

Abstract
The intergenic region internal ribosome entry site (IGR IRES) of the Dicistroviridae viral family can directly assemble 80S ribosomes and initiate translation at a non-AUG codon from the ribosomal A-site. These functions are directed by two independently folded domains of the IGR IRES. One domain, composed of overlapping pseudoknots II and III (PKII/III), mediates ribosome recruitment. The second domain, composed of PKI, mimics a tRNA anticodon–codon interaction to position the ribosome at the ribosomal A-site. Although adopting a common secondary structure, the dicistrovirus IGR IRESs can be grouped into two classes based on distinct features within each domain. In this study, we report on the modularity of the IGR IRESs and show that the ribosome-binding domain and the tRNA anticodon mimicry domain are functionally interchangeable between the Type I and the Type II IGR IRESs. Using structural probing, ribosome-binding assays, and ribosome positioning analysis by toeprinting assays, we show that the chimeric IRESs fold properly, assemble 80S ribosomes, and can mediate IRES translation in rabbit reticulocyte lysates. We also demonstrate that the chimeric IRESs can stimulate the ribosome-dependent GTPase activity of eEF2, which suggests that the ribosome is primed for a step downstream from IRES binding. Overall, the results demonstrate that the dicistrovirus IGR IRESs are composed of two modular domains that work in concert to manipulate the ribosome and direct translation initiation.
Keywords: translation, IRES, dicistrovirus, ribosome, tRNA
INTRODUCTION
Cap-dependent translation involves >12 translation initiation factors to mediate the recruitment of the 40S subunit to the 5′ end of the mRNA, subsequent scanning, and 80S assembly at the AUG codon (Pestova et al. 2007). Alternatively, a subset of cellular and viral mRNAs can utilize an internal ribosome entry site (IRES) to initiate translation in a cap-independent manner (Hellen and Sarnow 2001; Kieft 2008). IRESs are RNA elements that recruit the ribosome generally without the full complement of initiation factors. Thus, IRES-containing RNAs can be translated during times of cellular stress or viral infection when specific initiation factors are compromised and overall cap-dependent translation is inhibited. IRESs often adopt specific RNA secondary and tertiary structures that recruit canonical initiation factors and trans-acting factors to direct recruitment, positioning, and activation of the ribosome for translation initiation.
The simplest IRES to date is found within the intergenic region (IGR) of the Dicistroviridae viral family (Jan 2006; Kieft 2008; Nakashima and Uchiumi 2008). The dicistroviruses contain a positive single-stranded RNA genome encoding the nonstructural and structural proteins within two open reading frames, which are separated by the IGR IRES. Without the aid of initiation factors or initiator Met-tRNAi, the IGR IRES can bind directly to 40S subunits and 80S ribosomes, but not 60S subunits (Wilson et al. 2000a; Jan et al. 2001; Jan and Sarnow 2002; Nishiyama et al. 2003). The IRES then sets the reading frame by occupying the ribosomal P-site to position a non-AUG codon in the ribosomal A-site (Sasaki and Nakashima 1999, 2000; Wilson et al. 2000a,b; Jan et al. 2003; Pestova and Hellen 2003). After the delivery of the first aminoacyl-tRNA to the A-site by elongation factor 1A (eEF1A), the IRES undergoes a translocation event without peptide bond formation, which is mediated by eEF2 (Jan et al. 2003; Pestova and Hellen 2003). Biochemical and structural studies have revealed that the IRES recruits, positions, and sets the ribosome in an elongation mode, indicating that this ∼200-nucleotide (nt) RNA acts as an all-RNA translation factor (Sasaki and Nakashima 1999, 2000; Wilson et al. 2000a,b; Jan et al. 2003; Nishiyama et al. 2003; Pestova and Hellen 2003).
Phylogenetic analyses have revealed that all Dicistroviridae IGR IRESs adopt a similar secondary structure consisting of three overlapping pseudoknots, PKI, PKII, and PKIII (Fig. 1; Jan 2006; Kieft 2008; Nakashima and Uchiumi 2008). PKII and PKIII form one domain that folds into a compact core that is responsible for ribosome binding (Wilson et al. 2000a; Jan and Sarnow 2002; Nishiyama et al. 2003; Pfingsten et al. 2006, 2007), whereas PKI mimics an anticodon tRNA stem–loop to mediate ribosome positioning such that the start non-AUG codon of the IRES occupies the ribosomal A-site (Wilson et al. 2000a,b; Kanamori and Nakashima 2001; Costantino et al. 2008). Previous reports indicate that the domains are functionally independent. First, disruption of PKI does not affect ribosome assembly on the IRES (Jan and Sarnow 2002; Nishiyama et al. 2003). Second, the PKII/PKIII domain alone can fold independently and bind to ribosomes (Nishiyama et al. 2003; Costantino and Kieft 2005). This has led to the hypothesis that distinct domains of the IRES interact with specific regions of the ribosome to direct IRES translation. Consistent with this, structural and biochemical studies have revealed that SLIV and SLV interact with rpS5 and rpS25 to mediate 40S binding, whereas the conserved L1.1 region is predicted to interact with the L1 stalk of the 60S subunit to direct 80S assembly (Pfingsten et al. 2006, 2010; Schuler et al. 2006; Nishiyama et al. 2007; Costantino et al. 2008; Jang et al. 2009; Landry et al. 2009). The L1.1 region is disordered in the unbound IRES, suggesting that this region is dynamic (Jan and Sarnow 2002; Pfingsten et al. 2006, 2007; Schuler et al. 2006). Mutations within the L1.1 region disrupt IRES function and 80S assembly, indicating that specific nucleotides interact with the L1 stalk, thus contributing to 80S affinity (Pfingsten et al. 2006, 2010; Jang et al. 2009).
FIGURE 1.

The secondary structure of the (A) Type I CrPV IGR IRES and the (B) Type II TSV IGR IRES. Conserved nucleotide positions are shown in uppercase and nonconserved nucleotides are in lowercase. Numbering refers to the nucleotide position within the respective viral genome. Helical regions are indicated by a black dash between nucleotides. Underlined nucleotides represent the first two amino acid residues in the viral capsid protein. Properly positioned 40S and 80S ribosomes on the IGR IRES produces a toeprint, denoted Toeprint A, shown by the arrows. The point at which the two classes of IGR IRES were swapped is indicated by a black line. The ΔSLIII deletion within the Type II TSV IGR IRES is marked by a blue box. L1.1A and L1.1B are indicated by red and blue letters, respectively.
Structural studies show that the IRES primarily occupies the P- and E-sites of the ribosome, confirming that the IRES mimics a tRNA (Spahn et al. 2004b; Pfingsten et al. 2006; Schuler et al. 2006). The CrPV PKI domain resembles a tRNA anticodon stem–loop and appears tilted at an angle that is similar to that of a deacylated tRNA in a P/E hybrid state on the ribosome (Yamamoto et al. 2007; Costantino et al. 2008). It has been proposed that the P/E hybrid conformation of the IRES may help mediate the next steps of translation, such as the delivery of aminoacyl-tRNA and translocation (Yamamoto et al. 2007; Costantino et al. 2008). In support of this, the IRES can stimulate the ribosome-dependent GTPase activity of eEF2 (Yamamoto et al. 2007), an effect that is also observed when a deacylated tRNA occupies the ribosome in a P/E hybrid state (Lill et al. 1989; Valle et al. 2003; Zavialov and Ehrenberg 2003; Sergiev et al. 2005). It is unclear whether the proposed P/E hybrid conformation contributes to IRES activity.
Although adopting an overall similar secondary structure, the IGR IRESs can be grouped into two classes based on distinct features within the PKII/III and PKI domains (Jan 2006; Nakashima and Uchiumi 2008). The Type II IRESs, such as the Taura syndrome virus (TSV) IGR IRES, contain a longer L1.1 region and an extra stem–loop, SLIII, within the PKI domain, when compared with the Type I IRESs, such as the Cricket paralysis virus (CrPV) IGR IRES (Fig. 1). We and others have shown that mutations within the L1.1 region of both CrPV and TSV IGR IRESs can inhibit IRES activity and disrupt 80S assembly, suggesting that some of these distinct features have common functions (Pfingsten et al. 2006, 2010; Jang et al. 2009). Although both types of IRESs can mediate factor-independent translation initiation, the significance of these distinct features is poorly understood (Hatakeyama et al. 2004; Cevallos and Sarnow 2005). In this study, we have used a chimeric mutagenesis approach to address whether the distinct domains of the Type I and Type II IRESs are modular. Furthermore, we have explored the biochemical properties of the distinct structural elements within each IRES type. Our results demonstrate that the IGR IRES is composed of functional modular domains that independently direct distinct ribosomal interactions and functions.
RESULTS
80S ribosome assembly on the chimeric IGR IRES
Previous studies have indicated that the PKII/III and PKI domains of the IGR IRES are functionally independent and mediate distinct ribosomal activities (Kanamori and Nakashima 2001; Jan and Sarnow 2002; Costantino and Kieft 2005; Pfingsten et al. 2006; Costantino et al. 2008; Jang et al. 2009). To investigate this further, we constructed chimeric IRESs where the PKI domain was swapped between the Type I (CrPV) and Type II (TSV) IGR IRESs (Fig. 1). The point at which the domains were swapped is a single-stranded variable linker, which is not predicted to be structured. For this study, we will refer to the wild-type IGR IRESs as CrPV and TSV IRESs and the chimeric IRESs as CrPVII/III-TSVI (PKII/PKIII of CrPV IRES fused with PKI of TSV IRES) and TSVII/III-CrPVI (PKII/PKIII of TSV IRES fused with PKI of CrPV IRES).
To investigate ribosome-binding affinities, we used a competition filter-binding assay to monitor 80S assembly on wild-type or chimeric IRESs (Fig. 2A). Specifically, we asked whether the addition of excess unlabeled IRES can compete with radiolabeled wild-type CrPV IRES for 80S ribosomes. As shown previously, increasing amounts of unlabeled wild-type CrPV or TSV IRES, but not ΔPKI/PKIII CrPV IGR IRES, competed with radiolabeled wild-type CrPV IGR IRES for 80S ribosome binding (Fig. 2A; Table 1; Jang et al. 2009). The mutant ΔPKI/PKIII CrPV IGR IRES contains mutations that disrupt PKI and PKIII (Jan and Sarnow 2002). Curve-fitting analysis yielded an apparent dissociation constant (KD) of 8 ± 1 nM for the wild-type CrPV IRES and 25 ± 3 nM for the wild-type TSV IRES, which is consistent with previously published results (Table 1; Nishiyama et al. 2003; Jang et al. 2009). The chimeric IGR IRESs also competed for 80S binding with an apparent dissociation constant of 14 ± 2 nM for the TSVII/III-CrPVI IRES and 20 ± 2 nM for CrPVII/III-TSVI IRES (Table 1). In general, these results demonstrated that 80S binding to the wild-type and chimeric IRESs is relatively tight (KD 8–25 nM range), thus suggesting that the PKII/III domain of the chimeric IRESs is folded properly for 80S assembly and supports previous findings that the PKII/PKIII domain primarily mediates ribosome recruitment.
FIGURE 2.

80S assembly and structural probing of the chimeric IGR IRESs. (A) 80S-IGR IRES binding was monitored by competition titration experiments. The fraction of radiolabeled wild-type CrPV IGR IRES bound to 80S ribosomes is plotted against the log of the indicated unlabeled IRES RNA. Representative curves and quantitations are shown from at least three independent experiments. (B,C) Enzymatic probing of the chimeric IGR IRESs. Dicistronic RNAs containing the wild-type and chimeric IGR IRESs were treated with RNase T1 as indicated. Primer extension was performed as described in Materials and Methods using PrEJ69. The reaction products were separated on a denaturing polyacrylamide gel. The nucleotides that are cleaved 3′ by RNase T1 are indicated to the right. A sequencing ladder of the dicistronic construct using the appropriate primer is shown on the left.
TABLE 1.
Properties of mutant IGR IRESs

It is interesting to note that small differences in 80S binding affinities for the chimeric IRES were observed. These differences display a trend that followed that of the wild-type IRESs. For instance, given that the TSV IRES has a approximately threefold lower affinity for 80S ribosomes than the CrPV IRES (KD 25 versus 8 nM), the chimeric TSVII/III-CrPVI IRES showed a higher 80S binding affinity than the TSV IRES (KD 14 versus 25 nM), whereas the chimeric CrPVII/III-TSVI IRES had a lower affinity than the CrPV IRES (KD 20 versus 8 nM). This suggests that the PKI domain has a role in 80S assembly on the IGR IRES. The influence of the PKI domain on ribosome binding has been suggested in a previous report showing that disruption of both PKIII and PKI domains are required to completely abolish 80S assembly on the IGR IRES (Jan and Sarnow 2002). How the PKI domain may affect 80S assembly on the IGR IRES remains to be investigated.
PKI of the chimeric IGR IRESs fold independently
To determine whether the PKI domain of the chimeric IRESs is folded properly, we probed the structure of the IRESs by using RNase T1. RNase T1 cleaves 3′ of unpaired G residues and can thus detect single-stranded regions within the IGR IRES. The positions of the cleavage sites within the IRES were detected by primer extension analysis using reverse transcriptase. The numbering in Figure 2, B and C, refers to the nucleotide whose 3′ phosphodiester bond is enzymatically cleaved.
Overall, the cleavage patterns observed for wild-type and chimeric IRESs were similar (Fig. 2B,C). Specifically, we observe cleavage sites at G6183, GG6188–6189, G6192, G6195, and G6206 within PKI of both the wild-type CrPV IRES and the chimeric TSVII/III-CrPVI IRES, suggesting that the PKI domain is folded similarly. All RNase T1 cleavages observed were consistent with previously published results (Jan and Sarnow 2002). Furthermore, we also observed a cleavage site at GGG6905–6907 within PKI of both the wild-type TSV IRES and the chimeric CrPVII/III-TSVI IRES. If PKI was not folded properly within this chimera, we would have expected cleavages at GG6937–6936 and GG6932–6933, which are normally in paired regions. In summary, the probing data of the chimeric IRES indicates that PKI folds independently and similarly to PKI of the wild-type IRESs.
Translational activity of the chimeric IRES
Since our data showed that the chimeric IRESs fold properly and can bind to 80S ribosomes with relatively similar affinities as the wild-type IRESs (Fig. 2A,B; Table 1), we next investigated if IRES activity is affected. Dicistronic luciferase RNAs containing either the wild-type or the chimeric IRESs were incubated in rabbit reticulocyte lysates (RRL) in the presence of [35S]-methionine. Translation reactions were analyzed by SDS-PAGE, and the amount of radioactivity incorporated into Renilla (scanning-dependent translation) and firefly (IRES translation) luciferase was quantified by PhosphorImager analysis. As shown previously, translation directed by the TSV IRES was weaker (45%) when compared with that by the CrPV IRES (100%) (Fig. 3A,B; Table 1; Hatakeyama et al. 2004; Cevallos and Sarnow 2005; Jang et al. 2009). The chimeric CrPVII/III-TSVI IRES activity was slightly less active (93%) compared with the wild-type CrPV IRES (Fig. 3B; Table 1), indicating that the PKI domain of TSV can be swapped into the CrPV IRES without significantly affecting IRES translation. In contrast, the TSVII/III-CrPVI IRES activity was significantly reduced (29%) compared with the wild-type CrPV IRES. However, when compared with the weaker TSV IRES activity (here given as 100%), the TSVII/III-CrPVI IRES activity was only reduced to ∼64% activity, demonstrating that swapping in the PKI domain of the CrPV IRES into the TSV IRES still retained IRES activity (Table 1). Interestingly, although having relatively high affinity for the ribosome, the CrPVII/III-TSVI IRES displayed ∼2.5-fold lower affinity for the ribosome compared with the wild-type CrPV IRES, yet both IRESs displayed similar IRES activities (Table 1, KD 8 nM versus 20 nM). Furthermore, the reduced translation of the TSVII/III-CrPVI IRES is not attributed to differences in 80S affinities, as the chimeric TSVII/III-CrPV IRES showed higher 80S binding affinity compared with that of the TSV IRES (Table 1, KD 14 nM versus 25 nM). Thus, the differences in 80S binding affinities between these IRESs do not correlate with their translational activities. In summary, these results indicate that the PKI domains are functionally interchangeable between the CrPV and TSV IRESs.
FIGURE 3.

Translational activities of the chimeric IGR IRESs. (A) Uncapped dicistronic RNAs containing wild-type or chimeric IGR IRESs were incubated in RRL at 30°C for 60 min in the presence of [35S]methionine. The first cistron, encoding Renilla luciferase (Rluc), measures scanning-mediated translation, and the second cistron, firefly luciferase (Fluc), measures IGR IRES-mediated translation. Shown is a representative gel of radiolabeled Fluc and Rluc protein products detected by autoradiography. Where applicable, the amount of the ternary complex inhibitor NSC119889 added to the reactions is shown above the gel. (B) Quantitations of translational activities of the chimeric IRESs. The ratios of firefly to Renilla luciferase are shown and are normalized to the ratio of the dicistronic RNA containing the wild-type CrPV IGR IRES. (C) Normalized quantitation of the chimeric IRES translation under NSC119889 treatment. Firefly (top) and Renilla (bottom) luciferase activities were normalized to the translational activity of each dicistronic RNA in the absence of NSC119889. The data shown are the averages of at least three independent experiments ±SD. (D) Translational activities of the chimeric IRESs in the presence of edeine. Shown is a representative gel of radiolabeled Fluc and Rluc detected by autoradiography. The bottom panel shows quantitations of Rluc and Fluc, normalized to the amount of luciferase produced by each dicistronic RNA in the absence of edeine. The average value from three independent experiments ±SD is shown.
A previous report showed that CrPV IRES translation is resistant to the compound NSC119889, which disrupts the interaction between eIF2 and Met-tRNAi (Robert et al. 2006). This compound can be used to distinguish IRESs like the EMCV IRES that mediate internal initiation in an eIF2-dependent manner from ones that are eIF2 independent (Novac et al. 2004). We tested whether the chimeric IRES activities are resistant to the NSC119889 compound. Addition of NSC119889, as expected, reduced the translation of Renilla luciferase, indicating that scanning-dependent translation is inhibited (Fig. 3C, bottom panel). In contrast, the activities of the wild-type CrPV IRES, TSV IRES, and CrPVII/III-TSVI IRESs were relatively resistant to the effects of the compound at 10 and 20 μM (Fig. 3A; Fig. 3C, top panel). Interestingly, TSVII/III-CrPVI IRES translation was not resistant to the effects of NSC119889 (Fig. 3C). To examine this further, we tested the effects of the antibiotic edeine on IRES activity. It has been shown that the IGR IRES is also insensitive to edeine, which inhibits AUG codon recognition by the 40S•eIF2•GTP/Met-tRNAi complex (Kozak and Shatkin 1978; Wilson et al. 2000a; Dinos et al. 2004). As expected, addition of edeine inhibited scanning-dependent translation of Renilla luciferase (Fig. 3D). In contrast, translation by both wild-type and chimeric IRESs were resistant to the effects of edeine (Fig. 3D). In summary, these data indicate that the chimeric IRESs can direct internal initiation in an eIF2-independent manner similar to the wild-type IRESs.
Positioning of the IGR IRES chimeras correlates with their translational activities
After 80S assembly on CrPV and TSV IRESs, the ribosome is positioned on the IGR IRES such that the GCU alanine start codon and the preceding CCU triplet occupy the ribosomal A- and P-sites, respectively (Fig. 1; Wilson et al. 2000a; Jan and Sarnow 2002; Cevallos and Sarnow 2005). To determine whether the 80S ribosomes are positioned properly, we performed toeprinting assays (i.e., primer extension assays) on dicistronic RNAs containing chimeric IRESs bound to purified 40S or 80S ribosomes. Purified ribosomes bound to the wild-type CrPV IRES produced two toeprints, A and B, that are observed at nucleotides CA6226-7 and AA6161-2, respectively (Fig. 4A, left panel). Likewise, toeprints A and B were observed for ribosome-bound wild-type TSV IRES at UU6963–6964 and A6876, respectively (Fig. 4A, right panel). The presence of toeprint A is indicative of proper ribosome positioning, which is +13–14 nt downstream from the CCU triplet of both IRESs, given that the first C is +1. Thus, the ribosome is properly positioned on the wild-type IRESs with the CCU triplet and GCU alanine triplet occupying the ribosomal P- and A-sites, respectively (Wilson et al. 2000a; Jan and Sarnow 2002). Toeprint B represents a contact within the core PKII/PKIII domain of the IRES with the ribosome that impedes the reverse transcriptase in the primer extension analysis (Jan and Sarnow 2002). Mutations that disrupt PKI base pairing (i.e., ΔPKI) eliminated toeprint A but not that of toeprint B (Fig. 4A, left panel) in accordance with the previous finding that disruption of PKI abolishes ribosome positioning but not recruitment (Jan and Sarnow 2002).
FIGURE 4.

Ribosome positioning on the chimeric IGR IRESs by toeprinting analysis. Dicistronic RNAs containing wild-type or mutant IGR IRES were incubated with (A) 40S alone or 40S and 60S subunits (100 nM), or (B) in RRL in the presence of edeine as described in the Materials and Methods. Reactions were analyzed by primer extension analysis using oligo PrEJ69. Reaction products were separated by denaturing polyacrylamide gels. The gels were dried and exposed by autoradiography. The location of major toeprint A and toeprint B, are shown on the right. Sequencing ladders for the wild-type IRESs are shown on the left, with their respective nucleotide numbers as indicated. The intensity of toeprint A in B is shown as a percentage of the total radioactivty in each lane, normalized to the wild-type CrPV IGR IRES (100%).
For the chimeric IRESs, the presence of toeprint A, in general, correlated with their translational activities using purified ribosomal subunits. Specifically, the chimeric TSVII/III-CrPVI lacked toeprint A, whereas CrPVII/III-TSVI IRES produced toeprint A (Fig. 4A). The presence of toeprint B was not significantly affected in both chimeras, suggesting that the ribosome is bound to the chimeric IRES and supports the conclusion that these mutations do not affect ribosome recruitment (Fig. 2A). It is interesting to note that although toeprint A is not produced, the TSVII/III-CrPVI IRES translation is weakly active in RRL (Figs. 3B, 4A). It is possible that toeprint A cannot be detected by the toeprinting assay when the translational activity is weak. To further examine this, we investigated the position of the ribosome on these IRESs in RRL under identical conditions in which the IRES activities were measured in Figure 3. Incubation of dicistronic RNAs containing the wild-type CrPV IRES in RRL in the presence of edeine produced a strong toeprint at CA6226–6227 (toeprint A), which represents a properly positioned ribosome on the IRES (Fig. 4B). The addition of edeine at this concentration (10 μM) inhibits the delivery of an aminoacyl-tRNA to the ribosomal A-site and can inhibit IGR IRES-dependent translocation (Carrasco et al. 1974; Wilson et al. 2000a). Incubation of dicistronic RNAs containing the TSV IRES produced a toeprint at UU6963–6964 (toeprint A), which is similar to that observed when purified 80S ribosomes are bound (Fig. 4A,B). However, the TSV IRES produced a weaker toeprint A (39%) compared with that on the CrPV IRES (100%), which correlates with the reduced translational activity of the TSV IRES compared with the CrPV IRES (Figs. 3B, 4B). As expected, disruption of PKI significantly reduced toeprint A on the TSV and CrPV IRESs (Fig. 4B, ΔPKI lanes). For the chimeric IRESs, incubation of these dicistronic RNAs in RRL produced the same toeprint A as their wild-type counterparts (Fig. 4B). However, the intensity of toeprint A observed for the chimeric TSVII/III-CrPVI IRES was reduced when compared with that of the CrPVII/III-TSVI IRES (38% versus 121%), which is the same trend observed with their translational activities (Fig. 4B; Table 1). Thus, given the relatively similar 80S binding affinities between the chimeric IRESs, it appears that the intensity of the toeprint A reflects the translational activity of the IRESs. In support of this conclusion, similar intensities of toeprint A are observed on the CrPVII/III-TSVI and the wild-type CrPV IRESs, which display similar IRES activities (Figs. 3B, 4B). In summary, our results show that the activity of the chimeric IRES correlates with the presence of toeprint A in RRL, and thus proper positioning of the ribosome.
Modularity of the conserved L1.1A/B region of the IGR IRESs
The conserved L1.1 region consisting of L1.1A and L1.1B of the IGR IRESs is conserved within each IRES type (Fig. 1, color coded in red and blue) (Jan 2006; Nakashima and Uchiumi 2008). Although distinct between the two classes, mutations within this region reduce IRES translation and can disrupt ribosome recruitment and positioning, suggesting that the L1.1 region of both IRES types mediate common functions (Pfingsten et al. 2006, 2010; Jang et al. 2009). To test this directly, we asked whether the L1.1 region is interchangeable by swapping the L1.1A or both L1.1A/B regions between the CrPV and TSV IRESs. When the L1.1A or L1.1A/B of the TSV IRES was inserted into the CrPV IRES (CrPV + TSV L1.1A and CrPV + TSV L1.1A/B), IRES translational activity was preserved, albeit reduced when compared with the wild-type CrPV IRES (Fig. 5A,B; Table 1). In contrast, translational activity was abolished in the chimeric TSV IRES containing the L1.1A or both L1.1A/B of the CrPV IRES (Fig. 5A,B; Table 1). When we assayed for 80S binding, the TSV IRES containing the swapped CrPV L1.1A or L1.1A/B had significantly lower 80S affinity (approximately four- to sixfold lower affinity) as compared with the CrPV IRES containing the swapped TSV L1.1A or L1.1A/B regions (Fig. 5C; Table 1). Since the CrPV IRES with the TSV L1.1 region is still functional and can bind to 80S ribosomes, this chimeric CrPV + L1.1A/B IRES is likely folded properly to mediate IRES activity. In contrast, the TSV IRES cannot tolerate insertion of the CrPV L1.1 region, suggesting that the TSV + CrPV L1.1 chimeric IRES may not be folded properly.
FIGURE 5.

The activity of L1.1 IGR IRES chimeras. (A) Dicistronic RNAs containing wild-type or chimeric IGR IRESs were incubated in RRL at 30°C for 60 min in the presence of [35S]methionine. The first cistron, encoding Renilla luciferase (Rluc), measures cap-mediated translation, and the second cistron, firefly luciferase (Fluc), measures IGR IRES-mediated translation. Shown is a representative gel of radiolabeled Fluc and Rluc protein products detected by autoradiography, and the IRES mutants tested are labeled at the top of the gel. (B) Amounts of Fluc and Rluc made are quantitated below the gel by PhosphorImager analysis, shown as a ratio between Fluc and Rluc. Results were normalized against wild-type CrPV IGR IRES. (C) The fraction of radiolabeled wild-type CrPV IGR IRES bound to 80S ribosomes is plotted against the log of the indicated unlabeled IRES concentration. Representative quantifications are shown from at least three independent experiments.
The IGR IRES chimeras stimulate the ribosome-dependent GTPase activity of eEF2
The GTPase activity of EF-G can be stimulated when a deacylated-tRNA occupies the ribosome in a P/E hybrid state (Lill et al. 1989; Zavialov and Ehrenberg 2003; Sergiev et al. 2005). It has been shown that the related dicistrovirus Plautia stali intestine virus (PSIV) IRES can also stimulate the GTPase activity of eEF2 when bound to the ribosome, thus suggesting that the IGR IRES may mimic a P/E tRNA hybrid (Yamamoto et al. 2007; Costantino et al. 2008). Here, we examined whether the chimeric IGR IRESs can stimulate the ribosome-dependent GTPase activity of eEF2. Toward this, we monitored the multiple turnover of [γ-32P]GTP by incubating an excess of IRES with purified 80S ribosomes and eEF2. The fraction of released phosphate was resolved by thin-layer chromatography and analyzed by PhosphorImager analysis. Incubation of wild-type CrPV or TSV IRES with ribosomes and eEF2 stimulated GTP hydrolysis to similar levels (Fig. 6). This stimulation was specific, as reactions containing only ribosomes (-IRES/-eEF2) did not stimulate GTPase activity and reactions containing only ribosomes and eEF2 (-IRES) or ribosomes/eEF2 with the mutant ΔPKI/PKIII IRES, which does not bind to ribosomes (Jan and Sarnow 2002), stimulated GTP hydrolysis to half the level of the wild-type IRESs (Fig. 6). This trend was also observed with varying amounts of eEF2, indicating that the GTPase stimulation of eEF2 is dependent on IRES binding to the ribosome (data not shown). Similar to that observed for the PSIV IRES (Yamamoto et al. 2007), the ΔPKI mutant of the CrPV IRES stimulated GTPase activity to the same extent of the wild-type IRESs, indicating that an anticodon–codon interaction within PKI is not required for GTPase activation (Fig. 6). Collectively, these data point to PKII/III and the PKI stem of the IRES as the main determinants for stimulating ribosome-dependent eEF2 GTPase activity.
FIGURE 6.

Ribosome-dependent GTPase stimulation of eEF2 by the chimeric IGR IRES. GTP hydrolysis was monitored by incubation of 80S, eEF2, and the indicated IRES with radiolabeled [γ-32P]GTP. Aliquots of the reaction were quenched over time and then resolved by thin-layer chromatography. The slope of the linear regression lines, which represents the mean percent hydrolysis of radiolabeled GTP per minute, is shown in a bar graph. These values were derived from three independent experiments, and the error bars correspond to a 95% confidence interval. Note that ribosomes alone (-IRES/-eEF2) do not stimulate GTPase activity.
Similar to the wild-type IRESs, the chimeric IRESs also stimulated the GTPase activity of eEF2, but to varying degrees (Fig. 6). Specifically, the chimeric CrPVII/III-TSVI IRES stimulated GTPase activity to the same extent as the wild-type CrPV and TSV IRESs. In contrast, the chimeric TSVII/III-CrPVI IRES only slightly stimulated GTPase activity over background (Fig. 6). This reduced GTPase activity may be due to the chimeric TSVII/III-CrPVI IRES not being in the optimal P/E hybrid state on the ribosome. Alternatively, because the multiple turnover of GTP was monitored, the lower GTPase activity may be due to inhibition of another step in the eEF2 translocation cycle (i.e., release of eEF2-GDP from the ribosome).
Previous reports have indicated that the L1 stalk of the 60S subunit may interact with the deacylated tRNA in the P/E hybrid state and may assist in the translocation of the deacylated tRNA (Valle et al. 2003; Spahn et al. 2004a; Fei et al. 2008). Because the L1.1 region of the IGR IRES is predicted to interact with the L1 stalk, one hypothesis is that this region may direct the GTPase activity of eEF2 and thereby assist in the first translocation step. A mutant CrPV IRES containing a GACU6038–6041CUAG mutation (Jang et al. 2009), which has been shown to inhibit IRES translation, displayed similar GTPase stimulation as the wild-type IRESs (Fig. 6B), thus suggesting that the L1.1 region is not required for stimulating the GTPase activity of eEF2.
Deletion of SLIII in the TSV IGR IRES abolishes 80S positioning
One main difference between the two classes of IGR IRESs is an extra stem–loop, SLIII, within PKI of the Type II IRESs. Deletion of SLIII or mutations within SLIII inhibit IRES-driven translational activity, but these mutants can still assemble 80S ribosomes in RRL, suggesting that SLIII may mediate a step downstream from 80S binding (Hatakeyama et al. 2004; Pfingsten et al. 2007). To explore this further, we assayed whether deletion of SLIII within the TSV IRES affected ribosome binding, positioning, and ribosome-dependent eEF2 GTPase activity. As shown previously, translation by the ΔSLIII TSV IRES was severely inhibited, but this mutant IRES still bound to 80S ribosomes in RRL (Fig. 7A,B; Table 1; Pfingsten et al. 2007). Moreover, the addition of NSC119889 did not affect 80S binding on the mutant IRES (Fig. 7B). To examine this more closely, we monitored 80S binding by using the competition binding assay. The ΔSLIII IRES bound to purified 80S ribosomes with similar affinity as the wild-type TSV IRES, with a KD of 25 ± 2 nM, which is consistent with the observation that this mutation does not affect 80S assembly (Table 1; Pfingsten et al. 2007). By toeprinting analysis, 40S and 80S ribosomes assembled on the wild-type TSV IRES produced toeprint A at UU6963–6964, but not on the ΔSLIII TSV IRES (Fig. 7C), indicating that the presence of SLIII is required for proper ribosome positioning. When we monitored the ribosome-dependent GTPase activity of eEF2, the stimulation of GTP hydrolysis by the ΔSLIII TSV IRES was found to be similar to that of the wild-type TSV IRES (Fig. 6). Therefore, despite a lack of proper ribosomal positioning, the ΔSLIII mutant can still activate the GTPase activity of eEF2, further supporting that the GTPase stimulation is independent of the PKI domain of the IGR IRES.
FIGURE 7.

Characterization of SLIII within the TSV IGR IRES. (A) Dicistronic RNAs containing wild-type or ΔSLIII TSV IGR IRESs were incubated in RRL at 30°C for 60 min in the presence of [35S]methionine. The first cistron, encoding Renilla luciferase (Rluc), measures scanning-mediated translation, and the second cistron, firefly luciferase (Fluc), measures IGR IRES-mediated translation. Shown are radiolabeled firefly (Fluc) and Renilla (Rluc) luciferase protein products detected by autoradiography and quantitated by PhosphorImager analysis. (B) 80S assembly on TSV and ΔSLIII IGR IRESs was assessed in RRL by sucrose gradient analysis. Radiolabeled wild-type or ΔSLIII IGR IRES (100 nM) was incubated in RRL with 0.1 mg/mL cycloheximide in the presence (+NSC) or absence (−NSC) of 25 μM NSC119889 for 15 min at room temperature. Reactions were loaded on a 10%–30% sucrose gradient and shown are the percent of total radioactive counts in each fraction. The top and bottom of the gradient is represented from left to right, respectively. Fractions containing free IRES, 40S, and 80S ribosomes are indicated. (C) Toeprint analysis of assembled 40S and 80S ribosomes on TSV and ΔSLIII IGR IRESs. 40S alone or 40S and 60S subunits (100 nM) were incubated with dicistronic RNAs containing wild-type or mutant IGR IRES and analyzed by primer extension analysis using oligo PrEJ69. Reaction products were separated in denaturing polyacrylamide gels. The gels were dried and exposed by autoradiography.
DISCUSSION
The Type I and Type II IGR IRESs are comprised of two independently folded domains: a PKII/III ribosome binding domain and a PKI tRNA anticodon mimicry domain that recruit, position, and set the ribosome in an elongation-competent mode. While structurally similar, each domain of the Type I and Type II IRESs possesses distinct features, of which their functions are poorly understood. This study demonstrates that the ribosome-binding domain and the tRNA anticodon mimicry domain of the CrPV and TSV IGR IRESs are, in general, functionally interchangeable and thus modular. The results suggest that these modular domains mediate similar interactions and functions within the ribosome to direct factorless IRES translation.
Many biological systems often use modularity to realize functionality (Tang and Breaker 1998; Wierenga 2001). Modularity also allows recombination of domains to evolve new RNAs or proteins with novel functions. An example of this is found in the universally conserved RNase P RNA, which processes the precursor tRNA to produce the mature tRNA. RNase P RNA consists of two independently folded modular domains: the C- and S-domains, which are responsible for catalysis and substrate recognition, respectively (Loria and Pan 1996; Torres-Larios et al. 2006). In the case of the dicistrovirus IGR IRES, it is attractive to envision that the IRES itself evolved or is “constructed” from the recombination of a ribosome-binding RNA and a tRNA anticodon-like domain. Previous structural and biochemical studies are consistent with this idea. PKII/PKIII and PKI domains can fold independently and direct specific interactions with the ribosome (Jan and Sarnow 2002; Nishiyama et al. 2003; Costantino and Kieft 2005; Nishiyama et al. 2007; Pfingsten et al. 2007). Moreover, it has been shown that the PKII/PKIII domain alone can bind to the ribosome (Nishiyama et al. 2003; Costantino and Kieft 2005). In this study, our results provide further support to this idea as the chimeric IRESs are translationally active and the PKII/PKIII and PKI domains are functionally interchangeable (Figs. 2A, 3; Table 1).
The chimeric CrPVII/III-TSVI IRES is translationally more active than the TSVII/III-CrPVI IRES (Fig. 3; Table 1). This is not attributed to significant differences in ribosome binding (Fig. 2A; Table 1) or defects in RNA folding (Fig. 2B,C). It is possible that the overall structure of the hybrid TSVII/III-CrPVI IRES may not allow for maximal IRES translation. Alternatively, because the CrPV IGR IRES and the chimeric IRES containing the CrPVII/III domain are translationally more active than the TSV IGR IRES and the chimeric IRES containing the TSVII/III domain, this suggests that the CrPV PKII/III domain may confer higher translational activity (Fig. 3B). It has been proposed that an interaction of the PKII/III domain with the 60S subunit induces a structural change that propagates through the IRES to the PKI domain to properly position the ribosome (Yamamoto et al. 2007; Jang et al. 2009; Pfingsten et al. 2010). In support of this, mutations in the conserved L1.1 region of PKII/III can reduce the anticodon–codon interaction within PKI, suggesting a functional linkage between the two domains (Jang et al. 2009). Furthermore, our data indicates that 80S ribosome positioning is disrupted in the translationally weaker TSVII/III-CrPVI IRES chimera (Fig. 4), which is consistent with the idea that a conformational structural change induced by an IRES/ribosome interaction is transmitted from PKII/PKIII to PKI. Thus, an interdomain signal within the CrPVII/III-TSVI chimera may activate IRES translation, whereas structural incompatibilities within the TSVII/III-CrPVI chimera may result in uncoupling of the two domains, which prevents the successful transduction of this signal, thus resulting in weaker IRES activity. However, although RNase T1 probing analysis suggests that the CrPV PKI is folded properly in the unbound state (Fig. 2B,C), we cannot rule out that the CrPV PKI domain does not fold properly when the hybrid TSVII/III-CrPVI IRES is bound to the ribosome.
One domain that may be important for this allosteric interdomain signal is the L1.1 region, which is conserved within each type of IGR IRES and predicted to interact with the L1 stalk of the 60S subunit (Fig. 1; Jan and Sarnow 2002; Pfingsten et al. 2006, 2007; Schuler et al. 2006). We have previously demonstrated that mutations within the L1.1 domain of several IGR IRESs disrupt 80S binding and IRES translation, suggesting that all L1.1 domains of this viral family may function similarly (Jang et al. 2009). Interestingly, only the chimeric CrPV IRES containing the swapped L1.1A/B region of the TSV IRES retained IRES activity (Fig. 5A,B). Because the L1.1 region is flanked by two helices, the TSV L1.1A/B region inserted into the CrPV IRES likely interacts with the L1 stalk in the same manner as in the wild-type IRES. In contrast, swapping the L1.1A/B region of the CrPV IRES into the TSV IRES inhibited translational activity, suggesting that this smaller CrPV L1.1A/B region cannot be accommodated within the TSV IRES, possibly constraining the overall structure of the PKII/III domain (Fig. 5A,B). Indeed, 80S binding affinity for this hybrid IRES (TSV + CrPV L1.1A/B) was significantly (approximately fourfold) inhibited, as compared with that for the wild-type IRES and the hybrid CrPV + L1.1A/B IRES (Fig. 5C; Table 1).
Our results are consistent with the idea that the IRES mimics a P/E hybrid tRNA to stimulate the ribosome-dependent GTPase activity of eEF2 (Yamamoto et al. 2007; Costantino et al. 2008). Specifically, wild-type and mutant IRESs that can bind to 80S ribosomes with high-affinity stimulated GTPase activity (Figs. 3, 6; Table 1). This supports the observation that disruption of PKII or PKIII, which inhibits ribosome binding, does not stimulate GTPase activity (Yamamoto et al. 2007). The PKII/PKIII domain is not the only determinant for GTPase stimulation, as a PKII/PKIII domain alone partially stimulates GTPase activity (Yamamoto et al. 2007). Finally, our results are in agreement that the anticodon–codon interaction of PKI is not required for GTPase activity (Fig. 6; Yamamoto et al. 2007). In summary, it is likely that an IRES with an intact PKII/PKIII and helical stem of PKI, but not the anticodon–codon interaction of PKI, mimics a P/E tRNA hybrid that can stimulate the GTPase activity of eEF2.
How does the IRES stimulate the ribosome-dependent GTPase activity of eEF2? Cryo-EM studies have shown that the IGR IRES induces a conformational change on the ribosome where the stalk region of the 60S subunit consisting of the ribosomal P proteins becomes extended (Spahn et al. 2004b). This stalk region has been shown to be important for recruitment and stimulation of GTPase activation on elongation factors (Diaconu et al. 2005). Therefore, this IRES-induced structural change may be facilitating GTPase activation on eEF1A and/or eEF2 and thereby mediates the delivery of the next aminoacyl-tRNA and/or translocation. Previous reports point to the 3′ end of a tRNA in a P/E hybrid state that is responsible for ribosomal GTPase activation (Lill et al. 1989). Since the IRES occupies the ribosomal P- and E-sites, it will be interesting to determine whether a specific element within the IRES may be coupling the structural conformation of the stalk region and the ribosome-dependent GTPase activity of eEF2. In this study, our results show that mutations in the L1.1 region do not inhibit the GTPase activity of eEF2, suggesting that the interaction between L1.1 of the IRES and the L1 stalk of the 60S is not required for this activity (Fig. 6).
The functional role of SLIII in the TSV IGR IRES is unknown, although it has been shown previously that deletion of SLIII does not affect 80S assembly on the IRES (Pfingsten et al. 2007). We have expanded on this and showed that SLIII is predominantly responsible for proper ribosome positioning, which explains why the ΔSLIII mutation is translationally inactive (Fig. 7). It remains to be investigated whether SLIII is directly mediating anticodon–codon interactions within PKI or whether deletion of SLIII simply disrupts proper folding of PKI.
In summary, this report demonstrates that the IGR IRES is composed of two independent modular domains. Although we have only tested the CrPV and TSV IRESs, it is likely that the PKII/III and PKI domains of the other Type I and II IGR IRESs are also interchangeable. The modularity of these domains within the Dicistroviridae family implies that these IGR IRESs may have evolved from the recombination of distinct functional RNA domains.
MATERIALS AND METHODS
DNA constructs and reagents
The dicistronic and the monocistronic luciferase plasmids containing the CrPV (gi: 21321708) and TSV (gi: 14701764) IGR IRESs have been described previously (Jan and Sarnow 2002; Cevallos and Sarnow 2005). NSC119889 was provided by the NCI/DTP Open Chemical Repository (http://dtp.nci.nih.gov).
In vitro transcription and translation
Plasmids containing dicistronic and monocistronic luciferase plasmids were linearized with XbaI and EheI, respectively. The EheI restriction site cleaves 33 nt downstream from the start AUG codon of the firefly luciferase gene. RNAs were in vitro transcribed by a T7 RNA polymerase reaction and the resulting RNA was purified by RNeasy kit (Qiagen). The integrity and purity of the transcribed RNAs were confirmed by gel analysis. Uncapped dicistronic RNAs were incubated in RRL with 154 mM (final concentration) potassium acetate for 1 h at 30°C. Protein expression was measured by incorporation of [35S]methionine and analyzed by SDS-PAGE. Gels were dried and analyzed by PhosphorImager analysis (Typhoon, GE).
40S and 60S subunit purification
Ribosomal subunits were purified from HeLa cell pellets (National Cell Culture Center) as described (Jan and Sarnow 2002). In brief, HeLa cells were first lysed in a Triton X-100 lysis buffer (15 mM Tris–HCl at pH 7.5, 300 mM NaCl, 6 mM MgCl2, 1% [v/v] Triton X-100, 1 mg/mL heparin). Lysates were centrifuged to remove debris and the supernatant layered on a 30% [w/w] 0.5 M KCl sucrose cushion and centrifuged at 100,000g to pellet ribosomes. Ribosomes were resuspended in buffer B (20 mM Tris–HCl at pH 7.5, 6 mM magnesium acetate, 150 mM KCl, 6.8% [w/v] sucrose, 1 mM DTT), treated with puromycin to release ribosomes from mRNA, and KCl was added to a final concentration of 0.5 M. The dissociated ribosomes were then separated on a 10%–30% (w/w) sucrose gradient. The 40S and 60S peaks were detected at 260 nm, pooled, concentrated using Amicon Ultra spin concentrators (Millipore) in buffer C (20 mM Tris–HCl at pH 7.5, 0.2 mM EDTA, 10 mM KCl, 1 mM MgCl2, 6.8% sucrose). Western blot analysis verified the absence of eIF2. The purity of 40S and 60S was also examined by detecting 18S and 28S rRNA by ethidium bromide staining. The concentration of 40S and 60S subunits was determined by spectrophotometry, using the conversions 1 A260 nm = 50 nM for 40S and 1 A260 nm = 25 nM for 60S subunits.
Purification of eukaryotic elongation factor 2
A yeast strain expressing a histidine-tagged version of eEF2 was generously provided by Dr. Terri Kinzy (UMDNJ). A total of 4 L of yeast cells was grown in appropriate drop-out media to an optical density of 2.0. Cells were harvested by centrifugation at 5000 rpm for 5 min, and resuspended in 2 vol of lysis buffer (50 mM KPO4, 10 mM imidazole, 1 M KCl, 0.2 mM PMSF, and 1% v/v Tween-20 at pH 8.0). Cells were then lysed by three passes through a French press (Thermo) and centrifuged at 20,000g for 20 min. The supernatant was collected and centrifuged at 100,000g for 1.5 h. The supernatant was filtered through 3 MM Whatman paper and loaded onto a 1-mL Ni2+ chelating column that was pre-equilibrated with wash buffer (50 mM KPO4, 20 mM imidazole, 1 M KCl, 0.2 mM PMSF, 1% v/v Tween-20 at pH 8.0). The column was washed with 10 column volumes of wash buffer and eluted with a step gradient using 20 column volumes of elution buffer (50 mM KPO4, 500 mM imidazole, 1 M KCl, 0.2 mM PMSF, 1% v/v Tween-20 at pH 8.0). The fractions containing eEF2 were identified by SDS-PAGE, and these fractions were dialyzed in dialysis buffer (20 mM tris, 100 mM KCl, 0.1 mM EDTA, 10% glycerol, 1 mM DTT, 0.2 mM PMSF at pH 7.5) three times overnight, before storage at −80°C.
80S filter-binding competition assays
5′ end-labeled IRES RNAs (5 nM final concentration) were incubated in buffer E with increasing amounts of IRES competitor from 2 to 250 nM and 50 ng/μL of noncompetitor RNA (Jan and Sarnow 2002). RNAs were then incubated with preformed 80S ribosomes at a final concentration of 12.5 nM for 20 min at room temperature. Reactions were applied to a double membrane of nitrocellulose and nylon that was pre-wet with buffer E in a Bio-Dot filtration apparatus (Bio-Rad). Membranes were then dried, and the radioactivity was quantitated by PhosphorImager analysis (Typhoon, GE).
Obtained data was fit to the Linn–Riggs equation, which describes the competitive binding of two ligands to a protein. (Long and Crothers 1995) The fraction bound, θ, is described as:
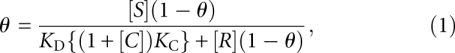 |
where [S], [R], and [C] are the concentrations of 80S ribosomes, radiolabeled wild-type CrPV IGR IRES, and competitor IRES, respectively. The dissociation constants are denoted as KD and KC for the wild-type CrPV IGR IRES and competitor IRES interaction with 80S ribosomes. Solving for θ in equation 1:
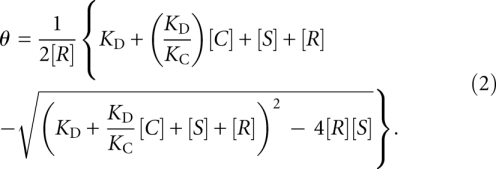 |
A competition curve, where θ = f[C], was fitted for the best value of KC. All KC values obtained are based upon at least three independent experiments.
Assembly and analysis of ribosomal complexes
Toeprinting analysis of ribosomal complexes using purified subunits was performed as described (Wilson et al. 2000a). A total of 500 ng of dicistronic IGR IRES RNAs was first annealed with primer PrEJ69 (5′-GCCCTGGTTCCTGGAACAATTGCTT-3′) in 40 mM Tris (pH 7.5) and 0.2 mM EDTA by slow cooling from 65°C to 37°C. Annealed RNAs were incubated in buffer E containing 40S subunits or 40S and 60S subunits (final 40 nM). Toeprinting analysis using rabbit reticulocyte lysates was conducted in the same manner, where annealed RNAs were incubated in 10 μL of RRL with 20 μM amino acid mix, 154 nM potassium acetate, and 10 μM edeine. Ribosomal complexes were analyzed by primer extension analysis using AMV reverse transcriptase in the presence of α-[32P]dATP (3000 Ci/mmol, PerkinElmer) as described (Jan and Sarnow 2002) cDNA products were analyzed on 6% (w/v) polyacrylamide/8 M urea gel. Gels were dried and analyzed by PhosphorImager analysis.
Ribosome assembly in rabbit reticulocyte lysates
5′ end-labeled RNAs (0.5 nM final concentration) were incubated with a final concentration of 154 mM potassium acetate, 0.1 mg/mL cycloheximide, 40 U of RiboLock (Fermentas), 40 μM of amino acids, and rabbit reticulocyte lysate (Promega). Reactions were incubated for 20 min at 30°C, and then layered onto a 10%–30% w/w sucrose gradient in buffer E with 5 mM Mg2+. Gradients were centrifuged for 3.5 h in an SW41 rotor at 36,000 rpm, and fractionated. The radioactivity in each fraction was measured by scintillation counting (PerkinElmer).
GTPase assays
A total of 11 pmol of IRES RNA was combined with 0.7 pmol of eEF2, 7 pmol of 40S, and 10 pmol of 60S in a buffer of 50 mM tris, 50 mM KCl, 1 mM MgCl2, 0.2 mM DTT (pH 7.6). A mixture of 12.5 nmol cold GTP and 0.415 pmol of [γ-32P]GTP (PerkinElmer) was added, and the volume of the reaction was made up to 50 μL. The reaction was incubated at room temperature and 5 μL aliquots at each time point were quenched with 1 μL of 6 M formic acid. Samples were centrifuged at 13,000 rpm for 10 min, and 0.25 μL of the sample was spotted on a prewashed PEI-cellulose thin layer chromatography plate (Sigma-Aldrich). Plates were developed in a solution of 0.75 M KH2PO4 (pH 3.5) and dried before PhosphorImager analysis.
ACKNOWLEDGMENTS
We thank Miranda Lo for her work on the in vitro translation assays. This work was supported by a Canadian Institute for Health Research grant (CIHR) (MOP-81244) and a National Sciences and Engineering Research Council of Canada (NSERC) Discovery grant (RGPIN 341459-07). E.J. is supported by career development awards from the Michael Smith Foundation for Health Research and from CIHR. C.J.J. is supported by an NSERC PGS D award.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2044610.
REFERENCES
- Carrasco L, Battaner E, Vazquez D 1974. The elongation steps in protein synthesis by eukaryotic ribosomes: Effects of antibiotics. Methods Enzymol 30: 282–289 [DOI] [PubMed] [Google Scholar]
- Cevallos RC, Sarnow P 2005. Factor-independent assembly of elongation-competent ribosomes by an internal ribosome entry site located in an RNA virus that infects penaeid shrimp. J Virol 79: 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino D, Kieft JS 2005. A preformed compact ribosome-binding domain in the cricket paralysis-like virus IRES RNAs. RNA 11: 332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino DA, Pfingsten JS, Rambo RP, Kieft JS 2008. tRNA-mRNA mimicry drives translation initiation from a viral IRES. Nat Struct Mol Biol 15: 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaconu M, Kothe U, Schlunzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC 2005. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 121: 991–1004 [DOI] [PubMed] [Google Scholar]
- Dinos G, Wilson DN, Teraoka Y, Szaflarski W, Fucini P, Kalpaxis D, Nierhaus KH 2004. Dissecting the ribosomal inhibition mechanisms of edeine and pactamycin: The universally conserved residues G693 and C795 regulate P-site RNA binding. Mol Cell 13: 113–124 [DOI] [PubMed] [Google Scholar]
- Fei J, Kosuri P, MacDougall DD, Gonzalez RL Jr 2008. Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol Cell 30: 348–359 [DOI] [PubMed] [Google Scholar]
- Hatakeyama Y, Shibuya N, Nishiyama T, Nakashima N 2004. Structural variant of the intergenic internal ribosome entry site elements in dicistroviruses and computational search for their counterparts. RNA 10: 779–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CU, Sarnow P 2001. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 15: 1593–1612 [DOI] [PubMed] [Google Scholar]
- Jan E 2006. Divergent IRES elements in invertebrates. Virus Res 119: 16–28 [DOI] [PubMed] [Google Scholar]
- Jan E, Sarnow P 2002. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J Mol Biol 324: 889–902 [DOI] [PubMed] [Google Scholar]
- Jan E, Thompson SR, Wilson JE, Pestova TV, Hellen CU, Sarnow P 2001. Initiator Met-tRNA-independent translation mediated by an internal ribosome entry site element in cricket paralysis virus-like insect viruses. Cold Spring Harb Symp Quant Biol 66: 285–292 [DOI] [PubMed] [Google Scholar]
- Jan E, Kinzy TG, Sarnow P 2003. Divergent tRNA-like element supports initiation, elongation, and termination of protein biosynthesis. Proc Natl Acad Sci 100: 15410–15415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang CJ, Lo MC, Jan E 2009. Conserved element of the dicistrovirus IGR IRES that mimics an E-site tRNA/ribosome interaction mediates multiple functions. J Mol Biol 387: 42–58 [DOI] [PubMed] [Google Scholar]
- Kanamori Y, Nakashima N 2001. A tertiary structure model of the internal ribosome entry site (IRES) for methionine-independent initiation of translation. RNA 7: 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS 2008. Viral IRES RNA structures and ribosome interactions. Trends Biochem Sci 33: 274–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M, Shatkin AJ 1978. Migration of 40 S ribosomal subunits on messenger RNA in the presence of edeine. J Biol Chem 253: 6568–6577 [PubMed] [Google Scholar]
- Landry DM, Hertz MI, Thompson SR 2009. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev 23: 2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Robertson JM, Wintermeyer W 1989. Binding of the 3′ terminus of tRNA to 23S rRNA in the ribosomal exit site actively promotes translocation. EMBO J 8: 3933–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KS, Crothers DM 1995. Interaction of human immunodeficiency virus type 1 Tat-derived peptides with TAR RNA. Biochemistry 34: 8885–8895 [DOI] [PubMed] [Google Scholar]
- Loria A, Pan T 1996. Domain structure of the ribozyme from eubacterial ribonuclease P. RNA 2: 551–563 [PMC free article] [PubMed] [Google Scholar]
- Nakashima N, Uchiumi T 2008. Functional analysis of structural motifs in dicistroviruses. Virus Res 139: 137–147 [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Yamamoto H, Shibuya N, Hatakeyama Y, Hachimori A, Uchiumi T, Nakashima N 2003. Structural elements in the internal ribosome entry site of Plautia stali intestine virus responsible for binding with ribosomes. Nucleic Acids Res 31: 2434–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama T, Yamamoto H, Uchiumi T, Nakashima N 2007. Eukaryotic ribosomal protein RPS25 interacts with the conserved loop region in a dicistroviral intergenic internal ribosome entry site. Nucleic Acids Res 35: 1514–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novac O, Guenier AS, Pelletier J 2004. Inhibitors of protein synthesis identified by a high throughput multiplexed translation screen. Nucleic Acids Res 32: 902–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Hellen CU 2003. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev 17: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Lorsch JR, Hellen CU 2007. The mechanism of translation initiation in eukaryotes. In Translational control in biology and medicine (ed. Mathews MB et al. ), p. 87 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Pfingsten JS, Costantino D, Kieft JS 2006. Structural basis for ribosome recruitment and manipulation by a viral IRES RNA. Science 314: 1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfingsten JS, Costantino DA, Kieft JS 2007. Conservation and diversity among the three-dimensional folds of the Dicistroviridae intergenic region IRESes. J Mol Biol 370: 856–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfingsten JS, Castile AE, Kieft JS 2010. Mechanistic role of structurally dynamic regions in Dicistroviridae IGR IRES. J Mol Biol 395: 205–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert F, Kapp LD, Khan SN, Acker MG, Kolitz S, Kazemi S, Kaufman RJ, Merrick WC, Koromilas AE, Lorsch JR, et al. 2006. Initiation of protein synthesis by hepatitis C virus is refractory to reduced eIf2•GTP•Met-tRNAiMet ternary complex availability. Mol Biol Cell 17: 4632–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki J, Nakashima N 1999. Translation initiation at the CUU codon is mediated by the internal ribosome entry site of an insect picorna-like virus in vitro. J Virol 73: 1219–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki J, Nakashima N 2000. Methionine-independent initiation of translation in the capsid protein of an insect RNA virus. Proc Natl Acad Sci 97: 1512–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M, Connell SR, Lescoute A, Giesebrecht J, Dabrowski M, Schroeer B, Mielke T, Penczek PA, Westhof E, Spahn CM 2006. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat Struct Mol Biol 13: 1092–1096 [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Lesnyak DV, Kiparisov SV, Burakovsky DE, Leonov AA, Bogdanov AA, Brimacombe R, Dontsova OA 2005. Function of the ribosomal E-site: A mutagenesis study. Nucleic Acids Res 33: 6048–6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CM, Gomez-Lorenzo MG, Grassucci RA, Jorgensen R, Andersen GR, Beckmann R, Penczek PA, Ballesta JP, Frank J 2004a. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J 23: 1008–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CM, Jan E, Mulder A, Grassucci RA, Sarnow P, Frank J 2004b. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: The IRES functions as an RNA-based translation factor. Cell 118: 465–475 [DOI] [PubMed] [Google Scholar]
- Tang J, Breaker RR 1998. Mechanism for allosteric inhibition of an ATP-sensitive ribozyme. Nucleic Acids Res 26: 4214–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Larios A, Swinger KK, Pan T, Mondragon A 2006. Structure of ribonuclease P—a universal ribozyme. Curr Opin Struct Biol 16: 327–335 [DOI] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J 2003. Locking and unlocking of ribosomal motions. Cell 114: 123–134 [DOI] [PubMed] [Google Scholar]
- Wierenga RK 2001. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett 492: 193–198 [DOI] [PubMed] [Google Scholar]
- Wilson JE, Pestova TV, Hellen CU, Sarnow P 2000a. Initiation of protein synthesis from the A site of the ribosome. Cell 102: 511–520 [DOI] [PubMed] [Google Scholar]
- Wilson JE, Powell MJ, Hoover SE, Sarnow P 2000b. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol Cell Biol 20: 4990–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Nakashima N, Ikeda Y, Uchiumi T 2007. Binding mode of the first aminoacyl-tRNA in translation initiation mediated by Plautia stali intestine virus internal ribosome entry site. J Biol Chem 282: 7770–7776 [DOI] [PubMed] [Google Scholar]
- Zavialov AV, Ehrenberg M 2003. Peptidyl-tRNA regulates the GTPase activity of translation factors. Cell 114: 113–122 [DOI] [PubMed] [Google Scholar]