

Abstract
Background
There is a considerable literature on the source of the thermostability of proteins from thermophilic organisms. Understanding the mechanisms for this thermostability would provide insights into proteins generally and permit the design of synthetic hyperstable biocatalysts.
Results
We have systematically tested a large number of sequence and structure derived quantities for their ability to discriminate thermostable proteins from their non-thermostable orthologs using sets of mesophile-thermophile ortholog pairs. Most of the quantities tested correspond to properties previously reported to be associated with thermostability. Many of the structure related properties were derived from the Delaunay tessellation of protein structures.
Conclusions
Carefully selected sequence based indices discriminate better than purely structure based indices. Combined sequence and structure based indices improve performance somewhat further. Based on our analysis, the strongest contributors to thermostability are an increase in ion pairs on the protein surface and a more strongly hydrophobic interior.
Background
Mesophiles, thermophiles, and hyperthermophiles
Organisms that thrive at very high temperatures have been actively studied since the discovery of Thermus aquaticus in the hot springs of Yellowstone in the 1960’s [1]. Heat tolerant organisms are often separated into two classes: thermophiles, which have optimum growth temperatures (OGT) in the range 45-80 °C, and hyperthermophiles with OGTs above 80 °C. Mesophilic organisms are defined as those with OGT’s between 15 °C and 45 °C, while psychrophiles, which we do not address here, have OGT’s no greater than 15 °C. Sometimes the break points between these classes are assigned slightly differently. Hyperthermophiles come mostly from the kingdom Archea, but there are two genera of hyperthermophilic Eubacteria, namely Thermotogales and Aquifex. Thermophiles are more phylogenetically diverse and include Eubacteria, Archea, and some fungi.
In addition to providing insights into the principles of protein folding and stability, understanding what makes some proteins more thermostable than others is of practical interest. Thermophilic proteins are more resistant to proteolysis and chemical denaturation, hence there is interest in engineering hyperstable biocatalysts relying on the same mechanisms that nature uses [2-4]. Thermophilic polymerases, proteases, and xylanases already have industrial applications [4,5].
The physical basis of thermophilic protein stability
The search for the physical basis of thermostability in proteins goes back 30 years to the work of Perutz [6]. Since then, a great many papers have been written on the subject. Some of the proposed mechanisms/indicators of increased thermostability include: a more highly hydrophobic core [7,8], tighter packing or compactness [9], deleted or shortened loops [10,11], greater rigidity [3,12,13] (for example through increased Proline content in loops), higher secondary structure content [14], greater polar surface area [15], fewer and/or smaller voids [14,16], smaller surface area to volume ratio [17], fewer thermolabile residues [16,18], increased hydrogen bonding [15], higher isoelectric point [19], and more salt bridges/ion pairs and networks of salt bridges [6,20-25].
Statistically significant changes in sequence composition between mesophilic and thermophilic proteins have been reported. The amino acids Asn, Gln, Met, and Cys are thermolabile—they are not stable at high temperatures and tend to undergo deamidation (Asn and Gln) or oxidation (Met and Cys) [22]. These amino acids are less common in thermophilic proteins and the thermolabile residues that do occur are usually buried [16]. Ile is preferred to Leu in hydrophobic regions of the structure because the side chain carbons can exist in all three χ rotameric states compared to only two for Leu which can result in tighter side chain packing [16]. Farias and Bonato [26] have reported that Gly, Lys, Tyr, and Ile are preferred in thermophilic organisms while Gln, His, Ala, and Cys are preferred in mesophiles. Camillau and Claverie [27] have reported that thermophilic proteins have less Gln, Ala, and His on their surfaces than mesophilic proteins do and more charged residues on their surfaces, particularly Lys and Glu. Haney et al. [28] have compared 115 proteins from Methanococcus jannaschii to mesophilic proteins from other Methanococcus species and found that the frequencies of Ile, Glu, Arg, Lys, Pro, and Tyr are significantly greater in the thermophile and the frequencies of Gly, Met, Gln, Thr, Asn, and Ser are smaller.
More ion pairs have been strongly and consistently linked with thermostability in the literature. Water has a dielectric constant of about 80 at 0°C, which drops to 55 at 100°C and is lower still at the extreme pressures near hydrothermal vents in the deep sea where some hyperthermophilic organisms live. A lower dielectric constant makes electrostatic interactions stronger and therefore ion pairs should have a greater stabilizing effect at high temperatures and pressures [21,29].
Evidence for some of these proposed mechanisms/indicators is equivocal. For instance, Karshikoff and Ladenstein found no significant difference in packing between thermophilic and mesophilic proteins [30] and salt bridges in a protein core have been reported to be destabilizing [31,32]. Das and Gerstein [33] have reported that the lengths of proteins from the eubacterium Aquifex aeolicus are greater than those of archeal hyperthermophilic orthologs and therefore hyperthermophilic proteins may be shorter than their mesophilic counterparts simply because most hyperthermophiles are archeal, not necessarily because shorter loops promote enhanced thermostability. Querol et al. [34] surveyed 122 references for 195 single point mutants which have been unambiguously linked to greater thermostability and found that greater rigidity, as measured by crystallographic B-factors, is not a good indicator of thermostability.
The overall view one comes away with from this body of work is that increased thermostability is due to relatively subtle differences in sequence and structure so that thermophilic and mesophilic orthologs are quite similar proteins (Fig. 1). They share the same catalytic mechanisms [35], although activity is typically lower at low temperatures for thermophilic enzymes [3]. The structures are similar, and sequence identity is usually, but not always, reasonably high.
Figure 1.

The CE structural alignment of 1aisA, a TATA-box-binding protein from the extreme thermophile Pyrococcus woesi , in red and 1ytbA, a TATA-box-binding protein from the mesophile Saccharomyces cerevisiae, in blue. The rmsd is 2Å and sequence identity is 40%. Clearly these are very similar structures.
Discrimination of thermophiles and mesophiles
Liang et al. have studied the proteomes of 15 thermophiles and 74 mesophiles using the tendencies of residue pairs separated by no more than 20 in primary sequence to occur together to discriminate mesophilic from thermophilic proteomes [36]. Farias and Bonato [26] have devised a sequence composition based index capable of correctly classifying organisms. The index ri is characteristic of a single protein and is defined as ri=(E+K)/(Q+H) where E, K, Q, and H are the percent compositions of these amino acid types in protein i. Those authors took the average of ri over all the proteins in an organism to give an average r that, without exception, fell in different ranges for the mesophiles (r < 2.5), thermophiles (3.2 < r < 4.6), and hyperthermophiles (r > 4.5) in their test set. Further, they showed that r is high in chaperonins (heat shock proteins) in both mesophiles and thermophiles [26] thereby concluding that the sequence signal is indicative of thermostability and not a phylogenetic artifact. Similarly, Claverie and colleagues [27,37] have devised the CvP bias (charged versus polar), defined as (D+E+K+R)-(N+Q+S+T), the sum of amino acid compositions, that they have also used to classify an organism as mesophilic, thermophilic, or hyperthermophilic by computing the average CvP for all the proteins from that organism. Zeldovich et al. [38] have reported a sequence composition based index defined as I+V+Y+W+R+E+L (abbreviated IVYWREL) that was an extremely good predictor of thermostability when averaged over whole proteomes, and even for just the membrane proteins from these proteomes. IVYWREL, again when averaged over proteomes, also correlates very well with OGT. None of these authors claim that their indices work well at discriminating individual pairs of thermophilic and mesophilic orthologs, however it is natural to ask if they can, and we will test this question here. Glyakina et al. have done one of the few large scale structure based discrimination studies [39].
Delaunay tessellation of protein structures and quantities derived from it
We will refer to some Delaunay tessellation based descriptors of protein structure, so a brief introduction is in order. Delaunay tessellation, a technique for decomposing a point set into non-overlapping tetrahedral subsets, has proven very versatile in the analysis of protein structures [40-57]. With this technique, the protein is abstracted to a set of points, here the α-carbons. These points are joined by edges in a unique way to form a set of non-overlapping, irregular, space-filling tetrahedra also known as Delaunay simplices (Fig 2) [58]. Residues joined by a Delaunay simplex edge are natural nearest neighbors in a well defined sense [58]. The analysis of statistical characteristics of the tessellation of proteins has been used in fold recognition [42-44], for structure alignment and comparison [45,46,56,59], as a way to identify cavities in the surface of a protein that could be potential binding pockets [48], to predict the stability and activity effects of point mutations [49,50], to define structural motifs [51-54], and to assign secondary structure [55].
Figure 2.

The all atom Van Der Waals spacefill representation (left) of phosphoglycerate kinase (PDB code 16pk), the Delaunay tessellation of 16pk with no simplex edge length cutoff (middle), and a view of the tessellation with a 10 Å cutoff (right). Notice that the surface of the tessellation with a cutoff corresponds more closely to that of the real molecule.
A Delaunay tessellation derived four body statistical contact pseudo-potential has been reported previously [42,43] which has been shown to contain more information than pairwise contact potentials [60]. Under this pseudo-potential, the score of some particular amino acid quadruplet (i, j, k, l), which corresponds to the residues at the vertices of a Delaunay simplex, is defined as:
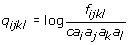 (1)
(1)
where: fijkl is the observed frequency of simplices with amino acid types i, j, k, and l at their vertices in a large non-redundant training set S; ai, aj, ak, and al are the observed frequencies of the individual amino acid types in S; and c is a combinatorial factor. Variations of this potential have been successfully applied to fold recognition [43,50] and the analysis of protein stability [44] and activity [49]. We will denote by Q the sum of the log-likelihoods qijkl from the residue quadruplets corresponding to all Delaunay simplices in the tessellation of a protein structure.
The radius of the sphere circumscribing a Delaunay tetrahedron gives a measure of its eccentricity. The small, nearly equilateral tetrahedra in the interior of the tessellation have small circumsphere radii, on the order of the size of the simplices. Radically skewed, nearly coplanar tetrahedra on the surface of the tessellated protein, on the other hand, can have circumsphere radii orders of magnitude larger than the diameter of the molecule. The tetrahedrality T is another measure of simplex eccentricity. Denote the length of the six edges of a simplex as L1 - L6 . The tetrahedrality is then defined by:
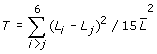 (2)
(2)
Protein contact graphs
A few protein structure descriptors we will use are based on molecule contact graphs, so a brief introduction to this is also in order. The residues in contact with one another in a protein can be thought of as a graph or network (Fig. 2) and analyzed using techniques from elementary graph theory and the theory of complex networks. In the literature, residues are typically defined to be in contact by a simple proximity cutoff, but in this work, graph nodes correspond to residues and graph edges join nodes when the corresponding residues are joined by a Delaunay simplex, shorter than some fixed cutoff, in the tessellation of the protein structure.
Several contact network derived quantities have been used before to analyze protein structures [61,62]. The degree k of a node in an undirected graph is the number of edges impinging on it. The average degree over all nodes in the contact graph will be referred to as the coordination number. A minimum path between nodes i and j is one for which the sum of weights of the edges along the path is smallest from among all possible paths. The minimum path length Lij between nodes i and j is the sum of the weights along a minimum path. In our case here, edges have weight one, and a minimum path is one for which the fewest edges are traversed. The characteristic path length L of a network is the average of the minimum paths between all node pairs i, j where i ≠ j. In general, there will be many paths between distinct nodes i and j that have the minimum path length. Some classes of networks have the clustering property, which means that two nodes which are both joined by edges to a third, are more likely to also be joined to each other than are two nodes picked at random [63]. In such networks, there are well defined neighborhoods with subsets of nodes tending to be connected to each other and tending not to be connected to nodes in other neighborhoods. The clustering property is measured by the clustering coefficient of a node Cn is the number of actual edges En between neighbors of node n divided by the number of possible connections between those neighbors: Cn = 2En/(k(k-1)), where k is the degree of node n. The clustering coefficient C for the entire network is the average of all the Cn.
Results
Sequences differences
Linear least square best fit lines of number of residues in hyperthermophilic (Nh) or thermophilic proteins (Nt) to the number of residues in their mesophilic counterparts (Nm) are: Nh=0.94 Nm + 5.00 and Nt=0.97 Nm + 5.15 where there are 122 pairs in the hyperthermophile pairs and 127 in thermophile pairs (see Additional files 1 and 2 for lists). Our data show that thermophilic proteins are usually somewhat shorter than their mesophilic counterparts, and hyperthermophilic proteins are shorter still. This observation is in line with the results of Eisenberg [11].
Tables 1 and 2 show the amino acid composition for a large nonredundant set of thermophilic, hyperthermophilic, and mesophilic proteins. T-tests were conducted to see if the average composition was different for the hyperthermophiles, thermophiles, and a control set of mesophiles. Table 1 is in broad agreement with previously published results: there are more charged residues in thermophilic proteins and fewer polar and thermolabile residues. Arg is preferred in thermophilic proteins but Lys is preferred in hyperthermophilic proteins [14].
Table 1.
t-tests of sequence percent compositions of mesophilc, thermophilic, and hyperthermophilic proteins.
| A | C | D | E | F | G | H | I | K | L | |
| mean_comp_meso: | 8.26 | 1.92 | 5.80 | 6.53 | 3.85 | 7.29 | 2.29 | 5.48 | 6.22 | 8.86 |
| sd_comp_meso: | 4.31 | 3.05 | 2.22 | 3.02 | 1.89 | 3.08 | 1.53 | 2.52 | 3.38 | 3.52 |
| mean_comp_therm: | 10.05 | 0.78 | 5.13 | 8.32 | 3.62 | 8.30 | 2.15 | 4.80 | 4.77 | 10.26 |
| sd_comp_therm: | 3.48 | 1.23 | 2.29 | 2.87 | 1.45 | 2.12 | 1.18 | 3.04 | 2.29 | 3.87 |
| t_therm_wrt_meso: | 6.03 | -9.08 | -3.57 | 7.49 | -1.83 | 5.42 | -1.35 | -2.72 | -7.21 | 4.40 |
| mean_comp_hyper: | 7.26 | 0.78 | 5.20 | 10.05 | 4.07 | 7.17 | 1.64 | 7.73 | 8.50 | 9.10 |
| sd_comp_hyper: | 2.95 | 1.05 | 1.76 | 2.51 | 1.64 | 2.23 | 1.05 | 2.44 | 2.60 | 2.39 |
| t_hyper_wrt_meso: | -4.09 | -10.19 | -4.17 | 17.47 | 1.72 | -0.65 | -7.37 | 11.72 | 10.74 | 1.21 |
| t_therm_wrt_hyp: | 7.99 | 0.00 | -0.32 | -5.93 | -2.71 | 4.83 | 4.22 | -9.80 | -14.19 | 3.30 |
| M | N | P | Q | R | S | T | V | W | Y | |
| mean_comp_meso: | 2.17 | 4.51 | 4.52 | 4.04 | 4.79 | 6.07 | 5.62 | 6.90 | 1.43 | 3.44 |
| sd_comp_meso: | 1.58 | 2.30 | 2.91 | 2.07 | 2.62 | 2.61 | 2.51 | 2.62 | 1.25 | 1.95 |
| mean_comp_therm: | 1.87 | 3.19 | 5.43 | 2.69 | 6.82 | 4.07 | 4.74 | 8.42 | 1.31 | 3.30 |
| sd_comp_therm: | 1.11 | 2.29 | 2.05 | 1.59 | 2.81 | 2.13 | 2.34 | 2.36 | 1.16 | 1.71 |
| t_therm_wrt_meso: | -3.14 | -6.94 | 5.10 | -9.87 | 8.77 | -11.07 | -4.52 | 7.65 | -1.20 | -1.00 |
| mean_comp_hyper: | 2.11 | 3.49 | 4.02 | 1.80 | 5.56 | 4.51 | 4.00 | 8.44 | 1.02 | 3.56 |
| sd_comp_hyper: | 1.14 | 1.62 | 1.57 | 1.16 | 2.27 | 1.72 | 1.48 | 2.28 | 1.07 | 1.71 |
| t_hyper_wrt_meso: | -0.59 | -7.65 | -3.60 | -22.12 | 4.23 | -10.82 | -12.71 | 8.45 | -4.78 | 0.87 |
| t_therm_wrt_hyp: | -1.98 | -1.39 | 7.11 | 5.88 | 4.55 | -2.10 | 3.46 | -0.08 | 2.41 | -1.41 |
t-tests of sequence percent compositions of mesophilc, thermophilic, and hyperthermophilic proteins Amino acids significantly over-represented in hyperthermophiles with respect to mesophiles are: Glu, Ile, Lys, Arg, and Val. Amino acids significantly under-represented in hyperthermophiles with respect to mesophiles are: Ala, Cys, Asp, His, Asn, Pro, Gln, Ser, Thr, and Trp. Amino acids significantly over-represented in thermophiles with respect to mesophiles are: Ala, Glu, Gly, Leu, Pro, Arg, and Val. Amino acids significantly under-represented in thermophiles with respect to mesophiles are: Cys, Asp, Lys, Met, Asn, Gln, Ser, and Thr. Amino acids significantly over-represented in hyperthermophiles with respect to thermophiles are: Glu, Ile, and Lys. Amino acids significantly under-represented in hyperthermophiles with respect to thermophiles are: Ala, Gly, His, Leu, Pro, Gln, Arg, and Thr. These statistics were tabulated from nonredundant sets of 184 hyperthermophilic structures (45419 residues), 162 thermophilic structures (41470 residues), and 1262 mesophilic structures (269799 residues).
Table 2.
Statistical significance of fractions in Table 1.
| Fraction | P for thermophile pairs | P for hyperthermophile pairs |
|---|---|---|
| 0.50 | 1.000 | 1.000 |
| 0.55 | 0.297 | 0.319 |
| 0.60 | 0.0328 | 0.0369 |
| 0.65 | 6.85 x 10-4 | 0.00143 |
| 0.70 | 6.97 x 10-6 | 1.65 x 10-5 |
| 0.75 | 1.94 x 10-8 | 4.99 x 10-8 |
| 0.80 | 3.12 x 10-12 | 8.34 x 10-12 |
| 0.85 | 2.66 x 10-16 | 6.78 x 10-16 |
| 0.90 | <2.20 x 10-16 | <2.20 x 10-16 |
| 0.95 | <2.20 x 10-16 | <2.20 x 10-16 |
Statistical significance of the fractions in Table 1 given by a two-sided binomial test. Fractions below ~0.6-0.65 should not be considered signficant.
Results of discrimination experiment
From Table 3 it can be seen that simplex geometry based indices are generally poor discriminators as are contact network based indices. Some compactness-based indices are good discriminators, for example Delaunay area/volume, a measure of general compactness, and van der Waals volume/Delaunay volume, a measure of void space. Secondary structure content, rigidity as measured by the mean B-factor, and sequence length are not very good discriminators. Sequence composition based indices, particularly IVYWREL and CvP are very good discriminators. Delaunay derived combined sequence-structure indices are very good discriminators as well, for example the 4-body potentials and the counts of over-represented residue quadruplets. Interestingly, even though the 4-body Delaunay threading potential works well as a discriminator, this is apparently not true for threading potentials in general. We have tested the ProsaII potential of Sippl et al [64], and found it to be a poor discriminator (Table 3).
Table 3.
Discriminatory power of structure and sequence derived quantities
| Numerical index | Thermophile (127 pairs) | Hyperthermophile (122 pairs) |
|---|---|---|
| Contact Network Derived Quantities | ||
| coordination number (no cutoff) | 0.559 | 0.689 |
| clustering coefficient (no cutoff) | 0.551 | 0.672 |
| characteristic path (no cutoff) | 0.520 | 0.631 |
| Combined Sequence and Structure Including Threading Potentials | ||
| total count 400 over-rep quads/residue | 0.850 | 0.943 |
| 4-body potential/residue (20Å cutoff) | 0.858 | 0.844 |
| 4-body potential/residue (no cutoff) | 0.843 | 0.852 |
| 4-body potential/res (hyper only, no cutoff) | ----- | 0.820 |
| 4-body potential/res (meso only, no cutoff) | 0.732 | 0.803 |
| 4-body potential/res (thermo only,no cutoff) | 0.866 | ----- |
| ProsaII combined score | 0.554 | 0.693 |
| Delaunay Simplex Geometry | ||
| median circumsphere radius(no cutoff) | 0.701 | 0.639 |
| mean tetrahedrality (no cutoff) | 0.598 | 0.574 |
| number simplices/residue (10Å cutoff) | 0.528 | 0.557 |
| number simplices/residue (no cutoff) | 0.567 | 0.697 |
| Volume/Surface Area/Compactness | ||
| Naccess solvent accessible area | 0.567 | 0.598 |
| Delaunay surface area (no cutoff) | 0.606 | 0.669 |
| van der Waals area | 0.559 | 0.549 |
| Delaunay volume (no cutoff) | 0.598 | 0.701 |
| Van der Waals volume | 0.528 | 0.598 |
| Delaunay area/volume (10Å cutoff) | 0.583 | 0.549 |
| Delaunay area/volume (no cutoff) | 0.669 | 0.803 |
| van der Waals area/volume | 0.512 | 0.557 |
| packing density | 0.543 | 0.549 |
| van der Waals volume/Delaunay volume | 0.685 | 0.779 |
| Rigidity | ||
| mean B-factor | 0.661 | 0.533 |
| Secondary Structure | ||
| secondary structure content (H+E 3 state DSSP) | 0.614 | 0.689 |
| Sequence Length | ||
| number of residues | 0.528 | 0.672 |
| Sequence Composition | ||
| total Kyte-Doolittle hydrophobicity | 0.575 | 0.549 |
| sd Kyte-Doolittle hydrophobicity | 0.677 | 0.836 |
| CvP bias | 0.803 | 0.918 |
| (E+K)/(Q+H) | 0.591 | 0.861 |
| IVYWREL | 0.827 | 0.926 |
A table showing the discriminatory power of sequence and structure based indices-the fraction of thermophile/mesophile pairs for which the quantity was systematically higher or lower by any amount. The contact network quantities are described in the introduction. The four body threading contact potentials are described in [1]. The cutoff indicates that simplices with at least one edge longer than the cutoff were omitted when frequencies are tallied during the calculation of the potential. "Hyper only" indicates that the potential was trained only on chains from hyperthermophilic organisms. The Delaunay simplex geometry indices are discussed in the introduction. The volume and surface area criteria are fairly self-explanatory except, perhaps, for packing density that is defined here as the ratio of the van der Waals volume of the protein divided by the all atom Voronoi volume. The sequence composition based indices CvP, (E+K)/(Q+H), and IVYWREL are described in the introduction.
The best discriminatory indices we tested were: one version of the 4-body Delaunay threading potential; the count of over-represented quadruplets; the ratio of Delaunay surface area to volume (for hyperthermophiles); the standard deviation of Kyte-Doolittle hydrophobicity (for hyperthermophiles); Delaunay area/volume=and van der Waals volume/Delaunay volume, particularly for hyperthermophiles; the CvP bias, and IVYWREL. Few of the tested indices (when computed for individual proteins not averaged over proteomes) correlate even moderately well with OGT. Those for which the correlation is strongest are the 4-body potentials, overrep400, CvP, IVYWREL, and E+K/Q+H which all have r~0.4-0.7.
The Delaunay simplex-based descriptors (overrep400 and the 4-body potentials) that work best for discrimination use large simplex edge length cutoffs ( >20 Å). This implies that there are important residue contacts on the surface of the proteins because that is invariably where simplices with very long edge lengths reside. This combined with threading potential data presented later leads us to believe that the presence of more charged residues on the protein surface is at least one of the things these descriptors pick up.
Since increased hydrophobicity of the protein core has been proposed as a mechanism for thermostability, one might expect the sum SKD of the Kyte-Doolittle hydrophobicities of all residues would be a good discriminator. SKD is not, but the variance of the Kyte-Doolittle hydrophobicities is (Table 3). Apparently, then, the increase in core hydrophobicity is accompanied by an increase in hydrophilic residues.
Why the Delaunay threading potential a good discriminator
We have studied the mean contribution to the 4-body potential score Q under a 7-letter reduced alphabet to each structure in the mesophilic, thermophilic, and hyperthermophilic subsets of 521nonredundant. The reduced alphabet was used to simplify the analysis by bringing down the possible number of residue quadruplets that can reside at the vertices of a Delaunay tetrahedron from 8855 for a 20 amino acid alphabet to 210. The reduced alphabet is D, E, K, R, I=(I,V), A=(A,F,G,L,P), N=(C,H,M,N,Q,S,T,W,Y. The four-body Delaunay threading score Q for a hyperthermophile with the average quadruplet composition is 36.3, for a thermophile Q is 28.2, and for a mesophile Q is 17.6. For hyperthermophiles, the ΔQ (with respect to mesophiles) attributable to quadruplets with at least two hydrophobic residues is +11.07, for quadruplets with at least two charged residues it is +12.78, and for quadruplets with at least two polar residues it is –2.53. For thermophilic quadruplets the ΔQ figures are: at least two hydrophobic residues +5.99, at least two charged residues +4.55, and at least two polar residues –0.34. We see ,therefore, that the 4-body potential goes up for (hyper)thermophiles both due to associations between charged residues but also due to quadruplets of more highly hydrophobic residues. The increase in charged residues produces a larger change than the stronger hydrophobics with hyperthermophiles, but the situation is reversed in thermophiles.
Conclusions
It is possible to accurately discriminate (hyper) thermophilic proteins from their mesophilic counterparts based on sequence and structural properties. Sequence based indices used to discriminate entire proteomes also work well on individual thermophile/mesophile ortholog pairs. Purely structure-based indices are, generally speaking, poor discriminators. Combined sequence structure indices like the threading potential are somewhat better than sequence alone.
The primary factors differentiating thermophilic from mesophilic proteins according to our analysis are surface ion pairs and more strongly hydrophobic core residues. The conclusion of previous authors that the basis for the thermostability of thermophiles and hyper-thermophiles is somewhat different is also borne out here (e.g. the preference of thermophiles for Arg and of hyperthermophiles for Lys).
Extensions of this work currently underway include compiling larger test sets and breaking them down by kingdom of origin as well as OGT. Heat shock proteins should be compared to regular proteins from the same organism and non-thermophilic archeal proteins should be compared to orthologs from thermophiles. Proteins from psychrophiles should be analyzed too. It would be a small step to use more sophisticated pattern recognition methods to discriminate or classify based on multiple indices.
Finally, it may be possible to design a thermostable protein from a non-thermostable one by an adaptive walk in sequence space, threading the altered sequences onto the structure of the non-thermostable protein, such that one or more of the good discriminators described here always increases.
Methods
Assembly of the test sets
In this paper, we have addressed the discrimination problem where given sequences or structures from a mesophilic protein and a thermophilic or hyperthermophilic counterpart, the objective is to determine which is which. This was done by assembling a large set of thermophilic protein chains from the PDB and their corresponding mesophilic analogs and another large non-redundant set of hyperthermophilic PDB protein chains along with their mesophilic analogs. They will be referred to as the pairs sets: pairs-thermophile and pairs-hyperthermophile. We have computed several structure and sequence based numerical indices, based on the quantities that other authors have reported are associated with thermostability, and tested their ability, individually, to successfully discriminate between the thermophile/mesophile pairs. One could apply more sophisticated classification or regression techniques to a combination of these quantities, but for now we have opted for a very simple test of each quantity in isolation in order to verify if it is indeed consistently associated with increased thermostability.
The pairs sets were constructed to contain pairs of high quality x-ray structures with high structural and functional similarity that differ only in that one is mesophilic and the other thermophilic or hyperthermophilic. They were assembled by compiling all PDB x-ray structures from a large list of organisms categorized as mesophile, thermophile or hyperthermophile using OGT’s obtained from the ATCC website (http://www.atcc.org/common/catalog/bacteria/bacteriaIndex.cfm). All structures with missing Cα coordinates, insertion codes, or alternate atoms were then eliminated. The resulting two lists of thermophilic and hyperthermophilic proteins were submitted separately to PISCES[65] to generate two much smaller non-redundant sets in which all members had crystallographic resolution no greater than 2.2 Å, an R-factor no greater than 0.23, and where no pair of structures had a sequence identity greater than 30%. The members of these non-redundant sets of chains from thermophilic organisms were each then submitted to structure comparison and alignment servers (CE[66] , SSM [67], DALI[68], VAST[69]) to obtain mesophilic structure neighbors with rmsd no greater than about 4 Å with respect to the thermophile where the structural alignment included ~80% or more of each structure, and where the thermophile and mesophile had identical or close EC numbers or functional annotation. In some cases, more than one mesophile per thermophilic protein was kept. When multiple mesophilic analogs to a single thermophilic protein were included in the pairs sets, no restriction was placed on their similarity with respect to each other except that the sequences not be identical and that they come from different organisms. Lists of the resulting structure pairs and structure alignment data can be found among the supplementary material. The structural alignment data in these tables were computed using CE [66].
To train threading potentials and compute sequence composition biases, we compiled a second set of PDB x-ray structures (521nonredundant), larger and more representative than the pairs sets. The set contained 175 mesophilic, 162 thermophilic, and 184 hyperthermophilic protein structures, none of which was in either pairs set. As with the pairs sets, 521nonredundant was assembled with the help of PISCES [65] All members had crystallographic resolution no greater than 2.2 Å, an r-factor no greater than 0.23, and no pair of structures had a sequence identity greater than 66%. The pairwise similarity threshold was set higher for this set than pairs in order to allow the possibility that it could contain mesophilic and thermophilic orthologs, however a lower similarity threshold would have made little difference—a 30% similarity cutoff would have eliminated only 41 structures.
Numerical discriminators tested
We have computed several structure and sequence based numerical indices to see if they can successfully discriminate between the related mesophilic and (hyper)thermophilic proteins in the pairs sets (Table 3). Among the quantities tested for discriminatory power were: the three contact-network derived quantities described in the introduction (coordination number, characteristic path length, and clustering coefficien), the ratio (E+K)/(Q+R) of Farias and Bonato [26], the CvP bias defined earlier [37], the sum and standard deviation of the Kyte-Doolittle hydrophobicities of all residues in the protein, several four-body Delaunay tessellation based threading Q-scores [42], the ProsaII two-body distance dependent threading score [64], the median circumsphere radius, the mean tetrahedrality, the surface area to volume ratio (both Delaunay and van der Waals), and the number of Delaunay simplices per residue. We have also tested the packing density defined as the van der Waals volume of the protein divided by the all-atom Voronoi volume. The algorithm of Gavezotti [70] was used to calculate van der Waals volumes and the Geometry Code Library 2.0 of Tsai et al. [71] to calculate Voronoi volumes.
We have defined discriminatory power of a given numerical index as simply the fraction f of (hyper)thermophile-mesophile pairs from a pairs set where that index is systematically larger or smaller for the (hyper)thermophilic protein, by any amount. For example, 86.1% of the time for structure pairs in the pairs hyperthermophile set, the ratio (E+K)/(Q+H) is greater for the hyperthermophilic than for the mesophilic protein, and so the discriminatory power is 0.861.
Analysis of the residue quadruplets occupying the vertices of the tetrahedra of tessellated hyperthermophilic and thermophilic protein structures shows that some are heavily over-represented (e.g. EEEK, AEER) or under-represented (e.g. ANQV, AELR) in (hyper)thermophilic proteins with respect to mesophilic (Table 4). Another discriminator index we have tested on the pairs sets, therefore, is the total count in a query protein of the top 400 most over-represented simplices in hyperthermophilic or thermophilic structures divided by the number of residues. These two indices are the most powerful discriminators of all those we tested. We will abbreviate them as overrep400-thermophile and overrep400-hyperthermophile.
Table 4.
Highly over-represented and highly under-represented residue quadruplets at the vertices of tessellated thermostable proteins and the factors by which they differ with respect to mesophiles.
| hyperthermophiles | thermophiles | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| over-represented | under-represented | over-represented | under-represented | |||||||
| quad | factor | quad | factor | quad | factor | quad | factor | |||
| EEEE | 7.473 | PQRT | 0.303 | EEER | 6.008 | AIQS | 0.380 | |||
| EEEK | 7.332 | GGNQ | 0.302 | ELWW | 5.520 | DLQS | 0.378 | |||
| EEER | 7.048 | FLLQ | 0.301 | RRRV | 5.222 | KLST | 0.377 | |||
| MRRR | 6.490 | AQVY | 0.301 | AEER | 5.106 | ILNQ | 0.373 | |||
| EEKR | 5.654 | AAAN | 0.299 | EEPR | 5.087 | ENQS | 0.370 | |||
| IRRR | 5.605 | AAEQ | 0.298 | EERR | 4.801 | KKLS | 0.370 | |||
| EEEF | 5.597 | DSTT | 0.296 | AERR | 4.538 | KNQR | 0.368 | |||
| EEKK | 5.282 | ADDT | 0.296 | RRRY | 4.508 | FKQS | 0.368 | |||
| EEEV | 4.936 | AENQ | 0.296 | EEEE | 4.508 | LNQS | 0.367 | |||
| EEIK | 4.889 | NSTT | 0.295 | ERRR | 4.458 | EFNS | 0.366 | |||
| EIKK | 4.881 | ALPQ | 0.292 | IRRR | 4.186 | DILQ | 0.365 | |||
| EEIV | 4.346 | ADQR | 0.292 | EEGP | 4.177 | LMSY | 0.364 | |||
| EIKR | 4.332 | ANRT | 0.292 | EEGR | 4.113 | KLNS | 0.362 | |||
| EKRR | 4.256 | DNQV | 0.292 | ERRV | 4.104 | DDQS | 0.361 | |||
| IKRR | 4.228 | FGLQ | 0.291 | EGPR | 4.092 | LNSS | 0.361 | |||
| EEKV | 4.225 | AQST | 0.291 | EERW | 4.015 | EHIK | 0.360 | |||
| EEIR | 4.169 | ANTY | 0.290 | GPRR | 3.963 | KKST | 0.358 | |||
| EEEN | 4.107 | INSY | 0.290 | EEPP | 3.951 | GKSS | 0.356 | |||
| EEEY | 4.092 | DNQT | 0.289 | EERV | 3.904 | CGLL | 0.354 | |||
| KRRR | 4.085 | ANQV | 0.289 | AELR | 3.792 | LNQT | 0.353 | |||
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
IIV originated the analysis of protein structure using Delaunay tessellation and the four-body pseudo potential. TJT assembled the test sets, determined which quantities to test, and wrote scripts to compute tessellation and sequence derived quantities. TJT drafted the paper, which was approved by IIV.
Supplementary Material
Contributor Information
Todd J Taylor, Email: todd.taylor@nih.gov.
Iosif I Vaisman, Email: ivaisman@gmu.edu.
Acknowledgements
The work was supported by NIST and NSF. We thank Lev Krayzman for his help with the compilation of datasets.
This article has been published as part of BMC Structural Biology Volume 10 Supplement 1, 2010: Selected articles from the Computational Structural Bioinformatics Workshop 2009. The full contents of the supplement are available online at http://www.biomedcentral.com/1472-6807/10?issue=S1.
References
- Brock TD, Freeze H. Thermus aquaticus gen. n. and sp. n., a Nonsporulating Extreme Thermophile. J Bacteriol. 1969;98(1):289–297. doi: 10.1128/jb.98.1.289-297.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scandurra R, Consalvi V, Chiaraluce R, Politi L, Engel PC. Protein thermostability in extremophiles. Biochimie. 1998;80(11):933–941. doi: 10.1016/S0300-9084(00)88890-2. [DOI] [PubMed] [Google Scholar]
- Van den Burg B, Vriend G, Veltman OR, Venema G, Eijsink VG. Engineering an enzyme to resist boiling. Proc Natl Acad Sci U S A. 1998;95(5):2056–2060. doi: 10.1073/pnas.95.5.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Burg B. Extremophiles as a source for novel enzymes. Curr Opin Microbiol. 2003;6(3):213–218. doi: 10.1016/S1369-5274(03)00060-2. [DOI] [PubMed] [Google Scholar]
- Persidis A. Extremophiles. Nat Biotechnol. 1998;16(6):593–594. doi: 10.1038/nbt0698-593. [DOI] [PubMed] [Google Scholar]
- Perutz MF, Raidt H. Stereochemical basis of heat stability in bacterial ferredoxins and in haemoglobin A2. Nature. 1975;255(5505):256–259. doi: 10.1038/255256a0. [DOI] [PubMed] [Google Scholar]
- Haney P, Konisky J, Koretke KK, Luthey-Schulten Z, Wolynes PG. Structural basis for thermostability and identification of potential active site residues for adenylate kinases from the archaeal genus Methanococcus. Proteins. 1997;28(1):117–130. doi: 10.1002/(SICI)1097-0134(199705)28:1<117::AID-PROT12>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Spassov VZ, Karshikoff AD, Ladenstein R. The optimization of protein-solvent interactions: thermostability and the role of hydrophobic and electrostatic interactions. Protein Sci. 1995;4(8):1516–1527. doi: 10.1002/pro.5560040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RJ, Hough DW, Danson MJ, Taylor GL. The crystal structure of citrate synthase from the thermophilic archaeon, Thermoplasma acidophilum. Structure. 1994;2(12):1157–1167. doi: 10.1016/S0969-2126(94)00118-9. [DOI] [PubMed] [Google Scholar]
- Russell RB, Sternberg MJ. Two new examples of protein structural similarities within the structure-function twilight zone. Protein Eng. 1997;10(4):333–338. doi: 10.1093/protein/10.4.333. [DOI] [PubMed] [Google Scholar]
- Thompson MJ, Eisenberg D. Transproteomic evidence of a loop-deletion mechanism for enhancing protein thermostability. J Mol Biol. 1999;290(2):595–604. doi: 10.1006/jmbi.1999.2889. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Hata Y, Kizaki H, Katsube Y, Suzuki Y. The refined crystal structure of Bacillus cereus oligo-1,6-glucosidase at 2.0 A resolution: structural characterization of proline-substitution sites for protein thermostabilization. J Mol Biol. 1997;269(1):142–153. doi: 10.1006/jmbi.1997.1018. [DOI] [PubMed] [Google Scholar]
- Bogin O, Peretz M, Hacham Y, Korkhin Y, Frolow F, Kalb AJ, Burstein Y. Enhanced thermal stability of Clostridium beijerinckii alcohol dehydrogenase after strategic substitution of amino acid residues with prolines from the homologous thermophilic Thermoanaerobacter brockii alcohol dehydrogenase. Protein Sci. 1998;7(5):1156–1163. doi: 10.1002/pro.5560070509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilagyi A, Zavodszky P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure Fold Des. 2000;8(5):493–504. doi: 10.1016/S0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Vogt G, Woell S, Argos P. Protein thermal stability, hydrogen bonds, and ion pairs. J Mol Biol. 1997;269(4):631–643. doi: 10.1006/jmbi.1997.1042. [DOI] [PubMed] [Google Scholar]
- Russell RJ, Ferguson JM, Hough DW, Danson MJ, Taylor GL. The crystal structure of citrate synthase from the hyperthermophilic archaeon pyrococcus furiosus at 1.9 A resolution. Biochemistry. 1997;36(33):9983–9994. doi: 10.1021/bi9705321. [DOI] [PubMed] [Google Scholar]
- Tanner JJ, Hecht RM, Krause KL. Determinants of enzyme thermostability observed in the molecular structure of Thermus aquaticus D-glyceraldehyde-3-phosphate dehydrogenase at 25 Angstroms Resolution. Biochemistry. 1996;35(8):2597–2609. doi: 10.1021/bi951988q. [DOI] [PubMed] [Google Scholar]
- Vieille C, Epting KL, Kelly RM, Zeikus JG. Bivalent cations and amino-acid composition contribute to the thermostability of Bacillus licheniformis xylose isomerase. Eur J Biochem. 2001;268(23):6291–6301. doi: 10.1046/j.0014-2956.2001.02587.x. [DOI] [PubMed] [Google Scholar]
- Kawashima T, Amano N, Koike H, Makino S, Higuchi S, Kawashima-Ohya Y, Watanabe K, Yamazaki M, Kanehori K, Kawamoto T. Archaeal adaptation to higher temperatures revealed by genomic sequence of Thermoplasma volcanium. Proc Natl Acad Sci U S A. 2000;97(26):14257–14262. doi: 10.1073/pnas.97.26.14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip KS, Stillman TJ, Britton KL, Artymiuk PJ, Baker PJ, Sedelnikova SE, Engel PC, Pasquo A, Chiaraluce R, Consalvi V. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure. 1995;3(11):1147–1158. doi: 10.1016/S0969-2126(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Elcock AH. The stability of salt bridges at high temperatures: implications for hyperthermophilic proteins. J Mol Biol. 1998;284(2):489–502. doi: 10.1006/jmbi.1998.2159. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tsai CJ, Nussinov R. Factors enhancing protein thermostability. Protein Eng. 2000;13(3):179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- Maes D, Zeelen JP, Thanki N, Beaucamp N, Alvarez M, Thi MH, Backmann J, Martial JA, Wyns L, Jaenicke R. The crystal structure of triosephosphate isomerase (TIM) from Thermotoga maritima: a comparative thermostability structural analysis of ten different TIM structures. Proteins. 1999;37(3):441–453. doi: 10.1002/(SICI)1097-0134(19991115)37:3<441::AID-PROT11>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Pace CN, Alston RW, Shaw KL. Charge-charge interactions influence the denatured state ensemble and contribute to protein stability. Protein Sci. 2000;9(7):1395–1398. doi: 10.1110/ps.9.7.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetriani C, Maeder DL, Tolliday N, Yip KS, Stillman TJ, Britton KL, Rice DW, Klump HH, Robb FT. Protein thermostability above 100 degreesC: a key role for ionic interactions. Proc Natl Acad Sci U S A. 1998;95(21):12300–12305. doi: 10.1073/pnas.95.21.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias ST, Bonato MC. Preferred amino acids and thermostability. Genet Mol Res. 2003;2(4):383–393. [PubMed] [Google Scholar]
- Cambillau C, Claverie JM. Structural and genomic correlates of hyperthermostability. J Biol Chem. 2000;275(42):32383–32386. doi: 10.1074/jbc.C000497200. [DOI] [PubMed] [Google Scholar]
- Haney PJ, Badger JH, Buldak GL, Reich CI, Woese CR, Olsen GJ. Thermal adaptation analyzed by comparison of protein sequences from mesophilic and extremely thermophilic Methanococcus species. Proc Natl Acad Sci U S A. 1999;96(7):3578–3583. doi: 10.1073/pnas.96.7.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN. Single surface stabilizer. Nat Struct Biol. 2000;7(5):345–346. doi: 10.1038/75100. [DOI] [PubMed] [Google Scholar]
- Karshikoff A, Ladenstein R. Proteins from thermophilic and mesophilic organisms essentially do not differ in packing. Protein Eng. 1998;11(10):867–872. doi: 10.1093/protein/11.10.867. [DOI] [PubMed] [Google Scholar]
- Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268(5214):1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Waldburger CD, Schildbach JF, Sauer RT. Are buried salt bridges important for protein stability and conformational specificity? Nat Struct Biol. 1995;2(2):122–128. doi: 10.1038/nsb0295-122. [DOI] [PubMed] [Google Scholar]
- Das R, Gerstein M. The stability of thermophilic proteins: a study based on comprehensive genome comparison. Funct Integr Genomics. 2000;1(1):76–88. doi: 10.1007/s101420000003. [DOI] [PubMed] [Google Scholar]
- Querol E, Perez-Pons JA, Mozo-Villarias A. Analysis of protein conformational characteristics related to thermostability. Protein Eng. 1996;9(3):265–271. doi: 10.1093/protein/9.3.265. [DOI] [PubMed] [Google Scholar]
- Vieille C, Zeikus GJ. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev. 2001;65(1):1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang HK, Huang CM, Ko MT, Hwang JK. Amino acid coupling patterns in thermophilic proteins. Proteins. 2005;59(1):58–63. doi: 10.1002/prot.20386. [DOI] [PubMed] [Google Scholar]
- Suhre K, Claverie JM. Genomic correlates of hyperthermostability, an update. J Biol Chem. 2003;278(19):17198–17202. doi: 10.1074/jbc.M301327200. [DOI] [PubMed] [Google Scholar]
- Zeldovich KB, Berezovsky IN, Shakhnovich EI. Protein and DNA sequence determinants of thermophilic adaptation. PLoS computational biology. 2007;3(1):e5. doi: 10.1371/journal.pcbi.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glyakina AV, Garbuzynskiy SO, Lobanov MY, Galzitskaya OV. Different packing of external residues can explain differences in the thermostability of proteins from thermophilic and mesophilic organisms. Bioinformatics. 2007;23(17):2231–2238. doi: 10.1093/bioinformatics/btm345. [DOI] [PubMed] [Google Scholar]
- Poupon A. Voronoi and Voronoi-related tessellations in studies of protein structure and interaction. Curr Opin Struct Biol. 2004;14(2):233–241. doi: 10.1016/j.sbi.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Vaisman I. Handbook of Computational Statistics. Springer; 2004. Statistical and computational geometry of biomolecular structure; pp. 981–1000. [Google Scholar]
- Singh RK, Tropsha A, Vaisman II. Delaunay tessellation of proteins: four body nearest-neighbor propensities of amino acid residues. J Comput Biol. 1996;3(2):213–221. doi: 10.1089/cmb.1996.3.213. [DOI] [PubMed] [Google Scholar]
- Tropsha A, Singh RK, Vaisman II, Zheng W. Statistical geometry analysis of proteins: implications for inverted structure prediction. Pac Symp Biocomput. 1996. pp. 614–623. [PubMed]
- Krishnamoorthy B, Tropsha A. Development of a four-body statistical pseudo-potential to discriminate native from non-native protein conformations. Bioinformatics. 2003;19(12):1540–1548. doi: 10.1093/bioinformatics/btg186. [DOI] [PubMed] [Google Scholar]
- Ilyin VA, Abyzov A, Leslin CM. Structural alignment of proteins by a novel TOPOFIT method, as a superimposition of common volumes at a topomax point. Protein Sci. 2004;13(7):1865–1874. doi: 10.1110/ps.04672604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach J, Sharma S, Kapustina M, Carter CW Jr.. Structure alignment via Delaunay tetrahedralization. Proteins. 2005;60(1):66–81. doi: 10.1002/prot.20479. [DOI] [PubMed] [Google Scholar]
- Bostick D, Vaisman II. A new topological method to measure protein structure similarity. Biochem Biophys Res Commun. 2003;304(2):320–325. doi: 10.1016/S0006-291X(03)00585-0. [DOI] [PubMed] [Google Scholar]
- Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shape computation of macromolecules: I. Molecular area and volume through alpha shape. Proteins. 1998;33(1):1–17. doi: 10.1002/(SICI)1097-0134(19981001)33:1<1::AID-PROT1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Masso M, Vaisman II. Comprehensive mutagenesis of HIV-1 protease: a computational geometry approach. Biochem Biophys Res Commun. 2003;305(2):322–326. doi: 10.1016/S0006-291X(03)00760-5. [DOI] [PubMed] [Google Scholar]
- Carter CW Jr., LeFebvre BC, Cammer SA, Tropsha A, Edgell MH. Four-body potentials reveal protein-specific correlations to stability changes caused by hydrophobic core mutations. J Mol Biol. 2001;311(4):625–638. doi: 10.1006/jmbi.2001.4906. [DOI] [PubMed] [Google Scholar]
- Tropsha A, Carter CW Jr., Cammer S, Vaisman II. Simplicial neighborhood analysis of protein packing (SNAPP): a computational geometry approach to studying proteins. Methods Enzymol. 2003;374:509–544. doi: 10.1016/S0076-6879(03)74022-1. full_text. [DOI] [PubMed] [Google Scholar]
- Cammer SA, Carty RP, Tropsha A. In: Proceedings of the 3rd Intl Workshop on Algorithms for Macromolecular Modeling. Schlick T, Gan HH, editor. Springer; 2000. Computational Methods for Macromolecules: Challenges and Applications; pp. 477–494. [Google Scholar]
- Wako H, Yamato T. Novel method to detect a motif of local structures in different protein conformations. Protein Eng. 1998;11(11):981–990. doi: 10.1093/protein/11.11.981. [DOI] [PubMed] [Google Scholar]
- Huan J, Wang W, Washington A, Prins J, Shah R, Tropsha A. Accurate classification of protein structural families using coherent subgraph analysis. Pac Symp Biocomput. 2004. pp. 411–422. [DOI] [PubMed]
- Taylor T, Rivera M, Wilson G, Vaisman II. New method for protein secondary structure assignment based on a simple topological descriptor. Proteins. 2005;60(3):513–524. doi: 10.1002/prot.20471. [DOI] [PubMed] [Google Scholar]
- Huan J, Bandyopadhyay D, Wang W, Snoeyink J, Prins J, Tropsha A. Comparing graph representations of protein structure for mining family-specific residue-based packing motifs. J Comput Biol. 2005;12(6):657–671. doi: 10.1089/cmb.2005.12.657. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay D, Snoeyink J. Almost-Delaunay simplices : Nearest neighbor relations for imprecise points. ACM-SIAM Symposium On Distributed Algorithms: 2004. 2004. pp. 403–412.
- Okabe A. Spatial tessellations : concepts and applications of Voronoi diagrams. 2. Wiley; 2000. [Google Scholar]
- Bostick DL, Shen M, Vaisman II. A simple topological representation of protein structure: Implications for new, fast, and robust structural classification. Proteins. 2004;56(3):487–501. doi: 10.1002/prot.20146. [DOI] [PubMed] [Google Scholar]
- Munson PJ, Singh RK. Statistical significance of hierarchical multi-body potentials based on Delaunay tessellation and their application in sequence-structure alignment. Protein Sci. 1997;6(7):1467–1481. doi: 10.1002/pro.5560060711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo M, Dokholyan NV, Paci E, Karplus M. Small-world view of the amino acids that play a key role in protein folding. Phys Rev E Stat Nonlin Soft Matter Phys. 2002;65(6 Pt 1):061910. doi: 10.1103/PhysRevE.65.061910. [DOI] [PubMed] [Google Scholar]
- Dokholyan NV, Li L, Ding F, Shakhnovich EI. Topological determinants of protein folding. Proc Natl Acad Sci U S A. 2002;99(13):8637–8641. doi: 10.1073/pnas.122076099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts DJ, Strogatz SH. Collective dynamics of 'small-world' networks. Nature. 1998;393(6684):440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17(4):355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- Wang G, Dunbrack RL Jr.. PISCES: a protein sequence culling server. Bioinformatics. 2003;19(12):1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11(9):739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233(1):123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6(3):377–385. doi: 10.1016/S0959-440X(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Gavezzotti A. The calculation of molecular volumes and the use of volume analysis in the investigation of structured media and of solid-state organic reactivity. J Am Chem Soc. 1983;105:5220–5225. doi: 10.1021/ja00354a007. [DOI] [Google Scholar]
- Tsai J, Taylor R, Chothia C, Gerstein M. The packing density in proteins: standard radii and volumes. J Mol Biol. 1999;290(1):253–266. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.