

Abstract
Esophageal atresia and tracheoesophageal fistula (EA/TEF) are major congenital malformations affecting 1:3500 live births. Current research efforts are focused on understanding the etiology of these defects. We describe well-known animal models, human syndromes, and associations involving EA/TEF, indicating its etiologically heterogeneous nature. Recent advances in genotyping technology and in knowledge of human genetic variation will improve clinical counseling on etiologic factors. This review provides a clinical summary of environmental and genetic factors involved in EA/TEF.
Keywords: Congenital anomaly, Foregut, VACTERL, Feingold syndrome, CHARGE syndrome, AEG syndrome, Genes
Introduction
Esophageal atresia (EA) and tracheoesophageal fistula (TEF) are congenital malformations that occur approximately in 1:3500 live-born infants [1]. Five subtypes are described, based on the location of the atresia and the type of connection between trachea and esophagus [2]. Associated anomalies occur in 50% of patients and include vertebral, anal, cardiovascular, tracheoesophageal, renal, and limb abnormalities (occurring together in the VACTERL association). Better surgical techniques and pre- and postoperative care have improved the prognosis of EA/TEF over the past decades, but patients still have significant short- and long-term morbidity [3•]. As with other congenital malformations, EA/TEF occurs at an increased rate in twins, but usually affects only one twin [1]. Once a couple has one child with EA/TEF, the risk of having a second child with this anomaly is increased to 1% [4].
The pathologic mechanism leading to EA/TEF is unknown. The trachea, esophagus, and lungs are foregut-derived structures. During the fourth week of embryonic life, the foregut divides into a ventral respiratory part and a dorsal esophageal part. The underlying mechanism of separation is not known.
EA/TEF is thought to be a multifactorial complex disease, with involvement of genetic and environmental factors. In 6% to 10% of patients a defined genetic syndrome can be diagnosed, leaving 90% of patients of unknown etiology [5]. This paper aims to give an overview of current knowledge and gaps in knowledge on the etiology of EA/TEF. In this review we distinguish between complex genetic syndromes and associations to facilitate targeted genetic follow-up and counseling.
Environmental Factors
Various environmental factors have been suggested as risk factors for the development of tracheoesophageal anomalies, including maternal exposure to methimazole [6], exogenous sex hormones [7], maternal alcohol and smoking [8], infectious diseases [9], and working in agriculture or horticulture [10]. Previous studies from our group implicate a possible role for maternal in utero exposure to diethylstilbestrol (DES) [11]. Observations in insulin-dependent diabetic mothers suggest that first trimester exposure to maternal diabetes is associated with the development of congenital anomalies, including EA/TEF and VACTERL-associated anomalies [12]. Very recently, studies from the European Surveillance of Congenital Anomalies (EUROCAT) birth registry network found that older mothers are at significantly greater risk of having a child with EA [13•].
Administration of the anthracycline antibiotic adriamycin to pregnant rats causes EA/TEF and other major congenital anomalies in the offspring [14]. However, no such association has been reported for humans [15]. A role for vitamin A deficiency in the development of EA/TEF has also been suggested. A vitamin A–deficient diet given to pregnant rats caused severe congenital anomalies in the offspring, including agenesis of the lung and TEF [16]. Ethylnitrosourea (ENU), an alkylating and mutagenic agent, induced a recessive mouse mutation with a phenotype that includes abnormal tracheoesophageal septation and VACTERL-associated anomalies [17]. So far, no specific environmental risk factor has consistently been identified.
Genetic Factors
Human Syndromes and Associations Involving EA/TEF
More than 50% of EA/TEF patients have associated anomalies. Certain anomalies, such as cardiovascular defects, renal agenesis, microcephaly, duodenal atresia, limb reduction defects, and polycystic kidney are especially prevalent in patients with EA/TEF [18, 19]. EA/TEF may be present in several syndromes and associations, as described in Table 1.
Table 1.
Genetic syndromes and associations involving esophageal atresia and tracheoesophageal fistula
| OMIM | Gene(s) | Locus | Major defectsa | ||
|---|---|---|---|---|---|
| Single gene disorders | |||||
| Feingold syndrome | 164280 | MYCN | 2p24.1 | Intestinal atresias, microcephaly, learning disability, CHD, limb defects, short stature | |
| CHARGE syndrome | 214800 | CHD7 | 8q12 | Coloboma, CHD, choanal atresia, GR, genitourinary and ear anomalies/deafness | |
| AEG syndrome | 206900 | SOX2 | 3q26.3-q27 | Clinical anophthalmia, GD, mesial temporal abnormalities of the brain | |
| Pallister-Hall syndrome | 146510 | GLI3 | 7p13 | Laryngotracheoesophageal cleft, hypothalamic hamartoblastoma, pituitary dysfunction, AA, limb defects | |
| Opitz G syndrome | 300000 | MID1 | Xp22 | Laryngotracheoesophageal cleft, hypertelorism, hypospadias, cleft lip/palate, CHD, AA, developmental delay | |
| Fanconi anemia | 607139 | FANCA | 16q24.3 | Anemia, abnormal skin pigmentation, short stature, microphthalmia, microcephaly, susceptibility to cancer, CHD, limb and renal defects | |
| VACTERL + hydrocephalus | 227645 | FANCC | 9q22.3 | VACTERL-associated defects, hydrocephalus, Arnold-Chiari malformation, cleft palate, incomplete lung lobation | |
| 605724 | FANCD1 | 13q12.3 | |||
| 227646 | FANCD2 | 3p25.3 | |||
| 602956 | FANCG | 9p13 | |||
| 300514 | FANCB | Xp22.31 | |||
| VACTERL + hydrocephalus | 276950 | PTEN | 10q23.31 | VACTERL-associated defects, macrocephaly, ventriculomegaly | |
| Chromosomal abnormalities | |||||
| Full trisomies | – | Chromosomes 13,18,21 | Major congenital anomalies, including MR, CHD, gastrointestinal atresias, Hirschsprung disease, dysmorphic features | ||
| 22q11 deletion (DiGeorge) syndrome | 188400 | TBX1 b | 22q11.2 | CHD, cleft palate, facial dysmorphism, hypocalcaemia, hypertelorism, hypospadias, thymic hypoplasia, and midline defects | |
| Opitz syndrome | 145410 | TBX1 b | 22q11.2 | Hypertelorism, laryngotracheoesophageal cleft, cleft lip/palate, GD, MR, CHD | |
| 13q deletion | – | ZIC2 b | 13q22-qter | Central nervous system malformations, intestinal atresias, GR, coloboma, genitourinary, midline and VACTERL-associated defects | |
| 17q deletion | – | RARα b | 17q21.3-q24.2 | MR, conductive hearing loss, impaired vision, craniofacial and skeletal defects | |
| NOG b | |||||
| TBX4 b | |||||
| 16q24 deletion | – | FOXF1 b | 16q24.1 | ACD, VACTERL-associated defects, urinary tract obstruction | |
| Associations | |||||
| VACTERL association | 192350 | – | – | Vertebral, anal, cardiovascular, renal and limb defects | |
| VACTERL + hydrocephalus | 314390 | – | X-linked | VACTERL-associated defects, hydrocephalus | |
| Oculo-Auriculo-Vertebral Spectrum (OAVS)/Goldenhar syndrome | 164210 | – | – | Hemifacial microsomia, CHD, vertebral and central nervous system anomalies | |
| Martinez-Frias syndrome | 601346 | – | – | Neonatal diabetes mellitus, intestinal atresias, hypoplastic pancreas and gallbladder, biliary atresia, hypospadias | |
aOther defects in combination with esophageal atresia and tracheoesophageal fistula
bGene(s) of interest on chromosomal locus
AA anal atresia/imperforate anus; ACD alveolar capillary dysplasia; AEG anophthalmia-esophageal-genital; CHARGE syndrome coloboma, heart anomalies, choanal atresia, growth and/or mental retardation, genital and ear anomalies; CHD congenital heart defects; GD genital defects, including cryptorchidism, hypospadias, genital hypoplasia; GR growth retardation; MR mental retardation; VACTERL vertebral, anal, cardiovascular, tracheoesophageal, renal, and limb abnormalities
The department of Pediatric Surgery at the Erasmus MC—Sophia Children’s Hospital admitted more than 300 EA/TEF patients from 1988 to 2009. In 29 patients, a chromosomal abnormality or a single gene disorder was causative to the EA/TEF phenotype. One in every 10 patients had a defined syndrome, which is in line with the literature. Many of the known genetic syndromes were seen in this cohort, including all full trisomies (Down syndrome, Edwards syndrome, Patau syndrome), single gene disorders (eg, CHARGE syndrome, Feingold syndrome, Opitz syndrome, and Fanconi anemia), and some less frequent syndromes (eg, Holt-Oram syndrome and Townes Brocks syndrome). The distribution of syndromes is according to the literature [5, 20].
More than 30% of EA/TEF cases in our cohort (syndromal cases excluded) were defined as VACTERL-associated. EA/TEF is a component of the VACTERL association, which includes vertebral, anal, cardiovascular, tracheoesophageal, renal, and limb anomalies, and is seen in 10% to 30% of EA/TEF patients [1, 19]. The use of this acronym as a clinical entity is still being debated, as is the minimum number of defects that must be present.
Of the VACTERL components, the vertebrae/ribs and the cardiovascular system are most commonly affected in combination with EA/TEF [23]. EA/TEF occasionally occurs in combinations with hemifacial microsomia, cardiac, vertebral, and/or central nervous system anomalies (Goldenhar syndrome), but no genetic basis has been described for this syndrome [21].
VACTERL associated patients are of interest in the search for new genetic factors underlying foregut-related and associated anomalies. However, the combinations of EA/TEF and its associated anomalies resemble and/or overlap phenotypically with defined syndromes, such as CHARGE syndrome, Feingold syndrome, and 22q11 deletion syndrome (discussed below). It is not easy, therefore, to discriminate between the causal syndromes, the more so as the etiology of VACTERL association is still unclear. Specific phenotypes, such as VACTERL-associated anomalies combined with hydrocephalus, did reveal causative mutations in the genes FANCB and PTEN [22, 23]. A recent study found deletions in the FOX gene cluster on chromosome 16q24, and mutations in the FOXF1 gene, in patients with alveolar capillary dysplasia combined with VACTERL-associated anomalies, including EA/TEF [24••].
Single Gene Disorders Involving EA/TEF
Feingold Syndrome
Feingold syndrome is caused by germline mutations in, or deletions of, the MYCN gene on chromosome 2p24.1. It is the most frequent cause of familial syndromic gastrointestinal atresias. About 30% to 40% of patients diagnosed with Feingold syndrome have EA/TEF [25]. Marcelis et al. [26••] reviewed the clinical features of Feingold syndrome in relation to the genotype; 23 different mutations of the MYCN gene and five deletions encompassing MYCN have been described over the years. The authors suggest that the presence of digital anomalies in combination with microcephaly is enough to justify MYCN analysis [26••]. Such analysis should also be considered in patients with EA/TEF in combination with microcephaly.
CHARGE Syndrome
About 10% of patients with CHARGE syndrome display EA/TEF. This well-defined syndrome—involving coloboma, heart anomalies, choanal atresia, growth and/or mental retardation, genital and ear anomalies—is caused by mutations in the chromodomain helicase DNA-binding (CHD7) gene, present in about 60% of CHARGE patients [27]. This gene is thought to have a role in early embryonic development affecting epigenetic regulation by chromatin organization and euchromatic gene expression. Still, its regulatory function and involvement in CHARGE syndrome and foregut development should be clarified in functional studies in humans and animal models.
AEG Syndrome
Deletions and mutations of the SOX2 gene are causative for the phenotype of clinical anophthalmia/optic nerve hypoplasia, esophageal atresia, and/or genital anomalies in the AEG syndrome [28]. Studies in chickens and Xenopus implicate a role for Sox2 in the developing foregut. Que et al. [29••,30] demonstrated EA/TEF in Sox2 mutant mice and thus provided evidence that down regulation of Sox2 plays a role in the etiology of EA. The proven relation between murine models and the human phenotype was a breakthrough in the knowledge about syndromic EA/TEF.
Pallister-Hall Syndrome
Pallister-Hall syndrome includes bifid epiglottis, hypothalamic hamartoblastoma, postaxial polydactyly, anal atresia, and occasionally laryngeal clefts. Mutations in the GLI3 gene can cause Pallister-Hall syndrome [31]. The foregut-related anomalies, such as laryngeal clefts and lobulation defects of the lungs, form a phenotypic link between human Pallister-Hall patients and the combined knockout mice for Gli2/Gli3. This provides evidence that the Gli-genes and their pathways are important in foregut development [32].
Opitz G Syndrome
Opitz G syndrome is characterized by midline abnormalities with mental retardation and agenesis of the corpus callosum. Even though EA/TEF is rare in Opitz G syndrome, the combination of EA/TEF with corpus callosum agenesis warrants testing for mutations in the MID1 gene, causing the X-linked form of Opitz syndrome. Laryngotracheoesophageal defects in general are present in most MID1-mutated males, but EA/TEF is rare [33]. In addition, deletions on chromosome 22q11.2 cause the autosomal-dominant form of Opitz syndrome, which has the same size of the deletion as observed in DiGeorge syndrome [34]. Also, several cases of EA/TEF have been described in patients with DiGeorge syndrome (see below).
Fanconi Anemia
Fanconi anemia (FA) is a genetically and phenotypically heterogeneous syndrome characterized by progressive bone marrow failure and early occurrence of acute myeloid leukemia, combined with several congenital malformations, including gastrointestinal malformations. Thirteen genetic subtypes have been described (A, B, C, D1, D2, E, F, G, I, J, L, M, and N), of which FANCA, FANCC, and FANCG are the three most common disease-causing genes [35]. Mutations in the FANCA, FANCB, FANCC, FANCD1 (BRCA2), FANCD2, and FANCG genes have been described in patients with EA/TEF with various combinations of defects, including microcephaly, short stature, pigment changes, and several VACTERL-associated anomalies, including heart, renal, and limb defects [22, 35–39]. EA/TEF is not a common feature of FA, but gastrointestinal malformations, including duodenal atresia, anorectal malformations, and EA/TEF, are seen in 14% of all FA patients [35]. In addition, mutations in the FANCB gene, on chromosome Xp22.31, were causes of X-linked VACTERL with hydrocephalus in patients who also had EA/TEF, lung lobulation defects, and confirmed central nervous system anomalies [22]. The diagnosis of Fanconi anemia calls for early therapeutic interventions and chromosome breakage studies. Faivre et al. [36] advised performance of breakage studies in patients with common VACTERL-associated anomalies, including EA/TEF, in combination with skin pigmentation abnormalities, growth retardation, microcephaly, and/or dysmorphism.
Chromosomal Abnormalities
Many well-known chromosomal aberrations are observed in EA/TEF patients, such as Down syndrome (trisomy 21), Edwards syndrome (trisomy 18), and Patau syndrome (trisomy 13) [1]. Deletions on several chromosomal loci, including 22q11 (DiGeorge syndrome), distal 13q, 17q21.3-q24.2, and 16q24.1 are found in some cases [24••, 40–43]. The paper by Felix et al. [42] reviewed the structural chromosomal anomalies reported in 30 patients. Some of the above-mentioned deletions are of interest, because the deleted loci include several candidate genes for EA/TEF, for example, the 17q21.3-q24.2 region including NOG and TBX4. Recently Stankiewicz et al. [24••] described deletions on the16q24.1 locus, including the forkhead genes FOXF1, FOXC2, and FOXL1. Of these transcription factors, FOXF1 is of particular interest, because heterozygote knockout mice for this protein have EA/TEF.
Better molecular cytogenetic techniques allow screening of large cohorts of patients with congenital anomalies; the copy number variations in microarray studies yield additional deletions and duplications. Such studies in EA/TEF patients are expected to reveal new candidate regions. We confirmed several chromosomal aberrations in patients with EA/TEF: Triple-X syndrome, Xp duplications, 22q11 microduplication syndrome, and a 5q11.2 deletion (unpublished data). Microarray studies will also provide more insight into the polymorphic regions of the human genome in relation to inherited aberrations in patients with congenital anomalies.
Murine Models
Several murine models are described to cause tracheoesophageal anomalies (Table 2) [17, 30, 44]. Genes of developmental pathways are involved, including vitamin A effectors (Rarα, Rarβ), effectors of the sonic hedgehog (SHH) pathway (Shh, Gli2, Gli3, Foxf1), other homeobox-containing transcription factors and their regulators (Hoxc4, Ttf-1, Pcsk5), and developmental transcriptional regulators (Tbx4, Sox2).
Table 2.
Overview of genes essential for tracheoesophageal development and their human homologues
| Gene | Mutant phenotype | Human homologue | Human locus |
|---|---|---|---|
| Shh −/− | EA; TEF; lungs form rudimentary sacs | SHH | 7q36 |
| Rarα −/− ;β2 −/− or Rarα1 −/− ;β −/− | TEF; lung hypoplasia or agenesis | RARα; RARβ | RARα: 17q21.1; RARβ: 3p24 |
| Gli2 −/− ; Gli3 +/− | EA; TEF; severe lung phenotype | GLI2; GLI3 | GLI2: 2q14; GLI3: 7p13 |
| Gli2 −/− ; Gli3 −/− | No formation of esophagus, trachea, and lungs | ||
| Foxf1 −/− | Lethal before embryonic day 10; extra-embryonic defects | FOXF1 | 16q24.1 |
| Foxf1 +/− | EA; TEF; lung immaturity/hypoplasia; lobulation defects | FOXF1 | 16q24.1 |
| Ttf-1 −/− | TEF; rudimentary peripheral lung primordial | TTF1 | 14q13 |
| Sox2 -/− | EA; TEF; lung branching defects | SOX2 | 3q26.3-q27 |
| Noggin −/− | EA; TEF; lung branching defects; abnormal notochord morphogenesis | NOG | 17q22 |
| Pcsk5 C470R mutanta | Abnormal tracheoesophageal septation; hypoplastic lungs | PCSK5 | 9q21.3 |
| Hoxc4 −/− | Partially or completely blocked esophageal lumen; disruption of esophageal musculature | HOXC4 | 12q13.3 |
| Tbx4 misexpression | TEF | TBX4 | 17q21-q22 |
Research into the human homologues of these genes has shed some light on the molecular basis underlying some human cases of EA/TEF. For example, Que et al. [30] found that 70% of null mutant mice for Noggin display EA/TEF. In several human TEF cases there is a deletion of the 17q21.3-24.2 locus, which includes the human orthologue NOG [30, 43]. Mice haploinsufficient for Foxf1 may show EA/TEF and other foregut-derived anomalies, including lung hypoplasia and lobulation defects [45]. A patient with a heterozygote deletion on chromosome 16q24, including the FOXF1 gene, displayed lung anomalies, EA/TEF and other VACTERL-associated features [24••]. The Gli2 and Gli3 genes are essential for formation of the trachea and the esophagus. Deletions of these genes are associated with congenital defects that resemble the VACTERL association, at least in mice [30, 32]. Szumska et al. [17] describe an ENU-induced recessive mouse mutation in the Pcsk5 gene, a regulator of Hox genes. Such mutations, in a heterozygote form, were also present in two patients with EA/TEF and features of VACTERL association. However, in these cases the mutations were inherited from a phenotypically normal parent. The etiologic role of these mutations remained unclear [17].
In conclusion, EA/TEF can be part of a spectrum of anomalies in specific syndromes with a known cause or it can be part of an association (Fig. 1). Clinicians should be aware of specific combinations of anomalies, and should these occur, consider counseling by a clinical geneticist.
Fig. 1.
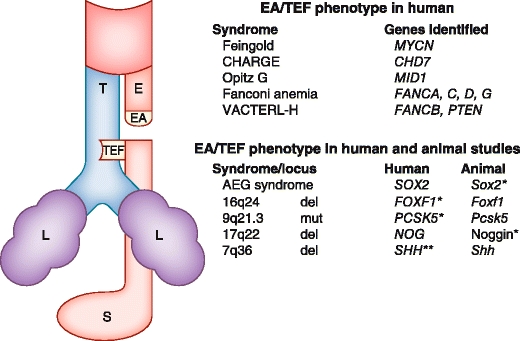
Schematic representation of the commonest type of EA/TEF and genetic syndromes and genes involved. The text boxes list the genetic syndromes and genes most frequently found to be involved in EA/TEF. EA —esophageal atresia; TEF —tracheoesophageal fistula; *—lung phenotype present; **—deletion in a single case; L—lung; E—esophagus; T—trachea; S—stomach; del—deletion; mut—mutation. AEG syndrome—Anophthalmia/optic nerve hypoplasia, Esophageal atresia, and/or Genital anomalies; CHARGE syndrome—Coloboma, Heart anomalies, choanal Atresia, growth and/or mental Retardation, Genital and Ear anomalies; VACTERL-H association—vertebral, anal, cardiovascular, tracheal, esophageal, renal, and limb abnormalities occurring together, with hydrocephaly
Strategies and Future Prospects
Phenotypic Approach
Patients presenting with esophageal atresia receive treatment by standardized clinical and surgical protocols. Adding standardized genetic protocols for counseling and research would provide a good opportunity to unravel the genetic background of EA and other congenital abnormalities. The clinical protocol for all patients with EA/TEF should include an echocardiogram and vertebral and limb radiographs, complemented with renal ultrasound evaluation if other features of the VACTERL association are present.
Genotypic Approach
Counseling by a clinical geneticist is advised, including standard karyotyping and screening for subtelomeric aberrations with multiplex ligation-dependent probe amplification. Clinical data should be reviewed and stored in comprehensive databases, together with genetic information, including pedigrees, DNA, tissue, and cell lines. With improvements in microarray technologies, genome-wide screening for copy number variations might replace standard karyotyping in the near future.
Epidemiologic Approach
Strategies that provide more insight into new etiologic factors should be in place to find more evidence for gene-environment interactions, to explore possible molecular mechanisms, and to find new genetic pathways. Environmental factors in families of patients with EA/TEF could be further explored by means of structured and validated questionnaires, adding to the knowledge gained from case-control studies [10]. Such studies require large cohorts, as have been used in population-based registries for surveillance of congenital anomalies, such as EUROCAT, the International Clearinghouse for Birth Defects Surveillance and Research, and the California Birth Defects Monitoring Program.
Experimental Approach
Studies in animal models implicate an essential role for sonic hedgehog signaling and its downstream effectors in the correct embryogenesis of the foregut. Better understanding of normal and abnormal development of the foregut could be achieved by focusing on the overlapping effectors of Shh-signaling, described in patients and animal models, for example, Gli2, Gli3, FOXF1, MID1, Noggin, Bmp4, and TBX1 [30, 46•].
Advances in genotyping technology and in knowledge of human genetic variation have enabled genome-wide association studies to identify susceptibility for common diseases, but also for congenital abnormalities, as proven in neural tube defects [47]. This approach requires large cohorts so as to provide statistical and clinical significance. Direct sequencing of candidate genes in patients and controls will provide an alternative approach that could reveal low-frequency alleles that influence disease susceptibility [48]. In addition, next generation sequencing will soon be an excellent tool in the search for new pathogenic mutations and copy number variations [49]. Also, there is increasing evidence that epigenetic modifications play an important role in developmental defects through DNA methylation and histone modifications [50]. A unifying approach seems to be the answer, therefore: analysis of multiple candidate genes should be done in large groups of well-genotyped individuals using next generation high-throughput genomic technology. This will be of great help in detecting possible gene-gene interactions as well as a possible role for copy number variations and regulatory mutations in patients with EA/TEF.
Conclusions
Many genetic pathways have been implicated in the development of EA/TEF. Patients with distinct phenotypes may be diagnosed with genetic syndromes such as Feingold syndrome, AEG syndrome, and CHARGE syndrome. There is a substantial gap in our knowledge of how environmental factors combined with genetic factors would disrupt foregut development. We would do well to “mind the gap” by performing clinical studies focusing on phenotyping, combined with targeted molecular genetic studies. In the near future, studies in large cohorts will lead to the discovery of new genes, genetic pathways, and perhaps environmental factors.
Acknowledgments
Disclosure
No potential conflict of interest relevant to this article was reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Elisabeth M. de Jong, Email: e.m.dejong@erasmusmc.nl
Janine F. Felix, Email: j.felix@erasmusmc.nl
Annelies de Klein, Email: a.deklein@erasmusmc.nl.
Dick Tibboel, Phone: +31-10-703 6567, FAX: +31-10-703 6288, Email: d.tibboel@erasmusmc.nl.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Torfs CP, Curry CJ, Bateson TF. Population-based study of tracheoesophageal fistula and esophageal atresia. Teratology. 1995;52:220–232. doi: 10.1002/tera.1420520408. [DOI] [PubMed] [Google Scholar]
- 2.Gross RE. Surgery of Infancy and Childhood. Philadelphia: WB Saunders; 1953. p. 76. [Google Scholar]
- 3.Spitz L. Oesophageal atresia. Orphanet J Rare Dis. 2007;2:24. doi: 10.1186/1750-1172-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMullen KP, Karnes PS, Moir CR, Michels VV. Familial recurrence of tracheoesophageal fistula and associated malformations. Am J Med Genet. 1996;63:525–528. doi: 10.1002/(SICI)1096-8628(19960628)63:4<525::AID-AJMG3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 5.Genevieve D, de Pontual L, Amiel J, et al. An overview of isolated and syndromic oesophageal atresia. Clin Genet. 2007;71:392–399. doi: 10.1111/j.1399-0004.2007.00798.x. [DOI] [PubMed] [Google Scholar]
- 6.Di Gianantonio E, Schaefer C, Mastroiacovo PP, et al. Adverse effects of prenatal methimazole exposure. Teratology. 2001;64:262–266. doi: 10.1002/tera.1072. [DOI] [PubMed] [Google Scholar]
- 7.Nora JJ, Nora AH, Perinchief AG, et al. Congenital abnormalities and first-trimester exposure to progestagen/oestrogen [letter] Lancet. 1976;1:313–314. doi: 10.1016/S0140-6736(76)91455-0. [DOI] [PubMed] [Google Scholar]
- 8.Wong-Gibbons DL, Romitti PA, Sun L, et al. Maternal periconceptional exposure to cigarette smoking and alcohol and esophageal atresia +/− tracheo-esophageal fistula. Birth Defects Res A Clin Mol Teratol. 2008;82:776–784. doi: 10.1002/bdra.20529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kyyronen P, Hemminki K. Gastro-intestinal atresias in Finland in 1970–79, indicating time-place clustering. J Epidemiol Community Health. 1988;42:257–265. doi: 10.1136/jech.42.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felix JF, van Dooren MF, Klaassens M, et al. Environmental factors in the etiology of esophageal atresia and congenital diaphragmatic hernia: results of a case-control study. Birth Defects Res A Clin Mol Teratol. 2008;82:98–105. doi: 10.1002/bdra.20423. [DOI] [PubMed] [Google Scholar]
- 11.Felix JF, Steegers-Theunissen RP, de Walle HE, et al.: Esophageal atresia and tracheoesophageal fistula in children of women exposed to diethylstilbestrol in utero. Am J Obstet Gynecol 2007, 197:38 e31–e35. [DOI] [PubMed]
- 12.Castori M, Rinaldi R, Capocaccia P, et al. VACTERL association and maternal diabetes: a possible causal relationship? Birth Defects Res A Clin Mol Teratol. 2008;82:169–172. doi: 10.1002/bdra.20432. [DOI] [PubMed] [Google Scholar]
- 13.Loane M, Dolk H, Morris JK. Maternal age-specific risk of non-chromosomal anomalies. Bjog. 2009;116:1111–1119. doi: 10.1111/j.1471-0528.2009.02227.x. [DOI] [PubMed] [Google Scholar]
- 14.Diez-Pardo JA, Baoquan Q, Navarro C, Tovar JA. A new rodent experimental model of esophageal atresia and tracheoesophageal fistula: preliminary report. J Pediatr Surg. 1996;31:498–502. doi: 10.1016/S0022-3468(96)90482-0. [DOI] [PubMed] [Google Scholar]
- 15.Germann N, Goffinet F, Goldwasser F. Anthracyclines during pregnancy: embryo-fetal outcome in 160 patients. Ann Oncol. 2004;15:146–150. doi: 10.1093/annonc/mdh009. [DOI] [PubMed] [Google Scholar]
- 16.Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92:189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- 17.Szumska D, Pieles G, Essalmani R, et al. VACTERL/caudal regression/Currarino syndrome-like malformations in mice with mutation in the proprotein convertase Pcsk5. Genes Dev. 2008;22:1465–1477. doi: 10.1101/gad.479408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoll C, Alembik Y, Dott B, Roth MP. Associated malformations in patients with esophageal atresia. Eur J Med Genet. 2009;52:287–290. doi: 10.1016/j.ejmg.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 19.de Jong EM, Felix JF, Deurloo JA, et al. Non-VACTERL-type anomalies are frequent in patients with esophageal atresia/tracheo-esophageal fistula and full or partial VACTERL association. Birth Defects Res A Clin Mol Teratol. 2008;82:92–97. doi: 10.1002/bdra.20437. [DOI] [PubMed] [Google Scholar]
- 20.Shaw-Smith C. Oesophageal atresia, tracheo-oesophageal fistula, the VACTERL association: review of genetics and epidemiology. J Med Genet. 2006;43:545–554. doi: 10.1136/jmg.2005.038158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendelberg A, Ariel I, Mogle P, Arad I. Tracheo-oesophageal anomalies in the Goldenhar anomalad. J Med Genet. 1985;22:149–150. doi: 10.1136/jmg.22.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holden ST, Cox JJ, Kesterton I, et al. Fanconi anaemia complementation group B presenting as X linked VACTERL with hydrocephalus syndrome. J Med Genet. 2006;43:750–754. doi: 10.1136/jmg.2006.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reardon W, Zhou XP, Eng C. A novel germline mutation of the PTEN gene in a patient with macrocephaly, ventricular dilatation, and features of VATER association. J Med Genet. 2001;38:820–823. doi: 10.1136/jmg.38.12.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stankiewicz P, Sen P, Bhatt SS, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–791. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Bokhoven H, Celli J, van Reeuwijk J, et al. MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in Feingold syndrome. Nat Genet. 2005;37:465–467. doi: 10.1038/ng1546. [DOI] [PubMed] [Google Scholar]
- 26.Marcelis CL, Hol FA, Graham GE, et al. Genotype-phenotype correlations in MYCN-related Feingold syndrome. Hum Mutat. 2008;29:1125–1132. doi: 10.1002/humu.20750. [DOI] [PubMed] [Google Scholar]
- 27.Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 28.Williamson KA, Hever AM, Rainger J, et al. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet. 2006;15:1413–1422. doi: 10.1093/hmg/ddl064. [DOI] [PubMed] [Google Scholar]
- 29.Que J, Okubo T, Goldenring JR, et al. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development. 2007;134:2521–2531. doi: 10.1242/dev.003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Que J, Choi M, Ziel JW, et al. Morphogenesis of the trachea and esophagus: current players and new roles for noggin and Bmps. Differentiation. 2006;74:422–437. doi: 10.1111/j.1432-0436.2006.00096.x. [DOI] [PubMed] [Google Scholar]
- 31.Johnston JJ, Olivos-Glander I, Killoran C, et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet. 2005;76:609–622. doi: 10.1086/429346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motoyama J, Liu J, Mo R, et al. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat Genet. 1998;20:54–57. doi: 10.1038/1711. [DOI] [PubMed] [Google Scholar]
- 33.De Falco F, Cainarca S, Andolfi G, et al. X-linked Opitz syndrome: novel mutations in the MID1 gene and redefinition of the clinical spectrum. Am J Med Genet A. 2003;120:222–228. doi: 10.1002/ajmg.a.10265. [DOI] [PubMed] [Google Scholar]
- 34.Robin NH, Feldman GJ, Aronson AL, et al. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2. Nat Genet. 1995;11:459–461. doi: 10.1038/ng1295-459. [DOI] [PubMed] [Google Scholar]
- 35.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faivre L, Portnoi MF, Pals G, et al. Should chromosome breakage studies be performed in patients with VACTERL association? Am J Med Genet A. 2005;137:55–58. doi: 10.1002/ajmg.a.30853. [DOI] [PubMed] [Google Scholar]
- 37.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44:1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levran O, Attwooll C, Henry RT, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 39.Yamada T, Tachibana A, Shimizu T, et al. Novel mutations of the FANCG gene causing alternative splicing in Japanese Fanconi anemia. J Hum Genet. 2000;45:159–166. doi: 10.1007/s100380050203. [DOI] [PubMed] [Google Scholar]
- 40.Digilio MC, Marino B, Bagolan P, et al. Microdeletion 22q11 and oesophageal atresia. J Med Genet. 1999;36:137–139. [PMC free article] [PubMed] [Google Scholar]
- 41.Walsh LE, Vance GH, Weaver DD. Distal 13q Deletion Syndrome and the VACTERL association: case report, literature review, and possible implications. Am J Med Genet. 2001;98:137–144. doi: 10.1002/1096-8628(20010115)98:2<137::AID-AJMG1022>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 42.Felix JF, Tibboel D, de Klein A. Chromosomal anomalies in the aetiology of oesophageal atresia and tracheo-oesophageal fistula. Eur J Med Genet. 2007;50:163–175. doi: 10.1016/j.ejmg.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 43.Puusepp H, Zilina O, Teek R, et al. 5.9 Mb microdeletion in chromosome band 17q22-q23.2 associated with tracheo-esophageal fistula and conductive hearing loss. Eur J Med Genet. 2009;52:71–74. doi: 10.1016/j.ejmg.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Felix JF, Keijzer R, van Dooren MF, et al. Genetics and developmental biology of oesophageal atresia and tracheo-oesophageal fistula: lessons from mice relevant for paediatric surgeons. Pediatr Surg Int. 2004;20:731–736. doi: 10.1007/s00383-004-1287-3. [DOI] [PubMed] [Google Scholar]
- 45.Mahlapuu M, Enerback S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001;128:2397–2406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- 46.Shaw-Smith C. Genetic factors in esophageal atresia, tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur J Med Genet. 2010;53:6–13. doi: 10.1016/j.ejmg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez CA, Northrup H, Lin JI, et al.: Genetic association study of putative functional single nucleotide polymorphisms of genes in folate metabolism and spina bifida. Am J Obstet Gynecol 2009, 201:394 e391–e311. [DOI] [PMC free article] [PubMed]
- 48.Furniss D, Kan SH, Taylor IB, et al. Genetic screening of 202 individuals with congenital limb malformations and requiring reconstructive surgery. J Med Genet. 2009;46:730–735. doi: 10.1136/jmg.2009.066027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoon S, Xuan Z, Makarov V, et al. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schnetz MP, Bartels CF, Shastri K, et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19:590–601. doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]