

Abstract
Arginase, a key metalloenzyme of the urea cycle that converts L-arginine into L-ornithine and urea, is presently considered a pharmaceutical target for the management of diseases associated with aberrant L-arginine homeostasis, such as asthma, cardiovascular diseases, and erectile dysfunction. We now report the design, synthesis, and evaluation of a series of 2-aminoimidazole amino acids inhibitors in which the 2-aminoimidazole moiety serves as a guanidine mimetic. These compounds represent a new class of arginase inhibitors. The most potent inhibitor identified in this study, 2-(S)-amino-5-(2-aminoimidazol-1-yl)-pentanoic acid (A1P, 10), binds to human arginase I with Kd = 2 μM and significantly attenuates airways hyperresponsiveness in a murine model of allergic airways inflammation. These findings suggest that 2-aminoimidazole amino acids represent new leads for the development of arginase inhibitors with promising pharmacological profiles.
Introduction
Arginase is a metalloenzyme that catalyzes the hydrolysis of L-arginine to form L-ornithine and urea. Two distinct isozymes with different tissue distributions, subcellular localizations, and metabolic functions have been identified in mammals.1 The cytosolic isozyme, arginase I, is found mainly in the liver where it catalyzes the final cytosolic step of the urea cycle. Here, arginase activity allows for the ultimate excretion of excess nitrogen resulting from protein catabolism in the form of urea.2 The mitochondrial isozyme, arginase II, is predominantly expressed in extrahepatic tissues3 where it serves to regulate L-arginine homeostasis and biosynthetic pathways dependent on L-arginine and L-ornithine. These pathways include nitric oxide (NO) biosynthesis, L-proline biosynthesis to support collagen production (as an alternative to the L-glutamate derived biosynthesis of L-proline), and polyamine biosynthesis to facilitate cellular proliferation (Figure 1).4–6 Arginase I is also expressed in extrahepatic tissues to regulate L-arginine homeostasis.7 For example, arginase I expression is significantly increased in the asthmatic lung.8 Arginase I is more strongly induced by Th2 cytokines such as IL-13 that have a critical role in the development of asthma in experimental models,9 while arginase I and II single-nucleotide polymorphisms may contribute to atopic asthma.10 Additionally, arginase inhibition or posttranscriptional gene silencing of arginase I significantly decreases airways hyperresponsiveness induced by IL-13 or aeroallergens in different animal models.11 The role of arginase in asthma has been recently reviewed.12 In addition to asthma, the biochemical interplay between arginase and nitric oxide synthase makes arginase an increasingly important pharmaceutical target for the management of various diseases associated with aberrant L-arginine homeostasis and/or NO biosynthesis, such as erectile dysfunction,13,14 pulmonary diseases such as cystic fibrosis,15 cardiovascular diseases including atherosclerosis,16 and cancer tumor growth.17
Figure 1.

Pathways of L-arginine metabolism.
In the active site of arginase, the binuclear manganese cluster and residues that interact with the α-amino and α-carboxylate groups of amino acid inhibitors constitute essential molecular recognition elements for high affinity binding. X-ray crystallographic studies of rat arginase I and human arginase I complexed with inhibitors such as 2-(S)-amino-6-boronohexanoic acid (ABH)13a,18,19 and S-(2-boronoethyl)-L-cysteine (BEC)13b,19 serve to guide continuing studies toward the structure-based design of new inhibitors that bear alternative functional groups targeting coordination interactions with the binuclear manganese cluster. New inhibitors with different chemical properties may exhibit alternative pharmacokinetic profiles that enable their study as potential lead compounds for drug development.
We have previously shown that the guanidine side chain of L-arginine is not a particularly effective metal ligand.20 The positively charged guanidinium group has a high pKa value of 12.5–13,21 so protonation significantly competes with potential metal coordination behavior. However, the incorporation of a guanidinium group within a 2-aminoimidazole heterocycle reduces the pKa by 4–5 units,22 which in turn can facilitate metal coordination interactions and improve the pharmacokinetic profile. Accordingly, 2-aminoimidazole derivatives display a broad range of biological properties and represent significant precursors for drug design and natural products synthesis. Interestingly, 2-aminoimidazoles have been isolated from marine sponges as secondary metabolites known as oroidins, which exhibit a wide range of biological activities.23–25
Although certain 2-aminoimidazole amino acids have been reported with potential applications in medicinal chemistry,26–28 such compounds have never been investigated as arginase inhibitors. Here, we report the structure-based design, synthesis, and evaluation of a series of 2-aminoimidazole amino acid inhibitors of human arginase I. The most active of these compounds, 2-(S)-amino-5-(2-aminoimidazol-1-yl)-pentanoic acid (A1P, 10), binds to human arginase I with low micromolar affinity and significantly reduces airways hyperresponsiveness in a murine model of allergic airways inflammation. Based on these findings, we advance that 2-aminoimidazole amino acids may serve as valuable leads for the development of a new class of arginase-directed drugs. Moreover, the utilization of a 2-aminoimidazole moiety to target metal coordination interactions is potentially generalizable toward the inhibition of other binuclear metallohydrolases.
Results
Chemistry and In Vitro Structure-Activity Relationships (SAR)
2-Aminoimidazole (Table 1) is a weak noncompetitive inhibitor of human arginase I with Ki = 3.6 mM. The X-ray crystal structure of the enzyme-inhibitor complex (Figure 2) reveals that 2-aminoimidazole binds in the active site but does not interact directly with the binuclear manganese cluster. Instead, the inhibitor is aligned parallel to the imidazole group of H126 with a mean inter-plane distance of ~4 Å. Additionally, the 2-amino group donates a hydrogen bond to a water molecule that in turn donates a hydrogen bond to the metal-bridging hydroxide ion. In studying the structure of this complex, we reasoned that the 2-aminoimidazole moiety could be targeted to interact directly with the binuclear manganese cluster by incorporating it within the side chain of an L-amino acid.
Table 1.
2-Aminoimidazole and Amino Acid Derivatives
| Synthesized as Inhibitors of Human Arginase I | |
|---|---|
| Compound | Ki (μM)a |
| 2-Aminoimidazole | |
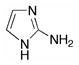 |
3600 ± 20 |
| 2AH | |
 |
300 ± 9 |
| AHH | |
 |
3000 ± 10 |
| A1P | |
 |
4.0 ± 0.2 2.0 ± 0.1b |
| A4P | |
 |
> 800000 |
| APP | |
 |
500 ± 8 |
Kinetic assay. Errors are standard deviations of experiments run in triplicate.
Surface plasmon resonance measurement of the dissociation constant, Kd.
Figure 2.

Stereoview of a simulated annealing gradient map showing 2-aminoimidazole (3.0 σ contour, cyan) bound to human arginase I. Dashed lines indicate manganese coordination (red) and hydrogen bond (green) interactions. Atom color codes: carbon (yellow), oxygen (red), nitrogen (blue), manganese (violet).
The synthesis of the simplest member of the series of 2-aminoimidazole amino acids shown in Table 1, L-2-aminohistidine (2AH), was achieved using a previously published procedure.26 This compound exhibits noncompetitive inhibition against human arginase I with a ~10-fold increase in inhibitor binding affinity (Ki = 0.3 mM) relative to 2-aminoimidazole.
The X-ray crystal structure of the human arginase I-2AH complex (Figure 3) reveals that the 2-aminoimidazole group is repositioned in the active site, but the side chain is too short to interact directly with the binuclear manganese cluster.
Figure 3.

Stereoview of a simulated annealing omit map showing the inhibitor 2AH and a sulfate ion bound to human arginase I. The inhibitor (3.0 σ contour, cyan) and sulfate ion (3.5 σ contour, red) were omitted from the structure factor calculation. The sulfate ion coordinates to the manganese ions and accepts hydrogen bonds from the side chain of 2AH. Dashed lines indicate manganese coordination (red) and hydrogen bond (green) interactions. Atom color codes: carbon (yellow), oxygen (red), nitrogen (blue), manganese (violet), sulfur (green).
However, inhibitor binding recruits a tetrahedral anion interpreted as sulfate to achieve manganese coordination. As observed for the binding of other amino acid inhibitors,19,29 the α-amino group donates hydrogen bonds to D183 and two water molecules, and the α-carboxylate group accepts hydrogen bonds from N130, S137, and two water molecules (Figure 3). Hydrogen bond interactions between the 2-aminoimidazole group and the metal-bound sulfate anion stabilize inhibitor binding and may contribute to the ~10-fold increase in affinity relative to 2-aminoimidazole. Although the binding affinity of 2AH is modest, the intermolecular interactions observed in the human arginase I-2AH complex illuminate a novel coordination mode to the binuclear manganese cluster.
Given that the side chain of 2AH is too short to allow the 2-aminoimidazole group to interact directly with the manganese ions, we synthesized the congener 2-aminohomohistidine (AHH, Table 1), which bears a side chain one carbon longer, using a previously published procedure.27 AHH is a competitive inhibitor with Ki = 3 mM for the racemic mixture, indicating no significant improvement in the binding affinity compared with 2AH.
The X-ray crystal structure of the human arginase I-AHH complex (Figure 4) reveals that the α-amino and α-carboxylate groups make the expected hydrogen bond interactions in the active site as observed for the binding of other amino acid inhibitors.19,29 Additionally, the 2-aminoimidazole group of AHH displaces the metal-bridging hydroxide ion of the unliganded enzyme and coordinates to Mn2+A with an average N---Mn2+A separation of 2.5 Å. The N---Mn2+B separation is 2.8 Å, which is too long to be considered an inner sphere coordination interaction. The imidazole group of AHH is sandwiched between the imidazole groups of H126 and H141 with mean inter-plane separations of 3.5 Å and 3.6 Å, respectively. The lengthy metal coordination interactions observed in this enzyme-inhibitor complex, which should be closer to 2.0 Å for optimal N---Mn2+ interactions, may be responsible for the lower binding affinity of AHH.
Figure 4.

Stereoview of a simulated annealing omit map showing the inhibitor AHH bound to human arginase I. The inhibitor (3.0 σ contour, cyan) was omitted from the structure factor calculation. Dashed lines indicate manganese coordination (red) and hydrogen bond (green) interactions. Atom color codes: carbon (yellow), oxygen (red), nitrogen (blue), manganese (violet).
Encouraged by these structural results, we opted to investigate 3 different derivatives bearing aminoimidazole side chains one carbon longer than that of AHH: 2-(S)-amino-5-(2-aminoimidazol-1-yl)-pentanoic acid (A1P), 2-amino-5-(2-aminoimidazol-4-yl)-pentanoic acid (A4P), and (S)-2-amino-5-(imidazol-2-ylamino)pentanoic acid (APP) (Table 1). We used the protected L-amino acid aldehyde 4 and the triprotected 2-aminoimidazole 5 as key intermediates in the syntheses of these derivatives (Scheme 1).
Scheme 1.

Synthesis of the Key Intermediates 4 and 6
The aldehyde 4 was synthesized from commercially available (S)-5-t-butoxy-4-(t-butoxycarbonylamino)-5-oxopentanoic acid (1) that was first esterified with methyl chloroformate, then further N-protected with a second t-butoxycarbonyl group (intermediates 2 and 3, respectively). Selective reduction of the less sterically hindered methyl ester group of 3 with di-i-butylaluminum hydride (DIBAH) at −78 °C provided the corresponding aldehyde 4 in good overall yield.30 The heterocyclic intermediates were generated by the treatment of 2-aminoimidazole with di-t-butyl pyrocarbonate, yielding the derivative 5 that was selectively deprotected with ammonia in methanol to form 2-[bis(t-butoxycarbonyl)amino]-imidazole (6).
While the synthesis of A1P has been previously reported,31 we developed an alternative synthetic route with a comparable overall yield. We reduced the aldehyde intermediate 4 with DIBAH to form alcohol 7 (Scheme 2).
Scheme 2.

Synthesis of A1P
Bromination of 7 with triphenyl phosphine and carbon tetrabromide led to the corresponding bromo derivative 8 (as previously reported by our group32), which was then coupled to the selectively protected 2-aminoimidazole 6 with potassium carbonate in dimethylformamide (DMF) to yield protected A1P (9); complete deprotection with trifluoroacetic acid (TFA) in methylene chloride generated A1P (10) in good overall yield. While it is conceivable that racemization of the amino acid could occur under these conditions, racemization of a related amino acid was blocked by the utilization of a bulky t-butyl protecting group on the α-carboxylate,33 as we have similarly utilized for the preparation of A1P.
Two different assays indicate that A1P binds to human arginase I with low micromolar affinity. The fixed-point kinetic assay using 14C-labeled L-arginine34 demonstrates that A1P is a competitive inhibitor with Ki = 4 μM (Figure 5A). Surface plasmon resonance analysis35 confirms low micromolar affinity with Kd = 2 μM in a 1:1 Langmuir binding model (Figure 5B). Unfortunately, we were unable to prepare crystals of human arginase I complexed with A1P by crystal soaking or co-crystallization approaches.
Figure 5.

A) Kinetic replot of the inhibition of human arginase I by A1P yields Ki = 4 μM. B) Surface plasmon resonance sensorgram showing the binding of A1P to human arginase I; Kd = 2 μM.
The N-alkylated derivative (APP, 12) was synthesized by the reductive coupling of aldehyde intermediate 4 with 2-aminoimidazole to yield protected APP (11); deprotection with TFA in methylene chloride generated APP (12) (Scheme 3).
Scheme 3.

Synthesis of APP
Enzymological measurements indicate that APP binds to human arginase I with modest affinity (Ki = 500 μM, Table 1). Alkylation via the 2-amino group increases the length of the amino acid side chain, which may contribute to decreased affinity relative to A1P.
The synthesis of A4P (Scheme 4) started from the commercially available 2,6-diaminoheptanedioic acid, which was diesterified with thionyl chloride in methanol, then N-protected with di-t-butyl pyrocarbonate in acetonitrile in the presence of dimethylamino pyridine (DMAP) to generate the protected intermediate 13.
Scheme 4.

Synthesis of A4P
One ester group of 13 was then selectively reduced to the corresponding aldehyde 14 by using 1 equivalent of DIBAH. Deprotection with 4N HCl and ethyl acetate (EtOAc) generated the diamino aldehyde 15 that was condensed with cyanamide in water36 at pH 4.5 to afford A4P (16). Enzymological assay indicates that A4P lacks inhibitory activity (Ki > 0.8 M).
Arginase Inhibition by A1P in a Murine Model of Allergic Airways Inflammation
To determine the effectiveness of the tightest binding 2-aminoimidazole amino acid, A1P, for reducing airways hyperresponsiveness in vivo, we used an acute (3-week) ovalbumin (OVA)-sensitization and -challenge murine model, as previously described.8a In this model, mice sensitized with OVA, but challenged with PBS (OVA/PBS) serve as controls, while OVA-sensitized and -challenged mice (OVA/OVA) develop lung-specific airways inflammation and airways hyperresponsiveness, as a model of asthma. We have previously shown that pharmacologic inhibition of arginase with BEC attenuates maximum central airways responsiveness (RN max) to methacholine in this model.8a Consistent with the effective in vivo inhibition of arginase, treatment with nebulized A1P (80 μg/g) attenuated significantly the total resistance of the respiratory system (R) following methacholine challenge in OVA/OVA mice (Figure 6A). Similarly, the maximum resistance of the central airways (RN) was significantly attenuated by A1P treatment in the same mice(Figure 6B).
Figure 6.

Impact of arginase inhibition by A1P in the acute ovalbumin (OVA)-sensitization and -challenge murine model of allergic airways inflammation. A) A1P attenuated significantly the maximum total lung resistance (R) evoked by methacholine challenge in mice sensitized to OVA and challenged with nebulized vehicle (OVA/PBS), or OVA (OVA/OVA and OVA/OVA A1P). B) A1P also notably decreased the maximum methacholine-induced increase in central airways responsiveness (RN) in the same animal model. Values are expressed as the means ± SE (n = 12/group) (*P < 0.05; ***P < 0.0001 to OVA/PBS; ##P < 0.01 to OVA/OVA).
Discussion
Imidazole and related azoles are well known ligands in metalloenzyme active sites, and ongoing explorations of azole derivatives as potential pharmacophores have inspired the current study. For example, spectroscopic studies on the binding of imidazole and its analogues 1,2,3-triazole, 1,2,4-triazole, and tetrazole to Co2+-substituted human carbonic anhydrase I and bovine carbonic anhydrase II suggest that 1,2,3-triazole forms a penta-coordinated metal complex, while 1,2,4-triazole and tetrazole bind in tetrahedral metal complexes.37 The crystal structure of human carbonic anhydrase I complexed with imidazole reveals the inhibitor bound via one of its nitrogen atoms as a fifth ligand to the Zn2+ ion,38 and the structure of human carbonic anhydrase II complexed with 1,2,4-triazole shows that the heterocycle coordinates to Zn2+ through the N4 atom.39 In other systems, the X-ray crystal structure of methionine aminopeptidase-2 in complex with a 1,2,4-triazole derivative reveals a novel binding mode that may account for nanomolar affinity: two of the heterocyclic N atoms coordinate the Co2+ ion, while the third one makes a hydrogen bond to a histidine residue in the active site.40 Finally, a common binding mode for imidazole-based heme oxygenase-1 inhibitors has been recently reported, revealing that the imidazole group coordinates to the heme iron.41
In contrast with imidazole and the azoles described above, 2-aminoimidazoles have had only limited study in the design of metalloenzymes inhibitors. To our knowledge, the only examples reported to date are those studied as inhibitors of NO synthase.28,42 Optical absorption, magnetic circular dichroism and electronic paramagnetic resonance analyses of endothelial nitric oxide synthase complexed with imidazole derivatives suggest the direct binding of 2-aminoimidazole to the guanidine binding subdomain near the catalytic heme group.42 Accordingly, such derivatives are considered promising ligands for exploring the active sites of different forms of nitric oxide synthase and for the development of isozyme-selective inhibitors. Some 2-amino-5-azolylpentanoic acids related to L-ornithine have also been reported as nitric oxide synthase inhibitors.28 However, we have determined that the 2-aminoimidazole amino acids shown in Table 1 are only weak or modest inhibitors of recombinant rat neuronal nitric oxide synthase (nNOS) or recombinant murine inducible nitric oxide synthase (iNOS), with IC50 values ranging 65–3300 μM (data not shown). Moreover, the new lead arginase inhibitor identified in the current study, A1P (10), is only a weak inhibitor of iNOS and nNOS, with IC50 values of 480 μM and 135 μM, respectively (data not shown). Such selectivity for arginase inhibition may facilitate the use of A1P and its derivatives for the selective modulation of L-arginine levels in the management of diseases associated with aberrant L-arginine homeostasis.
Notably, in vitro SAR based on biological assays and X-ray crystallographic studies show a clear correlation of inhibitor potency with the length of the carbon-linker separating the heterocycle and the amino acid moieties: the highest efficacy observed for A1P is associated with a side chain that is the same length as that of the natural substrate, L-arginine. Interestingly, 2-aminoimidazoles smaller than L-arginine (2AI and 2AH) are noncompetitive inhibitors, whereas inhibitors that are more isosteric with L-arginine (AHH and A1P) are competitive inhibitors. The orientation of the 2-aminoimidazole moiety as it is attached to the amino acid side chain is a critical affinity determinant, as shown by differences in the inhibitory potencies of A1P, A4P, and APP. Since the pKa of the 2-aminoimidazole moiety is relatively invariant to alkylation on imidazole N or C atoms,22 differences in pKa cannot contribute to differences in inhibitor affinity. Accordingly, the optimal affinity of A1P in the series of compounds shown in Table 1 correlates with a 2-aminoimidazole orientation that is most closely isosteric with the guanidinium moiety of the arginase substrate L-arginine. We speculate that this optimizes enzyme-inhibitor metal coordination interactions.
Human asthma patients have increased levels of expression and activity of arginase I in the airways,8 similar to the mouse model of OVA-induced airways inflammation43 used in this study. Recently, we have demonstrated that arginase inhibition by the boronic acid derivative BEC reduces airways hyperresponsiveness in murine models of allergic airways inflammation.8a The current study indicates that A1P is similarly effective in vivo as an arginase inhibitor in this acute asthma model.
Summary and Conclusions
This is the first investigation of arginase inhibition using the 2-aminoimidazole moiety as a “warhead” to target the binuclear manganese cluster. Importantly, A1P is a mimetic of the substrate L-arginine in which the side chain Nε and Nη atoms are covalently linked via the imidazole heterocycle:

Incorporation of the guanidinium nitrogen atoms within the imidazole moiety serves to depress the pKa of the side chain to ~7–8, which can also lead to improved bioavailability. Our SAR analysis demonstrates that the 2-aminoimidazole moiety can target metal coordination and hydrogen bond interactions in the enzyme active site. The most potent inhibitor studied, A1P, exhibits low micromolar binding affinity. Moreover, A1P inhibits arginase in vivo and significantly reduces airways hyperresponsiveness to methacholine in OVA-sensitized and -challenged Balb/C mice. Only three other types of amino acid inhibitors are known to bind to arginase with low micromolar affinity or better: those bearing boronic acid side chains,13b,18 one bearing an N-hydroxyguanidinium side chain,44 and one bearing an N-hydroxyamino side chain (N-hydroxy-L-lysine).45 Therefore, we now add the 2-aminoimidazole amino acid A1P to this important group of lead compounds for the development of new arginase inhibitors that may exhibit alternative and improved pharmacokinetic profiles.
Experimental Section
Synthesis of 2-Aminoimidazole Amino Acids. General Procedures
All reagents were of at least 95% purity, purchased from Sigma Aldrich Co., Fisher Scientific, or Alfa Aesar, and used as received. All solvents were of HPLC grade and purchased from Fisher Scientific or Sigma Aldrich Co. For anhydrous conditions, solvents were freshly distilled under N2 (Et2O from Na, CH2Cl2 from P2O5, and THF from Na/benzophenone). Reactions were monitored by TLC with Sigma-Aldrich aluminum plates (silica gel F254, 60 Å-0.25-mm), visualized by quenching under UV light, equilibration in a glass chamber containing iodine, and/or stained with ninhydrin solution. Flash column chromatography was performed using Fisher Scientific silica gel 60 (230–400 mesh). HPLC was performed on a Waters system with a 1525 binary pump and a 2487 dual λ absorbance UV detector (detection at 254 nm), using a Symmetry® (C18) 5 μM column (4.6 mm × 150 mm) (MeCN:H2O:TFA 10:90:0.1; isocratic elution). Mass spectrometry was carried out on Waters Acquity SQD UPLC-MS (low resolution) and on LCT Premier XE Micromass/Waters MS Technologies (high resolution). Purities of all synthesized and tested compounds were greater than 95 %.
1H and 13C NMR spectra were recorded on Bruker DMX 360 and DRX 500 spectrometers at 360 and 500 MHz for 1H, and at 90.6 and 125.6 MHz, respectively for 13C NMR. Assignments were made based on chemical shifts, signal intensity, COSY, and HMQC sequences. 1H and 13C NMR chemical shifts (δ) are reported in ppm relative to the residual solvent peaks. 1H NMR coupling constants (J) are reported in Hz, and multiplicities are denoted as follows: s, singlet; d, doublet; t, triplet; m, multiplet; bs, broad singlet.
2-Bis(t-butyloxycarbonyl)amino-1-t-butyloxycarbonylimidazole (5)
2-Aminoimidazolium sulfate (5.5 g, 41.5 mmol) was dissolved in water (25 mL) and treated with a solution of NaOH (1.7 g, 42.5 mmol) in water (3 mL). The water was removed under reduced pressure; the residue was extracted with MeOH (30 mL) and the methanolic extracts rotoevaporated to dryness. The resulting light brown oil was suspended in MeCN (30 mL), then NEt3 (20 mL) and DMAP (1 g) were added. The suspension was cooled to 0 °C, then di-t-butyl pyrocarbonate (27.6 g, 166 mmol) in MeCN (30 mL) was added dropwise. The mixture was stirred at 20 °C for 16 h, after which di-t-butyl pyrocarbonate (6.9 g, 41.5 mmol) in MeCN (15 mL) was added. The resulting suspension was refluxed for 8 h. The solvent was rotoevaporated and the residue partitioned between CH2Cl2 (200 mL) and water (100 mL). The aqueous layer was reextracted with CH2Cl2 (4 × 50 mL), and the combined organic extracts were washed with water (100 mL), sat. aq. NaHCO3 (100 mL), brine (100 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash column chromatography on SiO2 (hexane/EtOAc gradients) afforded 5 (10.1 g, 63%) pure product as colorless oil. 1H-NMR (CDCl3): δ 7.39 (d, J = 1.9, 1H, HC-4 of imidazole), 6.93 (d, J = 1.9, 1H, HC-5 of imidazole), 1.58 (s, 9H, N-1Boc), 1.43 (s, 18H, 2-NBoc2). 13C-NMR (CDCl3): δ 149.4 (2CO, 2-NBoc2), 146.4 (CO, N1-Boc), 138.3 (C-2 of imidazole), 126.7 (C-4/C-5 of imidazole), 118.5 (C-5/C-4 of imidazole), 86.1 (CMe3, N-1Boc), 83.6 (2CMe3, 2-NBoc2), 27.9 (9CH3, 3Boc). MS ES+ m/z 384.2 (M+H)+
2-Bis(t-butyloxycarbonyl)aminoimidazole (6)
To a solution of 5 (1.48 g, 3.9 mmol) in MeOH (20 mL) at room temperature NH3 in MeOH (3 mL, 7N) was added. The mixture was stirred at room temperature for 48 h, then the solvent was removed in vacuo. Purification by flash column chromatography on SiO2 (hexane/EtOAc gradient) yielded 6 (0.83 g, 56%) as white solid. 1H-NMR (DMSO-d6): δ 12.13 (s, 1H, NH of imidazole), 7.07 (s, 1H, HC-4/C-5 of imidazole), 6.80 (s, 1H, HC-5/C-4 of imidazole), 1.38 (s, 18H, 2Boc). 13C-NMR (DMSO-d6): δ 156.2 (2CO, 2Boc), 140.7 (C-2 of imidazole), 126.5 (C-4/C-5 of imidazole), 116.3 (C-5/C-4 of imidazole), 82.6 (2CMe3, 2Boc), 27.4 (6CH3, 2Boc). MS ES+ m/z 284.2 (M+H)+.
2-[Bis(t-butyloxycarbonyl)amino]-5-[2-bis(t-butyloxycarbonyl)aminoimidazol-1-yl]-pentanoic acid t-butyl ester (9)
Anhydrous K2CO3 (0.77 g, 5.6 mmol) was suspended in DMF (7.5 mL) and purged with N2 for 5 min. 6 (0.80 g, 2.8 mmol) was added in one portion under N2 purging, followed by the bromide 8 (1.3 g, 2.9 mmol) in DMF (3 mL). The mixture was warmed to 60 °C, while stirring vigorously and purging with N2, then the N2 line was removed and stirring was continued overnight at 60 °C under inert atmosphere. The reaction mixture was allowed to cool at room temperature, poured into cold water (50 mL), and extracted with CH2Cl2 (5 × 40 mL). The combined organic extracts were washed with water (50 mL), brine (50 mL), dried (Na2SO4), filtered, and concentrated. Flash chromatography on silica gel (hexane/EtOAc gradients) afforded 9 (1.1 g, 60%) as a light brown oil. 1H-NMR (CDCl3): δ 7.00 (d, J = 1.2, 1H, HC-4/C-5 of imidazole), 6.89 (d, J = 1.2, 1H, HC-5/C-4 of imidazole), 4.69 (dd, J = 8.4, 5.6, 1H, CHCOO), 3.73 (dd, J = 7.1, 5.8, 2H, CH2N), 2.1-1.81 (m, 4H, CHCH2 + CHCH2CH2), 1.48 (s, 18H, 2Boc), 1.42 (s, 18H, 2Boc), 1.41 (s, 9H, 3CH3 of COOtBu). 13C-NMR (CDCl3): δ 169.5 (CO, COOtBu), 152.9 (3CO, 3Boc), 150.1 (CO, Boc), 138.2 (C-2 of imidazole), 127.2 (C-4/C-5 of imidazole), 118.7 (C-5/C-4 of imidazole), 83.4 (2CMe3, 2Boc), 81.9 (2CMe3, 2Boc), 77.6 (CMe3, COOtBu), 58.5 (CHCOO), 45.3 (CH2N), 28.4 (6CH3, 2Boc), 28.2 (6CH3, 2Boc), 28.1 (3CH3 of COOtBu), 27.2 (CHCH2), 26.9 (CHCH2CH2). MS ES+ m/z 655.4 (M+H)+.
2-(S)-amino-5-(2-aminoimidazol-1-yl)-pentanoic acid ammonium salt (A1P, 10)
The Boc-protected derivative 9 (0.65 g, 1 mmol) was dissolved in CH2Cl2 (4 mL), cooled at 0 °C (water/ice bath) and treated dropwise with TFA (4 mL) with constant stirring. The flask was allowed to reach room temperature and the mixture stirred for another 4 h until TLC monitoring (MeOH:CH2Cl2 5:95) revealed complete deprotection. Evaporation, washing with CHCl3, and drying under vacuum yielded 10 (0.39 g, 92%) as a bis-trifluoroacetate salt. Further purification by flash column chromatography (CHCl3/MeOH/NH3 gradients) afforded 10 (0.31 g) as a colorless oil. 1H-NMR (D2O): δ6.80 (s, 1H, HC-4 of imidazole), 6.70 (s, 1H, HC-5 of imidazole), 3.86 (m, 2H, CH2N), 3.66 (m, 1H, CHCOO), 1.83 (m, 4H, 2CH2). 13C-NMR (D2O): d 176.2 (CO), 147.5 (C-2 of imidazole), 119.1 (C-4 of imidazole), 116.4 (C-5 of imidazole), 54.5 (CHCOO), 44.1 (CH2N), 28.2 (CHCH2CH2CH2N), 24.6 (CHCH2CH2CH2N). A1P−Na+ (pH = 14): 1H-NMR (D2O + NaOD): δ 6.61 (s, 1H, HC-4 of imidazole), 6.46 (s, 1H, HC-5 of imidazole), 3.63 (m, 2H, CH2N), 3.11 (m, 1H, CHCOO), 1.60 (m, 2H, CHCH2CH2CH2N), 1.44 (m, 2H, CHCH2CH2CH2N). A1P.2HCl (pH = 1): 1H-NMR (D2O): δ 6.75 (bs, 2H, HC-4, HC-5 of imidazole), 3.96 (t, J = 5.8, 1H, CHCOO), 3.84 (t, J = 6.4, 2H, CH2N), 1.84 (m, 4H). HRMS m/z 199.1188 (calcd for M+H, 199.1195).
2-[Bis(t-butyloxycarbonyl)amino]-5-(imidazol-2-ylamino)-pentanoic acid t-butyl ester (11)
2-aminoimidazolium sulfate (2 g, 15 mmol) was dissolved in water (10 mL) and treated with NaOH (0.6 g, 15 mmol) in water (3 mL). The solvent was removed under vacuum and the evaporation residue washed with EtOH (50 mL), then sonicated in EtOH (50 mL) for 10 min to ensure the complete extraction of 11 from the inorganic salt matrix. The resulting light brown solution was filtered and the inorganic precipitate washed with EtOH (3 × 10 mL). Rotoevaporation to dryness of the combined EtOH extracts yielded the 2-aminoimidazole free base (1.25 g, 99%) as brown oil. The crude product was retaken in MeOH (10 mL), aldehyde 4 (3 g, 7.75 mmol) was added, and the resulting mixture was treated with NaBH3CN (300 mg, 4.7 mmol) and activated molecular sieves (4 Å, 2 g). The suspension was stirred at room temperature until TLC indicated no further advancement of the reaction (48 h). Column chromatography of the residue (silica gel, hexane/EtOAc gradient) yielded 11 (1.05 g, 30%) as a purple oil (the alcohol 7 was the major by-product and could be recovered: 1.8 g, 60% conversion). 1H-NMR (CDCl3): δ 10.03 (s, 1H, NH of imidazole), 6.55 (s, 1H, HC-4/C-5 of imidazole), 6.49 (s, 1H, HC-5/C-4 of imidazole), 5.22 (t, J = 6.1, 1H, NH-2 of imidazole), 4.64 (dd, J = 9.8, 4.8, 1H, CHCOO), 3.30 (dd, J = 7.0, 6.5, 2H, CH2N), 2.10-1.67 (m, 4H, CHCH2 + CHCH2CH2), 1.50 (s, 18H, 2Boc), 1.43 (s, 9H, 3CH3 of COOtBu). 13C-NMR (CDCl3): δ 169.8 (CO, COOtBu), 153.4 (2CO, 2Boc), 148.1 (C-2 of imidazole), 121.8 (C-4 of imidazole), 111.0 (C-5 of imidazole), 84.0 (2CMe3, 2Boc), 82.1 (CMe3, COOtBu), 58.7 (CHCOO), 43.3 (CH2N), 28.3 (6CH3, 2Boc), 28.2 (3CH3 of COOtBu), 26.6 (CHCH2), 25.8 (CHCH2CH2). HRMS m/z 455.2856 (calcd for M+H, 455.2870).
(S)-2-amino-5-(imidazol-2-ylamino)pentanoic acid (APP) (12)
The same general procedure as for the synthesis of 10 afforded 12 as a bis-trifluoroacetate salt (90%). 1H-NMR (D2O): δ 6.87 (bs, 2H, HC-4, HC-5 of imidazole), 4.01 (m, 1H, CHCOO), 3.32 (m, 2H, CH2N), 2.05 (m, 2H, CHCH2), 1.80 (m, 2H, CHCH2CH2). 13C-NMR (D2O): δ 165.5 (CO), 146.7 (C-2 of imidazole), 113.4 (C-4 of imidazole), 112.9 (C-5 of imidazole), 60.6 (CHCOO), 42.5 (CH2N), 29.5 (CHCH2CH2CH2N), 26.5 (CHCH2CH2CH2N) (signals from TFA were omitted).
Dimethyl 2,6-bis(T-butoxycarbonylamino)heptanedioate (13)
2,6-diaminopimelic acid (6 g, 31.5 mmol) was dissolved in MeOH (50 mL) under N2 purging in an oven dried flask. The suspension was cooled to −5 – −10 °C (ice/acetone bath) and treated dropwise with thionyl chloride (5.1 mL, 69.4 mmol). The mixture was stirred at −5 – −10 °C for 1 h, then overnight at room temperature, when 1H-NMR and TLC monitoring (CHCl3:MeOH:NH4OH 2:8:1) indicated completion of the reaction. The solvent was rotoevaporated and the crude product washed with Et2O, then dried overnight under vacuum to yield the corresponding amino ester (8.5 g) as a dihydrochloride salt. NEt3 (12.3 mL) and DMAP (2.9 g) were added to a suspension of the amino ester in MeCN (80 mL). The suspension was cooled to 0 °C, then di-t-butyl pyrocarbonate (25.8 g, 118.1 mmol) in MeCN (30 mL) was added dropwise. The mixture was stirred at room temperature for 48 h. Solvent rotoevaporation and purification by flash column chromatography (silica gel, hexane/EtOAc gradients) afforded 13 as a light-yellow oil (88%). 1H-NMR (CDCl3): δ 5.09 (m, 2H, 2CHCOO), 4.26 (m, 2H, 2NH), 3.72 (s, 6H, 2COOCH3), 1.82 (m, 2H, CAH2CH), 1.63 (m, 2H, CBH2CH), 1.43 (s, 18H, 2Boc), 1.39 (m, 2H, CH2CH2CH). 13C-NMR (CDCl3): δ 173.5 (2CO, 2COOMe), 155.8 (2CO, 2Boc), 80.34 (2CMe3, 2Boc), 53.5 (CAH), 52.7 (CBH), 32.7 (CAH2CH), 32.6 (CBH2CH), 28.6 (6CH3, 2Boc), 21.8 (CAH2CH2CH), 21.6 (CBH2CH2CH). MS ES+ m/z 419.2 (M+H)+.
Methyl 2,6-bis(T-butoxycarbonylamino)-7-oxoheptanoate (14)
13 (1.2 g, 2.86 mmol) was dissolved in Et2O (30 mL) with stirring at room temperature and nitrogen purging, then cooled to −78 °C (acetone/dry ice bath). DIBAH 1M in hexane (1.86 mL, 1.86 mmol) was added dropwise. 1H-NMR monitoring indicated completion of the reaction after 1 h. The reaction mixture was quenched with water (5 mL) and filtered. The filtrate was rotoevaporated to dryness, washed with CHCl3, and dried overnight under vacuum to afford 14 (0.99 g, 89%) as a colorless oil. 1H-NMR (CDCl3): δ 9.57 (s, 1H, CHO), 5.09 (m, 2H, CHCHO + CHCOOMe), 4.27 (m, 2H, 2NH), 3.73 (s, 3H, COOCH3), 1.80 (m, 2H, CAH2CH), 1.64 (m, 2H, CBH2CH), 1.44 (s, 18H, 2Boc), 1.40 (m, 2H, CH2CH2CH). 13C-NMR (CDCl3): δ 200.0 (CHO), 173.4 (CO, COOMe), 155.9 (2CO, 2Boc), 80.3 (2CMe3, 2Boc), 59.8 (CHCHO), 53.4 (CAH), 52.6 (CBH), 32.8 (CAH2CH), 32.6 (CBH2CH), 28.6 (6CH3, 2Boc), 21.7 (CAH2CH2CH), 21.5 (CBH2CH2CH), 21.3 (CCH2CH2CH). MS ES+ m/z 389.2 (M+H)+.
2-amino-5-(2-aminoimidazol-4-yl)-pentanoic acid (A4P, 16)
14 (1.6 g, 4.12 mmol) was stirred in EtOAc (32 mL) and 4N HCl (30 mL) at room temperature for 2 h. The solvent was rotoevaporated and the resulting light brown oil was dried overnight under vacuum, then resolubilized in water (18 mL). The pH was adjusted to 4.5 with 2N NaOH, and cyanamide (1.04 g) was added in one portion. The reaction mixture was refluxed at 95 °C for 3 h, then rotoevaporated to dryness. Purification by flash column chromatography (CHCl3/MeOH/NH4OH gradients) generated 16 (0.23 g, 30%) as a light yellow oil. 1H-NMR (D2O): δ 6.77 (s, 1H, HC-4 of imidazole), 3.75 (m, 1H, CHCOO), 2.55 (m, 2H, CHCH2CH2CH2-imidazole), 1.88 (m, 2H, CHCH2CH2CH2-imidazole), 1.68 (m, 2H, CHCH2CH2CH2-imidazole). 13C-NMR (D2O): δ 175.1 (CO), 147.1 (C-2 of imidazole), 128.4 (C-4 of imidazole), 109.4 (C-5 of imidazole), 54.6 (CHCOO), 29.9 (CHCH2CH2CH2-imidazole), 23.9 (CHCH2CH2CH2-imidazole), 23.3 (CHCH2CH2CH2-imidazole). HRMS m/z 199.1189 (calcd for M+H, 199.1195).
Kinetic Assays
2-aminoimidazole amino acids affinities for human arginase I were determined using the fixed point assay with [14C-guanidino]-L-arginine34 as previously reported.46 Recombinant human arginase I was prepared as previously described.19 The final working concentrations in the reaction tubes were 1 mM for the unlabeled L-arginine and 0.07 μg/μL for human arginase I. Data analysis was based on the kinetic replot v0/vi as a function of inhibitor concentration, where v0 and vi were the observed velocities in the absence and presence of inhibitor, respectively. The mode of inhibition (i.e., noncompetive or competitive) was determined from Lineweaver-Burk plots of kinetic data at several different inhibitor concentrations.
Surface Plasmon Resonance
The binding affinity of A1P was determined on a Biacore 3000 instrument, following a previously reported procedure,32 except that all measurements were made at pH 8.5 and the analyte concentrations ranged 0–800 μM. Binding data were analyzed using a 1:1 Langmuir interaction model.47
Crystallography
Crystals of human arginase I complexed with 2AH were prepared by soaking 2AH into pre-formed crystals of the native enzyme, which were prepared by the hanging drop vapor diffusion method at 21 °C. Drops containing 3 μL of protein solution [3.5 mg/mL protein, 50.0 mM bicine (pH 8.5), 2 mM thymine, 100 μM MnCl2] and 3 μL of precipitant solution [0.1 M HEPES (pH 7.0), 28% Jeffamine] were equilibrated against a 1 mL reservoir of precipitant buffer. Crystals generally formed within 3–4 days. Crystals were harvested and soaked in a precipitant solution augmented with 20 mM 2AH for one week, and then cryoprotected in a precipitant solution containing 32% Jeffamine prior to flash cooling in liquid nitrogen.
Crystals of the human arginase I-AHH and human arginase I-2 aminoimidazole complexes were prepared by cocrystallization in hanging drops at 21°C. Drops containing 3 μL of protein solution [3.5 mg/mL protein, 50 mM bicine (pH 8.5), 2 mM AHH or 2 mM 2-aminoimidazole, 100 μM MnCl2] and 3 μL of precipitant solution [0.1 M HEPES (pH 7.0), 28% Jeffamine] were equilibrated against a 1 mL reservoir of precipitant solution. Crystals appeared overnight and grew with typical dimensions of 0.5 mm × 0.2 mm × 0.2 mm within one week. Crystals were soaked for 24 h in a precipitant solution augmented with 5 mM AHH or 2-aminoimidazole and cryoprotected as described above.
X-ray diffraction data from all crystals were collected at the National Synchrotron Light Source beam line X6A. Diffraction intensities measured from human arginase I-inhibitor complexes exhibited symmetry consistent with apparent space group P6 (unit cell parameters a = b = 90.1 Å, c = 69.7 Å). Intensity data integration and reduction were performed using the HKL2000 suite of programs.48 Data reduction statistics are recorded in Table 2. As with crystals of other human arginase I complexes,49 deviations from ideal Wilson statistics were observed with 〈I2〉/〈I〉2 = 1.5, indicating perfect hemihedral twinning.50 The structure of each enzyme-inhibitor complex was solved by molecular replacement using the program Phaser51 with chain A of the unliganded human arginase I (PDB accession code 2ZAV, less water molecules)49 used as a search probe against twinned data. In order to calculate electron density maps, structure factors amplitudes (|Fobs|) derived from twinned intensity data (|Iobs|) were deconvoluted into structure factor amplitudes corresponding to individual twin domains A and B (|Fobs/A| and |Fobs/B|, respectively) using the structure-based algorithm of Redinbo and Yeates50 implemented in CNS.52
Table 2.
Data Collection and Refinement Statistics
| Human arginase I-2AI | Human arginase I- 2AH | Human arginase I-AHH | |
|---|---|---|---|
| Data Collection | |||
| Resolution, Å | 50.0 – 1.90 | 50.0 – 1.47 | 50.0 – 1.90 |
| Total/Unique reflections measured | 91110/50007 | 196796/107754 | 95366/50344 |
| Rmergea,b | 0.061 (0.265) | 0.059 (0.616) | 0.086 (0.310) |
| I/σ(I)a | 13.9 (4.0) | 21.1 (2.3) | 15.4 (3.8) |
| Completeness (%)a | 99.1 (98.7) | 99.4 (99.6) | 99.8 (100.0) |
| Refinement | |||
| Reflections used in refinement/test set | 47507/2500 | 102365/5389 | 49288/2184 |
| Rtwinc | 0.168 | 0.149 | 0.141 |
| Rtwin/freec | 0.176 | 0.162 | 0.190 |
| Protein atomsd | 4772 | 4772 | 4762 |
| Water moleculesd | 246 | 460 | 338 |
| Inhibitor atomsd | 12 | 24 | 26 |
| Manganese ionsd | 4 | 4 | 4 |
| Sulfate ionsd | - | 2 | - |
| R.m.s. deviations | |||
| Bond lengths, Å | 0.007 | 0.011 | 0.008 |
| Bond angles, ° | 2.1 | 2.2 | 2.1 |
| PDB accession code | 2RJU | 3MFW | 3MVF |
Number in parentheses refer to the outer 0.1 Å shell of data.
Rmerge = Σ|I−〈I〉|/ΣI, where I is the observed intensity and 〈I〉 is the average intensity calculated for replicate data.
Rtwin = Σ|[|Fcalc/A|2+|Fcalc/B|2]1/2−|Fobs||/Σ|Fobs| for reflections contained in the working set. |Fcalc/A| and |Fcalc/B| are the structure factor amplitudes calculated for the separate twin domains A and B, respectively. Rtwin underestimates the residual error in the model over the two twin-related reflections by a factor of approximately 0.7. The same expression describes Rtwin/free, which was calculated for test set reflections excluded from refinement.
Per asymmetric unit.
Crystallographic refinement of each enzyme-inhibitor complex against twinned data was performed as previously described using CNS.49 In the later stages of each refinement, after the majority of water molecules were located, gradient omit maps revealed the inhibitor bound in the active site of each monomer in the asymmetric unit.
Inhibitors were refined with full occupancy and atomic B factors consistent with the average B-factor calculated for the entire protein. For the refinement of the human arginase I-2-aminoimidazole and human arginase I-2AH complexes, full-matrix least-squares refinement was performed against twinned data using the program Shelx53 in the final stages of refinement. Isotropic B factors were used in refinement with the exception of the manganese ions and sulfur atoms in the human arginase I-2AH complex, which were refined anisotropically (Table 2).
Disordered segments were excluded from the final models of the human arginase I-AHH complex (M1-S5 and N319-K322), the human arginase I-2-aminoimidazole complex (M1-S5 and N319-K322), and the human arginase I-2AH complex (M1-K4 and N319-K322). The quality of each refined model was assessed using PROCHECK.54
Acute murine OVA-Sensitization and Challenge Model of Allergic Airways Inflammation
These protocols were approved by the University of Toronto Faculty Advisory Committee on Animal Services, and were conducted in accordance with the guidelines of the Canadian Council on Animal Care. The acute model utilized a 3-week OVA-sensitization and -challenge protocol, as described previously.8a Briefly, female Balb/C mice (6–8 weeks old, Charles River Laboratories, Saint-Constant, PQ) were sensitized to chicken OVA on days 0 and 7 (i.p., 25 μg chicken OVA; Grade V > 98% pure: Sigma Chemical Company, Mississauga, ON), with 1 mg aluminum hydroxide gel (Invitrogen, Grand Island, NY). Beginning on day 14, animals were randomized to repeated exposure to nebulized 6% OVA in PBS or PBS alone for 25 min/day from days 14–20, using an AeroNebLab nebulizer (SciReq Inc., Montréal PQ).
In vivo Arginase Inhibition with A1P and Methacholine-Challenge
On day 21, twenty-four hours after the final OVA or PBS challenge, mice were anesthetized with 50 mg/kg ketamine (Bioniche, Belleville, ON) and 10 mg/kg xylazine (Bayer Inc., Toronto, ON) prior to intubation with an 18G stainless steel cannula (BD Biosciences Canada, Mississauga, ON) for in vivo, ventilator-based assessment of methacholine-responsiveness using the FlexiVent system Scireq. After determination of baseline pulmonary resistance parameters, A1P (80 μg/g body weight) was delivered via nebulization directly into the ventilatory circuit of a randomly selected subgroup of OVA/OVA mice. In a previous study we showed that arginase inhibition did not exhibit any effect on OVA/PBS mice. Fifteen minutes after delivery of the drug, baseline pulmonary resistance was re-assessed and the methacholine dose-response curve was initiated, as previously described.8a In brief, mice were challenged with methacholine (0–100 mg/mL in sterile PBS: Sigma) nebulized directly into the ventilatory circuit (AeroNebLab nebulizer). All data points were collected using the FlexiVent software (Scireq Inc.) and analyzed off-line using Excel (Microsoft Corporation, Redmond, WA). We have previously shown8a that this treatment-based protocol completely abrogates the allergic airways-induced hyperresponsiveness to methacholine in several murine models of allergic airways inflammation.
Acknowledgments
This work was supported by the America Asthma Foundation and NIH grant GM49758. We thank Dr. Steven Seeholzer, Dr. Hua Ding, and the Protein Core Facility at Children’s Hospital of Philadelphia for assistance with surface plasmon resonance measurements, and Dr. Rakesh Kohli for recording the HRMS spectra of the compounds. We also thank the National Synchrotron Light Source, beam time X6A, for access to X-ray crystallographic data collection facilities. D. W. C. is a Senior Fellow of the American Asthma Foundation. M. N. is a recipient of CIHR and Ontario Thoracic Society Doctoral Awards. J. A. S. is a Scientist in the Keenan Research Centre in the Li Ka Shing Knowledge Institute of St. Michael’s Hospital.
Abbreviations
- SAR
structure-activity relationships
- NO
nitric oxide
- ABH
2-(S)-amino-6-boronohexanoic acid
- BEC
S-(2-boronoethyl)-L-cysteine
- 2AH
L-2-aminohistidine
- AHH
2-aminohomohistidine
- A1P
2-(S)-amino-5-(2-aminoimidazol-1-yl)-pentanoic acid
- A4P
2-amino-5-(2-aminoimidazol-4-yl)-pentanoic acid
- APP
(S)-2-amino-5-(imidazol-2-ylamino)pentanoic acid
- DIBAH
di-i-butylaluminum hydride
- DMAP
dimethylaminopyridine
- DMF
dimethylformamide
- TFA
trifluoroacetic acid
References
- 1.(a) Borcic O, Straus B. Separation of arginase isoenzymes from human tissues by agar gel electrophoresis. J Clin Chem Clin Biochem. 1976;14:533–535. [PubMed] [Google Scholar]; (b) Grody WW, Dizikes GJ, Cederbaum SD. Human arginase isozymes. Isozymes Curr Top Biol Med Res. 1987;13:181–214. [PubMed] [Google Scholar]; (c) Jenkinson CP, Grody WW, Cederbaum SD. Comparative properties of arginases. Comp Biochem Physiol. 1996;114B:107–132. doi: 10.1016/0305-0491(95)02138-8. [DOI] [PubMed] [Google Scholar]; (d) Ash DE, Cox JD, Christianson DW. Manganese and its role in biological processes. In: Sigel A, Sigel H, editors. Metal Ions in Biological Systems. Vol. 37. M Dekker; New York: 2000. pp. 407–428. [PubMed] [Google Scholar]
- 2.Morris SM., Jr Regulation of enzymes of the urea cycle and arginine metabolism. Ann Rev Nutr. 2002;22:87–105. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- 3.(a) Morris SM, Jr, Bhamidipati D, Kepka-Lenhart D. Human type II arginase: sequence analysis and tissue-specific expression. Gene. 1997;193:157–161. doi: 10.1016/s0378-1119(97)00099-1. [DOI] [PubMed] [Google Scholar]; (b) Gotoh T, Araki M, Mori M. Chromosomal localization of the human arginase II gene and tissue distribution of its mRNA. Biochem Biophys Res Commun. 1997;233:487–491. doi: 10.1006/bbrc.1997.6473. [DOI] [PubMed] [Google Scholar]
- 4.(a) Christianson DW. Arginase: Structure, mechanism, and physiological role in male and female sexual arousal. Acc Chem Res. 2005;38:191–201. doi: 10.1021/ar040183k. [DOI] [PubMed] [Google Scholar]; (b) Dowling DP, Di Costanzo L, Gennadios HA, Christianson DW. Evolution of the arginase fold and functional diversity. Cell Mol Life Sci. 2008;65:2039–2055. doi: 10.1007/s00018-008-7554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Wu G, Morris SM., Jr Arginine metabolism: nitric oxide and beyond. Biochem J. 1998;336:1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morris SM., Jr Arginine metabolism: boundaries of our knowledge. J Nutr. 2007;137:1602S–1609S. doi: 10.1093/jn/137.6.1602S. [DOI] [PubMed] [Google Scholar]; (c) Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol. 2009;157:922–930. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Smith RJ, Phang JM. The importance of ornithine as a precursor for proline in mammalian cells. J Cell Physiol. 1979;98:475–482. doi: 10.1002/jcp.1040980306. [DOI] [PubMed] [Google Scholar]; (b) Albina JE, Abate JA, Mastrofrancesco B. Role of ornithine as a proline precursor in healing wounds. J Surg Res. 1993;55:97–102. doi: 10.1006/jsre.1993.1114. [DOI] [PubMed] [Google Scholar]; (c) Nieves C, Jr, Langkamp-Henken B. Arginine and immunity: a unique perspective. Biomed Pharmacother. 2002;56:471–482. doi: 10.1016/s0753-3322(02)00291-3. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kim PS, Iyer RK, Lu KV, Yu H, Karimi A, Kern RM, Tai DK, Cederbaum SD, Grody WW. Expression of the liver form of arginase in erythrocytes. Mol Gen Metab. 2002;76:100–110. doi: 10.1016/s1096-7192(02)00034-3. [DOI] [PubMed] [Google Scholar]; (b) Wei LH, Wu G, Morris SM, Jr, Ignarro LJ. Elevated arginase I expression in rat aortic smooth muscle cells increases cell proliferation. Proc Natl Acad Sci USA. 2001;98:9260–9264. doi: 10.1073/pnas.161294898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) North ML, Khanna N, Marsden PA, Grasemann H, Scott JA. Functionally important role for arginase 1 in the airway hyperresponsiveness of asthma. Am J Physiol Lung Cell Mol Physiol. 2009;296:L911–L920. doi: 10.1152/ajplung.00025.2009. [DOI] [PubMed] [Google Scholar]; (b) Bergeron C, Boulet LP, Page N, Laviolette M, Zimmermann N, Rothenberg ME, Hamid Q. Influence of cigarette smoke on the arginine pathway in asthmatic airways: increased expression of arginase I. J Allergy Clin Immunol. 2007;119:391–397. doi: 10.1016/j.jaci.2006.10.030. [DOI] [PubMed] [Google Scholar]; (c) Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, Hamid Q, Rothenberg ME. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–1874. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer – mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172:7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]; (b) Zimmermann N, Mishra A, King NE, Fulkerson PC, Doepker MP, Nikolaidis NM, Kindinger LE, Moulton EA, Aronow BJ, Rothenberg ME. Transcript signatures in experimental asthma: identification of STAT6-dependent and -independent pathways. J Immunol. 2004;172:1815–1824. doi: 10.4049/jimmunol.172.3.1815. [DOI] [PubMed] [Google Scholar]
- 10.(a) Li H, Romieu I, Sienra-Monge J-J, Ramirez-Aguilar M, Estela del Rio-Navarro B, Kistner EO, Gjessing HK, Lara-Sanchez IC, Chiu GY, London SJ. Genetic polymorphisms in arginase I and II and childhood asthma and atopy. J Allergy Clin Immunol. 2006;117:119–126. doi: 10.1016/j.jaci.2005.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Litonjua AA, Lasky-Su1 J, Schneiter K, Tantisira KG, Lazarus R, Klanderman B, Lima JJ, Irvin CG, Peters SP, Hanrahan JP, Liggett SB, Hawkins GA, Meyers DA, Bleecker ER, Lange C, Weiss ST. ARG1 is a novel bronchodilator response gene. Screening and replication in four asthma cohorts. Am J Respir Crit Care Med. 2008;178:688–694. doi: 10.1164/rccm.200709-1363OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Yang M, Rangasamy D, Matthaei KI, Frew AJ, Zimmermann N, Mahalingam S, Webb DC, Tremethick DJ, Thompson PJ, Hogan SP, Rothenberg ME, Cowden WB, Foster PS. Inhibition of arginase I activity by RNA interference attenuates IL-13-induced airways hyperresponsiveness. J Immunol. 2006;177:5595–5603. doi: 10.4049/jimmunol.177.8.5595. [DOI] [PubMed] [Google Scholar]; (b) Maarsingh H, Zuidhof AB, Bos IST, van Duin M, Boucher JL, Zaagsma J, Meurs H. Arginase inhibition protects against allergen-induced airway obstruction, hyperresponsiveness, and inflammation. Am J Respir Crit Care Med. 2008;178:565–573. doi: 10.1164/rccm.200710-1588OC. [DOI] [PubMed] [Google Scholar]
- 12.(a) Meurs H, Maarsingh H, Zaagsma J. Arginase and asthma: novel insights into nitric oxide homeostasis and airway hyperresponsiveness. Trends Pharmacol Sci. 2003;24:450–455. doi: 10.1016/S0165-6147(03)00227-X. [DOI] [PubMed] [Google Scholar]; (b) Zimmermann N, Rothenberg ME. The arginine-arginase balance in asthma and lung inflammation. Eur J Pharm. 2006;533:253–262. doi: 10.1016/j.ejphar.2005.12.047. [DOI] [PubMed] [Google Scholar]; (b) Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol. 2009;158:638–651. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Munder M. Role of arginase in asthma: potential clinical applications. Exp Rev Clin Pharmacol. 2010;3:17–23. doi: 10.1586/ecp.09.53. [DOI] [PubMed] [Google Scholar]
- 13.(a) Cox JD, Kim NN, Traish AM, Christianson DW. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat Struct Biol. 1999;6:1043–1047. doi: 10.1038/14929. [DOI] [PubMed] [Google Scholar]; (b) Kim NN, Cox JD, Baggio RF, Emig FA, Mistry SK, Harper SL, Speicher DW, Morris SM, Ash DE, Traish A, Christianson DW. Probing erectile function: S-(2-boronoethyl)-L-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry. 2001;40:2678–2688. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]; (c) Cama E, Colleluori DM, Emig FA, Shin H, Kim SW, Kim NN, Traish A, Ash DE, Christianson DW. Human arginase II: crystal structure and physiological role in male and female sexual arousal. Biochemistry. 2003;42:8445–8451. doi: 10.1021/bi034340j. [DOI] [PubMed] [Google Scholar]
- 14.(a) Bivalacqua TJ, Hellstrom WJG, Kadowitz PJ, Champion HC. Increased expression of arginase II in human diabetic corpus cavernosum: In diabetic-associated erectile dysfunction. Biochem Biophys Res Commun. 2001;283:923–927. doi: 10.1006/bbrc.2001.4874. [DOI] [PubMed] [Google Scholar]; (b) Bivalacqua TJ, Burnett AL, Hellstrom WJG, Champion HC. Overexpression of arginase in the aged mouse penis impairs erectile function and decreases eNOS activity: influence of in vivo gene therapy of anti-arginase. Am J Phys Heart. 2007;292:H1340–H1351. doi: 10.1152/ajpheart.00121.2005. [DOI] [PubMed] [Google Scholar]
- 15.Grasemann H, Schwiertz R, Matthiesen S, Racké K, Ratjen F. Increased arginase activity in cystic fibrosis airways. Am J Respir Crit Care Med. 2005;172:1523–1528. doi: 10.1164/rccm.200502-253OC. [DOI] [PubMed] [Google Scholar]
- 16.(a) Yang Z, Ming XF. Endothelial arginase: a new target in atherosclerosis. Curr Hypertens Rep. 2006;8:54–59. doi: 10.1007/s11906-006-0041-8. [DOI] [PubMed] [Google Scholar]; (b) Nelin LD, Stenger MR, Malleske DT, Chicoine LG. Vascular arginase and hypertension. Curr Hypertens Rev. 2007;3:242–249. [Google Scholar]; (c) Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, Balanson S, Ryoo S, Anderson M, Irani K, Khanday F, Di Costanzo L, Nyhan D, Hare JM, Christianson DW, Rivers R, Shoukas A, Berkowitz DE. Inducible NO synthase-dependent S-nitrosylation and activation of arginase 1 contribute to age-related endothelial dysfunction. Circ Res. 2007;101:692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]; (d) Santhanam L, Christianson DW, Nyhan D, Berkowitz DE. Arginase and vascular aging. J Appl Physiol. 2008;105:1632–1642. doi: 10.1152/japplphysiol.90627.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, Lim HK, Sohi J, Santhanam L, Soucy K, Tuday E, Baraban E, Ilies M, Gerstenblith G, Nyhan D, Shoukas A, Christianson DW, Alp NJ, Champion HC, Huso D, Berkowitz DE. Endothelial arginase II. A novel target for the treatment of atherosclerosis. Circ Res. 2008;102:923–932. doi: 10.1161/CIRCRESAHA.107.169573. [DOI] [PubMed] [Google Scholar]
- 17.(a) Singh R, Pervin S, Karimi A, Cederbaum S, Chaudhuri G. Arginase activity in human breast cancer cell lines: Nω-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000;60:3305–3312. [PubMed] [Google Scholar]; (b) Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 2001;61:1100–1106. [PubMed] [Google Scholar]; (c) Lind SD. Arginine and cancer. J Nutr. 2004;134:2837S–2841S. doi: 10.1093/jn/134.10.2837S. [DOI] [PubMed] [Google Scholar]; (d) Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nature Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]; (e) Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–3048. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]; (f) Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13:721s–726s. doi: 10.1158/1078-0432.CCR-06-2197. [DOI] [PubMed] [Google Scholar]; (g) Marx J. Cancer’s bulwark against immune attack: MDS cells. Science. 2008;319:154–156. doi: 10.1126/science.319.5860.154. [DOI] [PubMed] [Google Scholar]
- 18.Baggio R, Elbam D, Kanyo ZF, Carroll PJ, Cavalli RC, Ash DE, Christianson DW. Inhibition of Mn2+2-arginase by borate leads to the design of a transition state analog inhibitor, 2(S)-amino-6-boronohexanoic acid. J Am Chem Soc. 1997;119:8107–8108. [Google Scholar]
- 19.Di Costanzo L, Sabio G, Mora A, Rodriguez PC, Ochoa AC, Centeno F, Christianson DW. Crystal structure of human arginase I at 1.29-Å resolution and exploration of inhibition in the immune response. Proc Natl Acad Sci U S A. 2005;102:13058–13063. doi: 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Costanzo L, Flores LV, Jr, Christianson DW. Stereochemistry of guanidine-metal interactions: implications for L-arginine-metal interactions in protein structure and function. Proteins. 2006;65:637–642. doi: 10.1002/prot.21127. [DOI] [PubMed] [Google Scholar]
- 21.Creighton TE. Proteins: structures and molecular properties. WH Freeman; New York: 1992. [Google Scholar]
- 22.Storey BT, Sullivan WW, Moyer CL. The pKa values of some 2-amino imidazolium ions. J Org Chem. 1964;29:3118–3120. [Google Scholar]
- 23.Sullivan JD, Giles RL, Looper RE. 2-Aminoimidazoles from Leucetta sponges: synthesis and biology of an important pharmacophore. Curr Bioact Compd. 2009;5:39–78. [Google Scholar]
- 24.(a) Mourabit AA, Potier P. Sponge’s molecular diversity through the ambivalent reactivity of 2-aminoimidazole: a universal chemical pathway to the oroidin-based pyrrole-imidazole alkaloids and their palau’amine congeners. Eur J Org Chem. 2001;2:237–243. [Google Scholar]; (b) Hoffmann H, Lindel T. Synthesis of the pyrrole-imidazole alkaloids. Synthesis. 2003;12:1753–1783. [Google Scholar]; (c) Rogers SA, Bero JD, Melander C. Chemical synthesis and biological screening of 2-aminoimidazole-based bacterial and fungal antibiofilm agents. ChemBioChem. 2010;11:396–410. doi: 10.1002/cbic.200900617. [DOI] [PubMed] [Google Scholar]
- 25.(a) Benoit-Vical F, Salery M, Soh PN, Ahond A, Poupat C. Girolline: A potential lead structure for antiplasmodial drug research. Planta Medica. 2008;74:438–444. doi: 10.1055/s-2008-1034348. [DOI] [PubMed] [Google Scholar]; (b) Mancini I, Guella G, Debitus C, Waikedre J, Pietra F. From inactive nortopsentin D, a novel bis(indole) alkaloid isolated from the axinellid sponge Dragmacidon sp. from deep waters south of New Caledonia, to a strongly cytotoxic derivative. Helv Chim Acta. 1996;79:2075–2082. [Google Scholar]
- 26.Nagai W, Kirk KL, Cohen L. A. Synthesis of 2-amino-L-histidine and 2-aminohistamine. J Org Chem. 1973;38:1971–1974. doi: 10.1021/jo00951a002. [DOI] [PubMed] [Google Scholar]
- 27.Friedel M, Lindel T. Synthesis of L-aminohomohistidine (L-Ahh) Tetrahedron Lett. 2004;45:2779–2781. [Google Scholar]
- 28.Ulhaq S, Chinje EC, Naylor MA, Jaffar M, Stratford IJ, Threadgill MD. S-2-amino-5-azolylpentanoic acids related to L-ornithine as inhibitors of the isoforms of nitric oxide synthase (NOS) Bioorg Med Chem. 1998;6:2139–2149. doi: 10.1016/s0968-0896(98)00174-6. [DOI] [PubMed] [Google Scholar]
- 29.(a) Zakharian TY, Di Costanzo L, Christianson DW. Synthesis of (2S)-2-amino-7,8-epoxyoctanoic acid and structure of its metal-bridging complex with human arginase I. Org Biomol Chem. 2008;6:3240–3243. doi: 10.1039/b811797g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zakharian TY, Di Costanzo L, Christianson DW. (S)-2-Amino-6-nitrohexanoic acid binds to human arginase I through multiple nitro-metal coordination interactions in the binuclear manganese cluster. J Am Chem Soc. 2008;130:17254–17255. doi: 10.1021/ja807702q. [DOI] [PubMed] [Google Scholar]
- 30.Adamczyk M, Johnson DD, Reddy RE. Collagen crosslinks: a convenient synthesis of tert-butyl-(2S)-2-[(tert-butoxycarbonyl)amino]-4-(2-oxiranyl)butanoate. Tetrahedron: Asymmetry. 1999;10:775–781. [Google Scholar]
- 31.Ulhaq S, Naylor MA, Chinje EC, Threadgill MD, Stratford IJ. S-2-Amino-5-(2-nitroimidazol-1-yl)pentanoic acid: a model for potential bioreductively activated prodrugs for inhibitors of nitric oxide synthase (NOS) activity. Anti-Cancer Drug Design. 1997;12:61–65. [PubMed] [Google Scholar]
- 32.Cama E, Shin H, Christianson DW. Design of amino acid sulfonamides as transition-state analogue inhibitors of arginase. J Am Chem Soc. 2003;125:13052–13057. doi: 10.1021/ja036365b. [DOI] [PubMed] [Google Scholar]
- 33.Ciapetti P, Falorni M, Mann A, Taddei M. 2-Amino-4-bromobutanoic acid. A versatile reagent for the synthesis of nonnatural amino acids. Molecules Online. 1998;2:86–93. [Google Scholar]
- 34.Rüegg UT, Russell AS. A rapid and sensitive assay for arginase. Anal Biochem. 1980;102:206–212. doi: 10.1016/0003-2697(80)90340-1. [DOI] [PubMed] [Google Scholar]
- 35.Pearson JT, Hill JJ, Swank J, Isoherranen N, Kunze KL, Atkins WM. Surface plasmon resonance analysis of antifungal azoles binding to CYP3A4 with kinetic resolution of multiple binding orientations. Biochemistry. 2006;45:6341–6353. doi: 10.1021/bi0600042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lancini GC, Lazzari E. A new synthesis of alkyl and aryl 2-aminoimidazoles. J Heterocycl Chem. 1966;3:152–154. [Google Scholar]
- 37.(a) Alberti G, Bertini I, Luchinat C, Scozzafava A. A new class of inhibitors capable of binding both the acidic and alkaline forms of carbonic anhydrase. Biochim Biophys Acta. 1981;668:16–26. doi: 10.1016/0005-2795(81)90144-6. [DOI] [PubMed] [Google Scholar]; (b) Bertini I, Luchinat C. Cobalt(II) as a probe of the structure and function of carbonic anhydrase. Acc Chem Res. 1983;16:272–279. [Google Scholar]
- 38.Kannan KK, Chakravarty S, Satyamurthy P, Ramanadham M, Kumar V, Yadav VS. Structure and function of carbonic anhydrase: Synchrotron x-ray diffraction studies of human carbonic anhydrase I and inhibitor complexes. Synchrotron Radiat Biosci. 1994:86–95. [Google Scholar]
- 39.Mangani S, Liljas A. Crystal structure of the complex between human carbonic anhydrase II and the aromatic inhibitor 1,2,4-triazole. J Mol Biol. 1993;232:9–14. doi: 10.1006/jmbi.1993.1365. [DOI] [PubMed] [Google Scholar]
- 40.Marino JP, Jr, Fisher PW, Hofmann GA, Kirkpatrick RB, Janson CA, Johnson RK, Ma C, Mattern M, Meek TD, Ryan MD, Schulz C, Smith WW, Tew DG, Tomazek TA, Jr, Veber DF, Xiong WC, Yamamoto Y, Yamashita K, Yang G, Thompson SK. Highly potent inhibitors of methionine aminopeptidase-2 based on a 1,2,4-triazole pharmacophore. J Med Chem. 2007;50:3777–3785. doi: 10.1021/jm061182w. [DOI] [PubMed] [Google Scholar]
- 41.Rahman MN, Vlahakis JZ, Szarek WA, Nakatsu K, Jia Z. X-ray crystal structure of human heme oxygenase-1 in complex with 1-(adamantan-1-yl)-2-(1H-imidazol-1-yl)ethanone: a common binding mode for imidazole-based heme oxygenase-1 inhibitors. J Med Chem. 2008;51:5943–5952. doi: 10.1021/jm800505m. [DOI] [PubMed] [Google Scholar]
- 42.Berka V, Palmer G, Chen PF, Tsai AL. Effects of various imidazole ligands on heme conformation in endothelial nitric oxide synthase. Biochemistry. 1998;37:6136–6144. doi: 10.1021/bi980133l. [DOI] [PubMed] [Google Scholar]
- 43.(a) Kenyon NJ, Bratt JM, Linderholm AL, Last MS, Last JA. Arginases I and II in lungs of ovalbumin-sensitized mice exposed to ovalbumin: sources and consequences. Toxicol Appl Pharmacol. 2008;230:269–275. doi: 10.1016/j.taap.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bratt JM, Franzi LM, Linderholm AL, Last MS, Kenyon NJ, Last JA. Arginase enzymes in isolated airways from normal and nitric oxide synthase 2-knockout mice exposed to ovalbumin. Toxicol Appl Pharmacol. 2009;234:273–280. doi: 10.1016/j.taap.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Custot J, Moali C, Brollo M, Boucher JL, Delaforge M, Mansuy D, Tenu JP, Zimmermann JL. The new α-amino acid Nω-hydroxy-nor-L-arginine: a high-affinity inhibitor of arginase well adapted to bind to its manganese cluster. J Am Chem Soc. 1997;119:4086–4087. [Google Scholar]
- 45.Custot J, Boucher JL, Vadon S, Guedes C, Dijols S, Delaforge M, Mansuy D. Nω-hydroxyamino-α-amino acids as a new class of very strong inhibitors of arginases. J Biol Inorg Chem. 1996;1:73–82. [Google Scholar]
- 46.Di Costanzo L, Moulin M, Haertlein M, Meilleur F, Christianson DW. Expression, purification, assay, and crystal structure of perdeuterated human arginase I. Arch Biochem Biophys. 2007;465:82–89. doi: 10.1016/j.abb.2007.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schuck P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Ann Rev Biophys Biomol Struct. 1997;26:541–566. doi: 10.1146/annurev.biophys.26.1.541. [DOI] [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.Di Costanzo L, Pique ME, Christianson DW. Crystal structure of human arginase I complexed with thiosemicarbazide reveals an unusual thiocarbonyl μ-sulfide ligand in the binuclear manganese cluster. J Am Chem Soc. 2007;129:6388–6389. doi: 10.1021/ja071567j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeates TO. Detecting and overcoming crystal twinning. Methods Enzymol. 1997;276:344–358. [PubMed] [Google Scholar]
- 51.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read R. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 53.Sheldrick GM. A short history of SHELX. Acta Cryst A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 54.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]