

Abstract
Purpose
Sensitize melanomas to apoptosis and inhibit their growth and metastatic potential by compounds that mimic the activities of activating transcription factor 2 (ATF2)-driven peptides.
Experimental Design
Small-molecule chemical library consisting of 3,280 compounds was screened to identify compounds that elicit properties identified for ATF2 peptide, including (a) sensitization of melanoma cells to apoptosis, (b) inhibition of ATF2 transcriptional activity, (c) activation of c-Jun NH2-terminal kinase (JNK) and c-Jun transcriptional activity, and (d) inhibition of melanoma growth and metastasis in mouse models.
Results
Two compounds, celastrol (CSL) and acetyl isogambogic acid, could, within a low micromolar range, efficiently elicit cell death inmelanoma cells. Both compounds efficiently inhibit ATF2 transcriptional activities, activate JNK, and increase c-Jun transcriptional activities. Similar to the ATF2 peptide, both compounds require JNK activity for their ability to inhibit melanoma cell viability. Derivatives of CSL were identified as potent inducers of cell death in mouse and human melanomas. CSL and a derivative (CA19) could also efficiently inhibit growth of human and mouse melanoma tumors and reduce the number of lung metastases in syngeneic and xenograft mouse models.
Conclusions
These studies show for the first time the effect of CSL and acetyl isogambogic acid on melanoma. These compounds elicit activities that resemble the well-characterized ATF2 peptide and may therefore offer new approaches for the treatment of this tumor type.
The growth and metastasis of melanoma as well as its notorious resistance to therapy present major obstacles to its treatment (1, 2). A growing number of genetic and epigenetic changes in melanomas affect genes associated with melanocyte development and maintenance, cell cycle control, resistance to apoptosis, and angiogenic and metastatic capacity. Among the genetic changes commonly found in human melanomas are mutations in protein kinases of the mitogen-activated protein kinase (MAPK) family, indicating that signal transduction pathways have been largely modified in this tumor type (3, 4). Specifically, the prevalence of activating mutations in B-RAF and N-RAS has been largely associated with the activation of downstream targets—MAPK/extracellular signal-regulated kinase (ERK) kinase and in many cases ERK-MAPK (5–7). Constitutive activation of these kinases results in corresponding up-regulation of their targets, including genes implicated in the development and maintenance of melanoma, such as MITF, iNOS, and cyclin D1 (8–11).
In addition to changes in the MAPK signaling pathway, other pathways are deregulated in melanoma for reasons as yet unknown. For example, a growing fraction of melanomas is recognized as carrying an inactive form of PTEN, a protein phosphatase implicated in the regulation of AKT signaling pathways and their downstream mammalian target of rapamycin and p70S6 effectors (12, 13). Up-regulation of protein kinase C and c-Jun NH2-terminal kinase (JNK) has also been observed in melanoma (14–16). Other regulatory components that are modified in melanoma include cell cycle regulatory proteins, such as cyclin-dependent kinases (CDKN2A) CDK2, p16/CDK/pRb, and cyclin D1 (17–19).
Modifications were also recorded in matrix metalloproteinase, proteases associated with tumor cell metastatic capacity (20–22), and chaperones such as HSP90 (23). The antiapoptotic proteins, such as Bcl2 and the transcription factors activating transcription factor (ATF) 1, AP1, and ATF2, also cooperate in conferring resistance to apoptosis and metastatic capacity on melanoma (24–26). The complexity of changes that take place in melanoma are further illustrated in the rewiring of key signal transduction pathways; for example, ERK causes up-regulation of c-Jun and JNK activity (27).
In earlier studies, we showed that the transcription factor ATF2 serves as a good marker and possible target for this tumor type (28–32). Analysis of 544 human melanomas using tissue arrays revealed that nuclear localization of ATF2 is associated with poor prognosis (28), thereby pointing to the possibility that constitutively active ATF2 may contribute to the development and progression of human melanomas. Consistent with this possibility, inhibition of ATF2 function by means of a 50-amino acid peptide derived from ATF2 sensitized melanoma to apoptosis (29) and inhibited growth and metastasis in mouse models (30, 31). ATF2 peptides elicit such effects by virtue of their ability to inhibit ATF2 concomitant with an increase in JNK and consequently c-Jun activities (32). Given our understanding of the activities of the ATF2 peptide, we did a high-throughput screen to identify compounds that may mimic such activities. Characterization of two such compounds and newly generated derivatives in both in vitro and in vivo models suggests that they may have potential in the treatment of melanoma.
Materials and Methods
Cells
Mouse melanoma (SW1) and human melanoma (LU1205) cells were maintained in DMEM supplemented with 10% fetal bovine serum, l-glutamine, and antibiotics. The human melanoma cell lines MEWO and WM115 were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and antibiotics. Primary mouse melanocytes were cultured in F-12 medium supplemented with 10% fetal bovine serum, antibiotics, isobutylmethylxanthine, bovine pituitary extract, and 12-O-tetradecanoylphorbol-13-acetate.
Constructs and inhibitors
An ATF2 peptide (amino acids 51–100) was cloned into HA-tagged pcDNA3 vector as described previously (30). 5xJun2tk-Luc (marker for ATF2 transcriptional activities), 5xTREtk-Luc (marker for AP1/c-Jun transcriptional activities), and 2xNF-κB-Luc were described previously (31). Pharmacologic inhibitor of JNK (SP600125) was purchased (EMD Biosciences) and added (5 µmol/L) to cultured cells as indicated in Results.
Transcriptional analysis
Transient transfection of different reporter constructs into melanoma cells was done using Lipofectamine Plus (Invitrogen). Luciferase activity was determined as described previously (32).
Mitochondrial activity and apoptosis
Cells were exposed to the chemicals for 12 to 48 has indicated in Results. To measure changes in mitochondrial activity reflective of general cell viability, the ATPLite assay was used according to the manufacturer’s recommendations (Perkin-Elmer). Apoptosis analysis was done by flow cytometry of propidium iodide–stained cells. The percentage of cells to the left of the diploid G0-G1 peak, characteristic of hypodiploid cells that have partially lost DNA, was calculated as percentage of apoptotic cells (32). Analysis of changes in caspase-8 expression pattern was done using immunoblot analysis.
Chemical library screen
Cells (4,000) were seeded in a volume of 45 µL of medium per well using a Matrix WellMate bulk reagent dispenser. The cells were incubated for 24 h at 37°C in a humidified atmosphere containing 5% CO2. Subsequently, 5 µL of 0.1 mmol/L compounds in 10% DMSO were added to each well (final compound concentration was 0.01 mmol/L in 1% DMSO) using a Beckman Coulter Biomek FX automated liquid handler. Two small-molecule chemical libraries were used in the study. These libraries were the Library of Pharmacologically Active Compounds (LOPAC1280 from Sigma-Aldrich) as well as the 2,000 compounds that comprise the Spectrum Collection from MicroSource Discovery Systems, Inc. Both of these libraries are composed of small molecules that are pharmacologically active. The spectrum collection contains not only 1,000 small-molecule compounds but also 1,000 pharmacologically active purified natural products. These libraries have previously been used for screening campaigns (33, 34).
The treated cells were incubated for 24 h. After incubation, 25µL of ATPLite 1step (Perkin-Elmer) were added to each well. ATPLite uses a luciferase-based system to measure the relative quantity of ATP in a sample. It has been shown that ATP quantification can be used to measure cell proliferation and cytotoxicity (35). After the addition of ATPLite, the plates were read using the luminescence readout of a Molecular Devices Analyst HT multimode plate reader.
Compound synthesis
The celastrol (CSL) used for biological studies and to prepare amide and ester derivatives was obtained from Pay Pay Technologies as a dark red crystalline solid. The purity of this material was assessed by 1H nuclear magnetic resonance and liquid chromatography-mass spectrometry and found to be >98%. For the synthesis of esters and amides, all reactions were conducted in standard glassware without special regard to atmosphere or moisture. Reagents and anhydrous solvents were commercially purchased and used without purification. The purification of target compounds was done with a Shimadzu Discovery preparative high-performance liquid chromatography system or an ISCO Companion 4× flash chromatography system. Product analysis and identity confirmation was done using a Shimadzu liquid chromatography-mass spectrometer and a Varian 300 MHz nuclear magnetic resonance spectometer.
General procedure for the synthesis of amide derivatives of CSL
CSL (22 mg, 0.048 mmol) in dimethylformamide (2 mL) was treated with Hunig’s base (20 µL, 0.12 mmol), PyBOP (50 mg, 0.096 mmol), and the appropriate amine (0.05 mmol). After stirring for 20 h at room temperature, deionized water (15 mL) was added and the mixture was extracted by ethyl acetate twice. The combined organic layers were washed with brine, dried over MgSO4, and evaporated via rotary evaporator to yield a dark oil. The crude product was purified by flash chromatography (ethyl acetate/hexanes) and lyophilized (Labconco lyophilizer) to give a dark orange to red solid with yields ranging from 57% to 82%.
General procedure for the synthesis of ester derivatives of CSL
CSL (22 mg, 0.048 mmol) in dimethylformamide (2 mL) was treated with NaHCO3 (20 mg, 0.24 mmol) and the appropriate alkyl halide (0.05 mmol). After stirring for 36 h at room temperature, deionized water (15 mL) was added and the mixture was extracted by ethyl acetate twice. The combined organic layers were washed with brine, dried over MgSO4, and evaporated via rotary evaporator to yield a dark red oil. The crude product was purified by flash chromatography (ethyl acetate/hexanes) and lyophilized (Labconco lyophilizer) to give a dark red solid with yields ranging from 72% to 95%.
Immunoblot analysis
Proteins prepared from cells at the indicated time points using radioimmunoprecipitation assay buffer and 75 µg were separated on SDS-PAGE followed by immunoblot analysis using the antibodies indicated. Antibodies used were as follows: anti-JNK (Santa Cruz Biotechnology), phosphorylated JNK (Promega), phoshorylated c-Jun (Cell Signaling), c-Jun (Santa Cruz Biotechnology), caspase-8 (BD Biosciences), AKT and phosphorylated AKT (BD Biosciences), and ERK and phosphorylated ERK (Cell Signaling).
Tumor growth and metastasis in vivo
SW1 melanoma cells are metastatic derivative of 1735p cells originally developed by Dr. M. Kripke (M. D. Anderson Cancer Center, Houston, TX; ref. 31). Green fluorescent protein (GFP)-expressing SW1 cells were established and used as a means to follow melanoma progression in real time (30). SW1-GFP cells were trypsinized, resuspended in PBS, and injected s.c. (1 × 106) into the lower flank of 6-to 7-week-old mice. When tumors reached a size of ~50 mm3, the indicated compounds [100 µL of the vehicle—10% DMSO, 70% cremophor/ethanol (3:1), and 20% PBS and 1.0 mg/kg CSL or 0.5 mg/kg of the derivatives CA16 and CA19] were injected by i.p every alternate day. Body weight and tumor growth were monitored every 2 days. GFP-expressing SW1 tumors were monitored in vivo on shaved mice with a light box illuminated by blue light fiber optics (Lightools Research), and imaging was carried out with a digital camera (Nikon D100). Tumors were measured for up to 3 weeks. For metastases studies, LU1205 cells (1 × 106) were injected i.v. into the lateral tail vein of nude mice. At the end of the experiment, the lungs and tumors were excised and weighted. To detect metastatic lesions, lungs were fixed in formalin, embedded in paraffin, and subjected to H&E staining.
Immunohistochemistry
Tumor sections (5 µm in thickness) were prepared and deparaffinized using xylene. Tumor sections were incubated in DAKO antigen retrieval solution for 20 min in a boiling bath followed by treatment with 3% hydrogen peroxide for 20 min. Antibodies against phosphorylated JNK (1:400) was allowed to react with tumor sections at 4°C overnight. Biotinylated anti-rabbit IgG was allowed to react for 30 min at room temperature and diaminobenzidine was used for the color reaction. Hematoxylin was used for counter-staining. The control sections were treated with normal rabbit serum.
Results
screening for compounds that mimic the ATF2 peptide effect on melanoma cells
We screened a chemical library of 3,280 pharmacologically active small molecules (33, 34) to identify compounds that elicit the same major effects as the ATF2 peptide. The criteria for selection included the following: (a) the ability to elicit apoptosis of melanoma cells, (b) inhibition of ATF2 transcriptional activity, (c) increase in c-Jun transcriptional activity, and (d) dependency on JNK activity. Compounds that met all four criteria were further assessed against a panel of melanoma cultures and in syngeneic and xenograft mouse models.
The screening was stepwise; thus, only compounds that met the first criterion were taken forward for further analyses to determine their effects on ATF2 and c-Jun transcriptional activities. Using this approach, 26 of the 3,280 compounds (0.8%) elicited efficient apoptosis of melanoma cells, which was monitored using the ATPLite assay. Of the 26 compounds, only 3 (0.1%) affected transcriptional activities of ATF2 and c-Jun. Because one of the three compounds was an organomercurial derivative (thimerasol), it was omitted from further analysis. Instead, more extensive studies were focused on CSL and acetyl isogambogic acid (AIGA), the two compounds that met the initial criteria.
Characterization of CSL and AIGA effects on mouse melanoma cells
The structures of AIGA and CSL are depicted inFig. 1A and B. AIGA was tested for cytotoxicity at concentrations ranging between 0.1 and 2 µmol/L, whereas CSL was used at a concentration of 0.01 to 0.3 µmol/L. Treatment of SW1 melanoma cells with 1 µmol/L AIGA reduced the viability of melanoma cells to 10% (Fig. 1C). Treatment of the SW1 cells with 0.05 µmol/L CSL reduced the viability of melanoma cells to 50%, and 0.1 µmol/L reduced it to 10% (Fig. 1C). These data suggest that both compounds elicit efficient cytotoxicity at a low micromolar concentration. Normal melanocytes and fibroblasts were used as a control. At the concentrations that efficiently induced cell death in melanoma cells, CSL caused minimal toxicity (10% at 0.1 µmol/L), whereas AIGA was more toxic (40% toxicity at the dose of 0.1 µmol/L) on normal mouse melanocytes (Fig. 1D). Neither CSL nor AIGA elicited toxic effect on human diploid fibroblasts (IMR90) or on the normal human breast cultures MCF10 (data not shown). These data suggest that CSL exhibits potent toxicity toward melanoma cultures but not toward nontransformed cells.
Fig. 1.

AIGA and CSL reduces cell viability of SW1mouse melanoma cells. A, chemical structure of AIGA. B, chemical structure of CSL. C, SW1 cells were seeded on 96-well plates, and 24 h later, cells were treated with the indicated doses of AIGA or CSL for 20 h. ATF251–100 peptide (cloned in mammalian expression vector in frame with penetratin was transfected with the aid of Lipofectamine as described in ref. 31) was used as a positive control. ATP levels were used as an indicator of cell viability using the ATPLite kit. The number of surviving cells on AIGA or CSL treatment was normalized to the number of surviving cells under the control treatment, and the normalized number was designated as the “CellViability (%)”,which is represented by the Y axis. Results shown represent three experiments (P < 0.005 for AIGA and P < 0.0045 for CSL). D, experiment was done as indicated in C, except that normal melanocytes were used for the analysis (P < 0.01 for AIGA and P < 0.05 for CSL).
We next assessed the effects of the two compounds on c-Jun transcriptional activity in the mouse melanoma SW1 cells. Compared with the ATF2 peptide, which elicits an increase in TRE-luciferase, reflective of AP1/c-Jun transcriptional activity, AIGA and CSL elicited stronger activation of c-Jun transcription (Fig. 2A; Supplementary Fig. S1). AIGA induced c-Jun transcriptional activity at doses starting from 0.1 µmol/L (Supplementary Fig. S1). Compared with AIGA, CSL was more potent in the activation of AP1/c-Jun transcription, and as such, activation was seen in response to treatment at doses as low as 0.05 µmol/L (Fig. 2A). These compounds were next assessed for their effect on ATF2 transcriptional activity. About 0.5 µmol/L AIGA or 0.05 µmol/L CSL was required to cause ~ 50% inhibition of ATF2 transcriptional activity, monitored using a Jun2-luciferase construct (Fig. 2B; Supplementary Fig. S2). These data reveal that, similar to the changes seen after treatment with the ATF2 peptide, both compounds effectively alter the transcriptional activities of ATF2 and c-Jun, although CSL seems to be more potent compared with AIGA.
Fig. 2.

AIGA and CSL increase JNK and c-Jun and reduce ATF2 activities. A, SW1 cells were transfected with the TRE-luciferase plasmid, and 24 h later, cells were treated with the indicated doses of CSL for 8 h before proteins were prepared and level of luciferase activity was measured. ATF251–100 peptide was used as a positive control. Data represent results from three experiments (P < 0.005). B, the effect of CSL on Jun2-luciferase activity was assessed, as detailed in A. Data represent results from three experiments (P < 0.003). C, SW1 cells were pretreated (for 30 min) with the JNK inhibitor SP600125 (10 µmol/L) followed by addition of CSL for 20 h. ATP levels were used to measure the cell viability using the ATPLite kit. Calculation for altered viability was done as detailed in legend to Fig.1C. Representative results from two experiments (P < 0.02). D, top, Western blot analysis for JNK phosphorylation (pJNK) on residues 183/185 was done on protein extracts that were prepared from cells treated with CSL, as indicated in A; bottom, total level of JNK. SW1 cells were pretreated with the JNK inhibitor SP600125 followed by addition of AIGA or CSL for 20 h, as indicated in A and B. Total protein extracts were used to determine the phosphorylation of c-Jun (pc-Jun).
To assess whether CSL may affect the transcriptional activities of genes other than ATF2 and c-Jun, we monitored possible changes in transcriptional activities of nuclear factor-κB (NF-κB). Addition of either AIGA or CSL causes dose-dependent inhibition of NF-κB transcriptional activity, measured by the corresponding luciferase reporter construct (Supplementary Figs. S3 and S4). These findings are consistent with earlier reports about the effect of CSL on the transcriptional activities of NF-κB in HeLa cells (36).
To assess the possible effect of these compounds on other signaling pathways, we have monitored changes in the phosphorylation of AKT and ERK. CSL treatment of melanoma cells caused activation of AKT and ERK phosphorylation, suggesting a more global effect on stress-induced signaling (Supplementary Fig. S5).
Our earlier studies have revealed that JNK activity is central to the ability of the ATF2 peptide to elicit its effect on melanoma cells in culture and in vivo (inhibition of tumorigenicity). We therefore assessed whether the two compounds identified in the present study require JNK activity for their effects on melanoma cells. To this end, we first tested the ability of AIGA or CSL to affect the viability of melanoma cells in the presence of a JNK inhibitor. Inhibition of JNK activity by the pharmacologic inhibitor SP600125 attenuated the ability of AIGA or CSL to elicit the cell death of melanoma cells (Fig. 2C; Supplementary Fig. S6). Of note, inhibition of JNK activity was more pronounced at lower concentrations of CSL compared with AIGA. These findings suggest that AIGA and CSL require JNK activity to reduce the survival of melanoma cells.
To assess whether these compounds affect JNK activity, we monitored changes in JNK phosphorylation on residues 183/185, which are required for its kinase activity. Both AIGA and CSL induced phosphorylation of JNK on these residues, which was reduced on addition of the JNK inhibitor (Fig. 2D; Supplementary Fig. S7). The activation of JNK by both compounds was confirmed on analysis of its substrate c-Jun. Both compounds efficiently induced c-Jun phosphorylation, with CSL being more potent (Fig. 2D). These data indicate that CSL and AIGA induce JNK activity, which in turn results in the activation of its substrate c-Jun while attenuating the degree of ATF2 transcriptional activity.
The finding that CSL and AIGA reduced cell viability in melanoma was made using the ATPLite assay, which monitors the degree of mitochondrial ATP release, a marker for mitochondrial activity. To directly assess possible changes in the apoptosis of melanoma cells following their exposure to CSL or AIGA, we have done fluorescence-activated cell sorting analysis of the melanoma cells 48 h after treatment. Addition of AIGA or CSL to SW1 melanoma cells induced programmed cell death, with CSL exhibiting a more efficient effect. Treatment with 1 Amol/L AIGA caused 15% apoptosis in these melanoma cultures (Fig. 3A). Treatment with CSL caused 13% apoptosis at a dose of 0.5 µmol/L and 35% apoptosis at a dose of 1 µmol/L (Fig. 3A). Consistent with these finding is the activation of caspase-8, which was observed on addition of these compounds (Fig. 3B).
Fig. 3.

AIGA and CSL induce apoptosis in melanoma cells. A, SW1 cells were treated with CSL or AIGA at the indicated concentrations, and the degree of apoptosis, measured by fluorescence-activated cell sorting analysis, was determined 48 h later. Data represent three different experiments. B, SW1 cells were treated with AIGA or CSL at the indicated doses for 24 h. Staurosporine was used as a positive control. Immunoblot analysis using caspase-8 antibody monitored full-length and cleaved forms (active) of caspase-8. β-Actin was used as a loading control. The positions of the cleaved forms are indicated. C and D, human melanoma WM115 (C) or MEWO (D) cells were treated with the indicated doses of CSL or AIGA for 20 h. ATP levels were used to measure the cell viability using the ATPLite kit. Calculation was done as detailed in legend to Fig.1C. Representative results from three experiments.
Effects of CSL and AIGA on human melanoma cells
Given the effects of CSL and AIGA on mouse melanoma cells, we next assessed their effects on the human melanoma cell lines WM115 and MEWO. AIGA reduced the viability of the human melanoma cultures when added at a concentration of 0.5 to 2 µmol/L, whereas CSL reduced their viability at a concentration of 1 µmol/L (Fig. 3C and D). These data reveal that, similar to their effect on the mouse melanoma cells, AIGA and CSL efficiently reduced viability of human melanoma cells.
Generation and characterization of CSL derivatives that affect melanoma cells
Given the greater potency of CSL, compared with AIGA, on transcription and cell death, we have devoted efforts to further optimize the ability of CSL to elicit melanoma cell death. To this end, medicinal chemistry was initiated with the dual goals of (a) understanding the key structural features in CSL that affect its apoptotic activity and (b) identifying analogues of CSL that possess an improved pharmacologic profile compared with the parent compound. To this end, we first tested two commercially available analogues of CSL, the CSL methyl ester (pristimerin) and dihydrocelastrol, a reduced variant of CSL that lacks the quinone methide moiety in the parent compound. Pristimerin was equipotent or even slightly more potent than CSL (see Table 1) against SW1 cells. This finding suggests that the acidic proton present in CSL is not required for its ability to induce apoptosis in these cells. Dihydrocelastrol, however, was inactive in the assay, suggesting that the quinone methide moiety in CSL is crucial for apoptotic activity (see Discussion). With this information in hand, we focused on modification of the carboxylic acid moiety in CSL. We first synthesized a series of amide derivatives (CA01–CA08, CA13, and CA14) and these were tested in the cellular assays as shown in Table 1. This set of amides was designed to interrogate both the steric and electronic requirements of the carboxylate substitute needed for optimal cellular activity. The derivatives revealed some intriguing preliminary structure-activity relationship data (Table 1). The 4-methoxybenzyl amide (CA01) and the pyridin-3-ylmethanamine amide (CA03) were active in SW1 cells, whereas the unsubstituted benzyl amide (CA02) was inactive. Among the aliphatic amides, the isopropyl amide (CA04), the dimethyl amide (CA05), the pyrrolidine amide (CA06), and the methoxyethyl-amine (CA07) were all active in SW1 cells. However, the morpholine amide (CA08) and the N-methylpiperazine amide (CA14) were both inactive. Surprisingly, considering the dimethyl amide (CA05) result, the monomethyl amide (CA13) was also inactive (Table 1). This suggested that the presence of an amide N-H reduces cellular activity in this series.
Table 1.
Activity of CSL derivatives
| Code | 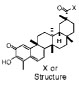 |
Activity or EC50 (µmol/L) in SW1 cells |
Activity or EC50 (µmol/L) in WM115 cells |
Activity or EC50 (µmol/L) in WM793 cells |
|---|---|---|---|---|
| CSL | ~0.1 | Active at 2 | Active at 2 | |
| Pristimerin | <0.1 | ND | ND | |
| Dihydrocelastrol | 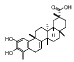 |
Inactive | ND | ND |
| CA01 | Active at 0.3 | Active at 2 | Active at 1 | |
| CA02 | Inactive | Inactive | Inactive | |
| CA03 | Active at 0.3 | Inactive | Inactive | |
| CA04 | Active at 0.3 | Active at 2 | Inactive | |
| CA05 | Active at 0.3 | Active at 0.5 | Inactive | |
| CA06 | Active at 0.3 | Active at 2 | Active at 0.5 | |
| CA07 | Active at 0.3 | Inactive | Active at 2 | |
| CA08 | Inactive | Inactive | Inactive | |
| CA13 | Inactive | Inactive | Inactive | |
| CA14 | Inactive | Inactive | Inactive | |
| CA15 | Inactive | Active at 2 | Active at 1 | |
| CA16 | Active at 0.3 | Active at 0.5 | Active at 0.5 | |
| CA18 | Active at 1* | Active at 2* | Active at 1* | |
| CA19 | Active at 0.3 | Active at 1 | Active at 1* |
NOTE: Active: >50% inhibition of melanoma cell viability. SW1 is a mouse melanoma cell. WM115 and WM793 are human melanoma cells.
Abbreviation: ND, not determined.
Represent single analysis.
These observations prompted us to synthesize and test some ester derivatives based on the rationale that, unlike primary amides, esters lack a heteroatom proton. Thus, the methyl 2-hydroxyacetate ester (CA15), the benzyl ester (CA16), the ethyl ester (CA18), and the isopropyl ester (CA19) were prepared and the cellular assay data are shown in Table 1. Among these four derivatives, two (CA16 and CA19) potently exhibited the ability to induce melanoma cell death. CA18 was not that efficient in the SW1 melanoma cells compared with CA19. Remarkably, the benzyl ester (CA16) was quite potent compared with the corresponding amide derivative (CA02), which was inactive. Taken together, these results suggest that the ester derivatives are more potent than the corresponding amide derivatives. Based on these data, the CA16 and CA19 derivatives were selected as representatives for further assessment in the syngeneic and xenograft mouse models.
Evaluation of CSL and its derivatives in melanoma tumorigenicity
To assess the ability of CSL and its derivatives to affect the tumorigenicity and metastasis of melanoma, we used the mouse SW1 cell lines in syngeneic models. Injection of SW1 melanoma cells s.c. into C3H mice was followed by i.p. injections of CSL on alternate days. Importantly, administration of CSL, CA16, or CA19 to mice at doses up to 1 mg/kg did not result in toxicity or any noticeable discomfort, indicating that these compounds are well tolerated and not toxic at these high doses. Significantly, CSL attenuated the growth of both mouse and human melanoma cells in vivo using immunocompetent and immunodeficient mouse models (Figs. 4 and 5). Comparison of CSL, CA16, and CA19 for their effects on the viability of SW1 cells in culture revealed that both derivatives were as potent as the parent CSL compound (Fig. 4A). Administration of these compounds, compared with vehicle control, revealed that both CSL and the derivatives attenuated growth of SW1 tumors in mice (Fig. 4B). Interestingly, CSL and CA19 were equally potent and reduced tumor growth 5-fold, whereas CA16 was less effective in inhibiting tumor size (~ 50% decrease; Fig. 4B). Representative figures of GFP-labeled tumors in animals treated with control or CSL show the effect of these compounds after 3 weeks of treatment (Fig. 4C). The effect of CSL on tumors that were excised from these animals is also shown (Fig. 4C). Consistent with our finding in cultured cells, CSL treatment also induced JNK activity in the tumors; such activity was not observed in the vehicle-treated tumors (Fig. 4D), thereby supporting the changes seen in the SW1 cultures. These data suggest that CSL and its derivatives efficiently reduce melanoma tumor growth in a syngeneic mouse model.
Fig. 4.

CSL and CSL derivatives, CA16 and CA19, reduce cell viability in vitro and attenuate tumor growth in vivo. A, SW1 cells were seeded on 96-well plates, and after 24 h, cells were treated with the indicated doses of CSL and the select derivatives CA16 and CA19 for 20 h. ATP levels were used to measure the cell viability using the ATPLite kit. Calculation of viability was done as detailed in Fig.1C. Representative results are from three independent experiments. B, SW1 cells that stably express GFP were injected s.c into C3H mice. When the tumor reached the size of 50 mm3, the vehicle, CSL (1mg/kg), CA16 (0.5 mg/kg), or CA19 (0.5 mg/kg) was given i.p. every second day for a period of 24 d. Tumor was measured at the indicated times. Data represent two experiments. Bars, SD. P < 0.005, t test. n = 7. C, representative figures of mice with GFP tumors formed in C3H mice (left panel) or of the tumors excised from the xenograft model in nude mice (right panel). D, tumors detailed in Fig. 5B were excised and paraffin sections were prepared and stained with phosphorylated JNK antibody. Bar, 50 µm. Arrows point to examples of phosphorylated JNK – positive cells.
Fig. 5.

CSL and CSL derivatives, CA16 and CA19, attenuate tumor metastasis in vivo. A, CSL, CA16, or CA19 inhibits lung lesions of human LU1205 cells. Tail vein injection of human melanoma LU1205 cells was followed by analysis of lung lesions. The effect of these compounds on the number and size of lesions obtained after H&E staining on serial sections of lungs was calculated. Average number of lesions was calculated within serial sections of the lungs from every mouse in two independent experiments. n = 10. B, CSL, CA16, or CA19 inhibits lung metastasis of SW1 tumor. S.c. injection of SW1 cells resulted in tumors (Fig. 4B) that within 6 to 8 wk metastasize to the lungs (29). Average number of metastatic lesions in the lungs and the mediastinal lesions was calculated based on analysis of several sections representing different longitudinal sections within the lungs. The change in metastatic lesions was divided based on the size of the lesions, as indicated in A. Data represent average of two experiments. n = 7
We next determined the possible effect of CSL and the selected derivatives on the metastatic capacity of melanoma cells. To this end, human melanoma LU1205 cells were subjected to tail vein injection followed by administration of the compounds every second day. This approach allows monitoring of changes in the number of lesions formed on the lungs, a primary organ for tumor cell growth under this experimental approach. Analysis done 60 days following injection determined changes in the number and size of metastatic lesions in each of the experimental groups. Significantly, CSL and CA19 were efficient in inhibiting growth of large-, medium-, and small-sized lesions in the lungs (Fig. 5A). CA16 had no effect on the development of the large lesions, although some activity was observed for 1 to 2 mm and for mediastinal lesions (Fig. 5A). These data suggest that the compounds are capable of inhibiting metastasis of human melanoma cells in a nude mouse model and that at least one of the two derivatives tested here, CA19, exhibits potent activity that is comparable with CSL.
To further assess the effect of these compounds on the development of metastatic lesions, we turned to the SW1 mouse melanoma model using the syngeneic C3H strain. S.c. injection of SW1 cells results in tumor formation at the site of injection followed by metastasis to different sites within 6 to 8 weeks (31). Similar to what was observed with the human melanoma cells in the nude mice model, CSL and CA19 potently exhibit the ability to inhibit large as well as small metastatic lesions, with the exception of the 1-to 2-mm range (Fig. 5B), whereas CA16 was somewhat active it elicited an opposite effect on the development of small-sized lesions (Fig. 5B). The fluctuations seen with CA16 imply that its activity may be limited due to stability or permeability. These data provide important support for the ability to attenuate tumorigenicity and metastatic lesion formation in melanoma xenograft and syngenic models using CSL and its isopropyl ester derivative CA19.
Discussion
The present study extends earlier reports that documented the ability of the ATF2 peptide to cause apoptosis in mouse and human melanoma cells in culture and in animal models (29–32). To identify compounds that mimic the activity of the ATF2 peptide, we screened a small-molecule chemical library consisting of 3,280 compounds. Two compounds, CSL and AIGA, at low micromolar concentrations, were able to (a) induce melanoma cell death, (b) inhibit the transcriptional activity of ATF2, and (c) induce the activity of JNK and transcriptional activity of c-Jun and required active JNK for their ability to affect melanoma viability.
Gambogic acid was originally isolated from the resin of the Garcinia hanburyi tree, whose cytotoxic properties are recognized (37). In recent studies, AIGA was shown to possess the capacity to potently induce apoptosis in a breast cancer cell line T47D (38), in a human hepatoma cell line SMMC-7721 (39), and in a gastric carcinoma cell line BGC-823, in part through G2-M phase arrest (40). Consistent with these findings, we show herein that AIGA elicits potent inhibition of melanoma cell growth in culture. We further show that AIGA elicits these effects in part through activation of JNK and c-Jun and inhibition of ATF2.
CSL, a quinone methide triterpenoid isolated from the Chinese “thunder of god vine” plant, was reported to exhibit immunosuppressive activities (41). CSL was also shown to induce the heat shock response and to elicit cytoprotection (42). Consistent with these findings, gene expression signature-based chemical genomic prediction identified CSL as a modulator of the HSP90 pathway (43). At micromolar concentrations, CSL was shown capable of inhibiting proteasome activity and the growth of prostate cancer cells in nude mice (44). At the concentrations used in the present studies (0.3–1 µmol/L), CSL did not affect the stability of short-lived proteins, including Mdm2 and p53 (data not shown). CSL was also shown to inhibit NF-κB activation through the inhibition of IκB kinase (36) and TAK1 activities (45). Previous studies showed that NF-κB inhibits JNK activities and that inhibition of NF-κB results in elevated JNK activities (46–49). Thus, it is likely that the ability of CSL to attenuate NF-κB activity would result in increased JNK activity, which is consistent with our findings. It is the increase in JNK activity, and consequently of its substrate c-Jun, in concert with inhibition of ATF2 activities that brings about the potent cytotoxic effects of both CSL and AIGA on melanoma cells, similar to what was previously shown for the ATF2 peptides (29–32, 50).
The greater apoptotic effects seen with CSL compared with AIGA in cultured cells prompted us to focus on CSL. Examination of the structure of CSL led us to postulate that the quinone methide functional group was responsible for its cytotoxic activity. Along these lines, we reasoned that the highly electrophilic nature of the quinone methide unit could potentially lead to irreversible or pseudo-irreversible binding to proteins, possibly through interaction with cysteinyl residues. Supporting this hypothesis is the fact that dihydrocelastrol, which lacks the quinone methide moiety, is inactive in the cellular assays (Table 1). On the other hand, the methyl ester derivative of CSL (pristimerin) is equally efficacious as CSL with respect to its ability to induce apoptosis, suggesting that the presence of the acidic carboxylate functional group in CSL is not required for apoptotic activity but that the quinone methide moiety is responsible for the observed cellular activity in our experiments. Data recently reported by Westerheide et al. (42) support this hypothesis and further suggest that the carboxylic acid moiety in CSL is responsible for the chemical chaperone activity observed in their experiments. Thus, in the study conducted by Westerheide et al. (42), three ester derivatives of CSL were inactive as regulators of the heat shock response, whereas dihydrocelastrol was active as a heat shock promoter. These results suggest that the quinone methide moiety is not responsible for the observed chemical chaperone activity. We therefore synthesized a series of CSL derivatives in which the carboxylic acid unit in CSL was converted to an amide or ester derivative with the quinone methide functional group retained intact in the molecules (see Results). Consistent with a role for the quinone methide unit in the cytotoxicity of CSL, some of the CSL derivatives exhibited improved efficacy (Table 1). These results led to the identification of the benzyl ester (CA16) and isopropyl ester (CA19) derivatives as potent inducers of apoptosis, which were profiled in detail in in vivo models of melanoma. The precise role for the carboxylic acid moiety in this modality of CSL remains to be further elucidated, however.
Consistent with its ability to induce apoptosis of melanoma cells in culture is the ability of CSL and its isopropyl ester derivative CA19 to attenuate the growth of melanoma in mouse models. Our analysis, which was done in syngeneic mouse (SW1 cells in C3H mice) and human xenograft (LU1205 in nude mice) models, shows the ability of these compounds to attenuate growth of metastatic lesions, which are the major clinical burden in treatment of this tumor type. The lower activity of CA16 compared with CA19 and CSL on tumor growth and metastasis development may be attributed to stability or permeability of this compound in vivo. Along these lines, in all cases, it was necessary to inject the compounds at high frequency; it is expected that improved formulation will allow prolonged half-life and more efficient delivery of these compounds. The low concentrations of the compounds used in our present studies would be expected to minimize effect on other organs, consistent with our initial maximum tolerated dose studies.
In conclusion, the effects of AIGA and CSL on melanoma cells in culture and in mouse models point to possible new treatment modalities for melanoma. In light of the notion that MAPK signaling is up-regulated in human melanoma, in many cases as a result of mutations in B-RAF or N-RAS, our findings suggest that these compounds could be also considered as a means of complementing the activities of pharmacologic inhibitors developed against MAPK/ERK kinase.
Supplementary Material
Acknowledgments
We thank M. Herlyn for providing us with the melanoma cell lines, members of the Ronai Lab for discussions, Dr. Marcia Dawson for critical comments, Buddy Charbono for help in injection in the mice, and Robbin Newlin for help in preparation of histologic samples.
Grant support: National Cancer Institute grant CA099961 (Z.A. Ronai).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–2182. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- 2.Reed JA, Medrano EE. Recent advances in melanoma research. Front Biosci. 2006;11:3003–3013. doi: 10.2741/2028. [DOI] [PubMed] [Google Scholar]
- 3.Meier F, Schittek B, Busch S, et al. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci. 2005;10:2986–3001. doi: 10.2741/1755. [DOI] [PubMed] [Google Scholar]
- 4.Halaban R. Rb/E2F: a two-edged sword in the melanocytic system. Cancer Metastasis Rev. 2005;24:339–356. doi: 10.1007/s10555-005-1582-z. [DOI] [PubMed] [Google Scholar]
- 5.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 6.Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126:154–160. doi: 10.1038/sj.jid.5700026. [DOI] [PubMed] [Google Scholar]
- 7.Gorden A, Osman I, Gai W, et al. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res. 2003;63:3955–3957. [PubMed] [Google Scholar]
- 8.Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Goding CR. Mitf from neural crest to melanoma: signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000;14:1712–1728. [PubMed] [Google Scholar]
- 10.Ellerhorst JA, Ekmekcioglu S, Johnson MK, Cooke CP, Johnson MM, Grimm EA. Regulation of iNOS by the p44/42mitogen-activated protein kinase pathway in human melanoma. Oncogene. 2006;25:3956–3962. doi: 10.1038/sj.onc.1209419. [DOI] [PubMed] [Google Scholar]
- 11.Bhatt KV, Spofford LS, Aram G, McMullen M, Pumiglia K, Aplin AE. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene. 2005;24:3459–3471. doi: 10.1038/sj.onc.1208544. [DOI] [PubMed] [Google Scholar]
- 12.Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev. 2005;24:273–285. doi: 10.1007/s10555-005-1577-9. [DOI] [PubMed] [Google Scholar]
- 13.Bedogni B, Welford SM, Cassarino DS, Nickoloff BJ, Giaccia AJ, Powell MB. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell. 2005;8:443–454. doi: 10.1016/j.ccr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Bergami P, Habelhah H, Bhoumik A, Zhang W, Wang LH, Ronai Z. RACK1 mediates activation of JNK by protein kinase C [corrected] Mol Cell. 2005;19:309–320. doi: 10.1016/j.molcel.2005.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillespie S, Zhang XD, Hersey P. Variable expression of protein kinase Cε in human melanoma cells regulates sensitivity to TRAIL-induced apoptosis. Mol Cancer Ther. 2005;4:668–676. doi: 10.1158/1535-7163.MCT-04-0332. [DOI] [PubMed] [Google Scholar]
- 16.Lahn MM, Sundell KL. The role of protein kinase C-α (PKC-α) in melanoma. Melanoma Res. 2004;14:85–89. doi: 10.1097/00008390-200404000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, Horner JW, 2nd, DePinho RA. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997;11:2822–2834. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 19.Carreira S, Goodall J, Aksan I, et al. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433:764–769. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- 20.Melnikova VO, Mourad-Zeidan AA, Lev DC, Bar-Eli M. Platelet-activating factor mediates MMP-2 expression and activation via phosphorylation of cAMP-response element-binding protein and contributes to melanoma metastasis. J Biol Chem. 2006;281:2911–2922. doi: 10.1074/jbc.M508683200. [DOI] [PubMed] [Google Scholar]
- 21.Hofmann UB, Houben R, Brocker EB, Becker JC. Role of matrix metalloproteinases in melanoma cell invasion. Biochimie. 2005;87:307–314. doi: 10.1016/j.biochi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 22.McGary EC, Lev DC, Bar-Eli M. Cellular adhesion pathways and metastatic potential of human melanoma. Cancer Biol Ther. 2002;1:459–465. doi: 10.4161/cbt.1.5.158. [DOI] [PubMed] [Google Scholar]
- 23.Grbovic OM, Basso AD, Sawai A, et al. V600EB-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci U S A. 2006;103:57–62. doi: 10.1073/pnas.0609973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jean D, Harbison M, McConkey DJ, Ronai Z, Bar-Eli M. CREB and its associated proteins act as survival factors for human melanoma cells. J Biol Chem. 1998;273:24884–24890. doi: 10.1074/jbc.273.38.24884. [DOI] [PubMed] [Google Scholar]
- 25.Ronai Z, Yang YM, Fuchs SY, Adler V, Sardana M, Herlyn M. ATF2 confers radiation resistance to human melanoma cells. Oncogene. 1998;16:523–531. doi: 10.1038/sj.onc.1201566. [DOI] [PubMed] [Google Scholar]
- 26.Berger AJ, Davis DW, Tellez C, et al. Automated quantitative analysis of activator protein-2α subcellular expression in melanoma tissue microarrays correlates with survival prediction. Cancer Res. 2005;65:11185–11192. doi: 10.1158/0008-5472.CAN-05-2300. [DOI] [PubMed] [Google Scholar]
- 27.Lopez-Bergami P, Huang C, Goydos S, et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berger AJ, Kluger HM, Li N, et al. Subcellular localization of activating transcription factor 2 inmelanoma specimens predicts patient survival. Cancer Res. 2003;63:8103–8107. [PubMed] [Google Scholar]
- 29.Bhoumik A, Ivanov V, Ronai Z. Activating transcription factor 2-derived peptides alter resistance of human tumor cell lines to ultraviolet irradiation and chemical treatment. Clin Cancer Res. 2001;7:331–342. [PubMed] [Google Scholar]
- 30.Bhoumik A, Gangi L, Ronai Z. Inhibition of melanoma growth and metastasis by ATF2-derived peptides. Cancer Res. 2004;64:8222–8230. doi: 10.1158/0008-5472.CAN-04-0714. [DOI] [PubMed] [Google Scholar]
- 31.Bhoumik A, Huang TG, Ivanov V, et al. An ATF2-derived peptide sensitizes melanomas to apoptosis and inhibits their growth and metastasis. J Clin Invest. 2002;110:643–650. doi: 10.1172/JCI16081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhoumik A, Jones N, Ronai Z. Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc Natl Acad Sci U S A. 2004;101:4222–4227. doi: 10.1073/pnas.0400195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamman BD, Pollok BA, Bennett T, Allen J, Heim R. Binding of a pleckstrin homology domain protein to phosphoinositide in membranes: a miniaturized FRET-based assay for drug screening. J Biomol Screen. 2002;7:45–55. doi: 10.1177/108705710200700107. [DOI] [PubMed] [Google Scholar]
- 34.Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, Caughey B. New inhibitors of scrapie-associated prion protein formation in a library of 2,000 drugs and natural products. J Virol. 2003;77:10288–10294. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kangas L, Grönroos M, Nieminen AL. Bioluminescence of cellular ATP: a new method for evaluating agents in vitro. Med Biol (Helsinki) 1984;62:338–343. [PubMed] [Google Scholar]
- 36.Lee JH, Koo TH, Yoon H, et al. Inhibition of NF-κB activation through targeting IκB kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Asano J, Chiba K, Tada M, Yoshii T. Cytotoxic xanthones from Garcinia hanburyi. Phytochemistry. 1996;41:815–820. doi: 10.1016/0031-9422(95)00682-6. [DOI] [PubMed] [Google Scholar]
- 38.Zhang HZ, Kasibhatla S, Wang Y, et al. Discovery, characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay. Bioorg Med Chem. 2004;12:309–317. doi: 10.1016/j.bmc.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 39.Guo QL, You QD, Wu ZQ, Yuan ST, Zhao L. General gambogic acids inhibited growth of human hepatoma SMMC-7721cells in vitro and in nude mice. Acta Pharmacol Sin. 2004;25:769–774. [PubMed] [Google Scholar]
- 40.Jun Y, Guo QL, You QD, et al. Gambogic acid induced G2/M phase cell cycle arrest via disturbing CDK7 mediated phosphorylation of CDC2/P34 in humangastric carcinomaBGC-823 cells. Carcinogenesis. 2007;28:632–638. doi: 10.1093/carcin/bgl168. [DOI] [PubMed] [Google Scholar]
- 41.Zhou BN. Some progress on the chemistry of natural bioactive terpenoids from Chinese medicinal plants. Mem Inst Oswaldo Cruz. 1991;86 Suppl 2:219–226. doi: 10.1590/s0074-02761991000600049. [DOI] [PubMed] [Google Scholar]
- 42.Westerheide SD, Bosman JD, Mbadugha BN, et al. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 43.Hieronymus H, Lamb J, Ross KN, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 45.Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-κB-regulated gene products and TAK1-mediated NF-κB activation. Blood. 2007;109:2727–2735. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 46.Javelaud D, Besancon F. NF-κB activation results in rapid inactivation of JNK in TNFα-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-κB. Oncogene. 2001;20:4365–4372. doi: 10.1038/sj.onc.1204570. [DOI] [PubMed] [Google Scholar]
- 47.Tang G, Minemoto Y, Dibling B, et al. Inhibition of JNK activation through NF-κB target genes. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 48.Eliseev RA, Zuscik MJ, Schwarz EM, O’Keefe RJ, Drissi H, Rosier RN. Increased radiation-induced apoptosis of Saos2 cells via inhibition of NFκB: a role for c-Jun N-terminal kinase. J Cell Biochem. 2005;96:1262–1273. doi: 10.1002/jcb.20607. [DOI] [PubMed] [Google Scholar]
- 49.Wullaert A, Heyninck K, Beyaert R. Mechanisms of crosstalk between TNF-induced NF-κB and JNK activation in hepatocytes. Biochem Pharmacol. 2006;72:1090–1101. doi: 10.1016/j.bcp.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 50.Ivanov VN, Fodstad O, Ronai Z. Expression of ring finger-deleted TRAF2 sensitizes metastatic melanoma cells to apoptosis via up-regulation of p38,TNF α and suppression of NF-κB activities. Oncogene. 2001;20:2243–2253. doi: 10.1038/sj.onc.1204314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.