

Abstract
The T cell co-stimulatory molecule CD28 plays an important role in the thymic generation of Foxp3+ regulatory T (Treg) cells essential for the maintenance of self-tolerance. Here, we show that a cell-intrinsic signal from CD28 is involved in the generation of cytokine-responsive Foxp3− precursors using studies of mixed bone marrow chimeras, as well as TCR-specific generation of Foxp3+ cells using intrathymic transfer of TCR transgenic thymocytes expressing a natural Treg TCR. Contrary to a previous report, the analysis of CD28 mutant knock-in mice revealed that this cell-intrinsic signal is only partially dependent on the Lck-binding PYAP motif. Surprisingly, even though the absence of CD28 resulted in a 6-fold decrease in thymic Treg cells, the TCR repertoires of CD28-deficient and sufficient cells were largely overlapping. Thus, these data suggest that CD28 does not operate by markedly enlarging the repertoire of TCRs available for Treg cell development, but rather by improving the efficiency of Treg cell development of thymocytes expressing natural Treg TCRs.
Introduction
Foxp3+ CD4+ regulatory T (Treg) cells are essential for the maintenance of self tolerance, as mice that are defective in the development or function of these cells develop spontaneous autoimmune disease (1, 2). The signals that lead to the development of uncommitted thymocytes to Treg cells have been the subject of substantial research. The seminal discovery that the expression of cognate antigen in the thymus could drive Treg cell development in TCR transgenic mice led to the hypothesis that Treg cells develop due to interactions with self-antigens at an avidity window between negative and positive selection (3-5). This was further supported by studies demonstrating that the Treg and non-Treg TCR repertoires mostly differed (6, 7). Recently, the use of Treg TCR transgenic mice revealed that TCR-specific natural Treg cell development is often restricted by a small developmental niche (8, 9). Thus, TCR-derived signals are important for thymic Treg differentiation.
Although TCR activation is essential for the selection of thymocytes into the Treg cell subset, additional signals are also important. In particular, co-stimulation by CD28 is required for efficient Treg cell development as mice deficient in CD28, or its ligands CD80/CD86, have a dramatic reduction of thymic and peripheral Treg cell numbers (10-13). While one function of CD28 is augmentation of IL-2 secretion, a potentially cell-extrinsic mechanism, the presence of normal thymocytes in mixed bone marrow chimeras was unable to rescue Treg differentiation in CD28 knockout (KO) cells (11). These data suggest that CD28 primarily regulates Treg development via a cell-intrinsic mechanism. Consistent with this observation, the frequency of Foxp3+ cells in hyperactive Stat5 (Stat5CA) transgenic CD28 KO mice was markedly lower than that in Stat5CA transgenic mice (14, 15), suggesting that enhanced cytokine signaling can only partially complement a deficiency in CD28 for thymic Treg cell development.
Recently, we and others proposed that thymic Treg cell development can be divided into at least two discrete steps (15-18). Consistent with studies demonstrating that CD25 and GITR can be upregulated in a Foxp3-independent manner during thymic Treg cell development (19-21), we observed that the CD25hiGITRhiFoxp3− CD4+CD8− subset is enriched in Treg cell precursors (16). Characterization of these cells suggested a model for thymic Treg development in which TCR-derived signals lead to the development of a cytokine responsive Treg cell precursor, which then responds to signals from IL-2 or IL-15 for the induction of Foxp3. Consistent with this model, Stat5CA diminished the requirement for TCR specificity in Treg cell generation (15). Visualizing the development of Treg cells after intrathymic injection of TCR transgenic cells into cognate antigen expressing hosts was also consistent with this model (17). Taken together, these data suggest that thymic Treg cell development is a multi-step process involving signals from TCR, cytokines, and other receptors including CD28.
Although CD28 is important in thymic Treg cell development, it is unclear whether it acts before or after the generation of Foxp3− Treg cell precursors. Note that we use “Treg cell precursors” in a different manner than in a recent study examining the role of CD28 and Lck in stabilizing Foxp3 mRNA, which used that term to refer to thymic Foxp3+ cells as precursors to peripheral Treg cells (22). CD28 may regulate Treg cell development by promoting cell-extrinsic IL-2 production and/or by generating cell-intrinsic signals. Co-stimulation might facilitate the generation of Treg cells via increasing the aggregate TCR signal, thereby recruiting thymocytes with lower TCR avidity to self-antigens into the Treg cell subset. Conversely, CD28 could provide a signal which increases the efficiency of Treg cell development of thymocytes expressing the natural repertoire of Treg TCRs. Thus, the process by which CD28 promotes Treg cell development is currently unknown.
Biochemically, the cell-intrinsic effect is primarily mediated by the C-terminal PYAP motif, as expression of a mutant CD28 transgene in which this motif is mutated failed to restore Treg cell development in CD28-deficient mice (11). However, previous studies of the structural basis for CD28 have been plagued by inconsistencies and conflicting results, in part due to variable levels of expression and the use of heterologous promoter systems (23). As CD28 expression varies throughout thymic development and as a consequence of T cell activation, faithful recapitulation of expression levels is essential to accurately determine the role played by CD28 in the development of Treg cells (24).
To address these issues, we utilized targeted knock-in mice expressing mutations of CD28 to definitively address the role of specific CD28 signaling motifs in Treg cell development. We found that the Lck-binding PYAP motif only partially accounts for CD28 signals, whereas the PI3K-binding Y170 motif is dispensable. We also determined that cell-intrinsic CD28 signals are required for the efficient generation of Foxp3− Treg cell precursors. Unexpectedly, analysis of the Treg cell TCR repertoire suggests that CD28 acts mostly by increasing the efficiency of TCR-instructed Treg cell selection rather than by dramatically expanding the TCR repertoire capable of Treg cell development.
Material and Methods
Mice
C57BL/6.SJL (CD45.1) mice were purchased from the National Cancer Institute (Frederick, MD). CD28 KO mice (25) were generated by C. Thompson (U. of Pennsylvania, Philadelphia, PA). Homozygous CD28-AYAA and -Y170F mice were previously described (23, 26). Foxp3gfp reporter mice (27) were provided by A. Rudensky (Memorial Sloan-Kettering, NY, NY). Mice were housed in a specific pathogen-free facility at Washington University and used under protocols approved by the Institutional Animal Care and Use Committee.
Reagents
Fluorescently conjugated monoclonal antibodies were purchased from eBioscience (San Diego, CA), Biolegend (San Diego, CA), and BD Biosciences (San Jose, CA). Human IL-2 (hIL-2) was obtained from NCI Biological Resources Branch (Frederick, MD) preclinical repository. Murine IL-7 (mIL-7) was purchased from Peprotech (Rocky Hill, NJ).
Thymocyte culture
CD25hiFoxp3− (5×103) or Foxp3− (1×105) CD4SP cells were FACS purified and cultured with hIL-2 (50 U/ml) in the presence of mIL-7 (5 ng/ml). Foxp3gfp expression was analyzed by flow cytometry at 24 hours.
Bone marrow chimeras
3-5×106 red-cell-lysed bone marrow cells were intravenously transferred into lethally irradiated hosts (10.5 Gys). Mice were analyzed 6-8 weeks after reconstitution.
Intrathymic transfer
G113 TCR-transgenic thymocytes largely devoid of Treg cells were mixed with WT Thy1.1+ cells to allow assessment of injection efficiency; labeled with DDAO-SE (Invitrogen, Carlsbad, CA); and intrathymically injected (8). Induction of Foxp3 in the donor cells was assessed by flow cytometry of the entire thymus.
TCR repertoire analysis
TRAV14 TCRα sequences from thymic Foxp3+ and Foxp3− Vβ6+ CD4SP thymocytes isolated from 6-10 week old mice were obtained as previously described (7). As before, the CDR3 amino acid sequence was used as unique identifiers for individual TCRs. Three of the WT thymus CD4SP datasets have been published (16).
Statistical analysis
Two-tailed Student’s t-test was used for calculation of statistical significance unless otherwise stated. Estimation of repertoire diversity by the abundance coverage estimator (ACE) was obtained using EstimateS (Version 7.5, R. K. Colwell, http://viceroy.eeb.uconn.edu/estimates).
Results
The CD28 Lck-binding PYAP motif is involved in thymic Treg cell development
A previous report that utilized transgenic expression of various mutated CD28 constructs in a CD28-deficient background suggested that the Lck-binding PYAP motif is important for the function of CD28 in Treg cell development (11). However, transgenic expression may not faithfully recapitulate the endogenous expression pattern of CD28 as it is developmentally regulated (Fig. S1A and (24)). Since studies of gene dosage suggested that the level of CD28 affects its function (23), we assessed the role of the PYAP motif using CD28-AYAA knock-in mice, which have targeted P187A and P190A mutations in the cytoplasmic tail (23). In addition, we analyzed the CD28-Y170F mutant, as PI3K-induced Akt can negatively regulate Treg cell development (26, 28). As expected, the mutant knock-in mice express CD28 at comparable levels to that of WT control (Fig. S1B).
The CD28 KO, AYAA, and Y170F strains were bred to the Foxp3gfp reporter for analysis of Treg cell development (16, 27). Overall CD4 and CD8 thymic development appeared normal in both CD28 mutant mice (Fig. 1A; Table 1). Flow cytometric analysis of the CD4SP subset revealed that the percentage of Foxp3+ cells was normal in Y170F mice (3.1 ± 0.5 %), but decreased two-fold in AYAA mice (1.4 ± 0.3 %, p<0.01) (Fig. 1B, C). However, contrary to the previous report showing that the mutation of the PYAP motif completely abolishes the function of CD28 for Treg cell development (11), we found that the frequency of Treg cells was significantly higher in AYAA than CD28 KO mice (1.4 ± 0.2 % versus 0.6 ± 0.1 %, p<0.01). These results imply that in addition to the PYAP motif, other sequences in the tail of CD28 contribute to thymic Treg cell development.
Figure 1.

The PYAP, but not Y170, motif of CD28 is required for thymic Treg cell development. (A) Normal CD4/CD8 thymic development in CD28-AYAA and -Y170F knock-in mice. Thymocytes from the indicated strains bred to the Foxp3gfp reporter were analyzed by flow cytometry. (B) Decreased Foxp3+ CD4SP cells in AYAA mice. CD4SP gated cells were assessed for CD25 and Foxp3 by flow cytometry. (C) Decreased CD25hiFoxp3− frequency in CD28 KO mice. The percentage of Foxp3− CD25hi and Foxp3+ subsets from each strain is summarized (mean ± S.D.; n=12, WT and KO; n=8 AYAA, n=6 Y170F for A, B, and C). (D) Decreased expression of CD122 in CD28 KO CD25hiFoxp3− cells. Representative FACS plots for CD122 (IL-2Rβ) is shown for CD25hiFoxp3− and Foxp3+ CD4SP cells. The mean percentage and S.D. of CD122hi cells in the CD25hiFoxp3− subset is indicated in the upper panels (n=8 WT and KO; n=7 Y170F; n=6 AYAA). ** p<0.01.
Table 1.
Cell count for total thymocytes and subsets from WT, CD28KO, Y170F, and AYAA mice.
| WT (n=13) | Y170F (n=7) | AYAA (n=9) | KO (n=13) | |
|---|---|---|---|---|
| Total cell number (106) | 142.0 ± 50.9 | 143.0 ± 75.8 | 121.4 ± 56.8 | 94.2 ± 28.7 |
| Total CD4SP (106) | 12.4 ± 5.0 | 14.0 ± 8.2 | 10.9 ± 5.2 | 10.1 ± 3.4 |
| CD4SP Foxp3− CD25hi (104) | 19.5 ± 9.8 | 22.9 ± 9.1 | 19.8 ± 15.0 | 12.2 ± 4.2 |
| CD4SP Foxp3+ (104) | 36.1 ± 14.2 | 43.5 ± 28.1 | 18.6 ± 15.7 | 6.1 ± 2.5 |
Data shown are the mean ± S.D. of thymocyte numbers from the various mutant mice.
The number of mice analyzed is indicated by “n”.
A role for CD28 in the generation of cytokine-responsive Treg cell precursors
Previously, we proposed that thymic Treg cell development can be divided into TCR-dependent and independent phases (16). CD28 could act coordinately with TCR in the first phase, or facilitate production of IL-2 for the latter step. To address this, we examined CD28 KO and mutant mice for the frequency of CD25hiFoxp3− CD4SP cells, which are enriched in Treg cell precursors (16). Flow cytometric analysis showed that the CD25hiFoxp3− CD4SP subset was decreased in CD28 KO mice (2.1 ± 0.3% versus 2.8 ± 0.4% in WT, p<0.01), whereas the frequency of the subset in Y170F and AYAA mice were comparable to that in WT mice (Fig. 1B, C). Consistent with the decreased number of CD25hiFoxp3− cells present in CD28 KO mice, there was also a reduction in the CD122hiCD25hiFoxp3− subset (Fig. 1D), which expresses the trimeric IL-2 receptor and contains the highest frequency of Treg cell precursors in normal mice (16). However, the cells which complete Treg cell development and express Foxp3 in CD28 KO mice express WT levels of CD122 and CD25 (Fig. 1D and S2). In contrast to CD28 KO mice, the percentage of CD122hi cells in the CD25hiFoxp3− subset in AYAA mice was comparable to that of WT mice (Fig. 1D). Also, we did not detect any difference in the level of Foxp3gfp reporter expression in the KO and mutant mice (Fig. S2), which is not consistent with a recent report associating the PYAP motif with stability of Foxp3 mRNA in thymic Treg cells (22). Altogether, these data demonstrate that CD28 is involved in the upregulation of IL-2 receptor chains via signaling motifs other than Y170 or PYAP.
Although the CD25hiFoxp3− cell subset is enriched in Treg cell precursors in normal mice, it was possible that CD28 could have differential effects on CD25 upregulation versus Treg cell development, thus affecting the enumeration of Treg cell precursors in CD28 KO mice. For example, CD28 could be more important for Treg cell precursor generation than CD25 upregulation. Conversely, the decreased percentage of CD25hi cells in the CD28 KO may reflect the dependence of CD25 expression on CD28 (25), rather than reflecting the percentage of Treg cell precursors. We therefore decided not to utilize CD25 to subset CD4SP cells, and instead functionally assessed the frequency of cytokine-responsive Treg cell precursors in the entire Foxp3− subset after IL-2 stimulation in vitro. We found that the percentage of cytokine-responsive Treg cell precursors in CD28 KO mice was decreased by half comparing to WT control (Fig. 2A). Mutation of the PYAP motif also reduced the precursor population by one-fourth. By contrast, the Y170F mutation had no effect on the Treg cell precursor (Fig. 2A). These results suggest that the PYAP motif only partially accounts for the involvement of CD28 in the generation of Treg cell precursors, and does so via a mechanism independent of IL-2 receptor upregulation.
Figure 2.

CD28 is involved in the generation of Foxp3− Treg cell precursors. (A) Decreased frequency of cytokine responsive Treg cell precursors in CD28 KO and AYAA mice. CD25hiFoxp3− CD4SP cells were FACS purified from WT and indicated strains, cultured in vitro with hIL-2 (50 U/ml), and analyzed at 24 hours for Foxp3 expression by flow cytometry. Data were normalized to that of WT from the same experiment to account for inter-experimental variation. Each dot represents data from an individual experiment. Bars indicate the average normalized percentage of Treg cell precursors (n=8 WT and KO; n=5 AYAA, n=6 Y170F). (B) The effect of CD28 on the generation of Treg cells is cell-intrinsic. Lethally irradiated congenic hosts (CD45.1) were reconstituted with an equal mixture of bone marrow cells from WT (CD45.1) and WT, KO, or AYAA (all CD45.2) mice. Foxp3 and CD25 expression in CD45.2+CD4SP cells were analyzed by flow cytometry six weeks post-reconstitution. (C) Percentages of CD25hiFoxp3− and Foxp3+ cells within the CD45.2+CD4SP subset are summarized (n=12 WT and KO; n=8 AYAA; n=6 Y170F). (D) The effect of CD28 on the generation of Treg cell precursors is cell-intrinsic. FACS purified CD45.2+ Foxp3− CD4SP cells were cultured in vitro with hIL-2 (50 U/ml) and Foxp3 expression was analyzed 24 hours later by flow cytometry. Data shown were normalized to that of WT cells in the same experiment. Each dot represents data from an individual mouse (n=6 mice, 4 independent experiments) and the bars indicate the average normalized percentage. ** p ≤0.01.
To address whether CD28 facilitated the generation of Treg cell precursors via a cell-intrinsic mechanism, we analyzed bone marrow chimeras generated with CD45.2 CD28 KO or AYAA bone marrow cells mixed in a 1:1 ratio with CD45.1 WT bone marrow. This approach has been utilized to provide a more normal cytokine environment during thymic Treg cell development (11). Consistent with previous results using CD25 as a Treg cell marker (11), we found that the generation of thymic Treg cells was not restored in CD28 KO cells despite the presence of WT thymocytes (Fig. 2B, C). However, we observed only a partial defect in Treg cell frequency in AYAA mutant cells (Fig. 2B, C), rather than a complete defect. We also analyzed the generation of Treg cell precursors by purifying CD45.2 Foxp3− CD4SP donor cells and culturing them with IL-2 in vitro. We found that the presence of WT thymocytes made essentially no difference in the generation of cytokine responsive AYAA or CD28 KO Treg cell precursors (Fig. 2A, 2D), even though Treg cell development of WT thymocytes appeared normal in the chimeras (Fig. S3). These data therefore suggest that CD28 delivers a cell-intrinsic signal involved in the generation of Treg cell precursors as it cannot be compensated by the presence of normal thymocytes.
CD28 ligands are required for efficient de novo Treg cell development
The previous set of experiments examined Treg cell development during the steady state. To evaluate de novo Treg cell generation, we utilized TCR transgenic line expressing the naturally arising Treg TCR G113 (8). As the Treg cell developmental niche for this TCR is quite small, G113 αβ-TCR transgenic thymocytes contain a very low frequency of Foxp3+ cells. Intrathymic transfer of the G113 thymocyte population can therefore be used to study Treg cell development (8). Three days after intrathymic transfer into WT recipients, G113 Foxp3+ cells can be readily found (8.3 ± 4.2 %; Fig. 3). However, the lack of CD28 ligands CD80/CD86 in the recipients inhibited the development of Foxp3+ cells 16-fold, with only a small frequency of Foxp3+ cells observed (0.5 ± 0.4 %, p<0.01; Fig. 3). This appeared to be related to cell-intrinsic CD28-derived signals rather than an environmental effect, as Treg cell development was not inhibited and perhaps slightly enhanced after transfer of G113 thymocytes into CD28 KO recipients. Thus, these data suggest that CD28 interaction with B7 is required for the efficient de novo generation of antigen specific Treg cells.
Figure 3.

Interaction with B7 is required for TCR-specific Treg cell development. (A) Diminished TCR transgenic Treg cell development in B7 KO mice. Total thymocytes from G113 TCR transgenic Foxp3gfp Rag1−/−, which are largely devoid of Treg cells, were mixed with Thy1.1+ filler cells, labeled with DDAO-SE and transferred intrathymically into WT, CD80−/− CD86−/− (B7 KO), or CD28−/− (CD28 KO) recipients. Expression of Foxp3 by DDAO+Vβ6+Thy1.1− donor cells was analyzed by flow cytometry at the indicated time. Each dot represents data from an individual mouse and the bar the average frequency. Data are pooled from at least two independent experiments (total mice: n=4, d1 WT and B7 KO; n=5, d2 WT; n=4, d2 B7 KO; n=13, d3 WT; n=10, d3 B7 KO; n=6, d3 CD28 KO). ** p<0.01.
CD28 signaling does not markedly increase the number of TCRs capable of facilitating Treg cell development
The above analysis of monoclonal T cell populations demonstrated that CD28-derived signals were important for G113-TCR-mediated Treg cell development. However, it remained possible that other TCRs would exhibit variable dependencies on CD28 signals for Treg cell development. One hypothesis is that CD28 signaling directly contributes to the overall TCR “signaling strength,” thereby greatly expanding the repertoire of TCRs which undergo Treg cell development resulting in the increased frequency of Treg cells in WT versus CD28 KO mice. An alternative, non-mutually exclusive possibility is that TCR alone dictates development into Foxp3+ cells, and that CD28 provides a parallel signal to improve the efficiency of this process. The former hypothesis would therefore predict that the Treg TCR repertoire in CD28 KO and WT mice would differ greatly, whereas the latter would show strong similarity in the TCR repertoires. Although there are some contested data suggesting that CD28 facilitates negative selection in TCR transgenic models (11, 29), it remains largely unknown whether CD28 alters the TCR repertoire.
Since the TCR repertoire of fully polyclonal mice is too diverse for experimental analysis at the individual TCR level, we analyzed the thymic TRAV14 (Vα2) TCR repertoires of CD28 KO and WT mice in a fixed TCRβ model (7). Similar to the fully polyclonal setting, TCliβ TCR transgenic Foxp3gfp Tcra+/− (TCliβ-tg) mice on a CD28 KO background have decreased frequencies of thymic CD4SP Foxp3+ cells (Fig. S4A). Furthermore, we did not observe skewing of Vα2 usage in CD28 KO and WT TCliβ-tg mice (Fig. S4B). Thus, the TRAV14 (Vα2) subset in TCliβ-tg mice appeared suitable for evaluating the role of CD28 on the thymic Treg TCR repertoire.
Thymic CD4SP Foxp3+ and Foxp3− cells were purified by flow cytometry and a total of five and six datasets were obtained from WT and KO TCliβ-tg mice, respectively (Table S1). Initial analysis showed that CD28 deficiency resulted in a decreased TCR diversity in the Foxp3+ subset, which may contribute to the decreased number of Treg cells in the CD28 KO (Table S1). Consistent with the similarity in Foxp3− CD4SP numbers between CD28 KO and WT mice, virtually all of the top 10 Foxp3− TCRs in the CD28 KO data set could be found in the WT data set (Fig. S5), suggesting that CD28 has no major impact on the Foxp3− TCR repertoire. As expected, the top 10 TCRs of Foxp3− and Foxp3+ subsets are largely distinct in both CD28KO and WT mice, similar to previously published results (Fig. 4A, B and (7)).
Figure 4.

Large overlap between the CD28 KO and WT TCR repertories. (A and B) The Foxp3+ and Foxp3− TCR repertoires are distinct in both WT and CD28 KO mice, with minor overlap. TRAV14 TCRα sequences from CD28 WT (A) and KO (B) mice were obtained as described in the Methods and Table S1. The frequencies of the top 10 Treg TCRs in the pooled data set, as well as their corresponding frequency in the Foxp3− data set, are shown. (C and D) Large overlap between the CD28 WT and KO Treg TCR repertoires. The top 10 Treg TCRs from the CD28 WT (C) or KO (D) data sets are shown, and their percentages in the Foxp3+ and Foxp3− data sets are shown. Data from individual data sets are shown in Fig. S5.
We next analyzed the effect of CD28 deficiency on the Treg cell TCR repertoires. The Treg TCR repertoire in CD28 KO mice appeared to more oligoclonal, as the top 10 TCRs comprised a greater portion of the total TCRs sequenced. Also, the individual frequencies of the top 10 Foxp3+ TCRs often differed between the CD28KO and WT data sets (Fig. 4C, D and S6), suggesting that CD28 signaling has variable effects on Treg cell development depending on the particular TCR specificity. Despite these observed differences between the CD28 KO and WT Treg TCR repertoires, almost all of the top 10 Treg TCRs in one data set could be found in the other. Thus, it appeared unlikely that CD28 co-stimulation enlarges the Treg TCR repertoire sufficiently to account for a 6-fold increase in Treg cell numbers (Fig. 1 and S4).
To obtain greater perspective regarding the similarity between the CD28 KO and WT mice, the frequencies of all TCRs greater than an average frequency of 0.25% in the KO or WT data sets were plotted in two dimensions (Fig. 5). This threshold frequency was chosen as 0.23% represents the minimal population frequency of a TCR that would be found in the data set at least once with 90% confidence, as calculated using the binomial distribution with a sample size of 1000. These data revealed that most of these Treg TCRs were present in both CD28 KO and WT mice. Remarkably, the overall pattern of TCR usage appeared similar regardless of whether Treg or Foxp3− TCRs were plotted. Although the overall pattern was similar between Treg and Foxp3− TCRs, we did observe 10 Treg TCRs that were found only in the CD28 KO and 4 only in the WT data sets (Fig. 5). By contrast, only 1 Foxp3− TCR was found exclusively in either the CD28 KO or WT data set. Since the use of the average TCR frequency between CD28 KO and WT data sets could favor overlapping TCRs, we also verified these observations using a threshold of 0.5% in either the CD28 KO or WT data sets (Fig. S7). Although infrequent TCRs cannot be easily analyzed via this approach, one interpretation of these results is that these Treg TCRs found in only the CD28 KO or WT data sets represent a small repertoire shift such that a few TCRs undergo negative selection in the presence of CD28 (points on the left edge), and are thus not found in the WT data set, and that a few others cannot facilitate Treg cell selection without CD28 co-stimulation (points on the bottom edge).
Figure 5.
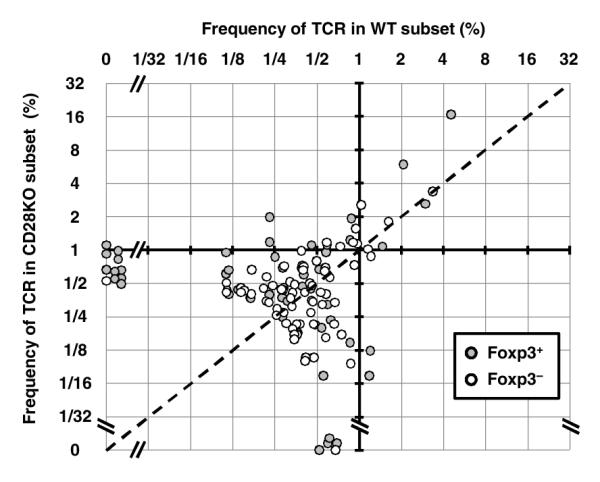
Differential requirement of CD28 for thymic selection. TCRs with an average frequency in the Foxp3+ CD28 WT and KO data sets of > 0.25% were plotted (58 TCRs, grey dots). The same process was also used to select Foxp3− TCRs (71 TCRs, unfilled dots). These TCRs represent 29.9%, 56.2%, 35.4%, and 39.1% of the total data set for WT Foxp3+, KO Foxp3+, WT Foxp3−, and KO Foxp3− subsets, respectively. The dashed line indicates equal frequency in the CD28 WT and KO data sets. Note that there are 4 and 10 TCRs found only in the CD28 WT or KO Foxp3+ data sets, respectively. By contrast, one TCR is found only in the CD28 WT and KO Foxp3− data sets. Using the non-parametric Wilcoxon Rank sum test to compare the TCRs above the 0.25% threshold, TCR usage in Foxp3+ CD28 WT and KO cells is different (p < 0.01), whereas TCR usage in Foxp3− cells is not (p = 0.7).
Taken together with statistical estimates of TCR diversity (Supplemental Table 1), these data suggest that the increased diversity of the Treg TCR repertoire from CD28 co-stimulation is primarily due to the inclusion of infrequent TCRs. While these infrequent TCRs can substantially increase the diversity of the Treg TCR repertoire, CD28-mediated recruitment of cells expressing these TCRs cannot, however, account for the 6 fold greater Treg cell number found in CD28 WT versus KO mice. In fact, it appears that some of these TCRs are inefficient at inducing Treg cell development, thereby requiring co-stimulation by CD28 (Fig. S8). Thus, although CD28 increases the pool of TCRs available for Treg cell selection, this does not appear to be the major mechanism by which CD28 facilitates the generation of the thymic Treg cell population.
Discussion
In this report, we examined the role of CD28 in thymic Treg cell development. Our results demonstrate that the number of Treg cell precursors is substantially decreased in the absence of cell-intrinsic signals provided by CD28 (Fig. 2). In contrast to previous reports, these CD28-derived signals are only partially dependent on the PYAP motif, as AYAA mutant knock-in mice have intermediate frequencies of both Treg cell precursors and mature Treg cells in comparison with CD28 KO mice (Fig. 1, 2). Finally, we infer that CD28 signaling primarily improves the efficiency of Treg cell development of thymocytes with Treg cell selecting TCRs, rather than markedly increase the repertoire of TCRs available for Treg cell development.
The identification that CD28 is involved in the generation of Treg cell precursors is perhaps not surprising as CD28 signaling is thought to be concomitant with TCR engagement. However, it remains difficult to determine whether CD28 is only involved in the generation of Treg cell precursors, or is also needed at latter stages of Treg cell development. First, the reduction of Treg cells in CD28 KO mice is greater than that observed for cytokine-responsive Treg cell precursors (Fig. 1, 2). One potential explanation for this is that the in vitro assay, while useful for identifying cells which can potentially become Foxp3+, over-represents the frequency of cells that become Foxp3+ after intra-thymic transfer (16). It is therefore possible that CD28 KO Treg cell precursors progress less efficiently to Treg cells in vivo than their WT counterparts, which could be explained by decreased levels of IL-2 in CD28 KO mice. However, we were unable to discern a clear requirement for environmental cytokines using mixed CD28 KO and WT bone marrow chimeras. In fact, we found that although the frequency of cytokine-responsive Treg cell precursors in these chimeras remained constant (Fig. 2), the percentage of CD28 KO Treg cells was actually 1/3rd lower in the presence of WT cells than in CD28 KO mice (Fig. 1, 2). This observation was not previously reported (11), perhaps related to the use of CD25 rather than Foxp3gfp as a Treg cell marker. One interpretation of these results is that CD28, in addition to facilitating the generation of Treg cell precursors themselves, also improves the competitive fitness of Treg cells or their precursors for cytokines or other factors during the generation of the Treg cell population.
Multiple signaling molecules can bind to CD28, including kinases PI3K and Lck that bind to YMNM and PYAP motifs, respectively. Our data shows that the Lck-binding motif is only partially involved in the generation of Treg cell precursors, whereas the PI3K-binding motif is dispensable (Fig. 1). This result suggests that prolonged or enhanced signaling by CD28-associated Lck is important for the development of Treg cell precursors. This data is consistent with a previous study which shows that the Lck-binding motif is important for the thymic Treg cell generation and also T cell activation in an autoimmunity model caused by CTLA-4-deficiency (11, 30). However, the decrease in Treg cell number is less than previously reported. One potential explanation is differences in CD28 expression between the two systems (23). Since the mutation of the PYAP motif only partially recapitulates the defect in Treg cell development observed in CD28 KO mice, this implies that additional signaling motifs are involved.
Thymic Treg cell selection is commonly attributed to TCRs with intermediate affinity to self-antigens in an avidity window between positive and negative selection (18). As CD28 signals are thought to be co-incident with TCR-derived signals (31), we hypothesized that the loss of CD28 would decrease the overall TCR signal, allowing only the highest affinity TCRs the ability to induce Treg cell development. The analysis of the Foxp3+ TCR repertoire of CD28 KO showed a lower diversity compared to that of WT. This may, in part, be due to the 6-fold decrease in the number of Treg cells in CD28 KO mice. Furthermore, individual TCRs exhibited variable dependence on CD28 signaling, with some TCRs found at higher or lower frequency in CD28 KO compared with WT mice. The variable importance of CD28 may reflect the level of CD80/CD86 amongst the various APCs capable of mediating Treg cell selection, such as cTECs, mTECs, and bone marrow-derived DCs. However, we did not observe a large number of Treg TCR sequences unique to the CD28 WT subset, suggesting that CD28 signaling itself does not dramatically increase the pool of TCRs available for Treg cell selection and thereby cannot account for the increased Treg cell number in CD28 WT mice relative to CD28 KO mice (Fig. 5). Although the molecular mechanism of CD28 in Treg cell development remains to be clearly defined, these data imply that Lck and other signals do not primarily enhance the TCR signals relevant for crossing the avidity threshold for Treg cell selection, but rather provide additional parallel signals which facilitate Treg cell development.
Supplementary Material
Acknowledgments
We would like to thank N. Santacruz and J. Hunn for expert technical assistance.
Abbreviations used in this paper
- ACE
abundance coverage estimator
- KO
knockout
- Treg
regulatory T
- WT
wild-type.
Footnotes
This work was supported by the Arthritis Foundation (C.S.H.), Burroughs Wellcome Fund (C.S.H.), and National Institutes of Health (C.S.H. and J.M.G) and the Tertiary Education Services Office, Macau S.A.R. (C.J.L.).
The authors have no conflicts of interest to report.
References
- 1.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nature Immunology. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Jordan MS, Boesteanu A, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+ CD25+ regulatory T cells induced by an agonist self-peptide. Nature Immunology. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 4.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nature Immunology. 2002;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 5.Maloy KJ, Powrie F. Regulatory T cells in the control of immune pathology. Nature Immunology. 2001;2:816–822. doi: 10.1038/ni0901-816. [DOI] [PubMed] [Google Scholar]
- 6.Wong J, Mathis D, Benoist C. TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J Exp Med. 2007;204:2039–2045. doi: 10.1084/jem.20070822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh C-S, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nature Immunology. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 8.Bautista JL, Lio CW, Lathrop SK, Forbush K, Liang Y, Luo J, Rudensky AY, Hsieh CS. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat Immunol. 2009;10:610–617. doi: 10.1038/ni.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung MW, Shen S, Lafaille JJ. TCR-dependent differentiation of thymic Foxp3+ cells is limited to small clonal sizes. J Exp Med. 2009;206:2121–2130. doi: 10.1084/jem.20091033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohr J, Knoechel B, Kahn EC, Abbas AK. Role of B7 in T cell tolerance. J Immunol. 2004;173:5028–5035. doi: 10.4049/jimmunol.173.8.5028. [DOI] [PubMed] [Google Scholar]
- 11.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 12.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 13.Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, Bluestone JA. Cutting Edge: CD28 controls peripheral homeostasis of CD4+ CD25+ regulatory T cells. Journal of Immunology. 2003;171:3348–3352. doi: 10.4049/jimmunol.171.7.3348. [DOI] [PubMed] [Google Scholar]
- 14.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 15.Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, Vegoe AL, Hsieh CS, Jenkins MK, Farrar MA. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wirnsberger G, Mair F, Klein L. Regulatory T cell differentiation of thymocytes does not require a dedicated antigen-presenting cell but is under T cell-intrinsic developmental control. Proc Natl Acad Sci U S A. 2009;106:10278–10283. doi: 10.1073/pnas.0901877106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liston A, Rudensky AY. Thymic development and peripheral homeostasis of regulatory T cells. Curr Opin Immunol. 2007;19:176–185. doi: 10.1016/j.coi.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 20.Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- 21.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Nazarov-Stoica C, Surls J, Bona C, Casares S, Brumeanu TD. CD28 signaling in T regulatory precursors requires p56lck and rafts integrity to stabilize the Foxp3 message. J Immunol. 2009;182:102–110. doi: 10.4049/jimmunol.182.1.102. [DOI] [PubMed] [Google Scholar]
- 23.Friend LD, Shah DD, Deppong C, Lin J, Bricker TL, Juehne TI, Rose CM, Green JM. A dose-dependent requirement for the proline motif of CD28 in cellular and humoral immunity revealed by a targeted knockin mutant. J Exp Med. 2006;203:2121–2133. doi: 10.1084/jem.20052230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turka LA, Ledbetter JA, Lee K, June CH, Thompson CB. CD28 is an inducible T cell surface antigen that transduces a proliferative signal in CD3+ mature thymocytes. J Immunol. 1990;144:1646–1653. [PubMed] [Google Scholar]
- 25.Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 26.Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Bricker TL, Russell JH, Green JM. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol Cell Biol. 2009;29:3710–3721. doi: 10.1128/MCB.01869-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor Foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walunas TL, Sperling AI, Khattri R, Thompson CB, Bluestone JA. CD28 expression is not essential for positive and negative selection of thymocytes or peripheral T cell tolerance. J Immunol. 1996;156:1006–1013. [PubMed] [Google Scholar]
- 30.Tai X, Van Laethem F, Sharpe AH, Singer A. Induction of autoimmune disease in CTLA-4−/− mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci U S A. 2007;104:13756–13761. doi: 10.1073/pnas.0706509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.