

Abstract
Membrane receptors for steroid hormones are currently a subject of considerable debate. One approach to selectively target these putative receptors has been to couple ligands to substances that restrict cell permeability. Using this approach, an analog of the estrogen receptor ligand 4-hydroxytamoxifen was attached to fluorescent dyes with differing degrees of predicted cell permeability. The conjugates bound to estrogen receptor in vitro, but all three conjugates, including one predicted to be cell-impermeable, inhibited estradiol-induced transcriptional activation. Fluorescence microscopy revealed cytoplasmic localization for all three conjugates. We further characterized a 4-hydroxytamoxifen analog conjugated to a BODIPY fluorophore in breast cancer cell lines. Those experiments suggested a similar, but not identical, mode of action to 4-hydroxytamoxifen, as the fluorescent conjugate was equally effective at inhibiting proliferation of both tamoxifen-sensitive and tamoxifen-resistant breast cancer cell lines. While these findings point to significant complicating factors in designing steroid hormone mimics targeted to the plasma membrane, the results also reveal a possible new direction for designing estrogen receptor modulators.
Introduction
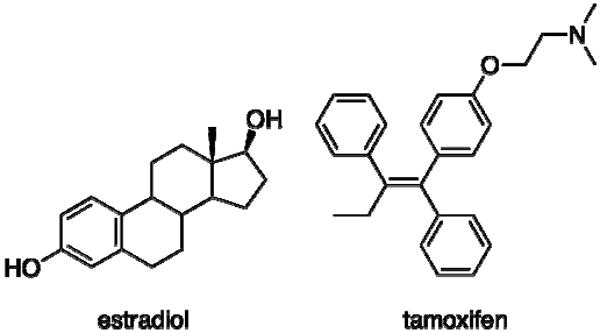
Estrogens, including 17β-estradiol (Scheme 1), are known to play key roles in female reproductive tissues such as the breast and uterus (1), and compounds that modulate estrogen signaling are clinically valuable for relieving menopausal symptoms or treating diseases such as breast cancer (2). However, current estrogen-based therapies have serious side effects in non-target tissues, presenting significant therapeutic challenges. Breast cancer therapies such as tamoxifen (Scheme 1) can cause hot flashes in patients and estrogen replacement therapies can increase the risk of breast cancer to unacceptable levels (3, 4). As a result, in order for progress in the field of estrogen drug design and discovery to continue to move forward, the key mechanisms of estrogen signaling and their roles in specific physiological responses to estrogen signaling modulators need to be identified.
Scheme 1.
There are multiple types of receptors in a cell that bind estrogens (5). The classical estrogen receptors and two major subtypes of the nuclear receptors are estrogen receptor (ER) alpha and ER beta (6, 7). Other receptors that can bind to estrogen ligands include the G-protein coupled receptor (GPCR) known as GPR30 (8) and variants of ER alpha (9). The possibility that some of these receptors may be associated with the plasma membrane has been an area of considerable debate, especially since certain cellular responses to estrogen such as the activation of kinase signaling cascades are typical downstream events of membrane receptor activation (10). Additional receptors have been identified based on homology with the classical estrogen receptors, but their activity does not appear to be directly regulated by physiological estrogens (11).
Membrane-associated ERs have been postulated for a long time (12), but identifying and characterizing those receptors has been difficult and controversial (13). One strategy to selectively target membrane ERs has been to increase the polarity of known ER ligands until they become cell-impermeable, but confirming the localization of these molecules has proven to be difficult (14, 15). Conjugating the ligand to proteins such as bovine serum albumin allows for tracking, since a fluorescent tag can also be attached to the protein (16, 17), but the use of the protein conjugates has been controversial for a number of reasons, including the purity of the conjugates and their unusual binding kinetics for ER (18, 19).
A potentially useful strategy that combines the ability to alter cell permeability with the ability to track localization is the conjugation of ER ligands to fluorescent dyes. Fluorescent ER ligand conjugates have been used for quite some time to try to determine the ER content of cell (20, 21), but the growing library of available fluorophores now includes compounds that are cell-impermeable, making it possible to generate cell-impermeable fluorescent conjugates to target membrane steroid hormone receptors. Toward this objective, a cell-impermeable fluorescent estradiol conjugate has recently been used to determine GPR30 localization the estrogen-binding GPCR (22). In the current study, we use a similar strategy with a well-known ER ligand, 4-hydroxytamoxifen to develop a novel set of fluorescent conjugates. These conjugates bind with good affinity to the estrogen receptor in vitro, and show remarkable differences in receptor localization and effects on estrogen signaling in cell-based assays when compared to the previously reported estradiol conjugate. We also show for the first time that even the ethinyl estradiol conjugate displays significant uptake over time, complicating the use of this type of chemical probe for longer-term studies of plasma membrane-initiated, estrogen signaling.
Experimental Procedures
General Procedures
All reagents were purchased from Sigma-Aldrich. Routine proton nuclear magnetic resonance spectra (1H NMR) were obtained on a Bruker DRX500 (500 MHz) instrument. 1H NMR chemical shifts are reported as δ values in parts per million (ppm) downfield from internal tetramethylsilane. NMR instruments were provided by the Shared Resource Center of the Purdue Cancer Center. The plasmids used in the reporter gene assays, pSG5-ERalpha, pSG5-ERbeta and estrogen response element (ERE)-luciferase, have been described elsewhere (23, 24). Analytical HPLC was performed using a 4.6 × 150 mm Agilent Eclipse XDB-C8 5 μm reverse phase column with UV signal detection at 280 nm and fluorescence detection at excitation/emission of 488/508 nm. Preparative HLC was performed using a 25 × 250 mm Vydac C8 15 μm semi-preparative column with signal detection at 280 nm. Nonlinear regression analysis of binding and reporter assay curves was performed using Prism 4 software. Images were acquired using either a Nikon TE2000 inverted microscope or a Zeiss LSM 710 upright microscope. Images on both microscopes were acquired sequentially using a 60× oil 1.4 NA lens. OHT-6C-BODIPY images were obtained by exciting with the 488 nm line of a 4-line argon laser. Alexa Fluor 546-labeled conjugates were excited at 543 nm using a green HeNe laser and emission wavelengths greater than 560 nm were collected.
Conjugate synthesis
The synthesis of OHT-6C has been described elsewhere (25). The ethinyl estradiol Alexa Fluor 546 was made by following a previously reported procedure (22). For the conjugation reactions, the commercially available N-hydroxysuccinimide ester of each fluorophore (5 mg for the carboxyfluorescein and BODIPY-FL esters and 2.5 mg for each of the Alexa Fluor esters) was dissolved in DMSO (0.25 mL). To each solution was added 10 mg of OHT-6C dissolved in DMSO (0.25 mL). The reaction proceeded in the dark at room temperature overnight. The sample was then purified by preparative HPLC with a water: acetonitrile eluent gradient starting at 20% acetonitrile and ending at 100% acetonitrile at 70 minutes. The eluent also contained 0.1% trifluoroacetic acid. The fractions containing the appropriate product were then freeze-dried using a lyophilizer. Characterization of each conjugate is included in the supplementary information.
Radiolabeled ligand receptor binding assay
In each well of a 96 well plate, 25 μL of a solution containing binding buffer (100 mM potassium phosphate (pH 7.4), 100 μg/mL bovine gamma globulin, and 0.02% sodium azide) plus 2 nM [2,4,6,7,16,17-3H] estradiol (GE Healthcare) and different concentrations of competitor were added to a 25 μL solution containing 30 nM of full length recombinant estrogen receptor alpha or beta (Invitrogen) in binding buffer. After a two-hour incubation at room temperature, the mixture was transferred to a MultiScreen™ HTS Filter Plate (Millipore) and washed with binding buffer to remove unbound ligand. Plates were allowed to dry overnight. Microscint 0™ scintillation fluid (Perkin Elmer) was added, and the radioactivity was counted on a Packard TopCount scintillation counter. Each drug concentration was tested three times each in three different experiments. Data were analyzed using Prism software.
Cell Culture
The ER-positive MCF-7 cell line was grown in phenol red free DMEM F-12 HAM medium supplemented with 0.876 g/L glutamine, 100 mg/L streptomycin sulfate, 100 units/mL of penicillin G and 10% fetal bovine serum (FBS) at 37 °C in an air/carbon dioxide (95:5) atmosphere. MDA-MB-231 and SKBR3 breast cell lines were grown in phenol red free RPMI 1640 medium supplemented with 0.876 g/L glutamine, 100 mg/L streptomycin sulfate, 100 units/mL of penicillin G and 10% fetal bovine serum (FBS) at 37 °C in an air/carbon dioxide (95:5) atmosphere. Transfection assays were run with the same media conditions except serum free media was used.
Luciferase Reporter Assays
Cells were plated in 24 well plates (2 × 106 cells per plate). Transfections were performed according to the protocol for Lipofectamine 2000™. In order to normalize for the transfection efficiency in each well, the dual luciferase system was used in which a constitutively expressed, chemically orthogonal luciferase expression vector was also transfected. The total amount of DNA/well for each plasmid was as follows: ERE-luciferase 0.3 μg/well, and Renilla-luciferase 0.1 μg/well. The ratio of total DNA/Lipofectamine 2000™ ratio was 1:3. After transfection, the plates were incubated at 37 °C for 6 hours before replacing the media with serum free media for 24 hours. The serum free media was then replaced with fresh serum free media and the ligands. All ligands were delivered in DMSO or ethanol and the total concentration of organic solvent in each was 0.1% or less. For competition experiments with 4-hydroxytmaoxifen compounds, the ligand was added to media already containing 10 nM estradiol. For the ethinyl estradiol conjugates, ligand was added to media containing 5 nM raloxifene, an antagonist of ER in the ERE assay. After 18 hours, the cells were lysed and assayed for dual luciferase activity in a Packard TopCount luminometer according to the protocol provided by Promega. The relative light units (RLU) were then calculated by dividing the output of the ERE-driven luciferase in each well by the output of the Renilla luciferase. For antagonist experiments, the percent transcriptional activation was calculated using the RLU value obtained with 10 nM estradiol as 100% and the RLU value obtained with 1 μM fulvestrant as 0%. Each drug concentration was tested in triplicate and each experiment was repeated at least three times.
Uptake studies
MCF-7 (ER-positive cell line) and MDA-MB-231 and SKBR3 (both ER-negative cell lines) were seeded on poly-L-lysine cover slips and grown to 90% confluency. Drugs were dosed in serum free medium for the appropriate time point. Cells were washed and fixed using 4% paraformaldehyde. Cover slips were washed and mounted using ProLong® Gold Antifade Reagent containing DAPI and then imaged.
Timed uptake studies
For the timed uptake studies in Figure 3, 3×105 MCF7 cells were seeded into each chamber of an 8-chambered, CC2-coated LabTek slide and grown to 90% confluency. The timed conjugate uptake assay was performed following a previously reported procedure (22).
Figure 3.
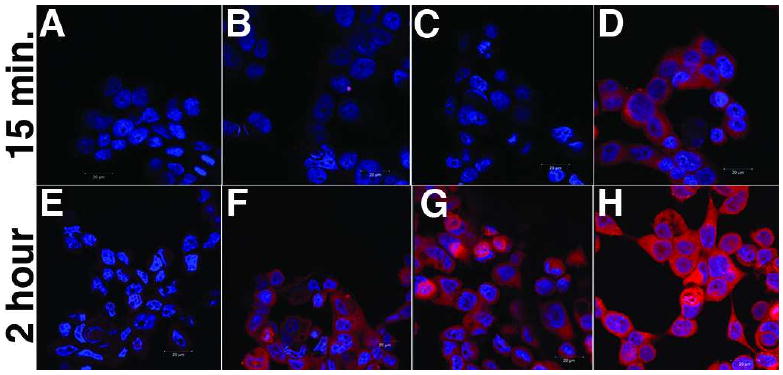
MCF7 cells were treated with different Alexa Fluor 546 conjugates for either 15 minutes (A-D) or 2 hours (E-H) and imaged as described in the experimental procedures. The conjugates used are as follows: images A and E: DMSO; Images B and F: 1 μM Alexa Fluor 546 dye alone; images C and G: 1 μM EE-Bn-AF; images D and H: 1 μM OHT-6C-AF The images represent merged images of the emission from the DAPI stain of the nucleus (blue) and the emission of the Alexa Fluor dye (red).
Proliferation studies
MCF-7 or tamoxifen-resistant MCF-7 cells (26) were plated in 96-well dishes (2000 cells per well) in steroid-free medium and treated with 1 nM of estradiol and various doses of 4-hydroxytamoxifen or OHT-6C-BODIPY. Cell numbers were determined by MTT assay after 7 days of treatment.
Receptor stability studies
MCF7 cells (5×105/dish) were plated in 60-mm dishes in steroid-free medium for 3 days prior to drug exposure. Whole cell extracts were prepared by suspending cells in 0.1 ml of lysis buffer (62 mM Tris, pH 6.8, 2% sodium dodecyl sulfate; 10% glycerol; 10 μl protease inhibitor cocktail set III). After sonication (3 × 10 sec), insoluble material was removed by centrifugation (15 min at 12,000 g), and protein concentration in the supernatant was determined using a protein assay kit (Bio-Rad Laboratories, Inc). The protein extracts were mixed with 1/4 vol of 5× electrophoresis sample buffer and boiled for 5 min at 90 C. Protein extract (50 μg per lane) was then fractionated by SDS-PAGE, transferred to polyvinylidene difluoride membrane, and probed with antibodies. Primary antibody was detected by horseradish peroxidase-conjugated second antibody and visualized using enhanced SuperSignal West Pico Chemiluminescent Substrate (Pierce Chemical Co., Rockford, IL). The band density of exposed films was evaluated with ImageJ software (http://rsb.info.nih.gov/ij/).
Results
Design and synthesis of the conjugates
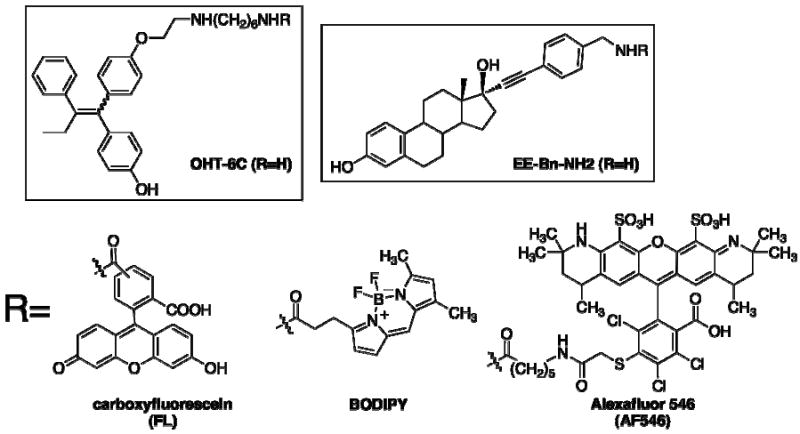
While there has been success in generating inherently fluorescent ER ligands (27), we opted for the traditional conjugation strategy of attaching the fluorescent dye to an ER ligand through a flexible linker. The keys to success with this strategy are 1) the choice of the ER ligand and 2) identifying a location on the drug where linker attachment is well tolerated. Tamoxifen, one of the most studied ER ligands, shows a mixed agonist/antagonist profile therapeutically (3) and has long been associated with possible plasma membrane binding sites, including some that might not bind estradiol itself (28). We have previously shown that potential linkers can be attached to the more potent tamoxifen metabolite, 4-hydroxytamoxifen (OHT), through the basic side chain with only small losses in binding affinity for ER and inhibitory potency in cell-based gene reporter assays (29). We have also shown that an analog with a 1,6-diaminohexane linker known as OHT-6C (Scheme 2), showed strong ER binding affinity when coupled to a poly (methacrylic acid) polymer (25). For the cell-impermeable fluorescent dye, Alexa Fluor 546 was chosen. For comparison, we also chose two dyes, carboxyfluorescein and BODIPY FL, that have been well characterized as cell-permeable fluorophores. Amine-reactive analogs of all three fluorophores were purchased as activated N-hydroxysuccinimide esters and linked to the primary amine of the OHT-6C analog to generate three different conjugates (Scheme 2). The resulting conjugates (and OHT-6C itself) are generated as mixtures of E and Z isomers– the two forms readily interconvert at room temperature. Previous work with 4-hydroxytamoxifen has shown that despite this interconversion, the Z isomer is almost exclusively bound by the receptor both in vitro and in vivo. (30, 31) All three of the conjugates showed nearly identical fluorescence spectra in solution as their parent fluorophores, although the overall fluorescence intensity of each conjugate was somewhat decreased (likely due to a change in the overall chemical environment of the fluorophore).
Scheme 2.
ER binding and effects on ER-mediated transcription
The conjugates were compared for their ability to bind to ER in solution using competition binding experiments with tritium-labeled estradiol. As predicted, all of the conjugates still retained their ability to bind to both ER alpha and ER beta in vitro at sub-micromolar concentrations, although there was some loss in affinity compared to the parent compound and to estradiol and OHT (Figure 1A and Table 1). In the published structures of the ER ligand binding domain bound to OHT, the basic side chain extends away from the binding pocket and into solution (32), which could explain the general tolerance in binding for the bulky OHT substitutions conjugates, although the weaker binding affinity suggests that there could be some somewhat unfavorable interactions with the fluorophore. This possibility seems likely considering that the largest fluorophore, Alexa Fluor 546, has the worst binding potency.
Figure 1.
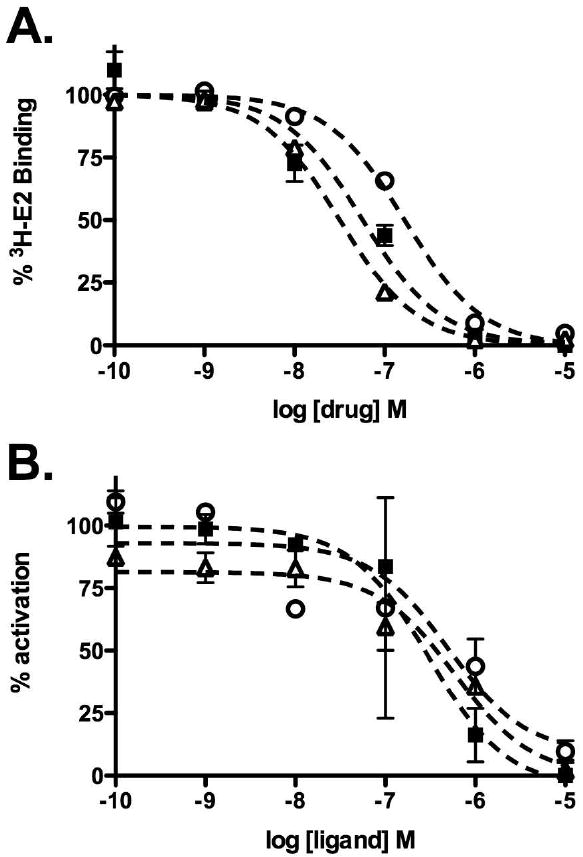
ER-modulatory activities of OHT conjugates. A. Effects of conjugates on the binding of purified ER alpha to 2 nM 3H-labeled estradiol. B. Effects of conjugates on ER-mediated transcription in MCF7 cells transiently transfected with a luciferase reporter plasmid under the control of ERE-containing promoter. The drugs were added in the presence of 10 nM estradiol. Luciferase activity was examined at 24 hours after drug treatment and percent activation calculated relative to cells treated with estradiol only. In both graphs, open circles= OHT-6C-AF; solid squares= OHT-6C-BODIPY; open triangles=OHT-6C-CF.
Table 1.
Estrogen receptor modulation properties of conjugates
| Compound | ER alpha IC50 (nM)a | ER beta IC50 (nM)a | ERE IC50 (nM)b |
|---|---|---|---|
| Estradiol | 2.5 ± 0.2 | 3.7 ± 1.0 | 16 ± 10 d |
| OHT | 8.5 ± 3.9 | 13 ± 7 | 40 ± 10 c |
| OHT-6C (R=H) | 15 ± 5 | 9 ± 5 | 85 ± 50 c |
| OHT-6C-FL | 25 ± 10 | 35 ± 20 | 900 ± 600 c |
| OHT-6C-BODIPY | 30 ± 10 | 40 ± 20 | 250 ± 110c |
| OHT-6C-AF546 | 180 ± 30 | 240 ± 70 | 600 ± 350 c |
| EE-Bn-NH2 | 7 ± 4 | 6 ± 4 | 150 ± 20d |
| EE-Bn-AF546 | 10 ± 2 | 35 ± 20 | 140 ± 90 d |
ER binding determined using competition experiments with purified ER and 3H-labeled estradiol as described in the experimental procedures
ERE activity determined using MCF7 cells transfected with luciferase reporter plasmid under control of consensus ERE as described in the experimental procedures.
IC50 values determined in the presence of 10 nM estradiol
IC50 values determined in the presence of 5 nM raloxifene
The conjugates were also tested for their ability to inhibit estradiol-induced activation of ER-mediated transcription using a cell-based luciferase reporter gene assay. In this assay, ER-positive MCF7 cells were transiently transfected with a reporter plasmid containing the luciferase gene under the control of a consensus estrogen response element-containing promoter. In this reporter system, dosing the cells with estradiol typically activates transcription of the reporter gene while OHT acts as an antagonist. Based on the predicted cell-permeability of the conjugates, we expected the carboxyfluorescein and BODIPY-labeled conjugates to still act as antagonists and the Alexa Fluor 546-labeled conjugate to have no effect due to its inability to enter the cell and interact with the ER. As shown in Figure 1B, all three conjugates antagonized ER-mediated transcription with roughly the same inhibitory IC50 (100-900 nM concentration range), 10 to 50 fold worse than OHT or OHT-6C. The conjugates were also rechecked for purity by analytical HPLC. No free OHT ligand was detected, suggesting that the conjugates enter the cell and modulate receptor function by an undetermined mechanism.
Conjugate uptake and localization
Confocal laser scanning microscopy was used to monitor the cellular uptake and localization of the conjugates at different time points. Previous studies using fluorescent ER ligands capable of modulating ER-mediated transcription reported observing the fluorescence in the nucleus (33). In addition, our work (34) along with multiple other studies have demonstrated that ER localizes to the nucleus in the presence of agonists or antagonists (reviewed in (35, 36)). Since all three conjugates were ER antagonists, we expected to see similar nuclear localization patterns; however, the fluorescence localized to the cytoplasm in a number of the ER-positive MCF7 cell lines and was clearly excluded from the nucleus at 100 nM and 10 μM (Figure 2). To our knowledge, this pattern of cellular localization has not been previously reported for any fluorescent ER ligand.
Figure 2.
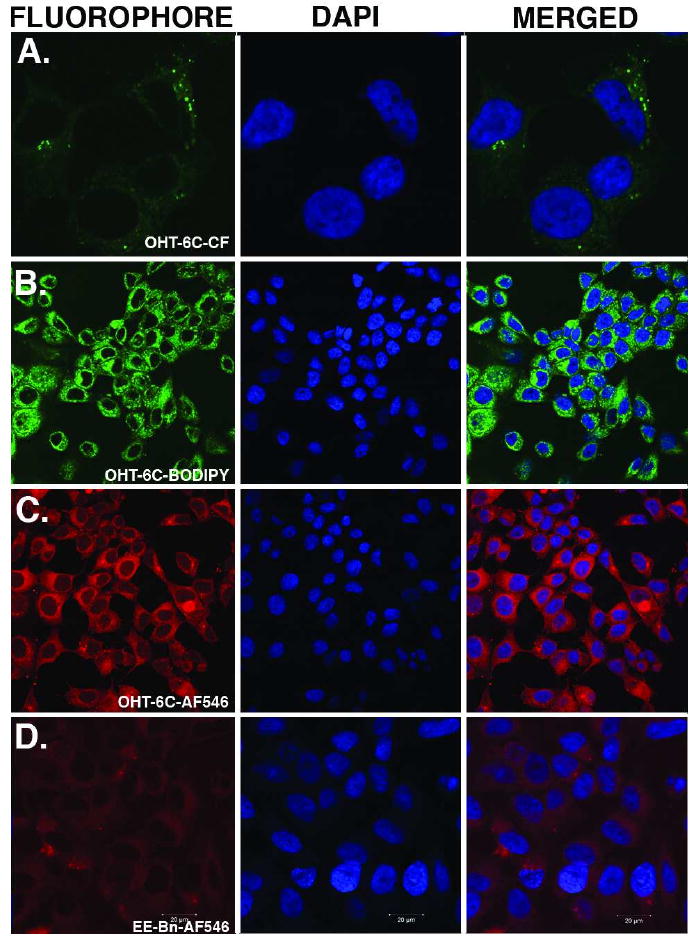
Images of uptake of conjugates by ER-positive MCF7 cell line after 6 hours of dosing. Row A. 10 μM OHT-6C-CF; Row B. 100 nM OHT-6C-BODIPY; Row C. 10 μM OHT-6C-AF546; Row D. 1 μM EE-Bn-AF546. The first image of each row is the emission of the fluorophore. The second image of each row is the image of the DAPI nuclear stain and the third image is the merged picture of the first two images. Due to the relatively low brightness of the carboxyfluorescein conjugate, we have shown an image at higher magnification than the other fluorophores.
Comparison with 17α-ethynyl estradiol conjugates
In order to compare the unusual localization of the OHT-derived conjugates to a conjugate with a known localization pattern, we also synthesized the previously reported 17α-ethinyl estradiol analog conjugated to Alexa Fluor 546. This conjugate which has been reported to be cell impermeable (22), was first tested for its ability to bind to ER alpha and ER beta in vitro. We observed sufficient binding affinity for both receptor subtypes (Table 1) and subsequently tested the conjugate for its effect on ER-mediated transcription in the ER-positive MCF7 breast cancer cell line. Surprisingly, the conjugate, at sub-micromolar concentrations, activated ER-mediated transcription from a consensus estrogen response element (Figure S1), in contrast to the original report of describing the 17α-ethinyl estradiol-Alexa Fluor conjugate as a cell-impermeable probe (22). It should be noted, however, that in the first report, the conjugate was tested at lower concentrations and for much shorter time periods than used for the transcriptional activation assays in the present study.
In order to attempt to reconcile the apparent differences in the permeability of our ethinyl estradiol conjugate compared to the previous report, we examined the cellular localization of both the OHT and ethinyl estradiol-Alexa Fluor 546 conjugates as well as the Alexa Fluor546 dye itself at different time periods using the original experimental conditions in the MCF7 breast cancer cell line ((22) and described in the Methods). After 15 minutes, no uptake was apparent for either the ethinyl estradiol conjugate or the Alexa Fluor dye; however, significant uptake of the OHT-6C conjugate was observed (Figure 3). Thus, the results for the ethinyl estradiol conjugate are consistent with a previous report (22), although after a 2 hour incubation, cytoplasmic localization was seen for all of the compounds (Figure 3).
Characterization of conjugate uptake and localization
Based on brightness and potency qualities, the OHT-6C-BODIPY conjugate was chosen to further examine the mechanisms of uptake and cytoplasmic localization. Similar to the OHT-6C-AF conjugate, uptake of the OHT-6C-BODIPY by ER positive MCF7 cells could be detected as soon as 15 minutes after dosing using confocal laser microscopy and uptake increased over time (Fig. S2). The fluorescence in the cytoplasm was stable, as no conspicuous change in fluorescence was observed 12 hours after the media containing the conjugate with conjugate-free media was replaced (data not shown).
Uptake of the BODIPY-labeled conjugate in the ER-positive MCF7 breast cell line was compared to the ER-negative MDA-MB-231 and SKBR3 breast cell lines using confocal laser microscopy. No difference in the degree of uptake was seen (Figure S3), suggesting that ER does not play a significant role in conjugate uptake or localization. Moreover, uptake and localization were not altered in the presence of the ER ligands estradiol or OHT (Figure 4), suggesting a “non-receptor mediated” mechanism of uptake. We also considered the possibility of an endocytic mechanism. However, even at the earliest time points, fluorescence from the conjugate was seen in the cytoplasm, and the fluorescence did not colocalize with a commercially available, fluorescent endosomal or lysosomal markers (data not shown). Taken together, these observations suggest that while there may be some endocytic conjugate uptake, the majority of the conjugate appears to enter the cell through either passive diffusion or pinocytosis.
Figure 4.
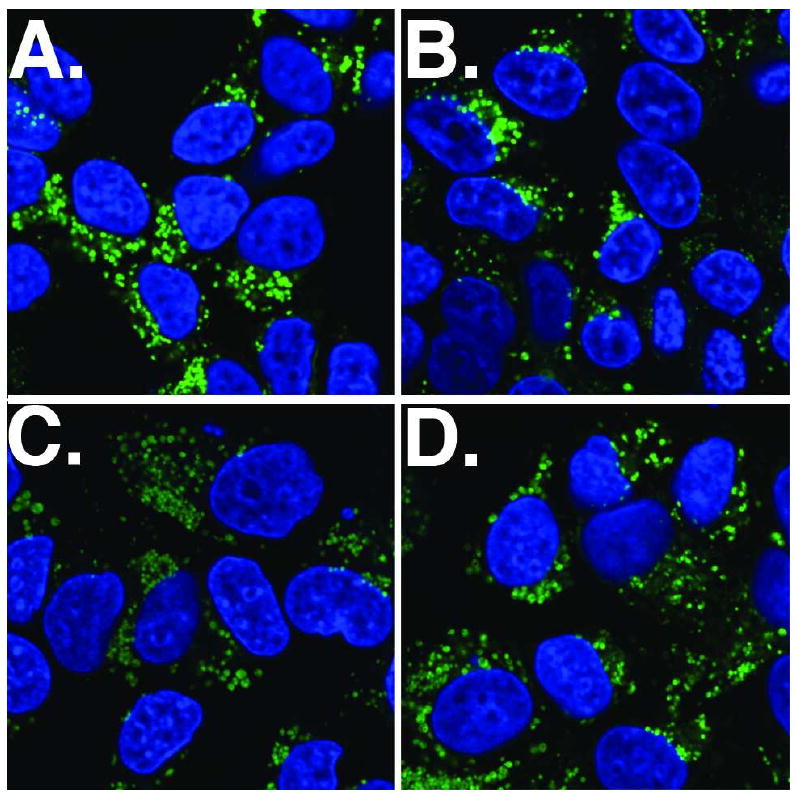
Uptake by MCF7 cells of 100 nM OHT-6C-BODIPY in presence of competing ER ligands as analyzed by confocal scanning laser microscopy. The competing drugs are as follows: A. DMSO; B. 1 μM estradiol; C. 1 μM 4-hydroxytamoxifen; D. 1 μM OHT-6C. Images represent merged fluorescent images of emission from DAPI nuclear stain (blue) and the emission from the BODIPY fluorophore (green)
Effects of the conjugate on ER activity
The unusual cytoplasmic localization of the OHT-6C-BODIPY conjugate prompted us to investigate whether this localization had any effect on ER function. To address this key question, we first compared classical ER responses to OHT-6C-BODIPY and tamoxifen. The effect of the conjugate on proliferation of both tamoxifen-sensitive and tamoxifen-resistant breast cancer cell lines were measured. The growth of ER-positive MCF7 breast cancer cells are stimulated by estradiol, and either tamoxifen or OHT block that stimulation. As shown in Figure 5, both OHT and OHT-6C-BODIPY were able to block estradiol-induced growth in tamoxifen-sensitive MCF7 cells, indicating that the conjugate is still capable of classic ER antagonism. The compounds were also tested against a sub-line of MCF7 cells that were grown in the presence of tamoxifen until they developed resistance to the drug (26). The conjugate, in contrast to OHT, showed equal effectiveness in the tamoxifen-resistant line as it did in the parental MCF7 cell line (Figure 5), suggesting that the conjugate has an overlapping, but not identical, mechanism of action to 4-hydroxytamoxifen.
Figure 5.
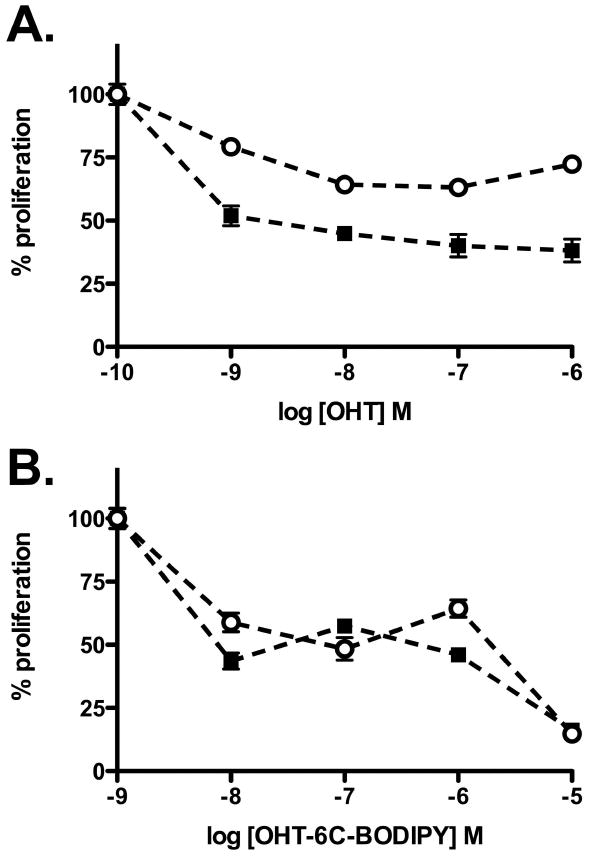
Effect of OHT (A) and OHT-6C-BODIPY (B) on proliferation of tamoxifen-sensitive, parental MCF7 cells (closed squares) and a tamoxifen-resistant MCF7 cell sub-line (open circles). Cells were seeded in hormone-free medium and treated in the presence of 1 nM E2. Seven days after treatment, cell number was determined by MTT assay. Experiments were performed twice in triplicate.
We next examined the effect of these conjugates on cellular ER levels, a key determinant of hormone sensitivity. One of the critical differences between the mechanism of action of selective estrogen receptor modulators (SERMs) like OHT and another class of drugs known as SERDs (selective ER down-regulators) such as fulvestrant is that OHT stabilizes cellular ER levels, while fulvestrant causes rapid receptor degradation (37, 38). Whereas both estradiol and fulvestrant induced marked receptor downregulation in MCF7 cells after a 24 hours incubation, no receptor degradation was observed after treatment with the OHT-6C-BODIPY conjugate (Figure 6), indicating that OHT-6C-BODIPY is more similar in mechanism of action to SERMs rather than compounds in the SERD class.
Figure 6.
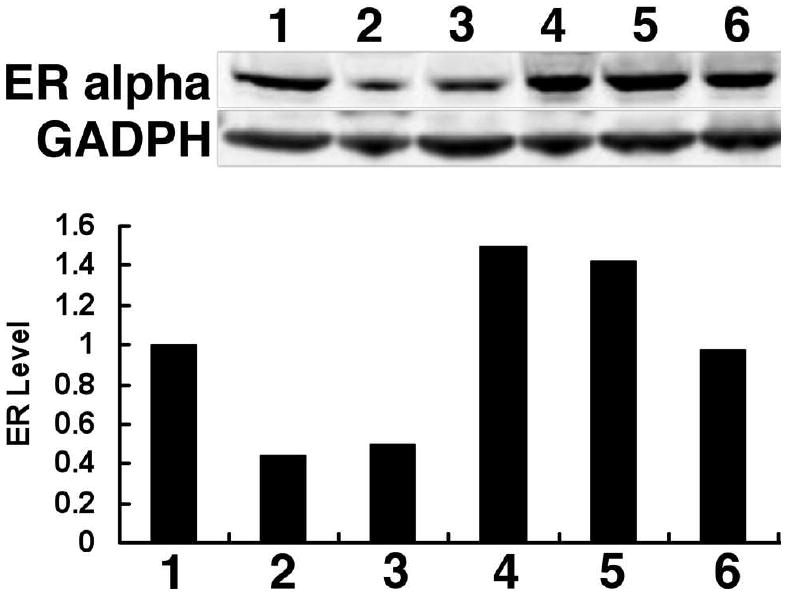
Effects of the OHT-6C-BODIPY conjugate on ER alpha stability. MCF7 cells were seeded in hormone-free medium for three days, then treated with different compounds for 24 hours. Lane 1: DMSO; Lane 2: 10 nM estradiol; Lane 3: 100 nM fulvestrant; Lane 4: 100 nM 4-hydroxytamoxifen; Lane 5: 1 μM OHT-6C; Lane 6: 10 μM OHT-6C-BODIPY. ER alpha levels in whole cell extracts were determined by immunoblotting with anti-ERα antibody. GAPDH was used as the loading control. Relative ER alpha levels (versus untreated cells) are shown in the corresponding histogram
Discussion
The design and synthesis of fluorescent conjugates for estrogen signaling modulators has previously focused on targeting activity either in the nucleus or the membrane. Although the compounds described in this paper were intended to target either of these locations, they instead localized to the cytoplasm. The only compounds for which specific cytoplasmic localization has been previously reported are a highly useful set of dendrimer-based estradiol analogs (39, 40). The uptake of those analogs appears to be mediated by ER and nuclear exclusion and seems to derive from the overall size and charge of the dendrimers (39, 40). For the fluorescent conjugates described in this current study, the mechanism of conjugate uptake and apparent exclusion from the nucleus is not as clear. ER does not appear to play a significant role in cellular uptake of the conjugates, which was similar in ER+/- breast cancer cell lines and not affected by the addition of competing ER ligands in ER+ cells (Figs. 2-4, Fig S3). Furthermore, it is unlikely that conjugate size is a contributing factor to the observed cytoplasmic accumulation, as all of the conjugates are small enough to easily enter the nucleus.
We also found that the ethinyl estradiol Alexa Fluor conjugate, previously reported to be cell-impermeable (22), showed similar uptake as the OHT conjugates, with accumulation time being the only difference between these conjugates (Fig. 3), indicating a slower mode of uptake. Although this observation would apparently contradict earlier claims regarding cell impermeability of the ethinyl estradiol conjugate, by using the reported concentrations and time intervals (22), we also found that no uptake was detected, consistent with those previous results. This lag in uptake has been indirectly seen with other highly polar ethinyl estradiol analogs. In ER alpha expressing COS7 cells, a longer time period was required to observe cellular uptake of compounds that did not stimulate rapid calcium mobilization, as these more polar ethinyl estradiol analogs showed a slower increase in mobilization (15).
It is difficult to exactly rationalize why the fluorescent conjugates all showed similar levels of cellular uptake and why the localization was almost entirely in the cytoplasm of the cells. One of the difficulties in trying to understand uptake is the fact that the conjugates are not particularly potent compared to their in vitro binding affinity. In addition, the fluorescence of the conjugates cannot be detected easily at lower nanomolar concetration ranges. As a result, localization only can be studied at higher concentrations where events such as aggregation could be playing a role. Aggregate formation would be consistent with the long-term stability and general punctate pattern of fluorescence seen in OHT-6C-BODIPY fluorophore treated cells, providing a possible reason for why the conjugate is excluded from the nucleus. Tamoxifen is known to interact with membranes (41), which could serves or initiate a nucleation event, leading to internalization of even the relatively more polar Alexa Fluor conjugates. Evidence for aggregation was not seen using standard fluorimetry of conjugates in solution, although we acknowledge that the level of sensitivity of the fluorimeters available to us does not approach that of confocal laser scanning microscopy.
A mechanism that also explains why the fluorescent conjugates are able to inhibit ER-mediated transcription is unclear. The dendrimer conjugates described by the Katzenellenbogen labs localize to the cytoplasm and have little to no effect on ER-mediated transcription (39, 42) The antagonist activity of the fluorescent OHT conjugates could arise from ER binding to the conjugate in the cytoplasm and being sequestered away from the nucleus. Alternatively, conjugate degradation within the cell could release free ligand from the fluorescent dye and subsequent transport of ligand to the nucleus. Toward these possibilities, we determined the hydrolytic susceptibility of the amide bond common to all of the conjugates by performing pH stability experiments in solution (various pHs at 37 °C for multiple days; data not shown). No release of OHT-6C was detected by HPLC, and no other non-fluorescent products were observed, but it is always possible with ligands as potent as OHT-6C that some level of hydrolysis undetectable by HPLC or any other standard analytical technique is still occurring and is responsible for the transcriptional inhibition. In addition, subcellular fractionation studies performed in ER-positive MCF7 cells dosed with OHT-6C-BODIPY revealed the presence of ER only in the nucleus, thereby contradicting the sequestration hypothesis. Another explanation is that, despite the apparent nuclear exclusion of the fluorescent conjugate, a small amount of non-aggregated conjugate is free to enter the nucleus. The concentration of this soluble conjugate is below detection sensitivity by microscopy in the presence of the much brighter cytoplasmic aggregates but able to inhibit ER transcription in a manner similar to the non-conjugated OHT6C ligand itself.
Overall, our findings indicate that a hydrophobic and planar drug portion of a conjugate can significantly complicate the use of fluorescent dyes to dictate subcellular localization. Considering that most drugs targeting steroid hormone receptors fit this description, care should be taken when using this type of approach to selectively target plasma membrane-initiated steroid hormone signaling. While it may be possible to obtain the desired localization by only studying very rapid signaling or by connecting the drug and fluorophore with a linker that prevents aggregation, we contend that thorough testing of the conjugates at different concentrations and time points is necessary before conclusions about selectivity can be drawn.
In addition to the unusual localization pattern, another unique property of the OHT conjugates is their ability to effectively inhibit proliferation of ER-positive, tamoxifen resistant breast cancer cells. Whether this activity is the result of altered ER function due to the added bulk of the fluorophore attachment in a manner similar but distinct from compounds like fulvestrant or to changes in the intracellular drug concentrations due to the aggregate is unknown at this time. At this point, ongoing work to improve the potency of the compounds is necessary before more detailed mechanistic work can be pursued. Nonetheless, either mechanism could have significant impact on the discovery and development of hormone therapies for ER-positive, antiestrogen resistant breast cancer, the focus of our ongoing studies.
Supplementary Material
Acknowledgments
The Army Breast Cancer Research Program (BC030507) and the National Institutes of Health (R01 DK075376) supported RVW, and National Cancer Institute CA113001 supported KPN. Purdue Research Foundation supported ELR with a predoctoral fellowship.
Footnotes
Supporting Information Available: Mass spectrometry data for all of the conjugates, further NMR characterization of the OHT-6C-BODIPY conjugate and additional graphs and images are included in the supporting information section. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Katzenellenbogen BS, Tsai TS, Tatee T, Katzenellenbogen JA. Estrogen and antiestrogen action: studies in reproductive target tissues and tumors. Adv Exp Med Biol. 1979;117:111–32. doi: 10.1007/978-1-4757-6589-2_6. [DOI] [PubMed] [Google Scholar]
- 2.Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell. 2004;5:207–13. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 3.Jordan VC. SERMs: meeting the promise of multifunctional medicines. J Natl Cancer Inst. 2007;99:350–6. doi: 10.1093/jnci/djk062. [DOI] [PubMed] [Google Scholar]
- 4.Jordan VC. Chemoprevention of breast cancer with selective oestrogen-receptor modulators. Nat Rev Cancer. 2007;7:46–53. doi: 10.1038/nrc2048. [DOI] [PubMed] [Google Scholar]
- 5.Weatherman RV. Untangling the Estrogen Receptor Web: Tools to Selectively Study Estrogen binding Receptors. In: Ottow E, Weinmann H, editors. Nuclear Receptors as Drug Targets. Wiley-VCH; Weinhem: 2008. pp. 47–64. [Google Scholar]
- 6.Hewitt SC, Harrell JC, Korach KS. Lessons in estrogen biology from knockout and transgenic animals. Annu Rev Physiol. 2005;67:285–308. doi: 10.1146/annurev.physiol.67.040403.115914. [DOI] [PubMed] [Google Scholar]
- 7.Koehler KF, Helguero LA, Haldosen LA, Warner M, Gustafsson JA. Reflections on the discovery and significance of estrogen receptor beta. Endocr Rev. 2005;26:465–78. doi: 10.1210/er.2004-0027. [DOI] [PubMed] [Google Scholar]
- 8.Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol Cell Endocrinol. 2009;308:32–8. doi: 10.1016/j.mce.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci U S A. 2003;100:4807–12. doi: 10.1073/pnas.0831079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wehling M, Losel R. Non-genomic steroid hormone effects: Membrane or intracellular receptors? J Steroid Biochem Mol Biol. 2006;102:180–3. doi: 10.1016/j.jsbmb.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 11.Giguère V, Yang N, Segui P, Evans R. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–4. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 12.Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. 1977;265:69–72. doi: 10.1038/265069a0. [DOI] [PubMed] [Google Scholar]
- 13.Levin ER. Membrane ER{alpha} signaling to cell functions. J Physiol. 2009 doi: 10.1113/jphysiol.2009.177097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarman M, Leung OT, Leclercq G, Devleeschouwer N, Stoessel S, Coombes RC, Skilton RA. Analogues of tamoxifen: the role of the basic side-chain. Applications of a whole-cell oestrogen-receptor binding assay to N-oxides and quaternary salts. Anticancer Drug Des. 1986;1:259–68. [PubMed] [Google Scholar]
- 15.Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, Sklar LA, Arterburn JB, Prossnitz ER. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem Biol. 2007;2:536–44. doi: 10.1021/cb700072n. [DOI] [PubMed] [Google Scholar]
- 16.Berthois Y, Pourreau-Schneider N, Gandilhon P, Mittre H, Tubiana N, Martin PM. Estradiol membrane binding sites on human breast cancer cell lines. Use of a fluorescent estradiol conjugate to demonstrate plasma membrane binding systems. J Steroid Biochem. 1986;25:963–72. doi: 10.1016/0022-4731(86)90330-4. [DOI] [PubMed] [Google Scholar]
- 17.Jansson SE, Gripenberg J, Karonen SL, Wasenius VM, Pantzar P, Kontula K, Karkkainen J, Adlercreutz H. Cytochemical demonstration of estrogen binding sites in breast cancer by estradiol covalently linked to horseradish peroxidase. Eur J Cancer Clin Oncol. 1985;21:625–30. doi: 10.1016/0277-5379(85)90091-4. [DOI] [PubMed] [Google Scholar]
- 18.Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology. 1999;140:5455–5458. doi: 10.1210/endo.140.11.7247. [DOI] [PubMed] [Google Scholar]
- 19.Taguchi Y, Koslowski M, Bodenner DL. Binding of estrogen receptor with estrogen conjugated to bovine serum albumin (BSA) Nucl Recept. 2004;2:5. doi: 10.1186/1478-1336-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlson KE, Coppey M, Magdelenat H, Katzenellenbogen JA. Receptor binding of NBD-labeled fluorescent estrogens and progestins in whole cells and cell-free preparations. J Steroid Biochem. 1989;32:345–55. doi: 10.1016/0022-4731(89)90206-9. [DOI] [PubMed] [Google Scholar]
- 21.Fisher B, Gunduz N, Zheng S, Saffer EA. Fluoresceinated estrone binding by human and mouse breast cancer cells. Cancer Res. 1982;42:540–9. [PubMed] [Google Scholar]
- 22.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 23.Weatherman RV, Carroll DC, Scanlan TS. Activity of a tamoxifen-raloxifene hybrid ligand for estrogen receptors at an AP-1 site. Bioorg Med Chem Lett. 2001;11:3129–31. doi: 10.1016/s0960-894x(01)00646-1. [DOI] [PubMed] [Google Scholar]
- 24.Weatherman RV, Clegg NJ, Scanlan TS. Differential SERM activation of the estrogen receptors (ERalpha and ERbeta) at AP-1 sites. Chem Biol. 2001;8:427–36. doi: 10.1016/s1074-5521(01)00025-4. [DOI] [PubMed] [Google Scholar]
- 25.Rickert EL, Trebley JP, Peterson AC, Morrell MM, Weatherman RV. Synthesis and characterization of bioactive tamoxifen-conjugated polymers. Biomacromolecules. 2007;8:3608–12. doi: 10.1021/bm070413t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, Salisbury JD, Cheng AS, Li L, Abbosh PH, Huang TH, Nephew KP. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006;66:11954–66. doi: 10.1158/0008-5472.CAN-06-1666. [DOI] [PubMed] [Google Scholar]
- 27.Hwang KJ, Carlson KE, Anstead GM, Katzenellenbogen JA. Donor-acceptor tetrahydrochrysenes, inherently fluorescent, high-affinity ligands for the estrogen receptor: binding and fluorescence characteristics and fluorometric assay of receptor. Biochemistry. 1992;31:11536–45. doi: 10.1021/bi00161a035. [DOI] [PubMed] [Google Scholar]
- 28.de Medina P, Favre G, Poirot M. Multiple targeting by the antitumor drug tamoxifen: a structure-activity study. Curr Med Chem Anticancer Agents. 2004;4:491–508. doi: 10.2174/1568011043352696. [DOI] [PubMed] [Google Scholar]
- 29.Trebley JP, Rickert EL, Reyes PT, Weatherman RV. Tamoxifen-based probes for the study of estrogen receptor-mediated transcription. Ernst Schering Res Found Workshop. 2006:75–87. doi: 10.1007/978-3-540-37635-4_6. [DOI] [PubMed] [Google Scholar]
- 30.Katzenellenbogen JA, Carlson KE, Katzenellenbogen BS. Facile geometric isomerization of phenolic non-steroidal estrogens and antiestrogens: limitations to the interpretation of experiments characterizing the activity of individual isomers. J Steroid Biochem. 1985;22:589–96. doi: 10.1016/0022-4731(85)90210-9. [DOI] [PubMed] [Google Scholar]
- 31.Robertson DW, Katzenellenbogen JA, Hayes JR, Katzenellenbogen BS. Antiestrogen basicity--activity relationships: a comparison of the estrogen receptor binding and antiuterotrophic potencies of several analogues of (Z)-1,2-diphenyl-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1-butene (tamoxifen, Nolvadex) having altered basicity. J Med Chem. 1982;25:167–71. doi: 10.1021/jm00344a015. [DOI] [PubMed] [Google Scholar]
- 32.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 33.Miksicek RJ, Carlson KE, Hwang KJ, Katzenellenbogen JA. Studies using fluorescent tetrahydrochrysene estrogens for in situ visualization of the estrogen receptor in living cells. Mol Endocrinol. 1995;9:592–604. doi: 10.1210/mend.9.5.7565806. [DOI] [PubMed] [Google Scholar]
- 34.Long X, Nephew KP. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. J Biol Chem. 2006;281:9607–15. doi: 10.1074/jbc.M510809200. [DOI] [PubMed] [Google Scholar]
- 35.Kawata M, Matsuda K, Nishi M, Ogawa H, Ochiai I. Intracellular dynamics of steroid hormone receptor. Neurosci Res. 2001;40:197–203. doi: 10.1016/s0168-0102(01)00253-x. [DOI] [PubMed] [Google Scholar]
- 36.Leclercq G, Lacroix M, Laios I, Laurent G. Estrogen receptor alpha: impact of ligands on intracellular shuttling and turnover rate in breast cancer cells. Curr Cancer Drug Targets. 2006;6:39–64. doi: 10.2174/156800906775471716. [DOI] [PubMed] [Google Scholar]
- 37.Berry NB, Fan M, Nephew KP. Estrogen Receptor alpha (ER{alpha}) Hinge-Region Lysines 302 and 303 Regulate Receptor Degradation by the Proteasome. Mol Endocrinol. 2008 doi: 10.1210/me.2007-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan M, Rickert EL, Chen L, Aftab SA, Nephew KP, Weatherman RV. Characterization of molecular and structural determinants of selective estrogen receptor downregulators. Breast Cancer Res Treat. 2007;103:37–44. doi: 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]
- 39.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20:491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 40.Kim SH, Katzenellenbogen JA. Hormone-PAMAM dendrimer conjugates: polymer dynamics and tether structure affect ligand access to receptors. Angew Chem Int Ed Engl. 2006;45:7243–8. doi: 10.1002/anie.200601923. [DOI] [PubMed] [Google Scholar]
- 41.Engelk M, Bojarski P, Bloss R, Diehl H. Tamoxifen perturbs lipid bilayer order and permeability: comparison of DSC, fluorescence anisotropy, laurdan generalized polarization and carboxyfluorescein leakage studies. Biophys Chem. 2001;90:157–73. doi: 10.1016/s0301-4622(01)00139-9. [DOI] [PubMed] [Google Scholar]
- 42.Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22:2116–27. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.