

Abstract
Evidence suggests that West Nile virus (WNV) neuroinvasive disease occurs more frequently in both solid organ and human stem cell transplant recipients. The effect of concomitant anti-B-cell therapy with rituximab, a CD20+ monoclonal antibody, on WNV infection in this population, however, has not been reported. We describe a case of a patient with alpha-1-antitrypsin deficiency who underwent single lung transplantation in 2005 and was maintained on tacrolimus, cytoxan and prednisone. More recently, she had received two courses of rituximab for recurrent A2–A3 grade rejection with concomitant capillaritis and presented six months later with rapid, fulminant WNV meningoencephalitis. Her diagnosis was made by cerebrospinal fluid (CSF) PCR but serum and CSF WNV IgM and IgG remained negative. She received WNV-specific hyperimmune globulin (Omr-Ig-Am®) through a compassionate protocol. She experienced a rapidly progressive and devastating neurological course despite treatment and died three wk after onset of her symptoms. Autopsy revealed extensive meningoencephalomyelitis.
Keywords: rituximab, transplant, West Nile virus
West Nile virus (WNV) is a flavivirus that has been an important cause of neuroinvasive disease during the late summer and early fall in the United States since 1999. In the immunocompetent population, meningoencephalitis occurs in 1:150 individuals infected with WNV. However, in immunocompromised patients, particularly transplant recipients, the incidence of central nervous system disease has been estimated to be as high as 40%, although these calculations are based on relatively small numbers of cases (1).
Multiple case reports describing poor outcome in most, but not all, WNV-infected transplant recipients have been published (2–4), likely due to immunosuppressive agents. These drugs act primarily by inhibiting T-lymphocyte function (5, 6). While mechanisms to block cellular rejection have focused on T-cell pathways, B-cell-mediated transplant rejection has more recently been identified (7). Use of rituximab, a monoclonal antibody directed against the B-cell-specific antigen B1 (CD20) (8, 9) is thought to have antihumoral rejection activity and has been employed as a therapeutic in humoral transplant rejection (7).
We report a case of a lung transplant recipient who received two courses of rituximab for acute humoral rejection (capillaritis), followed by a rapidly progressive and fatal WNV meningoencephalitis. Furthermore, she was unable to mount serum WNV IgM or IgG antibody responses against WNV two wk following the onset of her symptoms. The diagnosis of WNV neuroinvasive disease was confirmed by positive WNV polymerase chain reaction identification in the cerebrospinal fluid (CSF). Meningoencephalomyelitis at autopsy was extremely extensive, involving all levels of spinal cord sampled, as well as cerebral subcortical gray matter, cerebellum, brainstem, hippocampi, cerebral white matter, and proximal ventral nerve roots. Our concern is that the use of both T-cell and B-cell immunosuppression may have significantly contributed to her rapid decline, as indicated by the rapidity of her course, lack of WNV antibody production, and the severity of tissue damage on histopathologic examination of the brain and spinal cord tissue.
Case history
The patient was a 57-yr-old white woman who underwent a single left lung transplant for alpha-1-antitrypsin-induced chronic obstructive pulmonary disease in August of 2005 at the University of Colorado Health Sciences Center. In addition to standard lung transplant prophylaxis medications, her allograft was maintained on tacrolimus 1.5 mg twice daily, prednisone 10 mg daily and cytoxan 25 mg daily. Her course was previously complicated by recurrent A2–A3 rejection with capillaritis for which she required pulse-dose intravenous solumedrol 10 mg/kg/d for three d in addition to five d of total plasma exchange, followed by two doses of rituximab 700 mg intravenously (375 mg/m2/d) one wk apart, on April 12 and April 19, 2007. At the time of presentation, she resided in Las Vegas, Nevada and was employed in a casino warehouse.
She initially presented to a local hospital emergency room in Las Vegas on November 17, 2007 with a five d history of fevers, fatigue, malaise and weakness, and discharged with a diagnosis of viral syndrome. Two d later, she was admitted with a temperature of 102.2°F and altered mental status. Her initial diagnosis was sepsis, and she was empirically treated with vancomycin and meropenem. An admission chest X-ray was unremarkable, with subsequent development of a left lung infiltrate and peribronchial thickening on November 11, 2007. A magnetic resonance imaging scan (MRI) of the brain was non-diagnostic due to motion artifact. Due to increasing confusion and altered mental status, an infectious disease consultation was obtained and acyclovir and voriconazole were added. A lumbar puncture was recommended but not performed. The patient's mental status deteriorated further, and she was placed on ventilatory support for airway protection on November 21, 2007. The following day, she was transferred to the University of Colorado for further care. By report, she was following commands and moved all four extremities prior to her transfer.
Upon arrival, the patient was minimally responsive to commands. Soon thereafter, she developed diffuse flaccid paralysis and areflexia. A computerized tomographic (CT) scan of the brain without contrast was negative. A lumbar puncture performed on November 11, 2007 showed four white blood cells (WBC), with 48% lymphocytes, 45% monocytes, 3% polymorphonuclear leukocytes, 2% bands, and 1% eosinophils in the first tube, and eight WBC with 48% lymphocytes, 45% monocytes, 3% polymorphonuclear leukocytes, and 4% bands in the fourth tube, protein 125 mg/dL and glucose 125 mg/dL (peripheral glucose 288 mg/dL), gram stain showed no organisms and routine cultures were negative. Stains of the CSF were negative for acid fast bacilli. Her CSF cryptococcal antigen was negative. CSF qualitative PCR studies were negative for herpes simplex virus 1 and 2, cytomegalovirus, parovirus B19, Epstein–Barr virus and human herpes virus 6. CSF for WNV RNA by qualitative RT-PCR (ARUP Laboratories, Salt Lake City, UT, USA) was sent. Serum was concomitantly sent to ARUP laboratories for WNV IgM and IgG analysis. ELISA studies were negative for WNV IgG at 0.25 international units (IU) (negative <1.30 IU) and IgM at 0.01 IU (negative <0.90 IU). Serum coccidiomycosis antibody by complement fixation was negative. Bronchoalveolar lavage on November 22, 2007 was negative for respiratory viral PCR studies.
Abnormal studies included electromyogram (EMG) and nerve conduction studies. Compound motor action potential amplitudes were severely reduced in the right arm, with milder reductions in the legs. Sensory nerve action potential (SNAP) amplitudes were only mildly reduced with normal conduction velocities. No spontaneous activity or voluntary motor unit potentials were recordable with concentric needle EMG examination. This constellation of findings suggested early anterior horn cell disease affecting lower cervical myotomes most severely. The mild reduction in SNAP amplitudes was attributed to a superimposed mild sensory polyneuropathy or neuronopathy. Electroencephalography (EEG) showed only moderate, diffuse intermixed slowing. An MRI of the brain (Fig. 1) showed multiple non-enhancing lesions in the left anterior and posterior thalamus, left posterior brainstem, and right thalamus/posterior internal capsule, which is non-specific but could suggest a viral or demyelinating process. Several days later, an MRI of the cervical spine was also obtained, and was only significant for subtle meningeal enhancement surrounding the cerebellum and anterior to the pons. This non-specific finding was interpreted as possibility representing inflammation or infection. The clinical course of the patient continued to decline and she became totally unresponsive to verbal commands and then to painful stimuli.
Fig. 1.
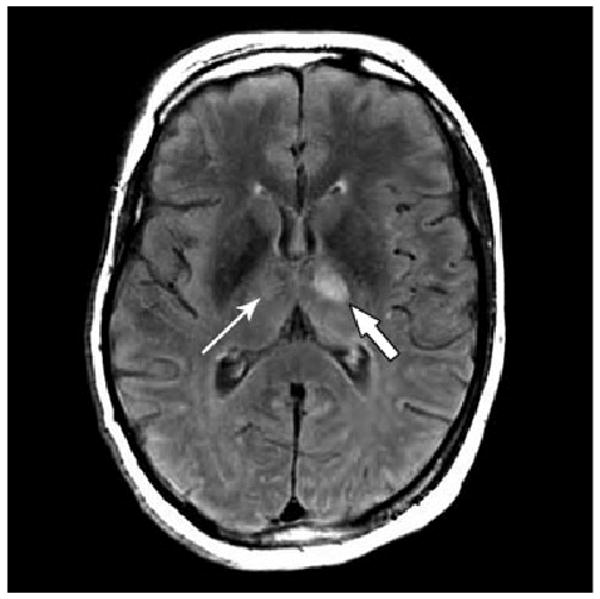
Axial T2-weighted FLAIR magnetic resonance image of the brain at the level of the thalamus. Thick arrow indicates 1.4 cm lesion showing T2/FLAIR bright signal in the ventral left thalamus. A smaller lesion was present in the left posterior thalamus and pulvinar. Thin arrow indicates another small area of increased T2/FLAIR signal in the right lateral thalamus. These lesions did not enhance with gadolinium.
Although the serum WNV IgM and IgG were negative, WNV remained high on the differential due to the EMG findings consistent with anterior horn cell disease, declining mental status with flaccid paralysis and residence in a warm temperate climate. Therefore, a repeat lumbar puncture was performed on November 27 showing 31 WBC with 53% lymphocytes and 47% monocytes, protein increased to 146 mg/dL and glucose 77 mg/dL. CSF studies sent for human herpes virus 6 and 8, Epstein–Barr virus, herpes simplex 1 and 2, poliovirus types 1, 2 and 3, mycoplasma, and varicella were all negative. Serum studies for WNV IgG and IgM were again negative (IgG 0.08 international units [IU] on November 27, 0.32 IV on November 28, and IgM 0.06 on November 27, 0.28 on November 27 and 0.36 on November 28). Repeat CSF for qualitative WNV RNA RT-PCR was sent (ARUP laboratories) while the initial CSF result was pending. On November 28, a positive WNV RNA RT-PCR result was obtained from the initial CSF sample sent out on November 22. The second CSF sample eventually returned with a positive result for WNV RNA by qualitative RT-PCR as well, confirming the diagnosis.
The infectious disease service was consulted and emergency use of WNV-specific intravenous immunoglobulin (Omr-Ig-am®, Omrix Biopharmaceuticals, ETHICON, Inc., Somerville, NJ, USA) IVIG was obtained on a compassionate use basis and administered on November 30, 2007. A repeat serum sample on December 2, 2007 showed a positive WNV IgG of 1.81 IV attributed to the use of WNV-specific IVIG, with a negative IgM at 0.43 IV. However, the patient remained unresponsive and per the patient's previously stated wishes, the family elected to withdraw care on December 4, 2007.
Permission for a limited autopsy was granted, with small samples only permitted from the brain and spinal cord The brain weighted 1205 g and showed no hemorrhages or grossly visible meningeal opacification, although edema and mild bilateral uncal grooving were evident. Except for ventricular narrowing due to the edema, coronal sections were unremarkable. Microscopically almost all sampled sections showed features of meningoencephalomyelitis with mild meningeal and perivascular CD3+ T-cell lymphocytic infiltrates in the cerebrum, but more extensive perivascular lymphocytic cuffing in thalami and spinal cord. Meningeal inflammation was particularly striking in the cerebellum, but also present in the spinal cord (Fig. 2D). In all areas, the meningeal infiltrates were accompanied by a significantly greater number of inflammatory cells within the parenchyma. Virtually every sampled tissue section demonstrated multifocal patches of tissue damage permeated by CD3+ T-cells (CD8+ T-cell suppressor cells greater than CD4+ helper T cells) and CD68+ activated microglia and macrophages. There was an absence of CD20+ B-cell lymphocytes.
Fig. 2.
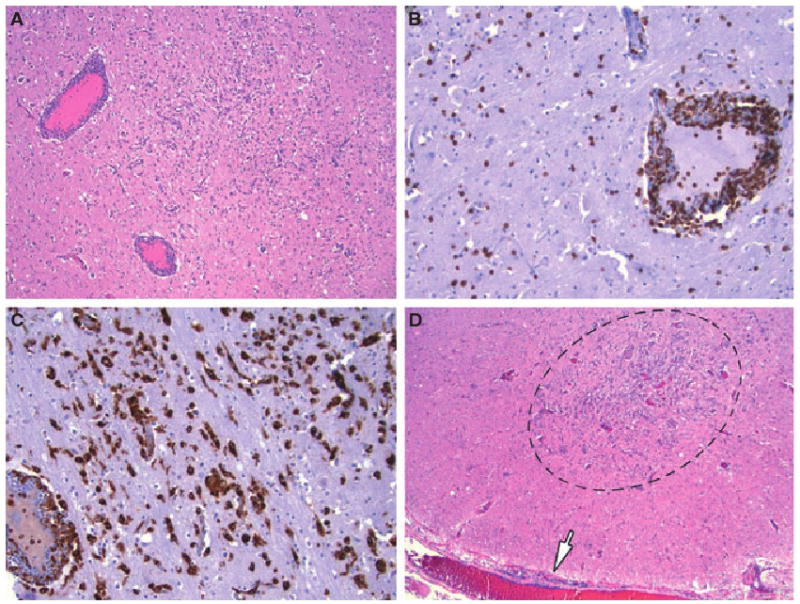
Photomicrograph of the left thalamus showing multifocal small regions of hypercellularity and prominent perivascular cuffing of blood vessels by T-cell lymphocytes and macrophages. (A) Shows these features on hematoxylin and eosin, with CD3 immunohistochemistry highlighting perivascular and parenchymal T-cell lymphocytes in (B) and CD68 immunohistochemistry demonstrating the intense activated microglial and macrophage response to the tissue damage in (C). (A) 40×, (B) 100×, and (C) 100× magnification. The low power photomicrograph in (D) highlights similar damage in the lumbar spinal cord that encompasses the entire cross-sectional area of the anterior horn (encircled); note leptomeningeal lymphocytes (arrow). Hematoxylin and eosin, 20×.
Severe involvement was present in bilateral thalami (Fig. 2A–C), basal ganglia, substantia nigra, red nuclei, hippocampi, anterior horn cell regions of cervical, thoracic, and lumbar spinal cord, proximal anterior motor nerve roots, and cerebellar cortex, especially the molecular layer. The spinal cord damage was the most notable for involvement of the entire cross-sectional area of the anterior horn (Fig. 2D); virtually all anterior horn cell regions examined were affected by the extensive neuronal loss, microglial and macrophage influx, and T-cell inflammation. Myelin stains (Luxol fast blue-periodic acid Schiff) showed myelin breakdown in the proximal anterior motor nerve roots with ovoid digestion chambers and swollen axons on Bielschowsky silver stain; some sensory nerve roots were also mildly inflamed.
Other sites in the nervous sytem with moderately severe damage included the cerebral cortex, brainstem tegmentum, midbrain tectum, inferior olivary nuclei, hypothalamus, and dorsal gray matter of the spinal cord. Cerebral white matter showed multifocal small clusters of macrophages and microglia superimposed on a background of diffuse microgliosis.
Discussion
This case serves to underscore the role of humoral immunity in clearing WNV infection. More specifically, in the solid organ transplant patient population, the combination of anti-rejection immunosuppression directly impairing T-cell function (and B-cell function indirectly) combined with therapy directed against primary humoral immunity (cytoxan and rituximab) may further worsen the ability to clear WNV viremia and meningoencephalitis. This may result in a rapidly fulminant course and even higher mortality than that seen in transplant patients not receiving specific anti-B-cell therapy.
This patient remained serumWNV antibody negative (IgM and IgG) from the onset of her symptoms until she received Omr-Ig-Am®. At that point, the serum WNV IgG became positive. We attribute this lack of antibody production presumably to a combination of T-cell directed immunosuppression (indirectly diminishing the humoral response via lack of B-cell help) and rituximab that directly abrogates the B-cell response. In fact, two additional patients with non-Hodgkin's lymphoma have been reported in the literature with history of rituximab therapy who succumbed to WNV infection with persistently negative serologies (10, 11). Another possibility is that the seroconversion of the serum WNV IgG could have been delayed, as there are a few case reports of delays in seroconversion up to 42 d (12, 13). However, most reported cases of WNV meningoencephalitis in transplant recipients indicate a time range of WNV RNA to IgG seroconversion of 4–14 d (2, 4, 14, 15).
Standard immunosuppression for solid organ transplantation utilizes both calcineurin inhibitors and cytotoxic agents that function to impair T-cell function directly and B-cell function indirectly by inhibiting B-cell help (5, 6). Specifically, calcineurin inhibition acts to block T-lymphocyte clonal proliferation by inhibiting IL-2R (5) and cytotoxic agents act to block de novo nucleoside synthesis of activated T-lymphocytes (6). The anti-CD20 monclonal antibody Rituximab, in contrast, binds to pre-B and mature B lymphocytes (8). Its activity is mediated presumably via direct depletion of pre-B and mature B-cells, therefore impairing the alloantibody response (7), although the exact mechanism has not been elucidated (6, 9). It is believed that rituximab directly depletes the B-cell response via complement and antibody-dependent T-cell-mediated cytotoxicity, but may also have direct antiproliferative effects against B-cells (8). In combination––calcineurin inhibitors, cytotoxics, and rituximab have the potential to severely inhibit both T and B-cell-mediated immunity.
Of note, early kinetic studies with rituximab demonstrated that B-cell immunity did not begin to recover until beyond six months post-treatment (8), which corresponds to the experience with this patient. It is well know in rodents that flavivirus infection is largely controlled by neutralizing antibodies directed against viral glycoprotein E and that these antibodies inhibit viral attachment, internalization, and replication (16). Interestingly, animal data also demonstrate that IgG/IgM humoral response act together with the cellular T-cell response to prevent dissemination and to resolve CNS involvement of WNV (16–19). While T-cell-mediated activity is considered a primary mode of defense against viral infections, the combination of T-cell and B-cell immunodeficiencies may have resulted in the rapid progression of this patient's WNV infection.
The severity of disease seen at necropsy was extraordinary compared to previous cases of WNV neuroinvasive disease in transplant recipients studied at our institution (2). There was particularly striking damage in the anterior horn cell areas of spinal cord bilaterally at all levels sampled and proximal ventral motor nerve roots. Together these findings corresponded to her striking pre-mortem abnormalities on EMG studies. Her very rapid neurological deterioration soon after admission, coupled with her severe obtundation until the time of death, likely are attributable to the severe, bilateral thalamic and hippocampal involvement, possibly in conjunction with her diffuse microgliosis, and macrophage influx in cerebral white matter
This case additionally highlights the importance of obtaining both serum and CSF WNV PCR studies. Measurement of antibody titers is the typical method to diagnose WNV infection, compared to PCR, due to the short duration of WNV viremia and neural tissue avidity of the virus. However, identification of viremia may be the only method of confirming the diagnosis in patients with impaired T- and B-cell immunity. Finally, given the important role of humoral-mediated immunity in limiting hematogenous spread of flavivirus infection and limitation of the extent of neuropathology, one must be cognizant of the potential risk of adding specific anti-B-cell therapy, such as cytoxan and rituximab, in solid organ transplant patients with humoral rejection during the late summer and early fall when the risk of WNV infection is at its highest.
Methods to prevent WNV infection by avoiding mosquito bites, particularly during the highest risk period of late summer and early fall when mosquitos are maximally viremic are critical. Patient education is encouraged in the use of insect repellents and limiting outside activities from dusk to dawn, particularly for immunocompromised individuals.
References
- 1.Kumar D, Drebot MA, Wong SJ, et al. A seroprevalence study of west nile virus infection in solid organ transplant recipients. Am J Transplant. 2004;4:1883. doi: 10.1111/j.1600-6143.2004.00592.x. [DOI] [PubMed] [Google Scholar]
- 2.Kleinschmidt-DeMasters BK, Marder BA, Levi ME, et al. Naturally acquired West Nile virus encephalomyelitis in transplant recipients: clinical, laboratory, diagnostic, and neuropathological features. Arch Neurol. 2004;61:1210. doi: 10.1001/archneur.61.8.1210. [DOI] [PubMed] [Google Scholar]
- 3.Wadei H, Alangaden GJ, Sillix DH, et al. West Nile virus encephalitis: an emerging disease in renal transplant recipients. Clin Transplant. 2004;18:753. doi: 10.1111/j.1399-0012.2004.00283.x. [DOI] [PubMed] [Google Scholar]
- 4.Ravindra KV, Freifeld AG, Kalil AC, et al. West Nile virus-associated encephalitis in recipients of renal and pancreas transplants: case series and literature review. Clin Infect Dis. 2004;38:1257. doi: 10.1086/383325. [DOI] [PubMed] [Google Scholar]
- 5.Vincenti F, Kirkman R, Light S, et al. Interleukin-2-receptor blockade with daclizumab to prevent acute rejection in renal transplantation. Daclizumab Triple Therapy Study Group. N Engl J Med. 1998;338:161. doi: 10.1056/NEJM199801153380304. [DOI] [PubMed] [Google Scholar]
- 6.Sollinger HW, Deierhoi MH, Belzer FO, Diethelm AG, Kauffman RS. RS-61443–a phase I clinical trial and pilot rescue study. Transplantation. 1992;53:428. doi: 10.1097/00007890-199202010-00031. [DOI] [PubMed] [Google Scholar]
- 7.Astor TL, Weill D, Cool C, Teitelbaum I, Schwarz MI, Zamora MR. Pulmonary capillaritis in lung transplant recipients: treatment and effect on allograft function. J Heart Lung Transplant. 2005;24:2091. doi: 10.1016/j.healun.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Maloney DG, Grillo-Lopez AJ, White CA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood. 1997;90:2188. [PubMed] [Google Scholar]
- 9.Nadler LM, Ritz J, Hardy R, Pesando JM, Schlossman SF, Stashenko P. A unique cell surface antigen identifying lymphoid malignancies of B cell origin. J Clin Invest. 1981;67:134. doi: 10.1172/JCI110005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mawhorter SD, Sierk A, Staugaitis SM, et al. Fatal West Nile virus infection after rituximab/fludarabine-induced remission for non-Hodgkin's lymphoma. Clin Lymphoma Myeloma. 2005;6:248. doi: 10.3816/CLM.2005.n.053. [DOI] [PubMed] [Google Scholar]
- 11.Huang C, Slater B, Rudd R, et al. First Isolation of West Nile virus from a patient with encephalitis in the United States. Emerg Infect Dis. 2002;8:1367. doi: 10.3201/eid0812.020532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar D, Prasad GV, Zaltzman J, Levy GA, Humar A. Community-acquired West Nile virus infection in solid-organ transplant recipients. Transplantation. 2004;77:399. doi: 10.1097/01.TP.0000101435.91619.31. [DOI] [PubMed] [Google Scholar]
- 13.Cushing MM, Brat DJ, Mosunjac ML, et al. Fatal West Nile Virus encephalitis in a renal transplant recipient. Am J Clin Pathol. 2004;121:26. doi: 10.1309/G23C-P54D-AR1B-CY8L. [DOI] [PubMed] [Google Scholar]
- 14.Busch MP, Kleinman SH, Tobler LH, et al. Virus and antibody dynamics in acute West Nile Virus infection. J Infect Dis. 2008;198:984. doi: 10.1086/591467. [DOI] [PubMed] [Google Scholar]
- 15.Iwamoto M, Jernigan DB, Guasch A, et al. Transmission of West Nile Virus from an organ donor to four transplant recipients. N Engl J Med. 2003;348:2196. doi: 10.1056/NEJMoa022987. [DOI] [PubMed] [Google Scholar]
- 16.King NJ, Getts DR, Getts MT, Rana S, Shrestha B, Kesson AM. Immunopathology of flavivirus infections. Immunol Cell Biol. 2007;85:33. doi: 10.1038/sj.icb.7100012. [DOI] [PubMed] [Google Scholar]
- 17.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003;77:2578. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med. 2003;198:1853. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyler KL, Mann MA, Fields BN, Virgin HWT. Protective anti-reovirus monoclonal antibodies and their effects on viral pathogenesis. J Virol. 1993;67:3446. doi: 10.1128/jvi.67.6.3446-3453.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]