

Abstract
Metal chelation-ligand interactions, such as occur between nitrilotriacetic acid (NTA)-nickel and multihistidines, enable the non-covalent attachment of histidine-modified proteins to liposomes and other particles. We compared three lipids: a mono-NTA lipid (circa 10 uM affinity) and two tris-NTA lipid derivatives (circa 3 nM and 0.2 nM affinity) in their ability to retain two different his6-containing proteins on NTA-liposomes in the presence of serum or plasma and after intravenous injection in mice. At nanomolar affinities the off-rate of a his6-ligand is sufficiently long so that his6-proteins attached to particle surfaces will remain with the particle for hours; thus, we hypothesized that the increased his6 affinity of multivalent NTA-modified liposomes would retain his6-proteins longer both in vitro and in vivo. For each of the three lipids, we found a robust association and complete activity retention of two his6-modified proteins: a far red-fluorescent protein, monomeric Katushka (mKate), and a prodrug-converting enzyme, yeast cytosine deaminase (yCD). Proteins associated more tightly in vitro with tris-NTA liposomes than with mono-NTA liposomes in the presence of refiltered fetal calf serum and mouse plasma. Free yCD exchanged with previously associated mKate for tris-NTA binding sites on the liposome surface. This exchange was due to the exchange of the proteins for NTA occupancy and not due to the exchange of tris-NTA lipid out of the liposome. The amount of yCD on the surface was similar if the proteins were co-associated or if mKate was pre-associated. This exchange confirms that NTA associated proteins are in a dynamic state and can exchange with multihistidine proteins in the biological milieu. There was no difference in circulation time of the protein when it was intravenously administered by itself or attached to any of the NTA-modified liposomes because in vivo the protein was rapidly released from the NTA liposomes. Upon recovery from blood, liposomes containing tris-NTA accumulated a different plasma protein profile than control liposomes, suggesting that Ni-NTA specifically interacts with some plasma proteins. The reason for the rapid protein dissociation from the liposome in vivo is not clear; it could be due to displacement by endogenous histidine-containing proteins or to natural chelators that remove nickel from the NTA. Regardless of the cause, improvements in chelator or ligand design are needed before metal chelation will be capable of retaining histidine-modified proteins on NTA liposomes after in vivo administration.
Introduction
Nickel chelation chromatography, and similar technologies that depend upon non-covalent interactions between a nickel chelator and a multihistidine-ligand, are widely used to purify proteins and associate proteins with surfaces (1, 2). The affinity of first generation nitrilotriacetic acid (NTAs) is about 10 micromolar whereas affinities of newer trivalent-NTAs (tris-NTAs) are in the nanomolar range (3–6).
NTA mediated protein association is a useful investigative tool that is mild enough to retain protein activity and requires only a simple modification of the amino acid sequence - the addition of a string of histidines (1, 7). This interaction is reversible by stripping the chelated nickel from the NTA or allowing molecules to compete with the protein for NTA association (2). Proteins remain in a lateral surface orientation (8–10) and free of surface involvement (11). The stable, well-defined orientation allows proteins to be imaged and their structure or function investigated (12, 13). NTA coated surfaces improve AFM-based protein imaging and have been used to measure real-time substrate degradation (14). Active, surface-immobilized proteins can also be used to exploit the activity of these proteins in microarrays and biosensors (7, 8, 15, 16).
Modifications to the linkage area of the NTA headgroups change the molecules affinity for his-tagged proteins. The addition of poly(ethyleneglycol) (PEG) increases protein capture efficiency (15, 16). Multivalent NTAs improve affinity by allowing multiple chelated nickels within the NTA cluster to interact with a single his-tag (6, 17–19).
Advances in protein production by recombinant techniques have largely removed protein availability as a limitation for protein related therapies (20). The stability and rapid clearance of proteins in vivo are barriers that can reduce their therapeutic potential (21). These limitations can be overcome, in part, by using nanocarriers to alter protein pharmacokinetics or increase protein stability (22–24). Additional problems arise as chemical conjugation of proteins to carrier surfaces can damage the protein and the percent of protein attached to the carrier can be low (25). Multivalent NTAs have the potential to increase the efficiency, simplify the modification of nanocarriers and enable a `toolkit' approach for in vivo therapeutic use. Coupling proteins to carriers using a non-covalent NTA also avoids harsh conjugation chemistries. At low nanomolar affinities, the off-rate of a his6 ligand from an NTA-nickel is sufficiently long so that the his6-proteins bound to particle surfaces will remain with the particle for hours. Thus, an appropriate NTA-lipid would enable the creation of NTA-modified liposomes that could be functionalized with any his6-containing protein.
The increased affinity of a multivalent NTA for a his-tagged protein is strong enough to make the interactions stable in vitro (3–5, 18, 19, 26–28). Immunoliposomes generated by associating single-chain antibody fragments to liposomes with mono-NTA lipids were found to be suitable for rapid in vitro screening (27, 28). However, incubation with human plasma as a surrogate for in vivo studies caused the protein to dissociate from the mono-NTA (29).
Other groups have shown success in vivo. His-tagged epitopes have been associated with both mono and multivalent-NTA nanoparticles to mediate immunotherapies (3, 26, 30). A his-tagged peptide designed to be membrane penetrating and immunostimulatory was shown to induce splenocytic immune stimulation in BALB/c mice when associated with NTA-DOGS-liposomes (26). Using the same NTA-DOGS-lipid, an NTA-wax nanoparticle induced immune response against an HIV Gag protein (30). Potential vaccines were also formulated from plasma membrane vesicles derived from tumor cells modified to contain tris-NTA lipid (3). When mice were immunized by subcutaneous injection of these vesicles, containing encapsulated cytokines, they exhibited decreased tumor growth when challenged two weeks after immunization (3). In all three studies, the nanoparticles were injected subcutaneously and were not exposed to blood. The stability of the protein-vesicle association in vivo was not monitored so the duration of protein retention is not known.
In this study, we compare the behavior of liposomes modified with three different NTA-lipids of increasing affinities for his-tagged proteins: mono-NTA-DOGS, tris-NTA-LYS-DOD, and tris-NTA-DAP-DOD (Figure 1). Liposome formulations containing NTA-lipids robustly associated both yCD and mKate without decreasing protein activity but association with the NTA liposomes did not increase the protein's circulation time.
Figure 1. Illustration of Nitrilotriacetic Acid (NTA)-Containing PEGylated Liposomes with an Associated His-Tagged Protein.

Reported equilibrium dissociation constants are given for a his6 peptide ligand (5, 18).
Experimental Procedures
Materials
All reagents and solvents obtained from commercial suppliers were used without further purification. Palmitoyloleoylphosphatidyl choline (POPC), palmitoyloleoylphosphatidyl glycerol (POPG), Methoxy polyethyleneglycol 2000 disteroylphosphatidyl ethanolamine (mPEG (2000)-DSPE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(Lissamine Rhodamine B Sulfonyl) (rhodamine-DOPE) and cholesterol were obtained from Avanti Polar Lipids (Alabaster, AL). Ni-NTA-DOGS (1,2-dioleoyl-sn-glycero-3-{(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl}) was purchased from Avanti Polar Lipids preloaded with nickel. Frozen, refiltered fetal calf serum (FCS) was obtained from the UCSF Cell Culture Facility and was prepared by filter-purifying serum, not heat inactivation, thus retaining active portions of the complement cascade.
Synthesis of tris-NTA-LYS-DOD and tris-NTA-DAP-DOD
The tris-NTA-lipids, were synthesized via the methods described by Huang et al (4, 5). Tris-NTA-lipids are constructed from a base of dioctadececyl (DOD) lipid chains and diaminopropionic acid (DAP) or lysine (LYS) for tris-NTA-DAP-DOD and tris-NTA-LYS-DOD, respectively (Figure 1). Lipids were stored dry and dissolved in organic solvents immediately prior to use. To load the NTA3 head groups with nickel, NiCl2 was dissolved in minimal H2O followed by methanol. NTA3-lipids were incubated with nickel in the methanol-water solution (3 molar equivalents NiCl2 per mol NTA3) at 60 °C for 15 min. The solution was allowed to cool to room temperature before adding it to the lipid formulation mixture. Preloading the nickel prior to liposome formation prevented dilution during removal of the excess nickel. Ni-NTA-DOGS was used as received from Avanti Polar Lipids.
Synthesis of FITC-DSPE
The fluorescent lipid was synthesized via direct coupling of FITC to DSPE. To a solution of DSPE (50 mg) in dry chloroform (5 mL) and triethylamine (37.3 μL) was added FITC (52.1 mg, 2 equiv.) in minimal dimethyl sulfoxide (DSMO). The mixture reacted overnight at room temperature in the dark. Crude product solution was concentrated by rotary evaporation and purified on a Biotage Horizon HPFC system with pre-packed silica columns (Biotage; Charlottesville, VA). The elution gradient consisted of 3 segments (solvent A:chloroform, B: methanol); 0%–20% B, 24 mL; 20%–20% B, 48 mL; 20%–30% B, 72 mL. Fractions of pure product were pooled, evaporated and dried under high vacuum overnight. Rf = 0.38 in chloroform:methanol (4:1 v/v). The product was dissolved and stored in chloroform at −40 °C. During synthesis, handling and use, FITC-DSPE was protected from photo-bleaching by minimizing exposure to light and heat. Structure of the product was confirmed with 1H NMR and MALDI mass spectrometry.
Formation of Liposomes
Liposomes were prepared by the sonication and extrusion method (31). Briefly, lipids dissolved in organic solvents were added to depyrogenated borosilicate glass tubes. Solvent was evaporated while rotating under reduced pressure at room temperature to form a lipid film. Residual solvent was removed under high vacuum overnight. Films were then rehydrated in 1 mL HEPES buffer (20 mM HEPES, 140 mM NaCl, pH 7.4) with intermittent vortexing and sonicated under argon for 10 minutes at 23 °C to form vesicles. Liposome diameter was reduced by serially extruding 11 times through 200 nm, 100 nm and 80 nm polycarbonate membranes with a hand-held extruder (Avestin; Ontario, Canada). Formulations contained either NTA3-LYS-DOD, NTA3-DAP-DOD, or NTA-Ni-DOGS (POPC/Chol/mPEG/NTA 55:40:5:n, molar ratio, where n corresponds to the mole percent NTA for each formulation). All liposomes contained 5 mol % mPEG(2000), unless noted. Control liposomes contained 1.5 mol % POPG to mimic the charge density of a 0.3% NTA3-containing liposome formulation.
In some experiments, a tris-NTA-containing formulation without nickel was also used as a negative control. Liposome diameter and zeta potential were measured using a Zetasizer 3000 (Malvern Instruments; UK) for 3 or more liposome preparations of each formulation. The fluorescent signal from FITC-DSPE lipid, incorporated into the formulation to allow for tracking of the liposome (at 0.05 mol % unless noted), was not significantly decreased once liposomes formed. Lipid concentration in solution was calculated from the FITC signal based on control liposomes containing the same NTA concentration. Inclusion of NTA lipids caused both a slight fluorescence decrease and a negative charge.
Transfer of Tris-NTA Lipids between Lipid Membranes
Liposomes were prepared as described above (20 μmol/mL total lipid) that contained 3% tris-NTA-DAP, POPC, cholesterol and 0.1 mol % FITC-DSPE (3:60:40:0.1 mol ratio) or POPC, cholesterol and rhodamine-DOPE (60:40:0.05 mol ratio). Liposomes containing tris-NTA-DAP were mixed with liposomes without NTA lipids (1:1 v/v) and incubated at 37 °C for 3 hours. Liposomes mixtures (3 μmol total lipid) were then passed over an AG 1-X2 anion exchange column (1 × 6 cm bed volume, Bio-Rad Laboratories, Inc.; Hercules, CA) pre-equilibrated with 20 mM HEPES, 140 mM NaCl (pH 7.4). Uncharged rhodamine-containing liposomes were not retained by the column and appeared in the void volume fractions. Negatively charged, tris-NTA-DAP, FITC-containing liposomes were retained by the column and eluted with 20 mM Tris-HCl, 1 M NaCl (pH 9.5). Presence of liposomes was determined based on rhodamine or FITC fluorescence (excitation/emission 544/590 nm and 490/520 nm respectively). The charge of neutral, rhodamine-containing liposomes was monitored (Zetasizer 3000, Malvern Instruments; UK) before and after incubation with NTA-liposomes.
Protein Production
Genes for thermostabilized yeast cytosine deaminase (yCD; 18 kDa) (32) and monomeric Katushka (mKate; 27 kDa) (33) were constructed from gene fragments. Briefly, an optimized gene sequence for E. coli K12 (high) codon bias was designed and 27 or 32 primers (for yCD and mKate respectively) were used to construct a complete gene sequence using PCR. PCR was performed with GoTaq DNA polymerase (Promega; Madison, WI) in a GeneAmp PCR System 2400 thermal cycler (Perkin Elmer; Waltham, MA). Synthesized genes were then ligated into pET15b vector for amplification and expression.
Protein was expressed in BL21-Codon Plus (DE3)-RIPL E.coli cells (Stratagene; La Jolla, CA). E. coli were grown in LB culture medium containing 30 μg/mL kanamycin and 50 μg/mL chloramphenicol at 37 °C until the OD600 nm reached 0.6–0.9. Protein expression was induced with 1 mM isopropyl α-D-1- thiogalactopyranoside (IPTG) with 0.5 mM zinc acetate. After overnight expression at room temperature, protein was purified from the cell supernatant by lysing the cells using freeze thaw followed by sonication. His-tagged proteins were purified from other cellular proteins on a HisTrapFF affinity column (Amersham Biosciences; Piscataway, NJ) and eluted with 300 mM imidazole. The remaining yCD protein contaminants were removed by size exclusion chromatography on a HiPrep Sepharose S-100HR 16/60 size exclusion column (Amersham Biosciences; Piscataway, NJ). Buffers were exchanged using a PD10 desalting column (GE Healthcare; UK). OD280 nm was measured to obtain the fractions containing protein.
Liposome-Protein Association
His-tagged proteins were associated with liposomes at room temperature. Liposomes (2 μmol total lipid) were incubated with 270 μg mKate-his6 or 180 μg yCD-his6 in 0.2 mL HEPES buffer. For a 1% NTA containing formulation, this solution corresponds to a 1:1 molar ratio of surface NTA to his6. To quantitate the extent of protein-liposome association, samples containing at least 2 μmol total lipid were passed through a pre-equilibrated 1 × 20 cm Sepharose CL-4B column under gravity flow. Fractions collected every 20 seconds (0.5 mL) were assayed for the presence of protein and liposomes. Liposomes eluted in the void volume and were measured by fluorescence (excitation/emission 490/520 nm). Protein concentration was quantified with a Bradford Protein Assay (Biorad Laboratories; Hercules, CA). This experiment was performed in triplicate for each protein/liposome formulation to ensure reproducibility.
Protein Activity
mKate activity was measured by fluorescence (excitation/emission 544/590 nm) in a Fluorostar 403 microplate reader (BMG Lab Technologies; Durham, NC). Enzymatic conversion of 5-FC to 5-FU per μg of yCD was measured using previously described methods (34). Briefly, 1 μg yCD or yCD on liposomes was incubated in 1 mL PBS containing 5 mM 5-FC at 37 °C. Aliquots of the reaction mixture (20 μL) were removed at 10, 20 and 30 minutes and enzymatic conversion quenched by dilution to 1 mL in 0.1 N HCl in PBS. The amount of 5-FU formed in the reaction was determined by measuring the UV absorbance at 255 and 290 nm. Concentration (mM) of 5-FU was calculated as 0.185 * (Abs 255) − 0.049 * (Abs 290) to adjust for presence of unconverted 5-FC. The activity of each sample was measured in triplicate.
Amount of enzyme activity per unit of lipid was measured after free protein was removed to ensure all active protein was associated with the surface of the liposome. Liposome concentration was estimated based on presence of a FITC-DSPE fluorophore within the membrane.
Protein Dissociation in Serum or Plasma
The dissociation of protein from liposomes was measured by challenging already associated protein with refiltered fetal calf serum (FCS). Liposomes with associated protein were mixed 1:1 in refiltered FCS and incubated at 37 °C for a predetermined time. After incubation, samples were passed through a 1 × 20 cm Sepharose CL-4B column under gravity flow to separate free from bound protein. Liposome-containing fractions were collected and yCD concentrations were measured based on conversion activity. mKate activity was measured by fluorescence. The experiment, including liposome formulation, was performed in triplicate.
For studies using mouse plasma, whole blood was collected using a heart puncture technique in BALB/c mice with a heparin (Sigma Aldrich; St. Louis, MO) loaded syringe. Plasma was clarified at 14,000 × g (Eppendorf Centrifuge 5415C, Brinkman Instruments, Inc.; Westbury, NY) for 20 minutes and stored at 4 °C for no longer than 2 days. Liposomes containing 1% tris-NTA-DAP were incubated with mKate for 1 hour prior to addition of mouse plasma. Liposomes, with pre-associated mKate, were mixed 1:1 with mouse plasma and incubated at 37 °C for 24 hours. Samples were prepared in triplicate.
His-Tagged Protein Competition
Liposomes were prepared as described above and incubated to give 1:1 surface NTA to mKate ratio. After 1 hour, surface association was challenged with yCD. Proteins were allowed to equilibrate for an additional hour at room temperature and then passed over a Sepharose CL-4B column. Total protein was measured using the Bradford protein assay. Fluorescence of mKate and liposomes were monitored as described above. The competition assay was performed in triplicate for each formulation.
Protein Half-Life in vivo
Liposomes were prepared at 20 mM lipid by hydrating films in sterile PBS, as described above. Once liposome diameter was reduced to approximately 80 nm, liposomes were sterilized with 0.2 micron filters. Each protein was iodinated following the Iodogen iodination protocol (Pierce Biotechnology, Rockford IL). Briefly, an iodination tube was washed with 1 mL PBS. To the tube was added 0.1 mL 0.2M HEPES, pH 7.4 and 10 μL NaI125 (≈ 1 mCi). Approximately 0.5 mg protein was added to the tube and the solution was reacted at room temperature for 10 minutes. The reaction mixture was diluted with non-radioactive protein and serially purified on a pre-packed PD10 desalting column followed by a 1 × 15 cm Sephadex G-50 column. Fractions containing radioactive protein were collected and reaction efficiency calculated. The protein was then further diluted to give one million counts per dose of protein. Diluted, iodinated protein was mixed with liposomes to give one protein per surface available NTA and incubated for 1 hour at room temperature prior to injection.
All animal procedures were conducted in accordance with the policies of the UCSF Institutional Animal Care and Use Committee. Mice (Jackson Laboratories; Bar Harbor, ME) were housed in an official UCSF facility. For periods of less than 48 hours, during experimentation, mice were housed in UCSF approved laboratory conditions. BALB/c or Swiss-Webster mice (6–8 weeks old) were divided into treatment groups of 3 animals. Each group received 10 mM total lipid with 1.35 mg/mL iodinated mKate or 0.89 mg/mL iodinate yCD in 200 μL PBS via tail vein injection (one million I125 counts per dose; injection accounts for approximately a 15% increase in total blood volume).
Radioactivity in the blood obtained from submandibular cheek pouch bleeds at 5 minutes and 1 hour (50 μL; less than 5% total blood volume) with a final bleed at 6 hours (0.5 to 1 mL; 30 to 60% total blood volume) was quantified by gamma scintillation counting (Wallac Wizard, Perkin Elmer; Waltham, MA). Liver, lung and spleen were collected and analyzed for accumulation of radioactivity.
Non-iodinated mKate and liposomes containing 0.5% FITC-DSPE were monitored in the blood by heart puncture at 15 minutes. Plasma was collected by centrifugation at 14,000 × g and liposomes were separated from the bulk free protein with a Sepharose CL-4B column. mKate fluorescence was measured (excitation/emission 544/590 nm) to quantify mKate remaining with the liposomes.
Association of Endogenous Proteins with NTA-Liposomes After Intravenous Administration
Liposomes containing either 1% tris-NTA-DAP or 1.5% POPG and 0.5% FITC-DSPE, 5% mPEG-DSPE were recovered from the blood of BALB/c mice. Whole blood was collected 10 minutes after liposome injection (2 μmol lipid in 200 μL PBS) by heart puncture using a heparin loaded syringe. Plasma was collected by sedimenting blood cells at 14,000 × g and liposomes were separated from unassociated protein using a Sepharose CL-4B column. Fractions containing liposomes were collected based on FITC fluorescence (excitation/emission 490/520 nm). Liposome concentration was calculated based on a FITC fluorescence standard curve constructed from serially diluted liposomes of the same lipid formulation. Protein concentration was measured using the Bradford Protein Assay (Biorad Laboratories; Hercules, CA).
Liposomes fractions containing liposome-associated proteins were mixed with SDS Gel-loading buffer (1:1 v/v) (35) and boiled for 10 minutes. Approximately 5 μg protein was loaded per well onto a 15-well, 4–15% Tris-Glycine, SDS-PAGE gel (Biorad Laboratories; Hercules, CA). Protein bands were separated at 120 V for approximately 1 hr and the gel was stained (Silver Stain Plus, Biorad Laboratories; Hercules, CA). Pre-stained protein ladders (10–230 kDa, New England Biolabs; Ipswich, MA) were included to estimate protein molecular weight. Relative protein quantity in each band was estimated using ImageJ gel analysis software (NIH ImageJ 1.42q; Bethesda, MD). For statistical comparisons, lanes were equalized to the 37 kDa protein band.
mKate Association with NTA-Liposomes Recovered After Intravenous Administration
BALB/c mice were injected with liposomes containing 1.0% tris-NTA-DAP and 5% mPEG-DSPE. Whole blood was collected and plasma was clarified via centrifugation as was described previously. Free protein was separated from liposome-bound protein and liposome containing fractions were collected based on fluorescence. Liposomes were concentrated using a microcon concentrator (YM10, 10,000 MWCO, Millipore; Bedford, MA) pre-equilibrated with untreated tris-NTA-DAP-containing liposomes. Liposome fractions were concentrated for 30 minutes at 14,000 × g. Final liposome concentration was estimated based on liposome fluorescence. Liposomes were then incubated with 5-fold excess of mKate (5 mKate per 1 surface available tris-NTA-DAP) for 3 hours at room temperature. Free mKate was separated from liposome-associated mKate on a Sepharose CL-4B column. mKate fluorescence was measured (excitation/emission 544/590 nm) in liposome-containing fractions.
Hemopexin Interactions with NTA-Liposomes in vitro
Hemopexin (HPX, Human, 70 kDa, Cell Sciences; Canton, MA) was dissolved in PBS (to 1 mg/mL) and incubated with liposomes (5 μmol/mL) containing 1.0% tris-NTA-DAP and 5% mPEG-DSPE for 3 hrs at room temperature. The final ratio of HPX to tris-NTA-DAP available on the liposome surface was 1:2 (a 2-fold excess tris-NTA-DAP). Following incubation, free HPX was separated from liposome-associated protein on a Sepharose CL-4B column pre-equilibrated in PBS. Liposome concentration was determined by FITC-fluorescence. Protein concentration in each fraction was determined by Bradford Protein Assay.
The disassociation of mKate from liposomes was measured by challenging liposomes containing pre-associated mKate with HPX. Liposomes were incubated with mKate for 1 hour prior to HPX addition. Liposomes containing pre-associated mKate (3 μmol/mL with 0.4 mg/mL mKate) were incubated with a 1.5 volume excess HPX (at 1 mg/mL). The final ratio of HPX to tris-NTA-DAP available on the liposome surface was 1.5:1. The molar ratio of HPX to mKate in solution was also 1.5:1. Samples were incubated at 37 °C for 3 hours. After incubation, samples were passed through a 1 × 20 cm Sepharose CL-4B column under gravity flow to separate free from bound protein. Fractions containing mKate were assayed based on fluorescence (excitation/emission 544/590 nm). The ratio of mKate fluorescence to μmol/lipid was based upon a FITC standard curve prepared from liposomes of the same lipid formulation. Samples were assayed in triplicate.
Results
Liposome Formulation and Construction
An illustration of a liposome and the NTA lipids used in this study is shown in Figure 1. The reported equilibrium dissociation constants for each lipid with a multihistidine peptide are also given (4, 18). Liposome diameter and stability was largely unaffected by the presence of NTA lipids (up to 3 mol %). We estimated, using the protein diameter (UCSF Chimera; San Francisco, California) and NTA-lipid surface density on the liposome (36), that steric hindrance between adjacent NTA binding sites should effect association around 3% NTA with a 1:1 surface NTA to protein loading ratio. The liposome diameter and charge as measured by the zeta potential were similar for all formulations (Table 1).
Table 1.
Liposome Diameter and Zeta-Potential of NTA-Containing Liposomes with 3, 1 or 0.3% NTA and 5 mol % mPEG or 1% NTA without mPEG. Control liposomes contained 1.5 mol % POPG to mimic the charge of 0.3% NTA-containing liposomes. (n=3)
| 3% NTA, 5% mPEG | 1% NTA, 5% mPEG | 1% NTA, 0% mPEG | 0.3% NTA, 5% mPEG | |
|---|---|---|---|---|
| tris-NTA-DAP | ||||
| Diameter (nm) | 88.0 ± 6.4 | 82.1 ± 2.5 | 88.3 ± 6.6 | 95.2 ± 15.1 |
| ζ potential (mV) | −21.0 | −18.3 ± 2.7 | −18.6 ± 1.3 | −19.3 |
| tris-NTA-LYS | ||||
| Diameter (nm) | 107.9 ± 3.8 | 96.0 ± 11.5 | 96.6 ± 12.1 | 82.9 ± 7.6 |
| ζ potential (mV) | −22.8 | −18.6 ± 0.2 | −20.1 | −15.3 |
|
| ||||
| NTA-DOGS | ||||
| Diameter (nm) | 78.7 ± 14.7 | 75.4 ± 12.3 | 85.0 ± 7.8 | 71.5 ± 1.0 |
| ζ potential (mV) | −18.9 | −16.9 ± 0.6 | −16.1 | −12.6 |
|
| ||||
| Control (1.5% POPG, 5% mPEG) | ||||
| Diameter (nm) | 80.5 ± 6.5 | |||
| ζ potential (mV) | −15.6 ± 1.6 | |||
His-Tagged Protein Association with Surface Displayed NTA on Liposomes
Liposomes containing any of the three NTA-lipids associated with protein to near the maximal expected amount. Free protein was separated from liposome bound protein to determine the extent of association. Liposomes, along with their associated protein, elute in the void volume of a Sepharose CL-4B size exclusion column, non-associated protein elutes at a larger volume. A representative example of protein separation in each fraction is shown in Figure 2. Liposomes with 3, 1 or 0.3% NTA3-DAP associated with yCD to near saturation, with no free yCD visible in later fractions. BSA did not associate with 1% NTA3-DAP liposomes.
Figure 2. Representative Association of Proteins with the Surface of NTA-Liposomes.
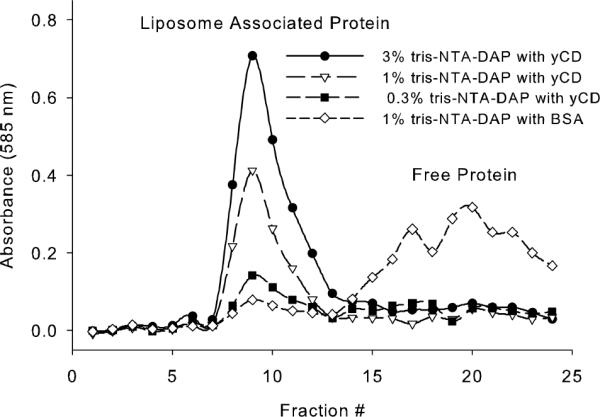
Liposomes bearing tris-NTA-DAP at 3% (circles), 1% (triangles) or 0.3% (squares) were associated with his-tagged yCD to saturate the available surface NTA. BSA was associated with 1% tris-NTA-DAP liposomes as a control (diamonds).
The concentration of liposomes in each fraction was calculated based on FITC-DSPE fluorescence. It was assumed that NTA lipids would distribute evenly between the inner and outer liposome surface. Hence, complete association was deemed to be a 1:1 ratio of his-tagged protein to expected surface available NTA (Table 2).
Table 2. yCD Association with NTA-Containing Liposomes.
Protein association was measured using the Bradford protein assay after unbound protein removal on a Sepharose CL-4B size exclusion column. Two variables are indicated for each NTA formulation: percent NTA present in the liposome is indicated by mol % NTA, protein added to formed liposomes is indicated as a ratio (NTA:protein). Data represents analysis of liposome-containing fractions.
| mol % NTA with 5% mPEG | tris-NTA-DAP | tris-NTA-LYS | NTA-DOGS | ||||
|---|---|---|---|---|---|---|---|
| (NTA:protein) ratio | mg/μmol lipid | % of expected | mg/μmol lipid | % of expected | mg/μmol lipid | % of expected | |
| 3.0% (1:1) | 0.28 ± 0.02 | 103 ± 7 | 0.28 ± 0.03 | 102 ± 12 | 0.27 ± 0.05 | 99 ± 16.8 | |
| 3.0% (3:1) | 0.09 ± 0.02 | 91 ± 16 | 0.10 ± 0.01 | 105 ± 9 | 0.09 ± 0.01 | 94 ± 7.3 | |
| 1.0% (1:1) | 0.10 ± 0.02 | 103 ± 16 | 0.12 ± 0.01 | 128 ± 11 | 0.10 ± 0.01 | 108 ± 12.2 | |
| 0.3% (1:1) | 0.04 ± 0.01 | 108 ± 15 | 0.04 ± 0.00 | 131 ± 1 | 0.03 ± 0.00 | 94 ± 12.9 | |
| 0.3% (1:3) | 0.03 ± 0.01 | 118 ± 21 | 0.04 ± 0.00 | 118 ± 3 | 0.06 ± 0.01 | 189 ± 16.4 | |
| 1.0% (1:1) | No mPEGa | 0.09 ± 0.01 | 104 ± 14 | 0.11 ± 0.01 | 119 ± 6 | 0.10 ± 0.02 | 104.2 ± 23.8 |
| 1.0% (1:1) | BSAb | 0.01 ± 0.00 | 11 ± 3 | 0.01 ± 0.00 | 12 ± 4 | 0.01 ± 0.00 | 8.6 ± 1.0 |
| 1.0% (1:1) | No nickelc | 0.02 ± 0.01 | 20 ± 6 | 0.02 ± 0.00 | 23 ± 2 | ||
| 1.5% POPG | No NTAd | 0.01 ± 0.00 | 13 ± 3 | ||||
Control liposomes of various formulations were used to validate NTA-nickel-his-tag interactions.
Liposomes formulated without a 5% mPEG coat associated similar amounts of yCD as those containing mPEG.
BSA, a control protein that does not contain a his-tag, had limited association with liposomes containing NTA-nickel.
Liposomes formulated with NTA, but without nickel, associated less yCD than those with nickel.
Liposomes containing POPG, to mimic the charge of NTA-containing liposomes, had minimal yCD association.
Liposomes that contained NTA but not nickel did not associate with protein. BSA, which does not contain a his-tag, did not associate with NTA-nickel-containing liposomes. Control liposomes, containing both mPEG and POPG and having a similar surface charge as 0.3% NTA liposomes, associated with a minor amount of protein. The addition of EDTA, a strong chelator for nickel, to any NTA-containing liposome eliminated protein binding. Thus a chelated nickel on the liposome surface and a his-tagged protein are necessary for this technology to be effective. When those elements are present, the amount of bound protein is stoichiometric with the amount of Ni-NTA on the liposome surface. yCD is a dimer so it might be expected that greater than 100% association would occur but this was rarely seen. This indicates that both his-tags of the dimer are engaged to the NTAs on the liposome surface. The amounts of mKate associated with 3, 1 and 0.3 mol % NTA containing liposomes are given in Figure 3.
Figure 3.
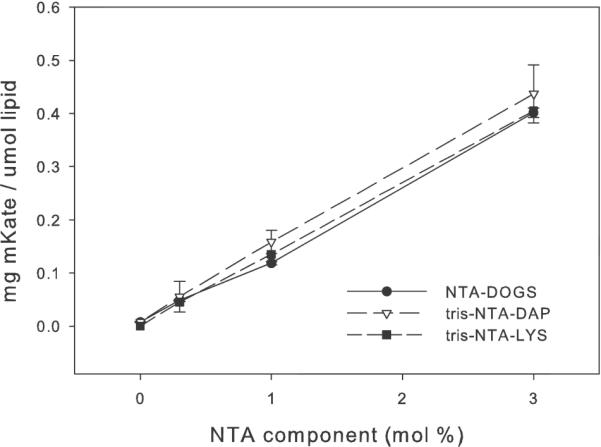
Association of mKate with the Surface of NTA-Liposomes The amount of protein per μmol liposomes containing 3, 1 and 0.3 mol % NTA-DOGS (circles), tris-NTA-DAP (triangles) or tris-NTA-LYS (squares). (n=3)
Activity Retention of Surface Associated Proteins
Covalent attachment methods often partially or fully inactivate proteins (25). We measured the retention of activity of both yCD and mKate after binding to the NTA liposome. yCD converts the prodrug 5-FC to the cytotoxic drug 5-FU and the association of yCD with the surface of liposomes does not hinder 5-FC prodrug conversion. yCD retains full enzymatic activity when on the surface of a liposome (Figure 4A). Some proteins exhibit increased function and experience less product inhibition (to which yCD is susceptible) when in close proximity to a lipid bilayer (22); a slight increase in activity is observed in the liposome bound yCD.
Figure 4. Retained Protein Activity of yCD (A) or mKate (B) on the Surface of NTA-Liposomes.

The protein activity per μmol liposomes containing 3, 1 and 0.3 mol % NTA-DOGS (circles), tris-NTA-DAP (triangles) or tris-NTA-LYS (squares). (n=3)
mKate also completely occupied all of the available sites on NTA-bearing liposomes at the NTA surface densities studied (Figure 4B). The activity of these liposomes was based on the fluorescence of the mKate. Unlike the observed increase in yCD activity when associated with the NTA-liposome there was no increase in fluorescence for mKate.
Protein Dissociation in the Presence of Serum or Plasma
Liposomes bearing NTA pre-associated with mKate or yCD were mixed at a 1:1 volume ratio with refiltered FCS. Figure 5 shows the retained protein on the surface of 1% NTA, 5% mPEG liposomes. The estimated amount of protein retained by the liposome is based on active protein and thermostabilized yCD has an activity half-life of over 50 hours (32). The decrease in fluorescent activity of mKate over 24 hours in FCS corresponds to approximately 5 – 10% of the total mKate fluorescence. This fluorescence decrease is equivalent to that seen in the loss of total fluorescence from free mKate in FCS at 37°C over the same time period.
Figure 5. Dissociation of yCD (A) or mKate (B,C) from NTA-Liposomes in Serum or Plasma.

Enzyme activity per unit of liposome (for yCD) or percent retained mKate fluorescence after unbound protein has been removed for each of four liposome types: 1% tris-NTA-DAP (circles), 1% tris-NTA-LYS (triangles), 1% NTA-DOGS (filled squares) and control liposomes containing 1.5% POPG (diamonds). Time-course studies were done in the presence of refiltered fetal calf serum (A,B). Disassociation in the presences of mouse plasma was studied at 24 hours (C). (yCD, n=1; mKate, n=3)
Disassociation of mKate from tris-NTA-DAP containing liposomes was also measured in the presence of mouse plasma. Plasma was prepared, stored at 4 °C and used within 2 days. More mKate disassociated from the surface of NTA-containing liposomes after 24 hours (Figure 5C) than disassociated from liposomes exposed to FCS (Figure 5B). Tris-NTA-DAP-liposomes exposed to mouse plasma retained 30 ± 4 % of the initial mKate after 24 hours.
NTA3-DAP has a higher affinity for histidine-tags than does NTA3-LYS (5). Tris-NTAs in turn have significantly higher affinity (0.2–10 nM) than mono-NTA (10 μM) (18). For both yCD and mKate, retention in refiltered FCS or mouse plasma is related, but not directly proportional to, the relative affinity for the NTA lipids.
Competition between NTA-Liposome Bound His6-Proteins and Subsequently Added His6-Proteins
mKate that was stably associated with the liposome surface was challenged with added his6-yCD. Despite the slightly higher affinity of tris-NTA-lipids for mKate (Kd=0.69 nM) than for yCD (Kd=3.02 nM) (5), yCD exchanged with pre-associated mKate for positions on the liposome surface (Figure 6). Upon addition of a competing protein, proteins redistributed to yield the stoichiometric distribution of proteins on the surface of the liposome regardless of their reported affinity. For example, if yCD and mKate are at the same final concentration in solution, there will be an equal amount of yCD and mKate on the surface of the liposome and in solution. This arrangement will occur regardless of which protein is first incubated with the liposomes. When pre-bound mKate was challenged with BSA, which lacked a his-tag, no mKate dissociated; however, BSA non- specifically associated with the liposome so that the total amount of liposome-bound protein increased.
Figure 6. Competition of Proteins with Pre-Associated mKate.
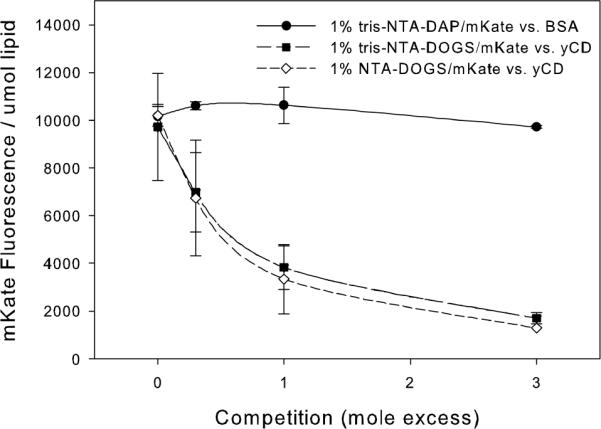
Competition of BSA for pre-associated mKate on 1% tris-NTA-DAP (circles) or yCD for pre-associated mKate on 1% tris-NTA-DAP (squares) and 1% NTA-DOGS (diamonds). (n=3)
Retention of Tris-NTA-lipids in Liposomes
Liposomes containing 3% tris-NTA-DAP (−23.6 mV) were retained on a highly charged anion exchange column while neutral liposomes (−4.1 mV) passed through the column in the void volume. The tris-NTA-DAP-DOD lipid did not transfer between labile lipid bilayers to a measurable degree. After 3 hours at 37 °C the neutral liposomes passed through the anion exchange column in the same fraction and retained the same surface charge (−4.8 ± 1.3) as neutral liposomes that were not incubated with NTA-containing liposomes (−3.8± 1.4). Neutral liposomes were fully recovered in the void volume (105,537 ± 13,546 a.u. fluorescence or 105 ± 14 % of the expected fluorescence based on untreated, neutral liposomes).
Half-Life of NTA-Liposome Associated Proteins Upon Intravenous Administration
The association of iodinated protein with NTA-liposomes did not increase the residence time of either protein in the blood after intravenous injection (Figure 7). To ensure that iodination was not responsible for protein loss from the liposome, non-iodinated mKate was associated with fluorescent tris-DAP-NTA-liposomes and injected into mice. After 15 minutes, blood was collected and the mKate-NTA liposomes remaining in plasma were separated on a Sepharose CL-4B column. A majority of the liposomes remained in circulation but only a small fraction of the injected mKate was associated with the liposome fractions from the column (Figure 7B, inset). This verified that the mKate rapidly dissociated from the liposome upon contact with blood.
Figure 7. Disappearance of Iodinated yCD (A) or mKate (B) after Intravenous Injection In Mice.

(filled triangles) on four liposome types: 1% tris-NTA-DAP (open triangles), 1% tris-NTA-LYS (diamonds), 1% NTA-DOGS (circles) or control liposomes containing 1.5% POPG (squares). Each animal was injected with one million I125 counts per dose. Non-iodinated mKate (B-inset) fluorescence remaining bound 15 minutes after injection on 1% tris-NTA-DAP liposomes. (n=3)
Endogenous Protein Binding to NTA-Liposomes Upon Intravenous Administration
When recovered from blood, tris-NTA-DAP containing liposomes had a different protein profile than liposomes with a similar charge (1.5% POPG, Figure 8A). In this experiment three mice were injected per group with each liposome formulation. Unassociated plasma proteins were removed from liposome fractions based on size exclusion on a Sepharose CL-4B column. Tris-NTA-DAP-containing liposomes associated more protein (420 ± 90 μg/μmol lipid) than did POPG-containing liposomes (320 ± 60 μg/μmol lipid); however, this difference was not significant (p=0.19). Lower molecular weight bands at approximately 12, 28, 58–60 and 75–90 kDa were enriched in tris-NTA-DAP-containing liposomes compared to POPG containing liposomes (Figure 8A).
Figure 8. Analysis of Protein Interactions with NTA-Modified and Control Liposomes.

Liposomes containing either 1% tris-NTA-DAP or 1.5% POPG with 5% mPEG were recovered from the plasma of mice after intravenous injection. Each lane represents recovery of liposomes from an individual mouse, which accounts for the variable amount of total protein per well. Liposomes were purified to remove unassociated proteins and run on a 4–15% Tris-Glycine SDS Page Gel. Circles (●) denote proteins that were substantially more visible in tris-NTA-DAP containing fractions than in control fractions after silver staining (A). Relative amount of protein in each band was quantified using ImageJ software. Association of the proteins at 60 kDa and 12 kDa are statistically greater for the tris-NTA-DAP-liposome than control liposomes (* p<0.05) (B). Competition to disassociate mKate from 1% tris-NTA-DAP, 5% mPEG liposomes by Hemopexin (HPX, light gray) or to associate mKate with liposomes after recovery from plasma (dark gray) (C). (HPX Competition n=2; mKate Addition n=3)
The quantity of protein present in the bands at approximately 12, 27, 37 and 60 (as a doublet) kDa was estimated using ImageJ gel analysis software and the area under the histogram peaks was calculated (Figure 8B). Equalizing the values based on the protein band at 37 kDa suggests that tris-NTA-DAP liposomes associate the 12 kDa and 60 kDa proteins significantly more than POPG containing liposomes (p=0.048, 0.040 respectively). Association of the 27 kDa protein was higher with tris-NTA-liposomes than POPG; however, the difference was not significant.
Liposomes recovered from the blood of mice associated mKate after separation from plasma proteins. Liposomes containing tris-NTA-DAP were purified from unassociated plasma proteins, concentrated and then mixed with a 5-fold excess of mKate. Liposomes associated approximately 70% of the mKate that would be expected based on liposome fluorescence (7000 units fluorescent/μmol lipid) compared to liposomes that were not exposed mouse blood. This confirms that the tris-NTA lipid was not removed from the liposome during circulation in mice (Figure 8C).
Hemopexin, a plasma protein which scavenges heme from the blood, was identified as a candidate for removing nickel from tris-NTA containing liposomes (37). Exposure of tris-NTA-DAP-liposomes to hemopexin (Figure 8C) did not effect mKate association. Hemopexin bound to tris-NTA-DAP-liposomes at approximately 25 μg/μmol lipid; this binding quantity (approximately 8% of 1:1 surface available NTA:HPX) is considered non-specific binding and is similar to the quantity seen for BSA association to NTA-liposomes or his-tagged protein association to POPG control liposomes.
Discussion
We characterized the effect of increasing affinity in a series of Ni-NTA/his6 pairs, on his6-protein-NTA liposome association, his6-protein activity and his6-protein retention on NTA-liposomes under a variety of conditions. The surprising finding is that despite the retention of the his6-proteins on the NTA liposomes in FCS and mouse plasma, the NTA-liposome-attached protein rapidly dissociated from the liposome upon intravenous administration and hence was eliminated from circulation as rapidly as the injected `free' protein. The rapid elimination occurred regardless if the liposome contained a low affinity NTA lipid or a high affinity NTA lipid and was not consistent with the measured off-rates of the same protein on a NTA Biacore chip.
Our experiments were enabled by the synthesis of a series of tris-NTA chelators with increasing affinity for the his6-tag (5). We synthesized tris-NTA-lipids with two different carbon lengths between each NTA headgroup to examine the effect of linker spacing on binding affinity. yCD and mKate retained activity when associated with the surface of NTA-containing liposomes, so the interaction holds forth the prospect of a modular system for easily constructing liposomes modified with any protein that has a his6-tag.
Retention time for the his6-proteins on the surface of a Biacore chip modified with the various NTA derivatives increased as NTA linker length decreased. Dissociation rates for the same NTA derivative also varied between his6-proteins; this was ascribed to differences in the accessibility of the his-tag (5). Assuming that the protein is rapidly eliminated when released from the liposome, the blood residence time of a his6-protein attached to an NTA-liposome after intravenous administration can be calculated from the dissociation rate. For the tris-NTA-LYS headgroup, with four carbons in the linker region, the half-time of release from the surface of a Biacore chip was 14 minutes with yCD (kd=0.83 * 10−3/sec). If the linker is decreased by one carbon, the half-time was 20 minutes with yCD (kd=0.59 * 10−3/sec) and 50 minutes with mKate (kd=0.22 * 10−3/sec). When the linker region was decreased by another carbon, the computed half-time with mKate increased to over 2 hours (kd=.089 * 10−3/sec) (5).
Proteins associated with the higher affinity NTA-liposomes were retained on the liposome much better when exposed to FCS then when on the mono-NTA modified liposomes (Figure 5). The magnitude of this increased association time is related to the affinity of each NTA-protein pair (Figure 5). At 24 hours, approximately 80% of the initially bound mKate remained associated with either the tris-NTA-LYS and tris-NTA-DAP liposomes, whereas only 20% mKate remained bound to liposomes containing NTA-DOGS. yCD dissociated to a greater extent from the surface of the liposome than did mKate which is consistent with the difference in affinity (Kd=0.69 and 3.02 nM) for mKate and yCD, respectively. When plasma was obtained from mice, approximately 30% of the initially bound mKate remained associated with the tris-NTA-DAP liposomes at 24 hours (Figure 5C). However, as expected for a reversible interaction, his6-proteins could exchange with proteins in solution even from the lipid with the highest affinity, the tris-NTA-DAP. This reflects the dynamic nature of this system (Figure 6).
While tris-NTA-DAP increased retention of both proteins as compared to NTA-DOGS in FCS, it did not improve protein association in vivo (Figure 7). All of the formulations studied exhibited a similar clearance rate as free protein. PEGylated liposomes of this diameter have a circulation half-life of half a day or more (38). If the disassociation rate was the prime determinant of protein loss from the liposome, tris-NTA association should have afforded some improvement in circulation time for the liposome bound protein over the free protein. The dynamic nature of non-covalent protein association with NTA may allow any multihistidine-containing protein in the body to actively compete with the associated protein, even if the affinity of the nascent, competing protein is much lower than that of the associated protein. However if loss of protein is a simple equilibrium phenomenon, the off-rate would still control the residence time of the protein on the liposome. Therefore there must be other factors that contribute to the rapid disassociation after intravenous injection.
Plasma proteins may play a crucial role in the disruption of his6-NTA association (Figure 9). Some plasma proteins may interact preferentially with the surface of NTA-liposomes (Figure 8A) (29) and may contribute to protein dissociation or loss of nickel from the chelator. Tris-NTA-DAP-containing liposomes recovered from mouse blood contained several proteins that were less present liposomes containing POPG. These proteins may include metal scavenging and histidine rich plasma proteins, such as thioredoxin (12 kDa), Hemopexin (HPX, approximately 60 kDa) and Histidine-rich glycoprotein (HRG, 75 – 95 kDa) (37, 39, 40). HPX scavenges heme within the blood, partially due to a high-affinity binding site containing eight histidines (37). This protein also binds divalent metal ions, including nickel (41) and has been shown to interact with immobilized metal-affinity columns (42). Although HPX did not bind tris-NTA-DAP liposomes or cause mKate disassociation under the conditions studied (Figure 8C), similar metal-scavenging plasma proteins may interact with liposome-displayed NTA in vivo. HRG, another abundant plasma protein, has also been shown to scavenge heme, interact with metal ions and bind to nickel-NTA with high affinity (39, 43). Endogenous bio-chelators may act as an additional destabilizing factor by removing bound nickel from the NTA (44). Although NTA-liposomes recovered from blood could associate mKate (Figure 8C) without addition of exogenous nickel, removal of nickel from the NTA in blood may act to partially disassociate proteins. Competition for NTA occupancy by endogenous histidine-rich proteins, along with loss of nickel, may cause the initial protein disassociation from the liposome upon exposure to blood.
Figure 9. Modes of Interaction of His-Tagged Proteins with NTA-Containing PEGylated Liposomes.
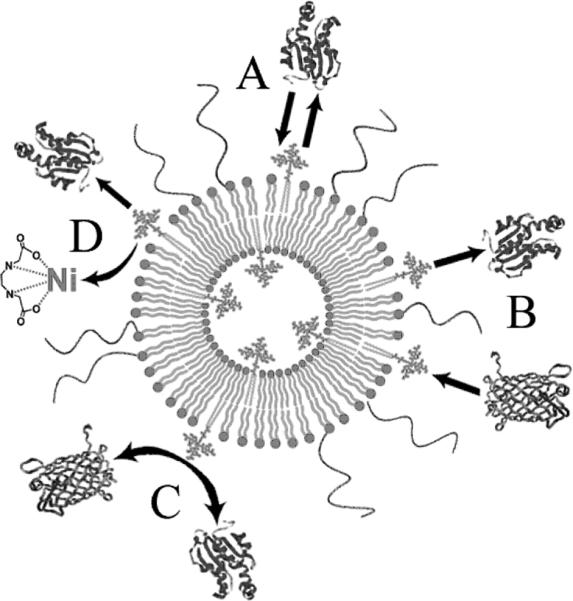
Proteins bearing a his-tag associate and dissociate from the liposome surface (A). Upon dissociation, other proteins may associate with the newly available NTAs (B), proteins may directly compete for NTA binding (C) or nickel may be removed by endogenous chelators (D).
Several groups have published the robust association of his-tagged proteins with NTA containing nanolipoparticles (3, 4, 26, 29, 30, 45). Our findings must be reconciled with a number of papers where proteins associated with liposomes or lipid particles containing mono-NTA or tris-NTA mediate a number of immune response (3, 26, 30). Protein-NTA particles generated a measurable immune response based on specific serum IgG levels (30) or an increase in splenocytes secreting cytokines (26). Mice immunized with tris-NTA containing plasma membrane vesicles showed decreased tumor growth (3). In these studies the NTA particle was injected via the subcutaneous route. After subcutaneous administration the particles are exposed to an order of magnitude lower concentration of endogenous proteins and the NTA liposome and its attached protein are eliminated more slowly than after intravenous administration. Second, the immune response is exquisitely sensitive to a protein antigen and even small quantities of protein that remained associated with the particle might have been responsible for the observed effects. Thus it is not possible to deduce the amount of protein that these various particles delivered from the observed immune responses.
Many in vitro cellular studies using protein-his6/NTA-lipooparticles are performed in the presence of FCS-rich cell culture media. Under these conditions, Herringson et al state that the his6-protein/tris-NTA association is sufficiently robust so that in vitro retention will translate to in vivo protein/particle stability (46). However, our results indicate that improvements in metal chelation methodologies are required if metal chelation technology is to be useful for in vivo particle applications. Our group is exploring methods to improve the affinity of the system and reduce loss of nickel chelation. One approach is to increase the shielding in the vicinity of the acetic acid moiety by incorporating branched methyl groups. This might provide a hydrophobic wall that impedes removal of the chelated metal. An alternative approach is to synthesize a “clamp” for the chelated nickel. Chemically, this can be easily accomplished by covalently attaching histamines to a triacetic acid scaffold, such as the building blocks used to synthesize tris-NTA. The imidazole groups would then form a triad complementary structure to interact with the chelated nickel on the lipids. Although, the triad histamine could not be incorporated into a recombinant protein, it could be used as a high affinity binding partner for the chelated nickel. It remains to be discovered whether such improvements will be sufficient to stabilize NTA-protein-his6 complexes in vivo.
Acknowledgements and Author Contributions
We gratefully thank Katherine Jerger for her assistance with animal experiments. This work was supported by grant NIH R01 GM061851. Protein production and the salary of Dr. Cao were supported by UC discovery grant Bio 60-10618 and Halozyme, Inc.
Our appreciation is extended to Greg Frost for suggesting hemopexin might interfere with his6/NTA interactions in plasma and Christopher Cullander for helpful discussions regarding this manuscript.
References
- (1).Sigal GB, Bamdad C, Barberis A, Strominger J, Whitesides GM. A self-assembled monolayer for the binding and study of histidine-tagged proteins by surface plasmon resonance. Anal Chem. 1996;68:490–497. doi: 10.1021/ac9504023. [DOI] [PubMed] [Google Scholar]
- (2).Ueda EK, Gout PW, Morganti L. Current and prospective applications of metal ion-protein binding. J Chromatogr A. 2003;988:1–23. doi: 10.1016/s0021-9673(02)02057-5. [DOI] [PubMed] [Google Scholar]
- (3).van Broekhoven CL, Altin JG. The novel chelator lipid 3(nitrilotriacetic acid)-ditetradecylamine (NTA(3)-DTDA) promotes stable binding of His-tagged proteins to liposomal membranes: potent anti-tumor responses induced by simultaneously targeting antigen, cytokine and costimulatory signals to T cells. Biochim Biophys Acta. 2005;1716:104–116. doi: 10.1016/j.bbamem.2005.09.003. [DOI] [PubMed] [Google Scholar]
- (4).Huang Z, Park JI, Watson DS, Hwang P, Szoka FC., Jr. Facile synthesis of multivalent nitrilotriacetic acid (NTA) and NTA conjugates for analytical and drug delivery applications. Bioconjug Chem. 2006;17:1592–1600. doi: 10.1021/bc0602228. [DOI] [PubMed] [Google Scholar]
- (5).Huang Z, Hwang P, Watson DS, Cao L, Szoka FC. Tris-Nitrilotriacetic Acids of Subnanomolar Affinity Toward Hexahistidine Tagged Molecules. Bioconjug Chem. 2009;20:1667–1672. doi: 10.1021/bc900309n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Altin JG, Banwell MG, Coghlan PA, Easton CJ, Nairn MR, Offermann DA. Synthesis of NTA(3)-DTDA - A chelator-lipid that promotes stable binding of His-tagged proteins to membranes. Australian Journal of Chemistry. 2006;59:302–306. [Google Scholar]
- (7).Balland V, Hureau C, Cusano AM, Liu Y, Tron T, Limoges B. Oriented immobilization of a fully active monolayer of histidine-tagged recombinant laccase on modified gold electrodes. Chemistry. 2008;14:7186–7192. doi: 10.1002/chem.200800368. [DOI] [PubMed] [Google Scholar]
- (8).Dietrich C, Schmitt L, Tampe R. Molecular organization of histidine-tagged biomolecules at self-assembled lipid interfaces using a novel class of chelator lipids. Proc Natl Acad Sci U S A. 1995;92:9014–9018. doi: 10.1073/pnas.92.20.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dorn IT, Pawlitschko K, Pettinger SC, Tampe R. Orientation and two-dimensional organization of proteins at chelator lipid interfaces. Biol Chem. 1998;379:1151–1159. doi: 10.1515/bchm.1998.379.8-9.1151. [DOI] [PubMed] [Google Scholar]
- (10).Tinazli A, Tang J, Valiokas R, Picuric S, Lata S, Piehler J, Liedberg B, Tampe R. High-affinity chelator thiols for switchable and oriented immobilization of histidine-tagged proteins: a generic platform for protein chip technologies. Chemistry. 2005;11:5249–5259. doi: 10.1002/chem.200500154. [DOI] [PubMed] [Google Scholar]
- (11).Schuy S, Treutlein B, Pietuch A, Janshoff A. In situ synthesis of lipopeptides as versatile receptors for the specific binding of nanoparticles and liposomes to solid-supported membranes. Small. 2008;4:970–981. doi: 10.1002/smll.200701006. [DOI] [PubMed] [Google Scholar]
- (12).Keller TA, Duschl C, Kröger D, Sévin-Landais A-F, Vogel H, Cervigni SE, Dumy P. Reversible oriented immobilization of histidine-tagged proteins on gold surfaces using a chelator thioalkane. Supramolecular Science. 1995;2:155–160. [Google Scholar]
- (13).Barklis E, McDermott J, Wilkens S, Schabtach E, Schmid MF, Fuller S, Karanjia S, Love Z, Jones R, Rui Y, Zhao X, Thompson D. Structural analysis of membrane-bound retrovirus capsid proteins. Embo J. 1997;16:1199–1213. doi: 10.1093/emboj/16.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dorn IT, Eschrich R, Seemuller E, Guckenberger R, Tampe R. High-resolution AFM-imaging and mechanistic analysis of the 20 S proteasome. J Mol Biol. 1999;288:1027–1036. doi: 10.1006/jmbi.1999.2714. [DOI] [PubMed] [Google Scholar]
- (15).Zhen G, Zurcher S, Falconnet D, Xu F, Kuennemann E, Textor M. NTA-Functionalized Poly(L-lysine)-g-Poly(Ethylene Glycol): A Polymeric Interface for Binding and Studying 6 His-tagged Proteins. Conf Proc IEEE Eng Med Biol Soc. 2005;1:1036–1038. doi: 10.1109/IEMBS.2005.1616595. [DOI] [PubMed] [Google Scholar]
- (16).Kang E, Park JW, McClellan SJ, Kim JM, Holland DP, Lee GU, Franses EI, Park K, Thompson DH. Specific adsorption of histidine-tagged proteins on silica surfaces modified with Ni2+/NTA-derivatized poly(ethylene glycol) Langmuir. 2007;23:6281–6288. doi: 10.1021/la063719e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Guignet EG, Hovius R, Vogel H. Reversible site-selective labeling of membrane proteins in live cells. Nat Biotechnol. 2004;22:440–444. doi: 10.1038/nbt954. [DOI] [PubMed] [Google Scholar]
- (18).Lata S, Reichel A, Brock R, Tampe R, Piehler J. High-affinity adaptors for switchable recognition of histidine-tagged proteins. J Am Chem Soc. 2005;127:10205–10215. doi: 10.1021/ja050690c. [DOI] [PubMed] [Google Scholar]
- (19).Lata S, Gavutis M, Tampe R, Piehler J. Specific and stable fluorescence labeling of histidine-tagged proteins for dissecting multi-protein complex formation. J Am Chem Soc. 2006;128:2365–2372. doi: 10.1021/ja0563105. [DOI] [PubMed] [Google Scholar]
- (20).Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- (21).Werle M, Bernkop-Schnurch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids. 2006;30:351–367. doi: 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- (22).Nagami H, Yoshimoto N, Umakoshi H, Shimanouchi T, Kuboi R. Liposome-assisted activity of superoxide dismutase under oxidative stress. J Biosci Bioeng. 2005;99:423–428. doi: 10.1263/jbb.99.423. [DOI] [PubMed] [Google Scholar]
- (23).Spira J, Plyushch OP, Andreeva TA, Andreev Y. Prolonged bleeding-free period following prophylactic infusion of recombinant factor VIII reconstituted with pegylated liposomes. Blood. 2006;108:3668–3673. doi: 10.1182/blood-2006-03-008276. [DOI] [PubMed] [Google Scholar]
- (24).Yatuv R, Dayan I, Carmel-Goren L, Robinson M, Aviv I, Goldenberg-Furmanov M, Baru M. Enhancement of factor VIIa haemostatic efficacy by formulation with PEGylated liposomes. Haemophilia. 2008;14:476–483. doi: 10.1111/j.1365-2516.2008.01741.x. [DOI] [PubMed] [Google Scholar]
- (25).Colletier JP, Chaize B, Winterhalter M, Fournier D. Protein encapsulation in liposomes: efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2002;2:9. doi: 10.1186/1472-6750-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chikh GG, Li WM, Schutze-Redelmeier MP, Meunier JC, Bally MB. Attaching histidine-tagged peptides and proteins to lipid-based carriers through use of metal-ionchelating lipids. Biochim Biophys Acta. 2002;1567:204–212. doi: 10.1016/s0005-2736(02)00618-1. [DOI] [PubMed] [Google Scholar]
- (27).Nielsen UB, Kirpotin DB, Pickering EM, Drummond DC, Marks JD. A novel assay for monitoring internalization of nanocarrier coupled antibodies. BMC Immunol. 2006;7:24. doi: 10.1186/1471-2172-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ruger R, Muller D, Fahr A, Kontermann RE. Generation of immunoliposomes using recombinant single-chain Fv fragments bound to Ni-NTA-liposomes. J Drug Target. 2005;13:399–406. doi: 10.1080/10611860500353328. [DOI] [PubMed] [Google Scholar]
- (29).Ruger R, Muller D, Fahr A, Kontermann RE. In vitro characterization of binding and stability of single-chain Fv Ni-NTA-liposomes. J Drug Target. 2006;14:576–582. doi: 10.1080/10611860600864018. [DOI] [PubMed] [Google Scholar]
- (30).Patel JD, O'Carra R, Jones J, Woodward JG, Mumper RJ. Preparation and characterization of nickel nanoparticles for binding to his-tag proteins and antigens. Pharm Res. 2007;24:343–352. doi: 10.1007/s11095-006-9154-7. [DOI] [PubMed] [Google Scholar]
- (31).Szoka F, Olson F, Heath T, Vail W, Mayhew E, Papahadjopoulos D. Preparation of unilamellar liposomes of intermediate size (0.1–0.2 umol) by a combination of reverse phase evaporation and extrusion through polycarbonate membranes. Biochim Biophys Acta. 1980;601:559–571. doi: 10.1016/0005-2736(80)90558-1. [DOI] [PubMed] [Google Scholar]
- (32).Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- (34).Senter PD, Su PC, Katsuragi T, Sakai T, Cosand WL, Hellstrom I, Hellstrom KE. Generation of 5-fluorouracil from 5-fluorocytosine by monoclonal antibody-cytosine deaminase conjugates. Bioconjug Chem. 1991;2:447–451. doi: 10.1021/bc00012a012. [DOI] [PubMed] [Google Scholar]
- (35).Sambrook J, Russell DW, Cold Spring Harbor, L. In: Molecular cloning : a laboratory manual. Sambrook Joseph, Russell David W., editors. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y.: 2001. [Google Scholar]
- (36).Scherer JR. Dependence of lipid chain and head group packing of the inverted hexagonal phase on hydration. Biophys J. 1989;55:965–971. doi: 10.1016/S0006-3495(89)82895-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Paoli M, Anderson BF, Baker HM, Morgan WT, Smith A, Baker EN. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat Struct Biol. 1999;6:926–931. doi: 10.1038/13294. [DOI] [PubMed] [Google Scholar]
- (38).Lasic DD, Martin FJ, Gabizon A, Huang SK, Papahadjopoulos D. Sterically stabilized liposomes: a hypothesis on the molecular origin of the extended circulation times. Biochim Biophys Acta. 1991;1070:187–192. doi: 10.1016/0005-2736(91)90162-2. [DOI] [PubMed] [Google Scholar]
- (39).Mori S, Takahashi HK, Yamaoka K, Okamoto M, Nishibori M. High affinity binding of serum histidine-rich glycoprotein to nickel-nitrilotriacetic acid: the application to microquantification. Life Sci. 2003;73:93–102. doi: 10.1016/s0024-3205(03)00261-3. [DOI] [PubMed] [Google Scholar]
- (40).Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct. 2001;30:421–455. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- (41).Mauk MR, Rosell FI, Lelj-Garolla B, Moore GR, Mauk AG. Metal ion binding to human hemopexin. Biochemistry. 2005;44:1864–1871. doi: 10.1021/bi0481747. [DOI] [PubMed] [Google Scholar]
- (42).Porath J, Olin B. Immobilized metal ion affinity adsorption and immobilized metal ion affinity chromatography of biomaterials. Serum protein affinities for gel-immobilized iron and nickel ions. Biochemistry. 1983;22:1621–1630. doi: 10.1021/bi00276a015. [DOI] [PubMed] [Google Scholar]
- (43).Morgan WT. The histidine-rich glycoprotein of serum has a domain rich in histidine, proline, and glycine that binds heme and metals. Biochemistry. 1985;24:1496–1501. doi: 10.1021/bi00327a031. [DOI] [PubMed] [Google Scholar]
- (44).Bell SG, Vallee BL. The metallothionein/thionein system: an oxidoreductive metabolic zinc link. Chembiochem. 2009;10:55–62. doi: 10.1002/cbic.200800511. [DOI] [PubMed] [Google Scholar]
- (45).Fischer NO, Blanchette CD, Chromy BA, Kuhn EA, Segelke BW, Corzett M, Bench G, Mason PW, Hoeprich PD. Immobilization of his-tagged proteins on nickel-chelating nanolipoprotein particles. Bioconjug Chem. 2009;20:460–465. doi: 10.1021/bc8003155. [DOI] [PubMed] [Google Scholar]
- (46).Herringson TP, Altin JG. Convenient targeting of stealth siRNA-lipoplexes to cells with chelator lipid-anchored molecules. J Control Release. 2009;139:229–238. doi: 10.1016/j.jconrel.2009.06.034. [DOI] [PubMed] [Google Scholar]