linoleic acid (LA) to 13S-hydroperoxy-9Z,11E-octadecadienoate (13-HPODE) by soybean LOX-1, were determined by monitoring the conversion of LA to 13-HPODE on a spectrophotometer at 234 nm. Briefly, to a solution of 20 μM LA in 100 mM borate buffer (pH 9.2) was added the inhibitor as a concentrated solution in ethanol (to final concentrations of 2.5 to 400 μM depending on the inhibitor and less than 5% ethanol, by volume), and then sLOX-1 to a final concentration of 3 nM. The initial rates were then plotted as a function of added inhibitor concentration relative to the initial rates measured under identical experimental conditions, but without added inhibitor.
Cytotoxicity assay with HepG2 cell line
In a 96-well microplate, four wells were used as blanks (medium alone). All other wells were dispensed with an equal number of cells. HepG2 cells were suspended at a density of 105 cells/mL in a volume of 100 μL per well. An additional 90 μL of medium was added to the wells that contained vehicle controls (ethanol or water) that comprised less than 0.5% of the total volume. Serial dilutions of each compound were used to construct the dose–response curves for each test compound. Two or three replicates were performed per dose of compound.
Cellular ATP content was measured using Cytolux L001-100® assay kits (EG&G Wallac, Finland) at room temperature. The number of cells plated per well and the amount of reagent used were optimized to produce a high signal/background ratio. Cells were exposed to the compounds for 24 h, after which 100 μL of culture was removed from each well, and 50 μL of Somalyze® reagent was added to lyse the cells. After 5 min of lysis, luciferase/luciferin reagent (50 μL/well) was added and the luminescence was measured after 10 min using a luminometer (Luminoskan RS, Labsystems, Finland).
Supplementary Material
Acknowledgments
We are grateful for the support of the Natural Sciences and Engineering Research Council of Canada, Ontario Ministry of Innovation, and Queen's University. D. A. P. also acknowledges the support of the Canada Research Chairs program. This work was also supported in part by grants from the NIH HL-17921, ES-013125, GM-15431, and by grants from Merck-Frosst Canada & Company and McNeil Consumer & Specialty Pharmaceuticals.
Footnotes
Electronic supplementary information (ESI) available: Copies of NMR spectra of new compounds. See DOI: 10.1039/b912528k
References and notes
- 1.Funk CD. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 2.Serhan CN, Chiang N, Van Dyke TE. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.See, e.g. Chisholm GM, Steinburg D. Free Radical Biol Med. 2000;28:1815–1826. doi: 10.1016/s0891-5849(00)00344-0.Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, Morrow JD, Montine TJ. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010.Marnett LJ. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361.
- 4.See, e.g. Upston J, Kritharides L, Stocker R. Prog Lipid Res. 2004;32:405–422. doi: 10.1016/s0163-7827(03)00024-9. and literature cited therein.
- 5.Rouzer CA, Marnett LJ. Chem Rev. 2003;103:2239–2304. doi: 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- 6.Brash AR. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 7.(a) Harvison PJ, Egan RW, Gale PH, Christian GD, Hill BS, Nelson SD. Chem Biol Interact. 1988;64:251. doi: 10.1016/0009-2797(88)90101-9. [DOI] [PubMed] [Google Scholar]; (b) Ouellet M, Percival MD. Arch Biochem Biophys. 2001;387:273. doi: 10.1006/abbi.2000.2232. [DOI] [PubMed] [Google Scholar]; (c) Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, Vyas SN, FitzGerald GA. N Engl J Med. 2001;345:1809. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]; (d) Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA. Proc Natl Acad Sci U S A. 2002;99:7130. doi: 10.1073/pnas.102588199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Aronoff DM, Oates JA, Boutaud O. Clin Pharmacol Ther. 2006;79:9. doi: 10.1016/j.clpt.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 8.See, e.g. Bromer MQ, Black M. Clin Liver Dis. 2003;7:351. doi: 10.1016/s1089-3261(03)00025-4.James LP, Mayeux PR, Hinson JA. Drug Metab Dispos. 2003;31:1499. doi: 10.1124/dmd.31.12.1499.Lee WM. Hepatology. 2004;40:6–9. doi: 10.1002/hep.20293.Larson AM, et al. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948.
- 9.Harvison PJ, Forte AJ, Nelson SD. J Med Chem. 1986;29:1737–1743. doi: 10.1021/jm00159a029. [DOI] [PubMed] [Google Scholar]
- 10.These are 5-LOX, 12S-LOX, 15-LOX-1, 12R-LOX, 15-LOX-2. A sixth isoform exists, eLOX-3, which has been shown to isomerize 12R-HPETE to a 11,12-epoxy-8-hydroxyeicosatrienoic acid; see: Yu Z, Schneider C, Boeglin WE, Marnett LJ, Brash AR. Proc Natl Acad Sci U S A. 2003;100:9162–9167. doi: 10.1073/pnas.1633612100.
- 11.Jobard F, Lefevre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, Ozguc M, Lathrop M, Prud'homme JF, Fischer J. Hum Mol Genet. 2002;11:107–13. doi: 10.1093/hmg/11.1.107. [DOI] [PubMed] [Google Scholar]
- 12.Ding XZ, Tong WG, Adrian TE. Int J Cancer. 2001;94:630–636. doi: 10.1002/ijc.1527. [DOI] [PubMed] [Google Scholar]
- 13.Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Arterioscler Thromb Vasc Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 14.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VM. Am J Pathol. 2004;164:1655–62. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kemal C, Louis-Flamberg P, Krupinski-Olsen R, Shorter AL. Biochemistry. 1987;26:7064–7072. doi: 10.1021/bi00396a031. [DOI] [PubMed] [Google Scholar]
- 16.Whitman S, Gezginci M, Timmermann BN, Holman TR. J Med Chem. 2002;45:2659–2661. doi: 10.1021/jm0201262. [DOI] [PubMed] [Google Scholar]
- 17.Katz M, Saibil F. J Clin Gastroenterol. 1990;12:203–206. [PubMed] [Google Scholar]; Sheikh N, Philen RM, Love LA. Arch Intern Med. 1997;157:913–919. [PubMed] [Google Scholar]; Sahu SC, Ruggles DI, O'Donnell MW. Food Chem Toxicol. 2006;44:1751–1757. doi: 10.1016/j.fct.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 18.(a) Pratt DA, DiLabio GA, Brigati G, Pedulli GF, Valgimigli L. J Am Chem Soc. 2001;123:4625–4626. doi: 10.1021/ja005679l. [DOI] [PubMed] [Google Scholar]; (b) Valgimigli L, Brigati G, Pedulli GF, DiLabio GA, Mastragostino M, Arbizzani C, Pratt DA. Chem Eur J. 2003;9:4997–5010. doi: 10.1002/chem.200304960. [DOI] [PubMed] [Google Scholar]; (c) Wijtmans M, Pratt DA, Valgimigli L, DiLabio GA, Pedulli GF, Porter NA. Angew Chem, Int Ed. 2003;42:4370–4373. doi: 10.1002/anie.200351881. [DOI] [PubMed] [Google Scholar]; (d) Wijtmans M, Pratt DA, Brinkhorst J, Serwa R, Valgimigli L, Pedulli GF, Porter NA. J Org Chem. 2004;69:9215–9223. doi: 10.1021/jo048842u. [DOI] [PubMed] [Google Scholar]; (e) Kim HY, Pratt DA, Seal JR, Wijtmans M, Porter NA. J Med Chem. 2005;48:6787–6789. doi: 10.1021/jm0507173. [DOI] [PubMed] [Google Scholar]; (f) Nam TG, Rector CL, Kim HY, Sonnen AFP, Meyer R, Werner WM, Atkinson J, Rintoul J, Pratt DA, Porter NA. J Am Chem Soc. 2007;129:10211–10219. doi: 10.1021/ja072371m. [DOI] [PubMed] [Google Scholar]; (g) Nara SJ, Jha M, Brinkhorst J, Zemanek TJ, Pratt DA. J Org Chem. 2008;73:9326–9333. doi: 10.1021/jo801501e. [DOI] [PubMed] [Google Scholar]
- 19.Panganamala RV, Miller JS, Gwebu ET, Sharma HM, Cornwell DG. Prostaglandins. 1977;14:261–271. doi: 10.1016/0090-6980(77)90171-x. [DOI] [PubMed] [Google Scholar]; Seeger W, Moser U, Roka L. Arch Pharmacol. 1988;338:74–81. doi: 10.1007/BF00168815. [DOI] [PubMed] [Google Scholar]; Mantri P, Witiak DT. Curr Med Chem. 1994;1:328–355. [Google Scholar]
- 20.Yasumoto K, Yamamoto A, Mitsuda H. Agric Biol Chem. 1970;34:1162–1168. [Google Scholar]; Thody VE, Buckle DR, Foster KA. Biochem Soc Trans. 1987;15:416–417. [Google Scholar]; Dailey LA, Imming P. Curr Med Chem. 1999;6:389–398. [PubMed] [Google Scholar]
- 21.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 22.DiLabio GA, Pratt DA, LoFaro AD, Wright JS. J Phys Chem A. 1999;103:1653. [Google Scholar]
- 23.DiLabio GA, Pratt DA, Wright JS. J Org Chem. 2000;65:2195. doi: 10.1021/jo991833e. [DOI] [PubMed] [Google Scholar]
- 24.See, e.g. Lucarini M, Pedulli GF, Cipollone M. J Org Chem. 1994;59:5063.Wayner DDM, Lusztyk E, Ingold KU, Mulder P. J Org Chem. 1996;61:6430. doi: 10.1021/jo952167u.Lucarini M, Pedrielli P, Pedulli GF, Cabiddu S, Fattuoni C. J Org Chem. 1996;61:9259. doi: 10.1021/jo960250r.
- 25.See, e.g. Dinis TCP, Madeira VMC, Almeida LM. Arch Biochem Biophys. 1994;315:161–169. doi: 10.1006/abbi.1994.1485.Nenseter MS, Halvorsen B, Rosvold O, Rustan AC, Drevon CA. Arterioscler Thromb Vasc Biol. 1995;15:1338–1344. doi: 10.1161/01.atv.15.9.1338.Chou T, Greenspan P. Biochim Biophys Acta. 2002;1581:57–63. doi: 10.1016/s1388-1981(02)00119-1.Ozsoy MB, Pabuccuoglu A. J Clin Biochem Nutr. 2007;41:27–31. doi: 10.3164/jcbn.2007004.
- 26.Ingold and co-workers have shown that this interaction depresses the rate, since the phenolic H-atom is no longer accessible for abstraction: Litwinienko G, Ingold KU. Acc Chem Res. 2007;40:222–230. doi: 10.1021/ar0682029.
- 27.See, e.g. Burton GW, Ingold KU. J Am Chem Soc. 1981;103:6472.Burton GW, Doba T, Gabe EJ, Hughes L, Lee FL, Prasad L, Ingold KU. J Am Chem Soc. 1985;107:7053.Amorati R, Pedulli GF, Valgimigli L, Attanasi OA, Filippone P, Fiorucci C, Saladino R. J Chem Soc, Perkin Trans 2. 2001:2142–2146.Amorati R, Ferroni F, Pedulli GF, Valgimigli L. J Org Chem. 2003;68:9654–9658. doi: 10.1021/jo0351825.
- 28.Hsuanyu Y, Dunford HB. J Biol Chem. 1992;267:17649–17657. [PubMed] [Google Scholar]
- 29.(a) Aronoff DM, Boutaud O, Marnett LJ, Oates JA. J Pharmacol Exp Ther. 2003;304:589–595. doi: 10.1124/jpet.102.042853. [DOI] [PubMed] [Google Scholar]; (b) Markey CM, Alward A, Weller PE, Marnett LJ. J Biol Chem. 1987;262:6266. [PubMed] [Google Scholar]
- 30.The activity assay for COX is usually performed in presence of 500 μM phenol to protect the enzyme from oxidative inactivation and to enhance its catalytic activity. However, as phenol is a reducing co-substrate, its presence in the reaction medium would confound the results, and therefore it was omitted for these experiments.
- 31.Szewczuk LM, Forti L, Stivala LA, Penning TM. J Biol Chem. 2004;279:22727–22737. doi: 10.1074/jbc.M314302200. [DOI] [PubMed] [Google Scholar]
- 32.See, e.g. Marcinkiewicz Z, Duniec E, Robak J. Biochem Pharmacol. 1985;34:148–149. doi: 10.1016/0006-2952(85)90115-7.Duniec Z, Robak J, Gryglewski R. Biochem Pharmacol. 1983;32:2283–2286. doi: 10.1016/0006-2952(83)90239-3.
- 33.Nelson MJ. Biochemistry. 1988;27:4273–4278. [Google Scholar]
- 34.Crouch SP, Kozlowski R, Slater KJ, Fletcher J. J Immunol Methods. 1993;160:81–88. doi: 10.1016/0022-1759(93)90011-u. [DOI] [PubMed] [Google Scholar]
- 35.Marnett LJ, Siedlik PH, Ochs RC, Pagels WR, Das M, Honn KV, Warnock RH, Tainer BE, Eling TE. Mol Pharamcol. 1984;26:328–335. [PubMed] [Google Scholar]
- 36.Galpin JR, Tielens LGM, Velkdink GA, Vliegenthart JFG, Boldingh J. FEBS Lett. 1976;69:179–182. doi: 10.1016/0014-5793(76)80681-3. [DOI] [PubMed] [Google Scholar]
- 37.Darley-Usmar VM, Hersey A, Garland LG. Biochem Pharm. 1989;38:1465–1469. doi: 10.1016/0006-2952(89)90186-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.