

Abstract
The beneficial effects of estrogens in multiple sclerosis are thought to be mediated exclusively by the classical nuclear estrogen receptors ERα and ERβ. However, recently many reports revealed that estrogens are able to mediate rapid signals through a G protein-coupled receptor (GPCR), known as GPR30. In the present study, we set out to explore whether effects mediated through this receptor were anti-inflammatory and could account for some of the beneficial effects of estrogen. We demonstrate that GPR30 is expressed in both human and mouse immune cells. Furthermore a GPR30-selective agonist, G-1, previously described by us, inhibits the production of lipopolysaccharide (LPS)-induced cytokines such as TNF-α and IL-6 in a dose-dependent manner in human primary macrophages and in a murine macrophage cell line. These effects are likely mediated solely through the estrogen-specific receptor GPR30 since the agonist G-1 displayed an IC50 far greater than 10 μM on the classical nuclear estrogen receptors as well as a panel of 25 other GPCRs. Finally, we show that the agonist G-1 is able to reduce the severity of disease in both active and passive EAE models of multiple sclerosis in SJL mice and that this effect is concomitant with a G-1-mediated decrease in proinflammatory cytokines, including IFN-γ and IL-17, in immune cells harvested from these mice. The effect of G-1 appears indirect, as the GPR30 agonist did not directly influence IFN-γ or IL-17 production by purified T cells. These data indicate that G-1 may represent a novel therapeutic agent for the treatment of chronic autoimmune, inflammatory diseases.
Keywords: Estrogen, GPCR, GPR30, multiple sclerosis, EAE, cytokine, chemokine
INTRODUCTION
There is an extensive literature centered on the beneficial effects of estrogens in multiple sclerosis. Overall, these studies support a role for estrogens as potent anti-inflammatory and neuroprotective agents. For example evidence from several studies in humans and animals (Abramsky, 1994; Damek and Shuster, 1997; Polanczyk et al., 2003; van Walderveen et al., 1994) has suggested that there is a reduction in the relapse rate of multiple sclerosis and in an experimental allergic encephalomyelitis (EAE) animal model during pregnancy. These initial observations were subsequently confirmed in the large Pregnancy In Multiple Sclerosis (PRIMS) study that demonstrated a 70% reduction in relapse rate during late pregnancy (Confavreux et al., 1998).
These and other studies performed by a number of different academic groups (Arnason and Richman, 1969; Jansson et al., 1994; Subramanian et al., 2003) have demonstrated independently and consistently that estrogen is a potent immunomodulator. There is some evidence that other steroids such as progesterone and testosterone also display immunomodulatory effects presumably through their cognate receptors (Shuster, 2008); however, the effects of estrogen have been far more extensively described. Nevertheless, both neurological and pathological findings have supported the notion that estrogens are potent anti-inflammatory as well as neuroprotective agents with a potential to serve as novel therapeutics for the treatment of autoimmune demyelination. In a disease such as multiple sclerosis, where the inflammatory process induces demyelination followed by secondary axonal damage and subsequent neurological disability, the potential neuroprotective therapeutic effect of estrogens can be considered an additional benefit to their anti-inflammatory properties.
Until now the beneficial effects of estrogens in multiple sclerosis were thought to be exclusively mediated by the classical nuclear estrogen receptors ERα (and possibly ERβ (Garidou et al., 2004; Polanczyk et al., 2003; Tiwari-Woodruff et al., 2007). However, in 2005 two reports demonstrated that estrogen is able to mediate rapid signals through the G protein-coupled receptor (GPCR) known as GPR30 (Revankar et al., 2005; Thomas et al., 2005). Together these studies revealed that GPR30 is selective for the physiologically active 17β isomer of estradiol as compared to the 17α isomer and furthermore does not bind other steroids such as progesterone, testosterone or cortisol. The discovery of an estrogen-responsive GPCR opened the possibility that such a receptor might play a beneficial role in multiple sclerosis and the present study set out to explore this notion. Here we show that GPR30 is expressed in both human and mouse immune cells. Furthermore a GPR30 specific agonist, previously described by us (Bologa et al., 2006), inhibits the production of LPS-induced cytokines such as TNF-α and IL-6 in a dose-dependent manner in human primary macrophages and in a murine macrophage cell line. These effects appear to be mediated through the estrogen-specific receptor GPR30 since the agonist G-1 displayed an IC50 far greater than 10 μM for the classical nuclear estrogen receptors as well as a panel of 25 other GPCRs. Finally, we show that the agonist G-1 is able to reduce the severity of disease in both active and passive EAE models of multiple sclerosis in SJL mice.
EXPERIMENTAL PROCEDURES
Materials
Estrogen and all laboratory chemicals were from Sigma unless otherwise noted. G-1 (1-[4-(6-bromobenzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone), the GPR30 specific agonist, was synthesized as previously described (Bologa et al., 2006).
Cell culture
RAW264.7 cells were cultured in DMEM supplemented with 10% FBS and 2 mM L-glutamine (no antibiotic) and plated in 24 well plates at a cell density of 2 × 105 cells/well. HL-60 cells were cultured in RPMI supplemented with 10% fetal bovine serum, 2mM L-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin and 10mM HEPES (pH 7.4). All cells were maintained at 37°C in a humidified incubator with 5% CO2.
Isolation of monocytes and macrophages
Peripheral blood mononuclear cells were isolated from human buffy coats by Ficoll gradient; CD14+ monocytes were positively selected using Miltenyi beads. The isolated monocytes were resuspended at approximately 1.5×106/mL with Invitrogen X-VIVO15 medium. GM-CSF (R&D Systems) was added at a concentration of 25 ng/mL. Cells were allowed to differentiate in plastic flasks for 6 days. At day 6, cells were washed with Hanks saline, and dissociated with dissociation buffer twice. Dissociated cells were collected, spun-down, resuspended in medium, and plated onto slides or plates. In general, 1×108 monocytes yielded about 2×107 macrophages. Mouse peritoneal macrophages were obtained after injection of mice with 5% thioglycollate broth as previously described (Correa et al., 2005).
Isolation and culture of microglia
Rat microglia were isolated from the brains of 2 day old rats. Briefly, after removal of meninges, brains were disaggregated in a 10 mL pipette to obtain a single cell suspension. The cell suspension was passed through 70 μm and 100 μm sieves and centrifuged at 800 rpm for 8 minutes. The pellet was resuspended in 16 mL of growth medium (DMEM/F12 supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, 250 ng/mL amphotericin B/mL, 10% fetal calf serum, 14 mM HEPES, 2 mM additional L-glutamine, 1 mM sodium pyruvate, 1 mM non-essential amino acids at pH 7.4) prewarmed to 37°C in a 75 mL culture flask. After 5 days of incubation. microglia, presenting as vacuolated cells that were loosely attached to the flask, were gently removed by swirling the flask in a circular motion 3 to 6 times. Microglia were harvested from the conditioned medium by centrifugation at 1200 rpm for 8 minutes, and then resuspended in growth medium for plating in 8-well chamber slides.
Immunostaining experiments
Human primary macrophages, RAW264.7 and rat primary microglia were plated at about 10,000 cells/well in 8-well chamber slides and fixed for 10 min at room temperature with 4% paraformaldeyde in 0.1 M sodium phosphate and 3% sucrose, pH 7.3. The cells were washed in Tris- buffered saline and incubated in blocking solution, 5% normal goat serum (Zymed) in PBS + 0.05% Tween 20, for 10 minutes at room temperature. After removing the blocking solution, slides were incubated with primary antibodies (3.2 μg/mL) diluted in blocking solution for 2 hours at room temperature. Anti-GPR30 antibody was raised in rabbits against a C-terminal peptide of GPR30, CAVIPDSTEQSDVRFSSAV (cysteine residue added for KLH conjugation) and affinity purified (Revankar et al., 2005). Purified rabbit immunoglobulin G (IgG) (Zymed) was used as a control. The slides were washed four times in Tris buffered saline and 100 μL of biotinylated secondary antibody (goat anti-rabbit) was added per well and incubated for 30 min at room temperature. The slides were washed with Tris-buffered saline and incubated with 100 μL of streptavidin-HRP conjugate for 30 min at room temperature. The slides were washed with Tris-buffered saline and incubated with 100 μL of 3,3′-Diaminobenzidine (DAB) per well for approximately 5 minutes. The slides were washed with distilled water and counter-stained with Mayer’s modified hematoxylin. Slides were washed several times in distilled water, dipped for 30 seconds in Lerner’s bluing reagent and washed several times in distilled water before being dehydrated in 95% ethanol and then in xylene. For immunofluorescence staining of HL60 cells, cells were fixed for 15 min in 2% paraformaldehyde (PFA), washed once in phosphate buffered saline (PBS) and resuspended in 20% Bovine serum albumin (BSA) in PBS. Cells (40,000) were spun onto poly-L-lysine-coated slides using a Shandon Cytospin 4 (Thermo Scientific) at 500 rpm for 8 min. Cells were blocked and permeabilized for 30 min using 3% BSA/0.1% Triton X-100 in PBS. Cells were stained with affinity purified rabbit polyclonal antibody directed against a GPR30 C-terminal peptide (or purified IgG as control) diluted in 3% normal goat serum (NGS) for 4 h at room temperature, washed three times with PBS and stained with Alexa568-conjugated anti-rabbit secondary antibody diluted in 3% NGS. Samples were mounted with Vectashield containing DAPI. Confocal images were collected using a Zeiss LSM 510 confocal microscope.
Inhibition of cytokine production by human macrophages
Primary human macrophages (3 × 105 cells) isolated as previously described were plated in 12 well plates. The cells were treated with increasing amounts of the GPR30-specific agonist G-1 or estrogen for 1 h followed by treatment with either 10 μg/mL poly I:C or LPS (100 ng/mL) overnight. At the end of the incubation the cellular supernatants were assayed for a variety of cytokines and chemokines by Luminex assay or by ELISA according to the manufacturer’s instructions.
Specificity of binding of the GPR30 agonist G-1 to GPCRs and nuclear receptors
G-1 at a concentration of 10 μM was tested in receptor binding assays on a number of GPCRs. All receptors were expressed in HEK293 cells except, β2-adrenergic receptor in NBR1 cells, bradykinin in HS729 cells, endothelin in CHO cells and muscarinic M1 and M2 in Sf9 insect cells. Binding assays were carried out according to the protocols of MDS Pharma Services.
Intracellular calcium mobilization
HL-60 cells (1 × 107/mL) were incubated in HBSS containing 5 μM indo1-AM and 0.05% pluronic acid for 30 min at room temperature. Cells were then washed three times with HBSS and resuspended in HBSS at a density of 108 cells/mL and incubated on ice until assay. For each sample, 2 × 106 cells were resuspended in 1mL HBSS containing 20mM HEPES, 1mM CaCl2, 1mM MgCl2, pH 7.4. Ca2+ mobilization was determined ratiometrically using λex 340 nm and λem 400/490 nm at 37°C in a spectrofluorometer (QM-2000-2, Photon Technology International) equipped with a magnetic stirrer.
Adoptively transferred experimental autoimmune encephalomyelitis in SJL mice
SJL mice (5–7 weeks old; Harlan, Indianapolis, IN) were immunized s.c. with 50 μg Proteolipid protein (PLP)139-151 and complete Freund’s Adjuvant (CFA) (400 μg Mycobacterium tuberculosis). Draining lymph node cells were harvested after 7 days and re-stimulated with 50 μg/mL PLP139-151 in the presence of 3 μM G-1 agonist or control DMEM medium with 5% FCS and 0.1% DMSO. Antigen-activated T cells were harvested after 72 hours of culture and 6×106 blasts were transferred i.v. to naïve SJL recipient mice. Clinical disease was scored as 0, asymptomatic; 1, tail atonia; 2, hind limb paresis; 3, unilateral hind limb paralysis; 4, bilateral hind limb paralysis; and 5, death (Miller and Karpus, 2007). All animal procedures were performed in accordance with USPHS policy and Northwestern University animal care and use committee approval.
Actively induced experimental autoimmune encephalomyelitis in SJL mice
SJL mice (5–7 weeks old) were immunized s.c. with 50 μg PLP139-151 and CFA (400 μg Mycobacterium tuberculosis). Mice were treated with 50 mg/kg/day G-1 daily for 21 days beginning at the day of disease induction (N=10). Control mice (N=7) were similarly treated with vehicle (5% Dimethyl sulfoxide (DMSO), 95% Polyethylene glycol (PEG)-300.
Inflammatory cytokine expression in lymph node cells from SJL mice
SJL mice (5–7 weeks old) were immunized s.c. with 50 μg PLP139-151 and CFA (400 μg Mycobacterium tuberculosis). Draining lymph node cells were harvested after 7 days and re-stimulated with 50 μg/mL PLP139-151 in the presence of 3μM G-1 agonist or control DMEM medium with 5% Fetal calf serum (FCS) and 0.1% DMSO. Culture supernatants were harvested after 48 h and assessed for the presence of cytokines using Beadlyte Mouse Multi-cytokine Flex Kit assay per manufacturer’s instructions (Upstate Cell Signaling Solutions, Temecula, CA).
G-1 effects on macrophage accumulation in SJL mice
SJL mice (5–7 weeks old) were immunized s.c. with 50 μg PLP139-151 and CFA (400 μg Mycobacterium tuberculosis). Draining lymph node cells were harvested after 7 days and re-stimulated with 50 μg/mL PLP139-151 in the presence of 3 μM G-1 agonist or control DMEM medium with 5% FCS and 0.1% DMSO. Antigen-activated T cells were harvested after 72 hours of culture and 6 × 106 blasts were transferred i.v. to naïve SJL recipient mice. When the control mice showed peak clinical disease, the central nervous system (CNS) was harvested from three representative mice in each of the control and G-1-treated groups. Leukocyte subpopulations were examined by flow cytometry.
Statistical Analysis
Sample median, mean, standard deviation, and statistical significance were calculated using SPSS 13.0 software. Comparisons of clinical median disease severity over the course of time were made and statistical significance was calculated using the Mann-Whitney test for ordinal data (Fleming et al., 2005). Single comparisons of two means were analyzed by Student’s t test. Comparisons of percent affected mice were performed using a χ2 test. P values < 0.05 were considered significant.
RESULTS
Although little is known regarding the expression of GPR30 within the immune system, preliminary studies in our laboratory to evaluate GPR30 expression by TaqMan demonstrated that the mRNA for GPR30 is expressed in human macrophages, although with some apparent donor-to-donor variability (data not shown). Based on these data, we studied the expression of the protein on a variety of cells using an anti-GPR30 antibody that we have previously described (Revankar et al., 2005). The antibody was raised against a C-terminal peptide of GPR30, which differs in human rat and mouse GPR30 sequences by only one conserved amino acid residue and thus cross-reacts with rodent receptors. Both human primary macrophages and a mouse macrophage cell line RAW264.7 showed reticular cytoplasmic expression of GPR30 (Figures 1A and 1B). In additional work (not shown), human dendritic cells also stained positive. Demonstrating cross-reactivity with the rat, and expression in a CNS cell lineage, the GPR30 antibodies stained positive in rat microglia (Figure 1C). In addition there was strong expression of GPR30 in human regulatory T cells (Figure 1D). Staining of the immune cells with the GPR30 antibody was specific since it could be blocked by pre-incubation with the C-terminal peptide of GPR30 that the antibody was raised against but not with a scrambled version of the same peptide (Figure 1D).
Figure 1.
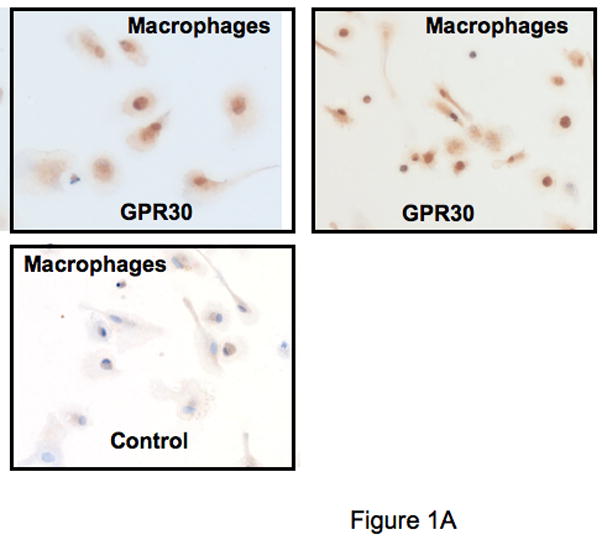
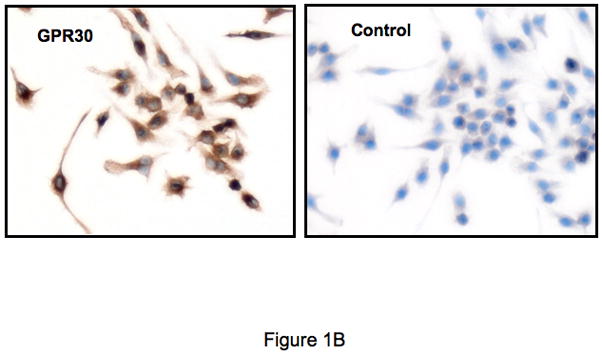
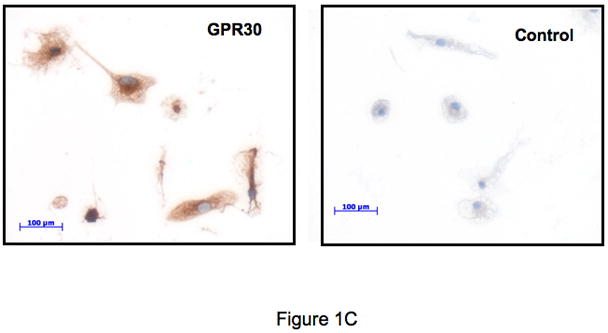
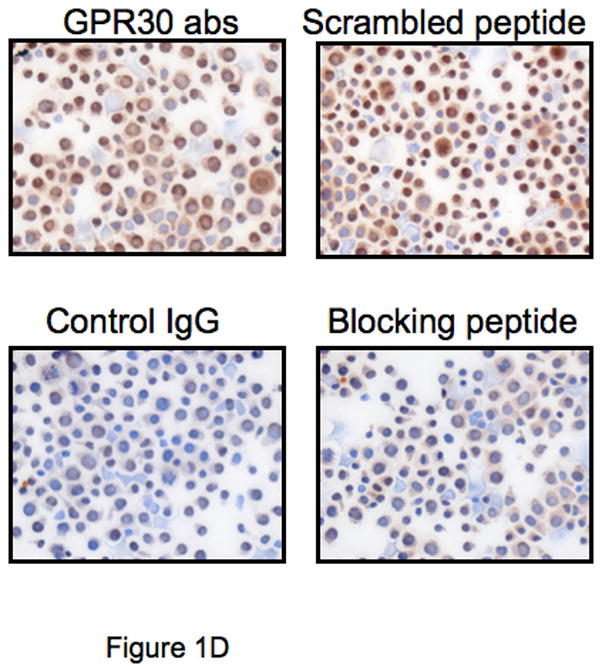
GPR30 expression by immunohistochemical staining (brown) in primary immune cells and cell lines. (A) Human macrophages, (B) Mouse RAW 264.7 cells, (C) Rat microglia and (D) Human regulatory T cells all stain with an antibody against GPR30. Staining is primarily cytoplasmic in the mouse and rat cells, while in the human macrophages and T cells it is both cytoplasmic and nuclear. There is little to no staining with a control antibody. In addition, staining is abolished by preincubation with a C-terminal peptide to GPR30 but not with a scrambled version of the same peptide (D). Nuclei are lightly counterstained with hematoxylin. All images taken with a 63- oil objective on a Zeiss Axioskop. Scale bars = 100 μm in D.
Previous studies to assign the cellular effects of estrogen to a specific receptor have been hampered by the lack of receptor-specific ligands. However, we have previously described a small molecule agonist of GPR30, G-1, which does not bind or activate the classical nuclear receptors but competes with estradiol for binding to GPR30 and also activates calcium transients in cells expressing GPR30 but not in cells expressing only nuclear estrogen receptors (Bologa et al., 2006). Furthermore, the agonist is also active on recombinant murine GPR30 (data not shown). To confirm the specificity of G-1 compound and extend these data, we tested G-1 in a receptor-binding screening panel against 25 known GPCRs together with the nuclear steroid receptors ERα and ERβ. At a concentration of 10 μM, the G-1 agonist showed no substantial binding to any of the eceptors tested (Table 1) and thus constituted a highly selective probe with which to examine GPR30 function in immune cells.
Table 1.
Specificity of G-1 binding. G-1 was tested in receptor binding assays on a number of receptors. All receptors were expressed in HEK293 cells except, β2-adrenergic receptor in NBR1 cells, bradykinin in HS729 cells, endothelin in CHO cells and muscarinic M1 and M2 in Sf9 insect cells. AKi of G-1 on test receptor/Ki G-1 on GPR30 (10 nM from Bologa et al., 2006).
| Receptor | Ligand | Tissue | % Inhibition of Binding |
|
|---|---|---|---|---|
| 10 μM | SelectivityA | |||
| Adenosine A1 | [3H]DPCPX | Human | 11 | >1000 |
| Adrenergic α1D | [3H]Prazosin | Human | −3 | >1000 |
| Adrenergic β1 | [125I]-cyanopindolol | Human | 5 | >1000 |
| Angiotensin AT1 | [125I]-Angiotensin II | Human | 32 | >1000 |
| Bradykinin B1 | [3H]des Arg10 Kallidin | Human | 8 | >1000 |
| Cannabinoid CB1 | [3H]CP-55,940 | Human | 24 | >1000 |
| Cannabinoid CB2 | [3H]WIN-55,212-2 | Human | 28 | >1000 |
| Chemokine CCR1 | [125I]-CCL3 | Human | −4 | >1000 |
| Chemokine CCR5 | [125I]-CCL4 | Human | 1 | >1000 |
| Dopamine D1 | [3H]SCH-23390 | Human | −9 | >1000 |
| Dopamine D2L | [3H]Spiperone | Human | 0 | >1000 |
| Dopamine D3 | [3H]Spiperone | Human | 21 | >1000 |
| Endothelin ETA | [125I]Endothelin-1 | Human | −1 | >1000 |
| Estrogen ERα | [3H] Estradiol | Human | 25 | >1000 |
| Estrogen ERβ | [3H] Estradiol | Human | −2 | >1000 |
| Histamine H1 | [3H] Pyrilamine | Human | 24 | >1000 |
| Leukotriene CysLT1 | [3H]LTD4 | Human | 5 | >1000 |
| Muscarinic M1 | [3H]NMS | Human | 13 | >1000 |
| Muscarinic M2 | [3H]NMS | Human | −13 | >1000 |
| Muscarinic M3 | [3H]NMS | Human | −4 | >1000 |
| Neuropeptide Y1 | [125I]Peptide YY | Human | −1 | >1000 |
| Neurotensin NT1 | [125I]Neurotensin | Human | −5 | >1000 |
| Opiate σ(OP1) | [3H]Natrindole | Human | 15 | >1000 |
| Serotonin 5-HT1A | [3H]8-OH-DPAT | Human | 12 | >1000 |
| Serotonin 5-HT15A | [3H]LSD | Human | 23 | >1000 |
| Somatostatin sst1 | [125I]Somatostatin-14 | Human | 43 | >1000 |
| Tachykinin NK1 | [3H]SR-140333 | Human | 11 | >1000 |
Since we have demonstrated the expression of GPR30 in immune cells we wanted to know whether these cells would respond to stimulation with the GPR30 agonist G1. Since these experiments are not trivial to carry out in primary cells we first used a promyelocytic cell line, HL-60, to ask whether G-1 could even elicit second messengers from before going on to the macrophages to examine cytokines. of GPR30 in a model human promyelocytic cell line, HL60. Staining with anti-GPR30 antibodies revealed a narrow region of cytoplasmic staining, consistent with localization in the endoplasmic reticulum (Figure 2A). Staining was absent when using purified IgG as a control. Subsequent analysis of intracellular calcium mobilization in response to estrogen and the GPR30-selective agonist G-1 revealed that both ligands mediated a calcium flux with EC50 values of 180 nM and 600 nM, respectively (Figure 2B and C). Together, these results indicate that promyelocytic immune cells express GPR30 and are capable mediating second messenger production in response to estrogen and G-1.
Figure 2.
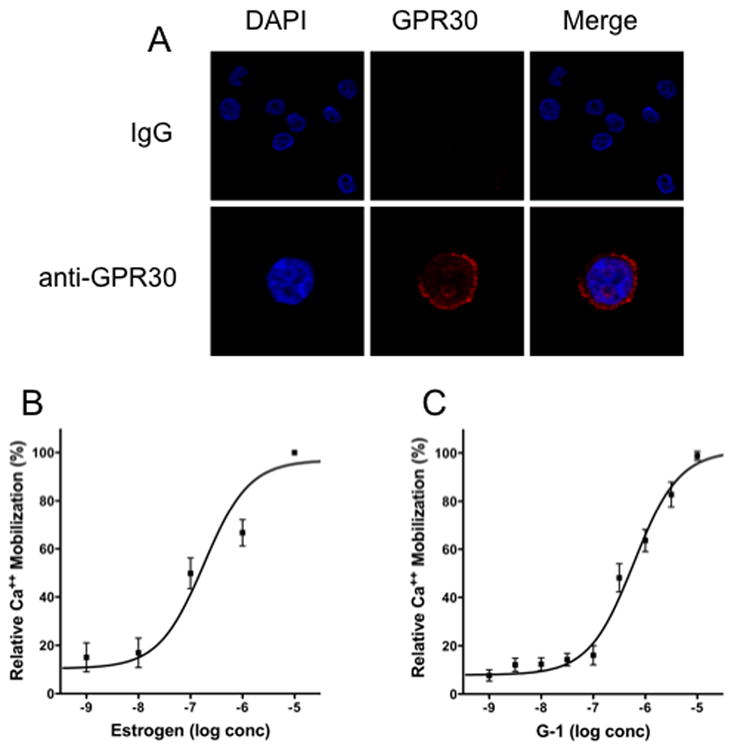
Expression and function of GPR30 in human promyelocytic HL60 cells. (A) HL60 cells were cytospun onto coated slides and stained with either IgG or GPR30 immune serum (red) and nuclei were counterstained with DAPI (blue). Calcium mobilization in indo-1-loaded HL60 cells was determined in response to estrogen (B) and the GPR30 agonist G-1 (C).
Given the expression of GPR30 on primary human macrophages and the function of GPR30 in a model promyelocytic cell line, we examined whether the receptor was functional in primary human macrophages. It is well known that macrophages respond to a poly IC challenge by secreting TNF-α and IL-6 (Hayes et al., 1991). Thus, we generated primary human macrophages derived from monocytes treated with GM-CSF over 6 days. Cells from two donors were treated with 100 nM G-1 for 1 h followed by, poly I:C for 18 h. At the end of the incubation we measured cytokine secretions in the cell supernatants using a multiplexed Luminex assay (Table 2). Both donors showed similar trends of cytokine inhibition by the GPR30 agonist. Following these preliminary experiments, we stimulated human macrophages with LPS and measured the ability of increasing doses of the G-1 agonist to inhibit TNF-α and IL-6 release. The agonist inhibited the induction of both cytokines with IC50 values of 209 nM and 317 nM, respectively (Figure 3). In addition to the inhibition of TNF-α and IL-6, G-1 was also able to inhibit LPS-induction of both IL-12 (p40) and the CCR1 ligand CCL5 (RANTES) but not the CCR1 ligand CCL3 (MIP-1α), thus revealing that the inhibition was specific and not simply due to non-specific or toxic effects on the cells (Table 3). G-1 was shown to be non-toxic at concentrations up to 10 μM against macrophages in an assay using the metabolic agent Wst-1 (data not shown). G-1 was also able to inhibit LPS induction of TNF-α in a mouse macrophage cell line, RAW 264.7 (Figure 4). These data confirmed that G-1 displayed agonist activity on murine GPR30 and opened the possibility to carry out experiments in animal models of disease, such as multiple sclerosis, most of which are in rodents.
Table 2.
The GPR30 agonist G-1 inhibits cytokine production by poly I:C stimulated macrophages (% inhibition is calculated based on dynamic range of the assay). Data shown are from two separate donors and represent a mean value +/− S.D. of triplicate points:; the table is representative of two separate experiments.
| TNF-α (pg/mL) | IL-6 (pg/mL) | |||
|---|---|---|---|---|
| Donor 1 | Donor 2 | Donor 1 | Donor 2 | |
| untreated | 8 +/− 1 | 5+/− 1 | 7+/− 0 | 7+/− 0 |
| I:C | 47 +/− 2 | 482 +/− 64 | 26 +/− 1 | 455 +/− 12 |
| I:C, 100 nM G-1 | 28 +/− 2 | 176 +/− 19 | 18 +/− 2 | 206 +/− 22 |
| % inhibition | 49 | 64 | 42 | 56 |
Figure 3.


Agonist properties of the GPR30 agonist G-1 on human primary macrophages. G-1 inhibits LPS-induced production of TNF-α (A) and IL-6 (B) in human macrophages in a dose-dependent manner. Data values are mean +/− S.D. of triplicate points within the same experiment; the data are representative of three separate experiments
Table 3.
The GPR30 agonist G-1 inhibits LPS-induced IL-12(p40) and CCL5 but not CCL3 from primary human macrophages. (Levels of untreated cytokines are normalized as 1, and levels induced by LPS are expressed as fold induction relative to untreated). Data are mean +/− S.D. of triplicate points within the same experiment; the table is representative of two separate experiments.
| IL-12 (p40) | CCL5 | CCL3 | |
|---|---|---|---|
| LPS (fold induction) | 3.67 +/− 0.11 | 4.78 +/− 1.56 | 7.18 +/− 0.11 |
| LPS + I μM G-1 (fold induction) | 2.22 +/− 0.27 | 1.78 +/− 0.33 | 7.29 +/− 0.31 |
| % inhibition by G-1 | 39% | 62% | 0% |
Figure 4.
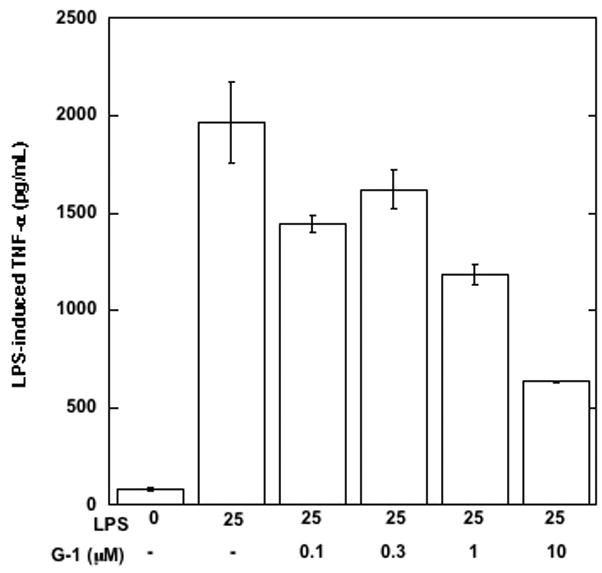
Agonist properties of the GPR30 agonist G-1 on the mouse macrophage cell line RAW 264.7 G-1 inhibits LPS-induced TNF-α in RAW 264.7 cells in a dose-dependent manner. Data values are mean +/− S.D. of triplicate points within the same experiment; the data are representative of four separate experiments
Previous studies by Fillmore et al. (Fillmore et al., 2004) demonstrated that ovariectomy resulted in enhanced clinical EAE. Moreover, the effects of estrogen on EAE have been well documented and have been largely explored using agonists and antagonists of the classical estrogen receptors (Elloso et al., 2005; Matejuk et al., 2001; Morales et al., 2006). To determine whether stimulation of the non-classical estrogen receptor GPR30 affected the development of EAE, an animal model for multiple sclerosis (Miller and Karpus, 2007), we immunized SJL mice with PLP, administered G-1 or vehicle control for 21 days and assessed the development of clinical signs of disease. The results in Figure 5A demonstrate that G-1 administration significantly reduced the severity of actively induced EAE (p<0.05, Mann-Whitney test) but not the incidence of disease. Additionally, when representative mice from the experiment were examined for histologic evidence of disease, we noted G-1 treatment reduced the qualitative degree of inflammation in the lumbar spinal cord (Figure 5B). Control- and G-1-treated mice from this experiment were also assessed for quantitative central nervous system (CNS) mononuclear cell infiltration using flow cytometry. Spinal cords were harvested and mononuclear cells isolated using a discontinuous Percoll gradient. The cells were stained with antibodies specific for CD45, CD11b, CD4 and CD8. The results shown in Figure 6 demonstrate that G-1 treatment reduced the fraction of CNS-infiltrating macrophages (CD45hiCD11b+) in three individually analyzed mice compared to three individually analyzed vehicle control-treated mice. Moreover, the mean percentage±SD of macrophages in the CNS of G-1-treated group was 11.84±1.75 which was significantly different (p<0.05, Student’s t test) compared to 21.54±7.01 in the control-treated mice. Collectively these data indicate that G-1 significantly inhibited clinical and histological EAE as well as the CNS accumulation of macrophages. Although there was a slight increase in CD4+ T cells in the CNS of G-1-treated mice, there was no increase in CD4+CD25+FoxP3+ T cells (data not shown) indicating that disease inhibition was not likely a result of G-1-mediated increases in regulatory T cells.
Figure 5.

GPR30 agonist treatment reduced the severity of actively induced experimental autoimmune encephalomyelitis. A) SJL mice (5–7 weeks old) were immunized s.c. with 50 μg PLP139-151 and CFA (400 μg Mycobacterium tuberculosis). Mice were treated with 50 mg/kg/day G-1 daily for 21 days beginning at the day of disease induction (Rx). Control mice were similarly treated with vehicle (5% DMSO, 95% PEG-300). Clinical disease was scored as 0, asymptomatic; 1, tail atonia; 2, hind limb paresis; 2, unilateral hind limb paralysis; 4, bilateral hind limb paralysis; and 5, death. The data are shown as the median clinical disease score as a function of days post immunization. Disease incidence for each treatment group is indicated in parentheses. The G-1-treated group showed significantly decreased acute clinical disease score, but not incidence, compared to control mice (*, p<0.05). B) The CNS of representative mice from each treatment group was evaluated for histologic EAE by standard H&E staining. The photomicrographs (100x magnification) indicate less severe mononuclear cell lesions in the G-1- compared to control-treated mice (heavy black arrows). These data are representative of three similar, independent experimental replicates.
Figure 6.

GPR30 agonist treatment reduced CNS macrophage accumulation. SJL mice (5–7 weeks old) were immunized s.c. with 50 μg PLP139-151 and CFA (400 μg Mycobacterium tuberculosis). Mice were treated with 50 mg/kg/day G-1 daily for 21 days beginning at the day of disease induction. Control mice were similarly treated with vehicle (5% DMSO, 95% PEG-300). When the control mice showed peak clinical disease (Figure 5), CNS was harvested from three representative mice in each of the control (disease scores=3, 3, 2) and G-1-treated (disease scores=1, 1, 1) groups. Leukocyte subpopulations were examined in each individual mouse by flow cytometry. The data show a decrease in CD45hiCD11b+ macrophage percentages in the CNS of G-1-treated mice. The data are representative of two similar, independent experimental replicates.
To further understand the mechanism of G-1-mediated disease inhibition, we analyzed cytokine expression patterns from autoantigen-stimulated lymphocyte cultures. Splenocytes were harvested from control- or G-1-treated mice and re-stimulated with PLP139-151 in vitro. Supernatants were harvested 48 hours after culture initiation and tested for inflammatory cytokine content using a bead-based assay. The results shown in Figure 7 demonstrate a significant decrease in antigen-specific IFN-γ, TNF, IL-17, CCL4, and CCL5 from G-1-treated lymphocytes. These data suggest that G-1 treatment either directly or indirectly affects the antigen-specific T cell inflammatory cytokine expression pattern, including IL-17 that is necessary for EAE development (Komiyama et al., 2006).
Figure 7.
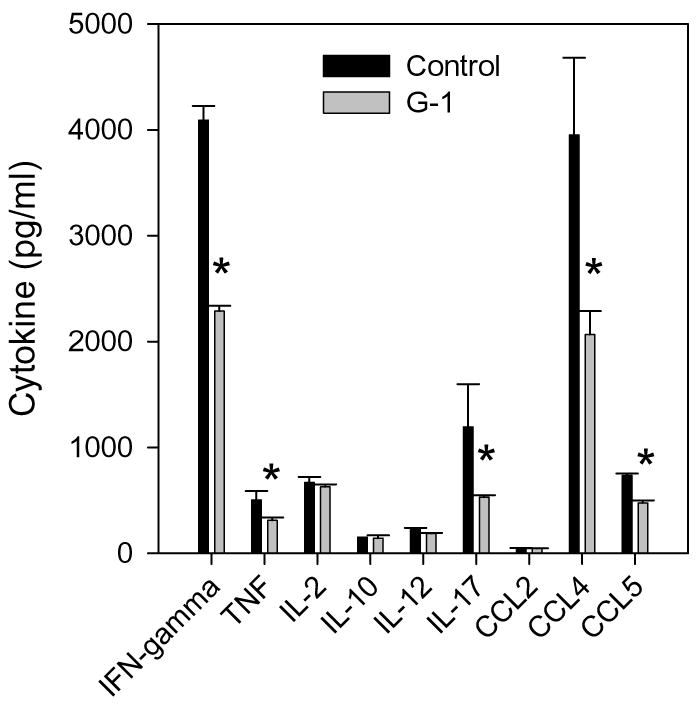
GPR30 agonist treatment inhibits inflammatory cytokine expression. Splenocytes from the individual mice analyzed in Figure 6 were re-stimulated with 50 μg/ml PLP139-151 peptide in vitro. Culture supernatants were harvested after 48 h and assessed for the presence of cytokines using Beadlyte Mouse Multi-cytokine Flex Kit assay. The data are shown as mean cytokine production (pg/ml from three individual mice ±SD) in cultures from G-1-treated mice compared to cells from control-treated mice. The results indicate significant (*, p<0.05) reductions in IFN-γ, TNF, IL-12, IL-17, CCL4, and CCL5 expression. The data are representative of two similar, independent experimental replicates.
To determine whether G-1 acted directly on disease-inducing T cells, we performed an experiment where SJL donor mice were primed with PLP139-151 in CFA, harvested the draining lymph node cells, and cultured the lymphocytes in the presence of antigen plus 3 μM G-1 or vehicle control. The lymphocytes were harvested after 3 days and adoptively transferred to naïve SJL recipient mice that were observed for the development of EAE. The results shown in Figure 8A demonstrate that recipients of G-1-treated, PLP139-151-specific T cells developed significantly less clinical disease than the recipients of control-treated cells. Lymphocytes were harvested from the CNS of representative recipient mice using a discontinuous Percoll gradient, re-stimulated with specific antigen in the presence of irradiated splenic APC, and the resulting culture supernatants were assessed for IFN-γ and IL-17 by ELISA. The results in Figure 8B indicate that antigen-specific T cells recovered from the CNS of mice that received G-1-treated lymphocytes produced less IFN-γ and IL-17 than those recovered from recipients of control-treated cells. Since GPR30 is expressed by T cells (Wang et al., 2008) and antigen presenting cells (present study) these data suggest that G-1 treatment could either directly or indirectly affect effector T cell cytokine production.
Figure 8.
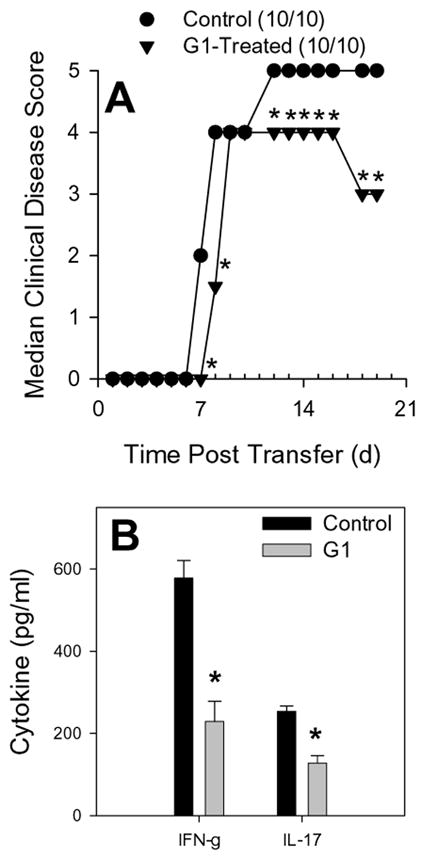
GPR30 agonist treatment reduced severity of adoptively transferred experimental autoimmune encephalomyelitis. SJL mice (5–7 weeks old) were immunized s.c. with 50 μg PLP139-151 and CFA (400 μg Mycobacterium tuberculosis). Draining lymph node cells were harvested after 7 days and re-stimulated with 50 μg/mL PLP139-151 in the presence of 3 μM G-1 agonist or control DMEM medium with 5% FCS and 0.1% DMSO. Antigen-activated T cells were harvested after 72 hours of culture and 6×106 blasts were transferred i.v. to naïve SJL recipient mice. Clinical disease was scored as 0, asymptomatic; 1, tail atonia; 2, hind limb paresis; 2, unilateral hind limb paralysis; 4, bilateral hind limb paralysis; and 5, death. A) The data are shown as the median clinical disease score as a function of days post lymphocyte transfer. The group that received G-1-treated cells showed significantly decreased clinical disease compared to recipients of control-treated cells (Mann-Whitney *, p<0.05). B) Lymphocytes were harvested from the CNS of representative recipient mice, restimulated with specific antigen and irradiated splenic APC, and the resulting culture supernatants were assessed for IFN-γ and IL-17 by ELISA. The results indicate that antigen-specific T cells recovered from the CNS of mice that received G-1-treated lymphocytes produced significantly less (*, p<0.05) IFN-γ and IL-17. The results are representative of two independent replicate experiments.
Since the GPR30 agonist G-1 appeared to regulate CNS macrophage accumulation (Figure 6), we investigated the CNS for the presence of CCL2, an important chemokine for macrophage accumulation and disease induction, in EAE (Kennedy et al., 1998). An experiment similar to the one described in Figure 5A was performed. Spinal cords from control- and G-1-treated mice were assessed by ELISA for the presence of CCL2 (Kennedy et al., 1998). The results in Figure 9A demonstrate that the CNS of G-1-treated mice contained significantly less CCL2 than control-treated mice. Both glia- and infiltrating macrophage-derived CCL2 has been shown to be important for development of EAE (Dogan et al., 2008). Therefore, it was possible that G-1 directly down-regulated CCL2 production by these cell types resulting in decreased CNS macrophage accumulation (Huang et al., 2001). Alternatively, it was possible that G-1 treatment directly affected T cell cytokine expression, which in turn resulted in diminished stimulation of glia to produce CCL2. In an effort to understand why G-1 treatment affected T cell cytokine expression, we assessed the same CNS material for the presence of IL-23 by ELISA. IL-23 has been shown to be a critical maintenance factor for pathogenic Th17 cells in EAE (Langrish et al., 2005). The results shown in Figure 9B indicate a significant decrease in IL-23 expression in the CNS of G-1-treated mice, suggesting that G-1 also induced a down-regulation of accessory cell-derived cytokines necessary to maintain the pathogenic IL-17 T cell phenotype. Although G-1 treatment affected EAE induced by the adoptive transfer of T cells, the majority of the data suggested that G-1 affects macrophages and other antigen presenting cells. In order to determine if G-1 had a direct effect on T cells, purified normal CD4+ T cells were stimulated in vitro with immobilized anti-CD3 and anti-CD28 in the presence or absence of 10 μM G-1 under normal and polarizing conditions. Polarizing conditions included IL-12 and anti-IL-4 for Th1 (IFN-γ) (Karpus et al., 1997) and TGF-β, IL-6 and IL-23 for Th17 (IL-17) (McGeachy et al., 2007). The results shown in Figure 9C indicate that G-1 treatment did not affect IFN-γ or IL-17 production under normal or polarizing conditions. Collectively, the data support the idea that G-1 down-regulated both active and adoptively induced EAE by affecting macrophages and other accessory cells (including dendritic cells and glia) and not by directly affecting the ability of T cells to either differentiate into effector cells or produce cytokines that are known to be involved in disease pathogenesis.
Figure 9.

GPR30 agonist treatment reduced CNS cytokine expression. SJL mice (5–7 weeks old) were immunized s.c. with 50 μg PLP139-151 and CFA (400 μg Mycobacterium tuberculosis). Draining lymph node cells were harvested after 7 days and re-stimulated with 50 μg/mL PLP139-151 in the presence of 3 μM G-1 agonist or control DMEM medium with 5% FCS and 0.1% DMSO. When the control group showed peak clinical disease, three representative mice from each group were harvested and the CNS tissue was analyzed for the presence of CCL2 (A) and IL-23 (B) by ELISA. The group that received G-1-treated cells showed significantly decreased CNS CCL2 and IL-23 compared to recipients of control-treated cells (*, p<0.05). C) Purified CD4+ T cells were stimulated in vitro with immobilized anti-CD3 and anti-CD28 in the presence or absence of 3 μM G-1 under normal and polarizing conditions. Polarizing conditions included IL-12 and anti-IL-4 for Th1 (IFN-γ) and TGF-β, IL-6 and IL-23 for Th17 (IL-17). The data indicate no effect of G-1 on T cell cytokine secretion.
DISCUSSION
The beneficial effects of estrogen in the autoimmune disease multiple sclerosis are well documented and have been thought to be a direct effect of the hormone acting through the classical nuclear receptors ERα and ERβ (Garidou et al., 2004; Polanczyk et al., 2003; Tiwari-Woodruff et al., 2007). However, not all of the activities of estrogen can be explained by activation of these receptors (Carmeci et al., 1997; Guo et al., 2002; Qiu et al., 2003). For example, estradiol can stimulate a rapid increase in intracellular free calcium in mouse macrophages. This effect is sensitive to pertussis toxin and cannot be blocked by the nuclear estrogen receptor antagonist’s raloxifene and tamoxifen, suggesting the involvement of a non-classical estrogen receptor with a profile very similar to the recently identified GPR30 (Guo et al., 2002). There is also one report describing expression of GPR30 within the immune system (monocytes, macrophages and B cells) (Kvingedal and Smeland, 1997). In addition, a report has demonstrated that estradiol can enhance the production of nerve growth factor in a THP-1-monocytic cell line and in peripheral blood monocytes (Kanda and Watanabe, 2003). These effects appeared to be mediated by GPR30 since antisense oligonucleotides against the receptor suppressed these estrogen-induced effects in both cell types. These expression patterns and activities could implicate GPR30 in mediating some of the immunomodulatory actions of estradiol.
To test this possibility, we examined whether GPR30 was expressed in immune cells. Using a specific antibody to GPR30 we were able to demonstrate that the receptor is expressed in a variety of immune cells including human macrophages. Elucidation of the physiological role of GPR30 in immune cells has been somewhat complicated bym the co-expression of the nuclear steroid receptors ERα and ERβ. However, we demonstrate here that a small molecule agonist of GPR30, G-1 (Bologa et al., 2006), has greater than 1000-fold specificity for GPR30 over 25 other receptors including the nuclear steroid receptors ERα and ERβ and thus is a highly selective probe with which to examine GPR30 function in immune cells.
We were able to demonstrate that GPR30 was functional in primary human macrophages and the GPR30 specific agonist G-1, in a dose-dependent manner, inhibited the LPS-induced secretion of the cytokines TNF-α, IL-6 and IL-12 (p40) together with the chemokine CCL5. The inhibitory effect of the GPR30 agonist G-1 on the secretion of the proinflammatory cytokines and chemokines TNF-α, IL-12, IL-6 and CCL5 by human macrophages is interesting because they, together with IFN-γ, have all been implicated in the cytokine storm that characterizes many autoimmune diseases such as multiple sclerosis (Link, 1998). The beneficial effects of 17β-estradiol in an EAE model of multiple sclerosis, for example, were shown to be linked to an inhibition of TNF-α, IFN-γ and IL-12 in mature dendritic cells (Liu et al., 2002). These findings raise the possibility that these inhibitory effects, mediated by G-1 through GPR30, could be beneficial in reducing the severity of multiple sclerosis and we were able to test for this possibility since we had shown that the GPR30-specific agonist was functional in a mouse macrophage cell line.
Studies presented here showed that the GPR30-specific agonist G-1 was therapeutic in the EAE model of multiple sclerosis in the mouse induced by either active immunization or passive T cell transfer. In the active model of EAE, dosing with G-1 significantly reduced the severity of disease and this was concomitant with a reduction of CNS-infiltrating macrophages. Our data further suggested that G-1 treatment either directly or indirectly affects the antigen-specific T cell inflammatory cytokine expression pattern, including IL-17 that is necessary for EAE development (Komiyama et al., 2006).
The effects of estrogen on the inhibition of EAE and the accompanying autoimmune response has been previously demonstrated to be linked to a direct upregulation of Foxp3, a regulatory T cell protein (Polanczyk et al., 2005), to regulation of T cell cytokine expression (Morales et al., 2006) as well as to non-T cell mediated mechanisms (Polanczyk et al., 2004) that may indirectly down-regulate T cell function through antigen presentation. Despite the apparent lack of direct effects of G-1 on T cells in EAE, GPR30 is expressed in regulatory T cells. Similar to our results, a recent study has also shown that G-1 protects against EAE without apparent effects on antigen-specific T cells (Wang et al., 2009). In this study GPR30 was not required for upregulation of FoxP3+ T cells, but was required for upregulation of PD-1 (programmed death-1) in regulatory T cells. A shift in regulatory T cells from PD-1- to PD-1+ appears to be involved in enhanced suppressive function (Alderson et al., 2008). GPR30 therefore may not be involved in direct suppression of T cells but in enhancement of regulatory T cell activity.
Distinct regulatory effects of estrogen on T cell development acting through traditional nuclear receptors or GPR30 have also been demonstrated (Wang et al., 2008), thus indicating that GPR30 may be a viable target for use in therapeutic intervention. The idea of using estrogen receptor stimulation as a therapy for multiple sclerosis is currently being explored in clinical trials (Offner and Polanczyk, 2006). Our present data indicate that the GPR30 agonist, G-1, has efficacy for the inhibition of EAE through indirect down-regulation of autoreactive T cell responses and thus may represent a clinically useful target to treat multiple sclerosis.
Acknowledgments
We wish to thank Dr. Hong-Tao Lu for providing human regulatory T cells.
BIBLIOGRAPHY
- Abramsky O. Pregnancy and multiple sclerosis. Ann Neurol. 1994;36(Suppl):S38–41. doi: 10.1002/ana.410360712. [DOI] [PubMed] [Google Scholar]
- Alderson KL, Zhou Q, Berner V, Wilkins DE, Weiss JM, Blazar BR, Welniak LA, Wiltrout RH, Redelman D, Murphy WJ. Regulatory and conventional CD4+ T cells show differential effects correlating with PD-1 and B7-H1 expression after immunotherapy. J Immunol. 2008;180:2981–2988. doi: 10.4049/jimmunol.180.5.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnason BG, Richman DP. Effects of estrogen, progestin and combined estrogen-progestin oral contraceptive preparations on experimental allergic encephalomyelitis. Trans Am Neurol Assoc. 1969;94:54–58. [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med. 1998;339:285–291. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- Correa F, Mestre L, Docagne F, Guaza C. Activation of cannabinoid CB2 receptor negatively regulates IL-12p40 production in murine macrophages: role of IL-10 and ERK1/2 kinase signaling. Br J Pharmacol. 2005;145:441–448. doi: 10.1038/sj.bjp.0706215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damek DM, Shuster EA. Pregnancy and multiple sclerosis. Mayo Clin Proc. 1997;72:977–989. doi: 10.1016/S0025-6196(11)63371-5. [DOI] [PubMed] [Google Scholar]
- Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J Immunol. 2008;180:7376–7384. doi: 10.4049/jimmunol.180.11.7376. [DOI] [PubMed] [Google Scholar]
- Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J Endocrinol. 2005;185:243–252. doi: 10.1677/joe.1.06063. [DOI] [PubMed] [Google Scholar]
- Fillmore PD, Blankenhorn EP, Zachary JF, Teuscher C. Adult gonadal hormones selectively regulate sexually dimorphic quantitative traits observed in experimental allergic encephalomyelitis. Am J Pathol. 2004;164:167–175. doi: 10.1016/S0002-9440(10)63107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming KK, Bovaird JA, Mosier MC, Emerson MR, LeVine SM, Marquis JG. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;170:71–84. doi: 10.1016/j.jneuroim.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Garidou L, Laffont S, Douin-Echinard V, Coureau C, Krust A, Chambon P, Guery JC. Estrogen receptor alpha signaling in inflammatory leukocytes is dispensable for 17beta-estradiol-mediated inhibition of experimental autoimmune encephalomyelitis. J Immunol. 2004;173:2435–2442. doi: 10.4049/jimmunol.173.4.2435. [DOI] [PubMed] [Google Scholar]
- Guo Z, Krucken J, Benten WP, Wunderlich F. Estradiol-induced nongenomic calcium signaling regulates genotropic signaling in macrophages. J Biol Chem. 2002;277:7044–7050. doi: 10.1074/jbc.M109808200. [DOI] [PubMed] [Google Scholar]
- Hayes MP, Enterline JC, Gerrard TL, KCZ Regulation of interferon production by human monocytes: requirements for priming for lipopolysaccharide-induced production. J Leukoc Biol. 1991;50:176–181. doi: 10.1002/jlb.50.2.176. [DOI] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson L, Olsson T, Holmdahl R. Estrogen induces a potent suppression of experimental autoimmune encephalomyelitis and collagen-induced arthritis in mice. J Neuroimmunol. 1994;53:203–207. doi: 10.1016/0165-5728(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Kanda N, Watanabe S. 17Beta-estradiol enhances the production of nerve growth factor in THP-1-derived macrophages or peripheral blood monocyte-derived macrophages. J Invest Dermatol. 2003;121:771–780. doi: 10.1046/j.1523-1747.2003.12487.x. [DOI] [PubMed] [Google Scholar]
- Karpus WJ, Lukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol. 1997;158:4129–4136. [PubMed] [Google Scholar]
- Kennedy KJ, Strieter RM, Kunkel SL, Lukacs NW, Karpus WJ. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1alpha and monocyte chemotactic protein-1. J Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Kvingedal AM, Smeland EB. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 1997;407:59–62. doi: 10.1016/s0014-5793(97)00278-0. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link H. The cytokine storm in multiple sclerosis. Mult Scler. 1998;4:12–15. doi: 10.1177/135245859800400104. [DOI] [PubMed] [Google Scholar]
- Liu HY, Buenafe AC, Matejuk A, Ito A, Zamora A, Dwyer J, Vandenbark AA, Offner H. Estrogen inhibition of EAE involves effects on dendritic cell function. J Neurosci Res. 2002;70:238–248. doi: 10.1002/jnr.10409. [DOI] [PubMed] [Google Scholar]
- Matejuk A, Adlard K, Zamora A, Silverman M, Vandenbark AA, Offner H. 17beta-estradiol inhibits cytokine, chemokine, and chemokine receptor mRNA expression in the central nervous system of female mice with experimental autoimmune encephalomyelitis. J Neurosci Res. 2001;65:529–542. doi: 10.1002/jnr.1183. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Miller SD, Karpus WJ. Experimental autoimmune encephalomyelitis in the mouse. Curr Protoc Immunol. 2007;Chapter 15(Unit 15):11. doi: 10.1002/0471142735.im1501s77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales LB, Loo KK, Liu HB, Peterson C, Tiwari-Woodruff S, Voskuhl RR. Treatment with an estrogen receptor alpha ligand is neuroprotective in experimental autoimmune encephalomyelitis. J Neurosci. 2006;26:6823–6833. doi: 10.1523/JNEUROSCI.0453-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H, Polanczyk M. A potential role for estrogen in experimental autoimmune encephalomyelitis and multiple sclerosis. Ann N Y Acad Sci. 2006;1089:343–372. doi: 10.1196/annals.1386.021. [DOI] [PubMed] [Google Scholar]
- Polanczyk M, Zamora A, Subramanian S, Matejuk A, Hess DL, Blankenhorn EP, Teuscher C, Vandenbark AA, Offner H. The Protective Effect of 17{beta}-Estradiol on Experimental Autoimmune Encephalomyelitis Is Mediated through Estrogen Receptor-{alpha} Am J Pathol. 2003;163:1599–1605. doi: 10.1016/s0002-9440(10)63516-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, Offner H. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J Immunol. 2004;173:2227–2230. doi: 10.4049/jimmunol.173.4.2227. [DOI] [PubMed] [Google Scholar]
- Polanczyk MJ, Hopke C, Huan J, Vandenbark AA, Offner H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J Neuroimmunol. 2005;170:85–92. doi: 10.1016/j.jneuroim.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Ronnekleiv OK, Kelly MJ. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–9540. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Shuster EA. Hormonal influences in multiple sclerosis. Curr Top Microbiol Immunol. 2008;318:267–311. doi: 10.1007/978-3-540-73677-6_11. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Matejuk A, Zamora A, Vandenbark AA, Offner H. Oral feeding with ethinyl estradiol suppresses and treats experimental autoimmune encephalomyelitis in SJL mice and inhibits the recruitment of inflammatory cells into the central nervous system. J Immunol. 2003;170:1548–1555. doi: 10.4049/jimmunol.170.3.1548. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proc Natl Acad Sci U S A. 2007;104:14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Walderveen MA, Tas MW, Barkhof F, Polman CH, Frequin ST, Hommes OR, Valk J. Magnetic resonance evaluation of disease activity during pregnancy in multiple sclerosis. Neurology. 1994;44:327–329. doi: 10.1212/wnl.44.2.327. [DOI] [PubMed] [Google Scholar]
- Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J Immunol. 2009;182:3294–3303. doi: 10.4049/jimmunol.0803205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Dehghani B, Magrisso IJ, Rick EA, Bonhomme E, Cody DB, Elenich LA, Subramanian S, Murphy SJ, Kelly MJ, Rosenbaum JS, Vandenbark AA, Offner H. GPR30 contributes to estrogen-induced thymic atrophy. Mol Endocrinol. 2008;22:636–648. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]