

Abstract
Actin-myosin contractility modulates focal adhesion assembly, stress fiber formation, and cell migration. We analyzed the contributions of contractility to fibroblast adhesion strengthening using a hydrodynamic adhesion assay and micropatterned substrates to control cell shape and adhesive area. Serum addition resulted in adhesion strengthening to levels 30–40% higher than serum-free cultures. Inhibition of myosin light chain kinase or Rho-kinase blocked phosphorylation of myosin light chain to similar extents and eliminated the serum-induced enhancements in strengthening. Blebbistatin-induced inhibition of myosin II reduced serum-induced adhesion strength to similar levels as those obtained by blocking myosin light chain phosphorylation. Reductions in adhesion strengthening by inhibitors of contractility correlated with loss of vinculin and talin from focal adhesions without changes in integrin binding. In vinculin-null cells, inhibition of contractility did not alter adhesive force, whereas controls displayed a 20% reduction in adhesion strength, indicating that the effects of contractility on adhesive force are vinculin-dependent. Furthermore, in cells expressing FAK, inhibitors of contractility reduced serum-induced adhesion strengthening as well as eliminated focal adhesion assembly. In contrast, in the absence of FAK, these inhibitors did not alter adhesion strength or focal adhesion assembly. These results indicate that contractility modulates adhesion strengthening via FAK-dependent, vinculin-containing focal adhesion assembly.
Keywords: actin-myosin, focal adhesion kinase (FAK), vinculin, contractility
Introduction
Integrin-mediated cell adhesion to the extracellular matrix (ECM) plays central roles in various cellular processes including survival, cell cycle progression, and the expression of tissue-specific phenotypes (Danen and Sonnenberg, 2003; Hynes, 2002). Cell adhesion comprises the coordinated evolution of binding of integrin receptors to adhesive domains in ECM ligands, integrin clustering, interactions with the actin cytoskeleton, and focal adhesion assembly (Geiger et al., 2001; Sastry and Burridge, 2000). Focal adhesions are discrete adhesive plaques that contain numerous structural (e.g., vinculin, talin, and α-actinin) and signaling elements (e.g., FAK, Src, paxillin, and p130CAS). Focal adhesions have emerged as putative mechanosensors for extracellular stimuli (Riveline et al., 2001; Wang et al., 2001). For example, external forces exerted on integrins enhance focal adhesion assembly and increase the strength and rigidity of the linkage between integrins and the actin cytoskeleton (Choquet et al., 1997; Geiger et al., 2009; Riveline et al., 2001; Wang et al., 1993). Moreover, focal adhesion assembly plays a key role in the generation of strong traction forces. Following initial integrin binding, recruitment of focal adhesion components, such as vinculin and talin, result in graded increases in traction forces (Balaban et al., 2001; Galbraith et al., 2002; Tan et al., 2003). Focal adhesion assembly also contributes to cell adhesion strengthening by distributing bond forces along the cell-substrate interface (Gallant et al., 2005; Lotz et al., 1989). Nevertheless, the specific contributions of focal adhesion size and distribution to adhesion strength, independently from integrin-ligand bond strength and cytoskeletal architecture, remain poorly understood.
Contractile forces generated inside the cell regulate migration, neurite extension, cytokinesis, muscle cell contraction, cell cycle progression, angiogenesis and differentiation (Griffin et al., 2004; Mammoto et al., 2009; Mammoto et al., 2004; McBeath et al., 2004; Parizi et al., 2000; Polte et al., 2004; Tanaka and Sabry, 1995; Wozniak et al., 2003). Contractility results from dynamic interactions between actin filaments and myosin, which are regulated via phosphorylation of myosin light chain (MLC) (Kaibuchi et al., 1999; Worthylake and Burridge, 2003). Rho GTPases control the formation of stress fibers and focal adhesion assembly by modulating MLC phosphorylation and generating actin-myosin contractility (Amano et al., 1997; Chrzanowska-Wodnicka and Burridge, 1996; Totsukawa et al., 2000). When activated by serum factors, such as lysophosphatidic acid (LPA), Rho acts through its effector Rho-kinase (also termed ROCK), to enhance the contraction of smooth muscle cells as well as nonmuscle cells by either inactivation of myosin phosphatase (Kimura et al., 1996) or direct phosphorylation of MLC (Totsukawa et al., 2000). Contractile forces can also be modulated by MLC kinase (MLCK), which promotes assembly of actin-myosin filaments and MLC phosphorylation (Gallagher et al., 1997).
The equilibrium of forces within a cell represents a balance of internal contractile forces and anchoring forces to the underlying substrate (Ingber, 2003; Zhu et al., 2000). This complex and dynamic balance is governed by the size and distribution of cell-substrate adhesive structures, cytoskeletal architecture, and spatiotemporally-regulated internal contractile forces. The contributions of actin-myosin contractility to adhesive interactions have been characterized primarily in spreading and migration assays. While these functional measurements have identified key roles for actin-myosin contractility in focal adhesion assembly, stress fiber formation, and migratory forces (Amano et al., 1997; Chrzanowska-Wodnicka and Burridge, 1996; Worthylake and Burridge, 2003), relatively little is known about how actin-myosin contractility and focal adhesion assembly regulate cell adhesive forces. This lack of quantitative understanding of adhesion strengthening limits the interpretation of functional studies of structural and signaling focal adhesion components. Cell spreading and migration assays do not provide direct measurements of adhesion strength and generally serve as implicit indicators of adhesion strength due to the complex spatiotemporal relationships between migration/spreading and adhesion strength. For instance, cell migration speed exhibits a biphasic dependence on adhesion strength (Palecek et al., 1997). In the present study, we used a robust quantitative cell adhesion assay in combination with micropatterned substrates to control adhesive area in order to examine the role of actin-myosin contractility in cell adhesion strengthening.
Materials and Methods
Reagents
Human plasma fibronectin and Dulbecco’s phosphate buffered saline (PBS) were obtained from Invitrogen (Carlsbad, CA). Bovine serum albumin, mouse anti-talin and anti-biotin antibodies, and H-7 were purchased from Sigma (St. Louis, MO). Mouse antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH), rabbit anti-paxillin, and rabbit anti-α5 antibodies were obtained from Chemicon (Temecula, CA). Biotin-conjugated donkey anti-rabbit and donkey anti-mouse antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). Rabbit anti-FAK and mouse anti-vinculin antibodies were purchased from Upstate Biotechnology (Lake Placid, NY). Rabbit anti-myosin light chain (MLC) 2 (Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit anti-phospho-MLC (Abcam, Cambridge, MA) antibodies were also used. Hoechst 33258, AlexaFluor 488-conjugated antibody against mouse IgG, ethidium homodimer, and rhodamine phalloidin were purchased from Molecular Probes (Eugene, OR). Poly(dimethylsiloxane) (PDMS) elastomers and curing agents were obtained from Dow Corning (Midland, MI). Inhibitors (Y-27632, HA-1077, blebbistatin, and cytochalasin D (CD)) were purchased from Calbiochem (La Jolla, CA). 3,3′-Dithiobis(sulfosuccinimidylpropionate) (DTSSP) was purchased from Pierce Chemical (Rockford, IL). Tri(ethylene glycol)-terminated alkanethiol (HO(CH2CH2O)3-(CH2)11SH) was purchased from ProChimia Surfaces (Sopot, Poland). All other reagents including hexadecanethiol (H3C(CH2)15SH) were purchased from Sigma.
Micropatterned Substrates
Micropatterned arrays of adhesive islands within a non-adhesive background were prepared by microcontact printing of self-assembled monolayers of alkanethiols on gold-coated glass coverslips (Gallant et al., 2002). A master template featuring circular holes (10μm diameter) with a 75 μm center-to-center spacing was prepared on a Si wafer. UV light was exposed to a photoresist-coated Si wafer through an optical mask with the desired pattern. The UB-exposed area was then etched away, leaving a template mold of recessed wells of the pattern. The template was coated with (tridecafluoro-1,1,2,2,-tetrahydrooctyl1)-1-trichlorosilane under vacuum for 30 minutes to facilitate removal of the template from a PDMS stamp following the curing process. PDMS precursors (Slygard 186:184, 10:1) and curing agents were vigorously mixed at 10:1 ratio, poured over the template in a 100 mm diameter glass Petri dish, placed under vacuum for 30 minutes to remove air bubbles, and cured overnight at 65°C. Following curing, the PDMS stamp was peeled from the master template and sonicated in 70% ethanol for 30 minutes. Glass coverslips were cleaned in O2 plasma using a plasma etcher (Plasmatic Systems, North Brunswick, NJ) for 4 min and coated with Ti (100 Å) followed by Au (150 Å) using an electron beam evaporator (Thermonics, San Leandro, CA). For stamping, the PDMS stamp was sonicated in 70% ethanol for 15 minutes, dried, and placed onto a glass slide for rigid backing. The Au-coated glass coverslip was cleaned under a stream of N2 and laid down on a glass slide. The patterned face of the PDMS stamp was brushed with 1.0 mM hexadecanethiol (in absolute ethanol) using a cotton swab, blown dry under a stream of N2 and overlaid onto the Au-coated glass coverslip for 20 seconds. Conformal contact of the stamp with the Au substrate generated CH3-terminated circular patterns on the glass coverslip that readily allow protein adsorption and cell adhesion. The coverslip featuring hexadecanethiol islands was subsequently incubated in 1.0 mM ethanolic solution of (HO(CH2CH2O)3-(CH2)11SH) for 4 hours to create non-fouling domains around the cell-adhesive islands. The micropatterned substrate was then rinsed twice with 70% ethanol and PBS, coated with fibronectin (20 μg/mL) for 30 minutes, then blocked with heat-inactivated serum albumin (1% w/v) for 30 minutes, and incubated in PBS overnight to elute non-specifically adsorbed fibronectin from non-adhesive regions (Capadona et al., 2003).
Cell Culture and Inhibitor Treatment
Murine NIH3T3 fibroblasts (CRL-1658) were purchased from the American Type Culture Collection (Manassas, VA) and cultured in DMEM containing 10% newborn calf serum, penicillin (100 unit/mL), and streptomycin (100 μg/mL). Vinculin-null and vinculin +/+ mouse embryo fibroblasts were a kind gift from Eileen Adamson and have been previously described (Xu et al., 1998). Vinculin−/− and vinculin+/+ mouse embryo fibroblasts were cultured in DMEM supplemented with 10% FBS, sodium pyruvate (1 mM), penicillin (100 units/mL), streptomycin (100 μg/mL), and non-essential amino acids (1 mM). Tet-FAK fibroblasts were engineered to express FAK under a tetracycline-regulated promoter have been previously described (Owen et al., 1999). In the presence of tetracycline, FAK expression is repressed, whereas in the absence of tetracycline, high FAK levels are induced. Tet-FAK cells were cultured in DMEM supplemented with 10% FBS, sodium pyruvate (1 mM), penicillin (100 unit/mL), streptomycin (100 μg/mL), amphotericin B (0.25 μg/mL), and non-essential amino acids (1mM) in the absence or presence of tetracycline (1.0 μg/mL) for two days prior to cell seeding. Cells were enzymatically lifted from the culture dish using trypsin/EDTA and seeded onto micropatterned substrates at 225 cells/mm2. Cell cultures were maintained for 16 hours in serum-containing media prior to analysis of steady state adhesion (Gallant et al., 2002). For pharmacological treatments, cultures were incubated for 30 minutes prior to analysis in Y-27632 (50 μM), H-7 (500 μM), HA-1077 (50 μM), blebbistatin (250 μM), and CD (1 μM). For serum-free studies, cells were cultured in DMEM supplemented with 1% serum albumin and 0.1% ITS.
Adhesion Assay
Mean cell adhesion strength was measured using a spinning disk as previously described (Gallant et al., 2002; Garcia et al., 1998). Briefly, a cell-seeded micropatterned substrate was mounted on the spinning platform and spun in 2 mM dextrose in PBS to apply well-defined hydrodynamic forces to adherent cells. The applied shear stress τ (force/area) increases linearly from the center of the sample to the periphery and is given by the following equation:
| (1) |
where r is the radial position along the sample, ρ and μ are the density and viscosity of the solution, respectively, and ω is the rotational speed. Following spinning, the remaining adherent cells were fixed in 3.7% formaldehyde, permeablized with 0.1% Triton X-100, and stained with ethidium homodimer. The number of adherent cells was counted using a fluorescence microscope equipped with a motorized stage and ImagePro image analysis system (Media Cybernetics, Silver Spring, MD). Sixty-one fields (0.5 mm2/field) were analyzed per substrate, and the fraction of adherent cells (f) was calculated by normalizing the number of cells in each field to the number of cells at the center of the array, where negligible forces are applied. The detachment profile (f vs. τ) was then fit to a sigmoidal curve (f =1.0/(1.0 + exp[b(τ−τ50)]) and the shear stress for 50% detachment (τ50) was used as the mean cell adhesion strength.
Protein Expression and Phosphorylation Levels
Cultures were rinsed in PBS and lysed for 20 min at room temperature in RIPA buffer (150 mM NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, 150 mM Tris, pH 7.2) containing Na3VO4 (0.04% w/v) and protease inhibitors (10 μg/mL leupetin, 10 μg/mL aprotinin, and 350 μg/mL PMSF). The protein content of total cell lysates was determined by microBCA assay (Pierce, Rockford, IL). Identical amounts of cell lysates were mixed in sample buffer (50 mM Tris-HCl pH 6.8, 100 mM DTT, 2% SDS, 10% glycerol, and 0.1% bromophenol blue) and separated by SDS-PAGE (8% or 16% gels). After transferring to nitrocellulose membranes, proteins were visualized by incubating in primary and secondary antibodies and ECF substrate (Pierce, Rockford, IL). Relative amounts of proteins were quantified by image analysis.
Focal Adhesion Assembly
For immunostaining of focal adhesion proteins, adherent cells were rinsed with PBS, fixed in ice-cold formaldehyde (3.7% in PBS) for 3 min, permeablized for 15 minutes in cold 0.5% Triton X-100 containing protease inhibitors (20 μg/mL aprotinin, 1 μg/mL leupeptin, and 350 μg/mL PMSF). After incubating in blocking buffer (5% FBS, 0.1% Tween 20, 0.02% NaN3 in PBS) for 1 h at 37°C, samples were incubated in primary antibodies for 1 h at 37°C, followed by AlexaFluor488-conjugated secondary antibody, rhodamine phalloidin, and Hoechst 33258 for 1 h at 37°C. For quantification of proteins localized to focal adhesions, micropatterned cells were analyzed by a modified wet cleaving method (Keselowsky and García, 2005). Briefly, cultures were rinsed with PBS (Ca2+/Mg2+ free) containing protease inhibitors. A dry nitrocellulose sheet (PROTRAN BA85, Schleicher & Schuell) was then overlaid onto the cells for 1 min and rapidly removed to isolate cell bodies from basal cell membranes containing focal adhesions. Remaining adhesive structures on surfaces were scraped into sample buffer (100 μL). Western blotting was used for quantitative analysis of recovered focal adhesion proteins.
Integrin Binding
Integrin binding was quantified via a cross-linking/extraction/reversal procedure (García et al., 1999; Keselowsky and García, 2005). After rinsing cultures three times with PBS, DTSSP (1.0 mM in cold PBS + 2 mM dextrose) was incubated for 30 minutes to cross-link integrins to their bound ligands. The cross-linking reaction was quenched by addition of Tris (50 mM in PBS) for 15 minutes. Uncross-linked cellular components were then extracted in 0.1% SDS containing 10 μg/mL leupeptin, 10 μg/mL aprotinin and 350 μg/mL PMSF. Cross-linked integrins to their bound ligands were visualized by immunostaining with α5 integrin-specific antibodies. Alternatively, bound integrins were quantified by Western blotting following cleaving of the cross-linker in 50 mM DTT and 0.1% SDS for 30 minutes at 37°C.
Statistics
Data are reported as mean ± standard deviation. ANOVA was used with a 95% confidence interval (SYSTAT 8.0, SPSS, Chicago, IL). If ANOVA detected significant differences, Tukey HSD multiple comparison tests were performed to establish treatment effects.
Results
Actin-myosin contractility modulates cell adhesion strength
Cells were plated on fibronectin-coated micropatterned substrates with dimensions smaller than a cell diameter in order to control adhesive area and cell shape. This approach allows isolation of focal adhesion assembly from changes in cell spreading/shape and provides for direct comparisons among experimental groups. We previously reported that NIH3T3 fibroblasts remained viable for several days when adhering to micropatterned circular islands with dimensions ranging from 2 to 20 μm diameter (Gallant et al., 2005). Cells maintained a round morphology (Fig. 1A), and their contact area and focal adhesions were constrained to the micropatterned domain. The 75 μm center-to-center spacing of islands restricted a single cell to occupy one adhesive island, preventing interactions with neighboring cells.
Fig. 1.

A, Phase contrast image of adherent NIH 3T3 cells cultured for 16 hours on micropatterned substrate (scale bar: 10 μm). B, Characteristic detachment profiles showing the fraction of adherent cells (f) vs. applied shear stress (τ). The shear stress for 50% detachment (indicated by arrows in the profile), τ50, represents the mean adhesion strength. Adhesion strength is 632 dyne/cm2 for untreated control cells, and is shifted left-ward to 451 dyne/cm2 for cell treated with Y-27632 (50μM).
We measured adhesion strength using a spinning disk device that applies a range of well-defined hydrodynamic shear forces to adherent cells (Garcia et al., 1998). For a particular sample, the fraction of adherent cells (f) is plotted as a function of the applied shear stress (τ). From this detachment profile, the shear stress for 50% detachment (τ50), which represents the mean cell adhesion strength, is determined. Fig. 1B depicts typical detachment profiles showing the fraction of adherent cells vs. applied shear stress. We chose a 16 hour culture time point for analysis of long-term adhesion strength because it was previously demonstrated that the adhesion strength of NIH3T3 fibroblasts on micropatterned substrates reached constant values by 4 h and remained constant for up to 16 hours (Gallant et al., 2005). Equivalent levels of adhesion strength were observed in serum- and LPA-treated cultures (~650 dyne/cm2) (Fig. 2A). These values are 30–40% higher than those for cells cultured under serum-free conditions (~450 dyne/cm2). Similar differences in adhesion strength between serum or LPA-treated and serum-free cultures were observed for unpatterned, spread cells (Fig. 2A), validating the use of micropatterned cells to analyze strengthening responses.
Fig. 2.

A, Adhesion strength for patterned and spread (unpatterned) NIH 3T3 fibroblasts cultured under serum-containing or serum-free conditions, and treated with LPA and serum supplemented with Y-27632 (50μM) for 30 minutes. Adherent cells were spun and analyzed to determine adhesion strength. *p<0.04 relative to serum containing controls (mean ± standard deviation, n = 3–5). B, Adhesion strength for micropatterned NIH 3T3 fibroblasts cultured under serum-containing or serum-free conditions, and treated with pharmacological agents for 30 minutes: Y-27632 (50μM), H-7 (500 μM), HA-1077 (50μM), blebbistatin (250μM), or CD (1μM). Adherent cells were spun and analyzed to determine cell adhesion strength. *p<0.05 and #p<0.01 relative to no treatment controls (mean ± standard deviation, n = 3–5). Dashed line represents adhesion strength for cells cultured under serum-free conditions. C, Western blotting (left) for MLC and phosphorylated-MLC in cells cultured in serum-containing media on micropatterned substrates for 16 hours and treated with Y-27632 (50μM), H-7 (500μM), and HA-1077 (50μM) for 30 minutes and quantification (right) of protein levels (n=3). D, Immunostaining for vinculin on adherent cells on micropatterned islands (10 μm diameter) of fibronectin located on the periphery of the disk following application of detachment forces. Cells on edge of disk are exposed to maximal detachment force. Cells treated with vehicle, Y-27632 (50 μM), or CD (1μM).
Because actin-myosin contractility is driven by phosphorylation of MLC, which is regulated by MLCK and Rho-kinase (Gallagher et al., 1997), we used pharmacological inhibitors that impair phosphorylation of MLC to examine the contributions of contractility to adhesion strength. Y-27632 is a specific inhibitor of Rho-kinase (Narumiya et al., 2000). H-7 inhibits phosphorylation of MLCK, which in turn potently blocks phosphorylation of MLC (Volberg et al., 1994). HA-1077 has a broad negative influence on both Rho-kinase and MLCK activity (Yanase et al., 2003). Inhibitor dosage and exposure time were selected from published reports and preliminary experiments showing no gross adverse effects. To examine the contributions of actin-myosin contractility inhibitors to cell adhesion strengthening, we measured adhesion strength of cells exposed to contractility inhibitors for 30 minutes prior to spinning. In general, inhibitors of Rho-kinase and MLCK significantly reduced adhesion strength for cells adhering in the presence of serum as demonstrated by a left-ward shift in the detachment profile (Fig. 1B). Adhesion strength values for each inhibitor (409 ± 845 dyne/cm2 for Y-27632, 468 ± 80 dyne/cm2 for H-7, and 473 ± 84 dyne/cm2 for HA-1077) were 30–35% lower than those for untreated controls in the presence of serum (640 ± 55 dyne/cm2) (Fig. 2B). Although Y-27632 and HA-1077 are known inhibitors of Rho-kinase and actin-myosin contractility, Rho-kinase has other targets and, therefore, may alter cell-matrix adhesion by a mechanism not directly related to contractility (Amano et al., 2000). As an alternative to inhibiting MLC phosphorylation, additional experiments were performed in the presence of blebbistatin, a specific inhibitor of myosin II activity that acts via a distinct mechanism (Kovács et al., 2004). Blebbistatin also reduced serum-induced cell adhesion strength to similar levels (480 ± 110 dyne/cm2) as the agents that reduce MLC phosphorylation via inhibition of Rho-kinase and MLCK (Fig. 2B). Additional experiments under serum-free conditions revealed that these contractility inhibitors do not reduce adhesion strength below levels for untreated, serum-free samples (Fig. 2B). Taken together, these results show that actin-myosin contractility accounts for the increases in adhesion strength resulting from serum stimulation. Finally, treatment with cytochalasin D (CD) was included for comparison as CD is expected to modulate adhesion strength via a different mechanism, namely disruption of actin filament polymerization. Cells treated with CD exhibited significantly lower adhesion strength (270 ± 36 dyne/cm2) than cells treated with any of the contractility inhibitors or cells cultured under serum-free conditions.
Following treatment with these inhibitors, phosphorylation of MLC was analyzed by Western blotting. For equivalent total MLC levels, relative amounts of phosphorylated MLC in cells treated with inhibitors were significantly decreased compared to untreated controls (Fig. 2C). There were no differences in MLC phosphorylation levels among inhibitor treatments as determined by quantification of the Western blots in Figure 2C. These results confirm essential roles of MLCK and Rho-kinase in MLC phosphorylation. In addition, since all inhibitor treatments reduced MLC phosphorylation to the same level despite distinct mechanisms of action in blocking actin-myosin contractility, this result suggests that cell adhesion strength is regulated by overall levels of phosphorylated MLC rather than specific effects of Rho-kinase or MLCK.
To elucidate the mechanism by which cell detachment occurred on 10μm islands of fibronectin under the applied forces, cells were spun and stained for vinculin, F-actin, and DNA. For control untreated samples, cells at the center of the disk, which experience low detachment forces, stained positive for vinculin and F-actin (not shown) in a manner similar to unstressed cells. However, at the periphery of the substrate where detachment forces are highest, minimal traces of vinculin (Fig. 2D) and F-actin were observed. This negative staining indicates that cell detachment took place at the junction between focal adhesions and the underlying substrate, resulting in removal of the entire cell without gross failure. Similar staining patterns were observed for cells treated with contractility inhibitors (Fig. 2D). In contrast, cultures treated with CD displayed significant cytoskeletal debris and vinculin at the periphery of the sample following detachment (Fig. 2D). This residual cytoskeletal debris indicates gross cell failure at points above focal adhesions due to loss of cellular integrity arising form impaired actin polymerization. Taken together, these results indicate that inhibition of MLC phosphorylation and contractility does not reduce adhesion strength by compromising cellular integrity and that cell detachment under these conditions occurs at the focal adhesion-substrate interface.
Reduction in cell adhesion strength correlates with dissolution of focal adhesions independently of changes in integrin binding
The reduction in adhesion strength as a result of inhibiting MLC phosphorylation is consistent with the role of MLC-mediated contractility in focal adhesion assembly and stress fiber formation. Two mechanisms have been proposed for the regulation of focal adhesion assembly by contractility (Balaban et al., 2001). Tension generated by actin-myosin contractility on adhesion sites may regulate recruitment and assembly of focal adhesion components. Alternatively, contractile forces may trigger changes in integrin binding affinity or bond density within the focal complex. To explore these two possibilities, we first determined whether actin-myosin contractility modulates integrin binding to ECM. We have previously shown that NIH3T3 adhesion to these micropatterned FN islands is mediated by integrin α5β1 (Gallant et al., 2005). Immunofluorescence staining confirmed that bound α5β1 integrins were present throughout the contact area with a preferential localization to the periphery of the adhesive area (Fig. 3A). Equivalent staining patterns were observed between control and cells treated with contractility inhibitors, suggesting no differences in integrin intensity or distribution. To confirm these observations, bound integrins were quantified using a biochemical cross-linking/extraction method that isolates integrins bound to fibronectin (García et al., 1999; Keselowsky and García, 2005). Following recovery of bound integrins by cleaving the cross-linker and analysis by Western blotting, no differences in bound α5β1 integrins were detected among control and experimental groups (Fig. 3B). These results indicate that integrin binding activity is not significantly altered by changes in actin-myosin contractility.
Fig. 3.
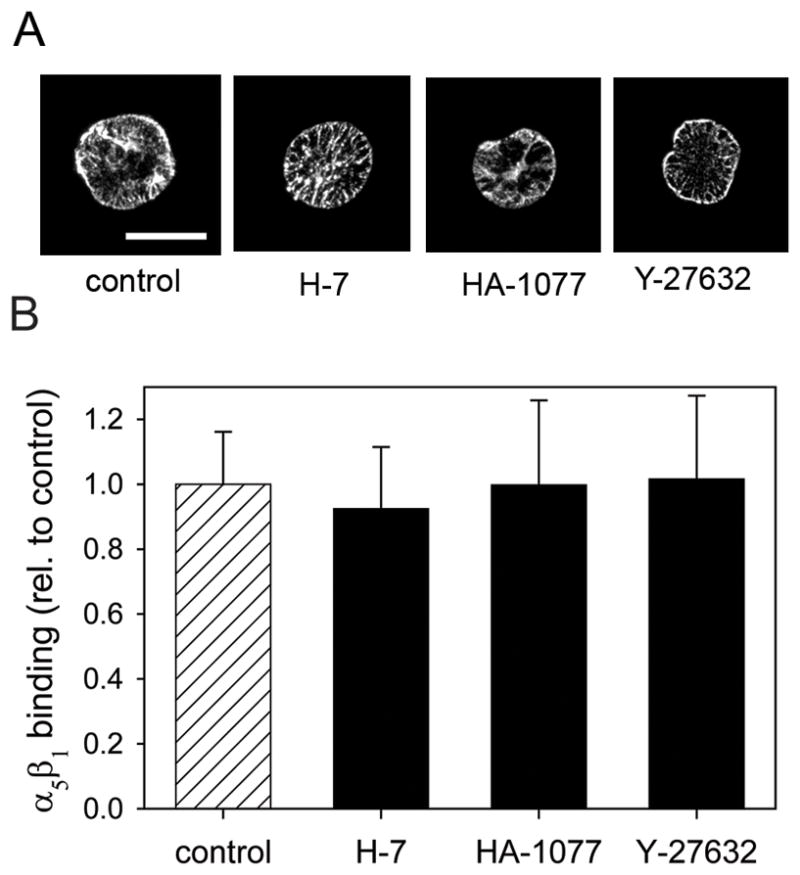
A, Inhibitors of MLC phosphorylation do not alter integrin binding to micropatterned substrates. Cells were cultured on micropatterned substrates for 16 hours and treated with inhibitors: Y-27632 (50μM), H-7 (500 μM), HA-1077 (50μM), blebbistatin (250μM) for 30 minutes. Bound integrins were cross-linked with DTSSP. After extracting cells, the bound integrins were recovered and analyzed by Western blotting. A, Immunofluorescence staining for α5 integrin subunit following the cross-linking/extraction showing localization of integrins mostly at the periphery of the substrate. Counter-staining with Hoechst 33258 and rhodamine-phalloidin confirmed complete extraction of unbound cellular components (not shown). B, Relative amounts of bound α5 were quantified by Western blotting and image analysis.
To investigate the recruitment and localization of focal adhesion components to adhesive plaques in response to changes in actin-myosin contractility, two independent but complementary approaches were used. Immunostaining for vinculin and talin showed that untreated cells on micropatterned substrates formed discrete focal adhesion clusters throughout the adhesive island (Fig. 4A). Vinculin was spatially segregated to discrete clusters throughout the circular adhesive domain of micropatterned islands, which was consistent with the localization of F-actin (data not shown). These adhesive structures were similar to those previously observed for the same cells cultured on micropatterned substrates (Gallant et al., 2005). In general, treatment of cells with inhibitors of MLC phosphorylation resulted in disassembly of focal adhesions, particularly in the center of the micropatterned adhesive area (Fig. 4A). Treatment with Y-27632 and HA-1077 resulted in minimal vinculin clustering in the center of the micropatterned adhesive area (Fig. 4A). Treatment with H-7 inhibited formation of vinculin-containing focal adhesions at both the center and the periphery of the adhesive area. Similar changes in the localization of the structural protein talin were also observed (Fig. 4A). In control, untreated cells, talin localized to punctuate structures constrained to the circular adhesive island with a preferential distribution toward the periphery of the adhesive domain. Inhibitors of MLC phosphorylation disrupted talin clustering to various degrees with the most significant effects elicited by Y-27632. Consistent with the dissolution of focal adhesions containing vinculin and talin, inhibitors of Rho-kinase and MLCK also blocked stress fiber formation. The dissolution of focal adhesions by inhibitors of MLC phosphorylation was also confirmed by internal reflection microscopy (IRM) in cells adhering to FN-coated glass substrates. In IRM, areas of ‘close’ (> 15 nm) cell-substrate contact appear as dark patches (Izzard and Lochner, 1976; Izzard and Lochner, 1980). Untreated NIH3T3 cells spread and formed close contacts with the substrate, as demonstrated by the dark line structures (Fig. 4B). In contrast, cells treated with Y-27632 for 30 minutes lacked areas of close contacts between the cell membrane and the glass substrate (Fig. 4B). These results indicate that, upon addition of inhibitors of MLC phosphorylation, focal adhesions disassemble. To further characterize the changes in focal adhesions, we used a wet-cleaving biochemical technique in which basal cell membranes containing focal adhesive structures are mechanically isolated and analyzed by Western blotting (Gallant et al., 2005; Keselowsky and García, 2005). Inhibitors of MLC phosphorylation significantly reduced the localization of vinculin and talin to focal adhesions compared to control cells (Fig. 4C and D). There were no differences in protein levels for either vinculin or talin in whole cell lysates, demonstrating that the decreases in vinculin and talin arise from dissolution of focal adhesions. These results are in excellent agreement with our immunostaining observations. These findings indicate that inhibitors of MLC phosphorylation reduce adhesion strength via dissolution of focal adhesions independently of changes in integrin binding.
Fig. 4.

A, Inhibitors of MLC phosphorylation modulate recruitment of vinculin and talin to adhesive structures on micropatterned substrates. Fibroblasts were cultured on micropatterned substrates for 16 hours and treated with Y-27632 (50μM), H-7 (500μM), and HA-1077 (50μM) for 30 minutes. Controls received fresh media without inhibitors. The cells were then fixed, extracted, and stained for vinculin, talin, or actin. B, Inhibition of MLC phosphorylation reduces focal adhesion formation. Fibroblasts were cultured overnight on glass substrates coated with 10 μg/ml FN and treated with indicated condition 30 minutes before IRM imaging. C, Inhibitors of contractility modulate recruitment of vinculin and talin to focal adhesions. Western blotting for vinculin and talin recruited to focal adhesions as analyzed by wet-cleaving method. D, relative amounts of localized vinculin and talin were analyzed by Western blotting (mean ± standard deviation, n = 3 from two independent experiments). *p<0.05 relative to no treatment controls which received fresh media without inhibitors.
We next examined the role of vinculin in contractility-induced increases in adhesion strength. Vinculin binds between the actin cytoskeleton and integrins and has been implicated in modulating focal adhesion turnover and transmitting mechanical forces (DeMali, 2004; Humphries et al., 2007; Mierke et al., 2007). Using vinculin-null (vinc−/−) and vinculin-expressing (vinc+/+) mouse embryonic fibroblast cells (MEFs), we investigated the role of vinculin in contractility-mediated adhesive force generation. First, we confirmed that the spinning disk detachment profiles for these cells were consistent with those of the NIH3T3 cell lines (Fig. 5A). Next, we analyzed the effect of the MLC phosphorylation inhibitor Y-27632 on adhesion strength. Consistent with our data for NIH3T3 cells, vinculin-expressing MEFs showed a 20% reduction in adhesion strength upon exposure to Y-27632 for 30 minutes compared to untreated controls (Fig. 5B). In contrast, vinculin-null MEFs showed no decrease in adhesion strength upon exposure to Y-27632 for 30 minutes (Fig. 5B). In addition, there were no differences in adhesive force between vinculin-expressing cells treated with Y-27632 and vinculin-null cells. Immunofluorescence staining showed that bound α5β1 integrins were present throughout the contact area with a preferential localization to the periphery of the adhesive area (Fig. 5C). Importantly, no differences in the intensity or localization of bound integrins could be detected between the treatment groups indicating that adhesion strength differences did not arise from differences in integrin binding. These findings indicate that vinculin expression is required for contractility-induced increases in adhesion strength.
Fig. 5.
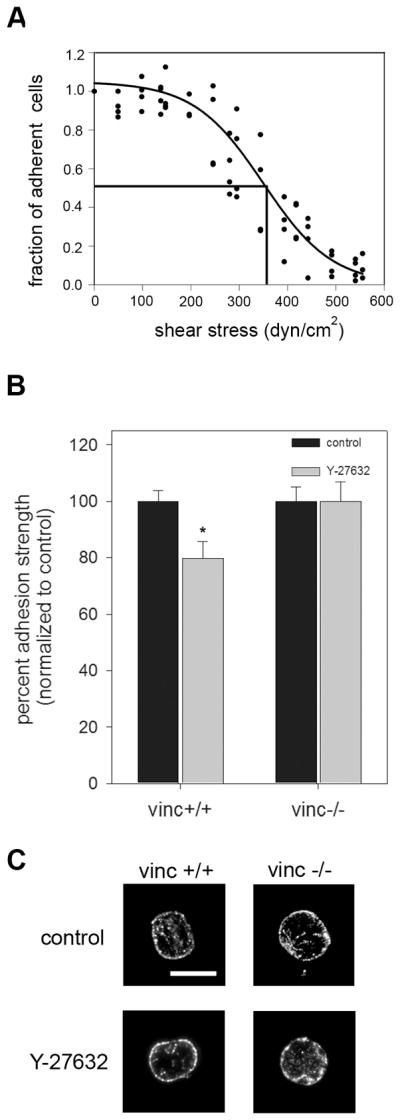
Loss of vinculin abolishes differences in adhesion strength due to contractility independent of bound integrin levels. A, Characteristic detachment profile for vinculin+/+ and vinculin−/− MEFs showing the fraction of adherent cells (f) vs. applied shear stress (τ). These cells exhibit the same detachment profiles as the NIH3T3 cells. B, vinculin +/+ and vinculin −/− MEFs were cultured for 16 hours on micropatterned surfaces and treated with Y-27632 (50μM) for 30 minutes prior to spinning. The no-treatment group received fresh media without inhibitor. Addition of inhibitor resulted in a 20% decrease in adhesion strength for the vinculin +/+ cells. No differences in adhesion strength were detected for the vinculin −/− cells. *p<0.03 relative to no treatment vinculin +/+ MEFs. C, Immunoflourescence staining for α5 integrin subunit following cross-linking/extraction showing localization of integrins mostly at the periphery of the substrate. Treatment with Y-27632 (50μM) for 30 minutes did not significantly alter integrin binding.
Effects of MLC phosphorylation on cell adhesion strength are FAK-dependent
FAK is a central regulator of focal adhesions and adhesive interactions (Hanks et al., 1992; Ilić et al., 1995). Furthermore, it is well established that inhibition of contractility reduces tyrosine phosphorylation of focal adhesion components, including FAK (Chrzanowska-Wodnicka and Burridge, 1996). In addition, we recently demonstrated that FAK regulates the early stages of the adhesion strengthening process via changes in integrin activation (Michael et al., 2009). To examine the effects of contractility on FAK phosphorylation in micropatterned cells, protein and phosphorylation levels in the presence of inhibitors of MLC phosphorylation were analyzed by Western blotting. Consistent with the effects on MLC phosphorylation and adhesion strength, all contractility inhibitors tested reduced phosphorylation of FAK at the autophosphorylation site Y397 compared to untreated controls with no differences in total FAK levels (Fig. 6). These results are in excellent agreement with previous observations for spread cells (Chrzanowska-Wodnicka and Burridge, 1996).
Fig. 6.
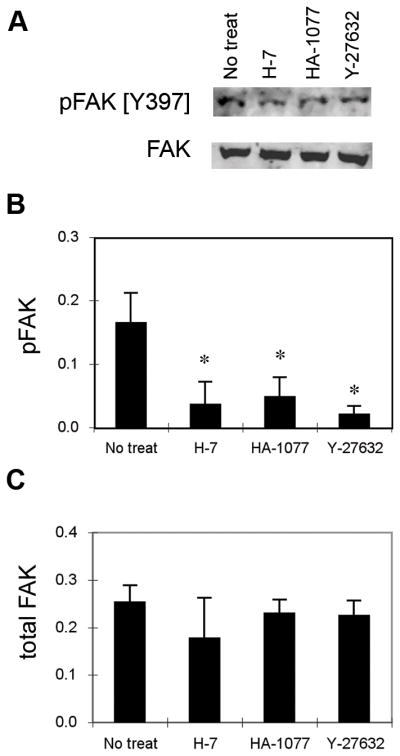
FAK phosphorylation is regulated by inhibitors of MLC phosphorylation. Cells were cultured on micropatterned substrates for 16 hours and treated with inhibitors for 30 minutes. A, Western blotting for pFAK[Y397] and total FAK. Relative amounts of (B) pFAK[Y397] and (C) total FAK. The results represent two independent experiments with mean ± standard deviation (n=3). *p<0.05 relative to no treatment controls that received fresh media without inhibitors.
The role of FAK in contractility-mediated adhesion strengthening was examined using Tet-FAK cells. Tet-FAK cells are stable clones derived from FAK−/− mouse embryo fibroblasts engineered to express FAK under a tetracycline-responsive promoter (Owen et al., 1999). In the presence of tetracycline, FAK expression is repressed and these cells have no FAK (FAK−); removal of tetracycline from the culture media results in expression of FAK (FAK+) to similar levels as wild-type fibroblasts (Fig. 7A). This inducible system allows examination of the effects of FAK in the same cell population, without non-specific effects from dominant-negative constructs or clonal variation. Tet-FAK cells, induced and non-induced to express FAK, adhered to fibronectin-coated micropatterned substrates in a similar fashion as NIH3T3 fibroblasts. Consistent with our observations with NIH3T3 fibroblasts, the Rho-kinase inhibitor Y-27632 reduced MLC phosphorylation for Tet-FAK cells in the presence and absence of FAK without changing levels of total MLC (Fig. 7B). Higher Y-27632 concentrations were required to reduce MLC phosphorylation in cells without FAK, consistent with higher expression levels of Rho-kinase in these cells (Chen et al., 2002). Adhesion strength in the presence and absence of inhibitors of contractility was determined using the spinning disk assay. In FAK+ cells, treatment with Y-27632 reduced adhesion strength by 30% compared to untreated cells (Fig. 7C). Furthermore, treatment with Y-27632 also reduced localization of vinculin to focal adhesions (data not shown). The decrease in adhesion strength and reduced localization of vinculin to focal adhesions are in excellent agreement with the 35% decrease in adhesion strength observed in NIH3T3 fibroblasts (Fig. 2A) and reductions in vinculin localization to focal adhesions (Fig. 4D). Remarkably, in FAK− cells, inhibition of MLC phosphorylation by Y-27632 did not reduce cell adhesion strength (Fig. 7C) or alter focal adhesion assembly (data not shown). We note that the FAK− and FAK+ control conditions are normalized values, not meant for direct comparison. These results indicate that the contractility-mediated cell adhesive forces require FAK expression.
Fig. 7.
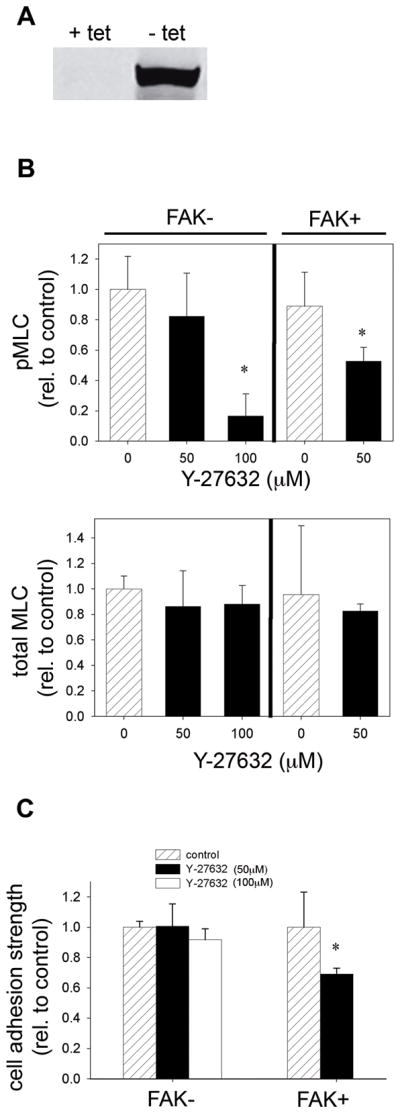
Rho-kinase modulates cell adhesion strength via FAK. A, Tet-FAK cells with inducible FAK expression. In the absence of tetracycline (tet), FAK expression is induced at high levels, while expression is repressed in the presence of tet. Tet-FAK cells expressing FAK (FAK+) and cells without FAK (FAK−) were cultured on micropatterned surfaces for 16 hours and treated with Y-27632 (50 or 100 μM) for 30 minutes. B, Inhibitor of Rho-kinase reduced adhesion strength in the presence of FAK, but adhesion strength was not altered in the absence of FAK. *p < 0.05 relative to untreated controls of corresponding FAK condition.
Discussion
We tested the hypothesis that actin-myosin contractility regulates cell adhesion strengthening via modulation of focal adhesion assembly. While previous reports have shown that Rho-mediated actin-myosin contractility drives focal adhesion assembly and regulates cells spreading and migration (Arthur and Burridge, 2001; Chrzanowska-Wodnicka and Burridge, 1996), the specific contributions of contractility to cell adhesion strengthening are not well understood. Recent evidence supports a complex relationship between the state of contractility in a cell and its adhesion to the underlying matrix (Griffin et al., 2004). Moreover, it is not clear how localized, directional traction forces involved in migration are integrated to regulate overall levels of adhesion strength. Although the size of focal adhesion structures corresponds to the level of traction force generated (Balaban et al., 2001; Galbraith et al., 2002; Tan et al., 2003), focal adhesion size does not solely control adhesion strength, as integrin-ligand bond strength, actin cytoskeleton architecture, and the spatiotemporal distribution of focal adhesions also regulate adhesion strength (Gallant et al., 2005). In order to control cell adhesive area and position as well as cell shape, we examined adhesion strength for cells cultured on micropatterned substrates. This approach allows for direct comparisons of adhesive force among experimental groups by eliminating differences in adhesive area/distribution and cell morphology. Adhesions strength was quantified using a hydrodynamic detachment assay that applies a wide range of forces and provides sensitive measurements of adhesion strength (García et al., 1999). Equivalent levels of adhesion strength were observed between serum- and LPA-treated cultures (data not shown). These values are 30% higher than those for cells cultured under serum-free conditions. Since LPA induces Rho-dependent contractility (Chrzanowska-Wodnicka and Burridge, 1996), this result suggests that activation of contractility enhances adhesion strength. To impair actin-myosin contractility, cells were treated with pharmacological agents that inhibit either Rho-kinase (Y-27632) or MLCK (H-7 and HA-1077). Due to their high specificity and our ability to precisely control timing and concentration of dosage, we use chemical inhibitors of contractility throughout this study. We note that several previous studies have relied exclusively on the use of these same inhibitors (Chen et al., 2002; Chrzanowska-Wodnicka and Burridge, 1996; Delanoe-Ayari et al., 2004; Totsukawa et al., 2000). Treatment with any of these inhibitors eliminated the serum-induced enhancements in adhesion strength by >95% to levels indistinguishable from serum-free cultures. Furthermore, these inhibitors did not reduce adhesion strength for cells cultured in the absence of serum, indicating that the effects of serum stimulation on adhesion strength were regulated by actin-myosin contractility. Moreover, blocking myosin II activity with blebbistatin, which acts through a different mechanism, had equivalent effects on adhesion strength. These results demonstrate that actin-myosin contractility is responsible for serum-induced enhancements in adhesion strength. Despite distinct mechanisms of action in blocking actin-myosin contractility (Rho-kinase vs. MLCK vs. myosin II activity), all inhibitors tested reduced adhesion strength to similar values. Since all inhibitor treatments reduced MLC phosphorylation to the same levels, this result suggests that cell adhesion strengthening arising from serum or LPA stimulation is regulated by overall levels of phosphorylated MLC. Consistent with our observations, Chrzanowska-Wodnicka and Burridge showed that inhibition of MLC phosphorylation via different mechanisms leads to inhibition of Rho-induced focal adhesion assembly and stress fiber formation (Chrzanowska-Wodnicka and Burridge, 1996).
Treatment with these inhibitors of contractility also resulted in the dissolution of focal adhesions as indicated by reduced localization of vinculin and talin to adhesion structures, in agreement with previous reports (Chrzanowska-Wodnicka and Burridge, 1996). Both inhibitors of Rho-kinase and MLCK reduced recruitment of talin and vinculin to adhesive plaques to similar levels, consistent with their effects on adhesion strength. Although distinct roles and spatial activities for Rho-kinase and MLCK in spread fibroblasts have been reported (Totsukawa et al., 2000), we observed no significant differences between these two regulators of MLC phosphorylation, possibly due to the constrained micropatterned adhesive domains. Because no differences in integrin binding in response to treatment with contractility inhibitors were observed, we attribute the contractility-mediated differences in adhesive force to changes in focal adhesion assembly. Indeed, experiments with vinculin-null cells demonstrated that vinculin was required for the contractility-dependent differences in adhesive force. Recruitment of vinculin and other structural components to adhesion plaques enhances adhesion strength by increasing the local membrane stiffness thereby modulating bond stressing in the contact area (Gallant and Garcia, 2007; Gallant et al., 2005; Goldmann and Ingber, 2002; Wang et al., 1993). Interestingly, inhibition of MLC-driven contractility did not alter integrin binding, in terms of bound density and distribution, to the ECM. In contrast to our observations, Friedland et al. recently reported that α5β1 integrins do not cross-link to FN when cells were serum-starved overnight and exposed to inhibitors of contractility (Friedland et al., 2009). The differences between this study and the present findings may be attributed to differences in available adhesive area (fully spread vs. micropatterned) and the duration of exposure to both serum-free conditions and inhibitors. Taken together, our results indicate that actin-myosin contractility modulates recruitment of cytoskeletal elements to adhesion plaques independently of integrin binding. A possible mechanism for this recruitment is that contraction-driven forces applied to the adhesion plaque concentrates or “focuses” cytoskeletal elements (e.g., vinculin, talin) into clusters (Palecek et al., 1997; Wolfenson et al., 2009). Alternatively, application of forces to integrins or integrin-associated elements may lead to conformational changes favorable to the recruitment of focal adhesion proteins, such as vinculin (Del Rio et al., 2009; Hytönen and Vogel, 2008). In contrast to our observations, Chrzanowska-Wodnicka and Burridge reported that contractility also modulates integrin distribution in spread fibroblasts (Chrzanowska-Wodnicka and Burridge, 1996). The discrepancy between this study and the present findings may be attributed to differences in available adhesive area (fully spread vs. micropatterned cells).
Inhibitors of contractility also down-regulated tyrosine phosphorylation of FAK, consistent with previous reports (Chrzanowska-Wodnicka and Burridge, 1996; Gallagher et al., 1997; Wozniak et al., 2003). Given the central role of FAK in focal adhesion turnover (Ilić et al., 1995; Webb et al., 2004), we examined the role of FAK in contractility-mediated adhesion strengthening using cells with inducible expression of FAK. In the presence of FAK, inhibitors of contractility reduced serum-induced adhesion strengthening as well as eliminated focal adhesion assembly, in excellent agreement with our observations for NIH3T3 fibroblasts. In the absence of FAK, however, these inhibitors did not alter adhesion strength or focal adhesion assembly. These results indicate that actin-myosin contractility modulates adhesion strengthening via FAK-dependent organization of focal adhesions. While it is well established that FAK plays central roles in modulating focal adhesion turnover and cell motility (Ilić et al., 1995; Ren et al., 2000; Webb et al., 2004), this is the first report demonstrating a direct role for FAK in serum-dependent increases in steady-state adhesive force. This finding contrasts with the view that modulation of FAK activity (phosphorylation) is a downstream event following contractility-driven focal adhesion assembly (Chrzanowska-Wodnicka and Burridge, 1996). Our results implicate FAK in regulating adhesion strengthening via focal adhesion assembly. This function is distinct from the role of FAK in modulating focal adhesion turnover (Webb et al., 2004) and promoting directional persistence in motility and reorientation in response to mechanical forces (Wang et al., 1993). Furthermore, the FAK-dependent effects of contractility on adhesion strengthening contrast the actions of FAK and contractility on cell spreading and migration. Chen et al. reported that inhibition of Rho-kinase by Y-27632 in FAK−/− fibroblasts induced spreading and enhanced cell motility with concomitant reorganization of focal adhesions (Chen et al., 2002). These authors concluded that Rho-kinase leads to formation of actin-myosin filaments at the periphery of FAK−/− cells, which generate contractile forces that reduce cell migration. These results are consistent with the observation that FAK suppresses Rho activity to promote focal adhesion turnover (Ren et al., 2000). The distinct effects of the interplay of contractility and FAK on migration and adhesion strengthening highlight the complex interactions in these two adhesive processes. Finally, we note that we recently established a role for FAK in the modulation of initial adhesive forces via changes in integrin activation (Michael et al., 2009). The role of FAK in the contractility-mediated changes in steady-state focal adhesion assembly and adhesion strengthening described in the present study appears to be distinct from these earlier adhesive processes.
In summary, we demonstrate that actin-myosin contractility controls serum-dependent cell adhesion strengthening. Phosphorylation of MLC, either via Rho-kinase or MLCK, was central to the strengthening process by modulating focal adhesion assembly and required vinculin to achieve maximum adhesion strength. Notably, the effects of MLC phosphorylation on adhesion strengthening were mediated by FAK, implicating this adhesion kinase in the generation of strong adhesive forces.
Acknowledgments
This work was supported by NIH (R01-GM065918) and an NSF Graduate Fellowship (KEM). The cells with FAK inducible expression were kindly provided by Steven K. Hanks (Vanderbilt University School of Medicine) and the vinculin−/− and vinculin+/+ cells were donated by Eileen Adamson (The Burnham Institute, La Jolla, CA).
References
- Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, Kaibuchi K. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science. 1997;275(5304):1308–1311. doi: 10.1126/science.275.5304.1308. [DOI] [PubMed] [Google Scholar]
- Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Exp Cell Res. 2000;261(1):44–51. doi: 10.1006/excr.2000.5046. [DOI] [PubMed] [Google Scholar]
- Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell. 2001;12(9):2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3(5):466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- Capadona JR, Collard DM, Garcia AJ. Fibronectin adsorption and cell adhesion to mixed monolayers of tri (ethylene glycol)-and methyl-terminated alkanthiols. Langmuir. 2003;19:1847–1852. [Google Scholar]
- Chen BH, Tzen JT, Bresnick AR, Chen HC. Roles of Rho-associated kinase and myosin light chain kinase in morphological and migratory defects of focal adhesion kinase-null cells. J Biol Chem. 2002;277(37):33857–33863. doi: 10.1074/jbc.M204429200. [DOI] [PubMed] [Google Scholar]
- Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88(1):39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133(6):1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danen EH, Sonnenberg A. Integrins in regulation of tissue development and function. J Pathol. 2003;201(4):632–641. doi: 10.1002/path.1472. [DOI] [PubMed] [Google Scholar]
- Del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez J, Sheetz M. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science. 2009;323(5914):638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delanoe-Ayari H, Al Kurdi R, Vallade M, Gulino-Debrac D, Riveline D. Membrane and acto-myosin tension promote clustering of adhesion proteins. Proc Natl Acad Sci U S A. 2004;101(8):2229–2234. doi: 10.1073/pnas.0304297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMali KA. Vinculin--a dynamic regulator of cell adhesion. Trends Biochem Sci. 2004;29(11):565–567. doi: 10.1016/j.tibs.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323(5914):642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol. 2002;159(4):695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. J Muscle Res Cell Motil. 1997;18(1):1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- Gallant ND, Capadona JR, Frazier AB, Collard DM, García AJ. Micropatterned surfaces to engineer focal adhesions for analysis of cell adhesion strengthening. Langmuir. 2002;18:5579–5584. [Google Scholar]
- Gallant ND, Garcia AJ. Quantitative analyses of cell adhesion strength. Methods Mol Biol. 2007;370:83–96. doi: 10.1007/978-1-59745-353-0_7. [DOI] [PubMed] [Google Scholar]
- Gallant ND, Michael KE, Garcia AJ. Cell adhesion strengthening: contributions of adhesive area, integrin binding, and focal adhesion assembly. Mol Biol Cell. 2005;16(9):4329–4340. doi: 10.1091/mbc.E05-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AJ, Huber F, Boettiger D. Force required to break α5β1 integrin-fibronectin bonds in intact adherent cells is sensitive to integrin activation state Integrin. J Biol Chem. 1998;273:10988–10993. doi: 10.1074/jbc.273.18.10988. [DOI] [PubMed] [Google Scholar]
- García AJ, Vega MD, Boettiger D. Modulation of cell proliferation and differentiation through substrate-dependent changes in fibronectin conformation. Mol Biol Cell. 1999;10(3):785–798. doi: 10.1091/mbc.10.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10(1):21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- Goldmann WH, Ingber DE. Intact vinculin protein is required for control of cell shape, cell mechanics, and rac-dependent lamellipodia formation. Biochem Biophys Res Commun. 2002;290(2):749–755. doi: 10.1006/bbrc.2001.6243. [DOI] [PubMed] [Google Scholar]
- Griffin MA, Sen S, Sweeney HL, Discher DE. Adhesion-contractile balance in myocyte differentiation. J Cell Sci. 2004;117(Pt 24):5855–5863. doi: 10.1242/jcs.01496. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci USA. 1992;89(18):8487–8491. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries JD, Wang P, Streuli C, Geiger B. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol. 2007;179:1043–1057. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Hytönen VP, Vogel V. How Force Might Activate Talin’s Vinculin Binding Sites: SMD Reveals a Structural Mechanism. PLoS Comput Biol. 2008;4(2):e24. doi: 10.1371/journal.pcbi.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilić D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377(6549):539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Tensegrity I. Cell structure and hierarchical systems biology. J Cell Sci. 2003;116(Pt 7):1157–1173. doi: 10.1242/jcs.00359. [DOI] [PubMed] [Google Scholar]
- Izzard CS, Lochner LR. Cell-to-substrate contacts in living fibroblasts: an interference reflexion study with an evaluation of the technique. J Cell Sci. 1976;21(1):129–159. doi: 10.1242/jcs.21.1.129. [DOI] [PubMed] [Google Scholar]
- Izzard CS, Lochner LR. Formation of cell-to-substrate contacts during fibroblast motility: an interference-reflexion study. J Cell Sci. 1980;42:81–116. doi: 10.1242/jcs.42.1.81. [DOI] [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- Keselowsky BG, García AJ. Quantitative methods for analysis of integrin binding and focal adhesion formation on biomaterial surfaces. Biomaterials. 2005;26(4):413–418. doi: 10.1016/j.biomaterials.2004.02.050. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273(5272):245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kovács M, Tóth J, Hetényi C, Málnási-Csizmadia A, Sellers JR. Mechanism of blebbistatin inhibition of myosin II. J Biol Chem. 2004;279(34):35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- Lotz MM, Burdsal CA, Erickson HP, McClay DR. Cell adhesion to fibronectin and tenascin: quantitative measurements of initial binding and subsequent strengthening response. J Cell Biol. 1989;109(4 Pt 1):1795–1805. doi: 10.1083/jcb.109.4.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto A, Connor K, Mammoto T, Yung C, Huh D, Aderman C, Mostoslavsky G, Smith LE, Ingber D. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009;457(7233):1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto A, Huang S, Moore K, Oh P, Ingber DE. Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2-p27kip1 pathway and the G1/S transition. J Biol Chem. 2004;279(25):26323–26330. doi: 10.1074/jbc.M402725200. [DOI] [PubMed] [Google Scholar]
- McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6(4):483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- Michael KE, Dumbauld DW, Burns KL, Hanks SK, Garcia AJ. FAK Modulates Cell Adhesion Strengthening via Integrin Activation. Mol Biol Cell. 2009;20(9):2508–2519. doi: 10.1091/mbc.E08-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierke CT, Kollmannsberger P, Zitterbart DP, Smith J, Fabry B, Goldmann WH. Mechano-coupling and regulation of contractility by the vinculin tail domain. Biophys J. 2007;94(2):661–670. doi: 10.1529/biophysj.107.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S, Ishizaki T, Uehata M. Use and properties of ROCK-specific inhibitor Y-27632. Methods Enzymol. 2000;325:273–284. doi: 10.1016/s0076-6879(00)25449-9. [DOI] [PubMed] [Google Scholar]
- Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385(6616):537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- Parizi M, Howard EW, Tomasek JJ. Regulation of LPA-promoted myofibroblast contraction: role of Rho, myosin light chain kinase, and myosin light chain phosphatase. Exp Cell Res. 2000;254(2):210–220. doi: 10.1006/excr.1999.4754. [DOI] [PubMed] [Google Scholar]
- Polte TR, Eichler GS, Wang N, Ingber DE. Extracellular matrix controls myosin light chain phosphorylation and cell contractility through modulation of cell shape and cytoskeletal prestress. Am J Physiol Cell Physiol. 2004;286(3):C518–528. doi: 10.1152/ajpcell.00280.2003. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci. 2000;113:3673–3678. doi: 10.1242/jcs.113.20.3673. [DOI] [PubMed] [Google Scholar]
- Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153(6):1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry SK, Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp Cell Res. 2000;261(1):25–36. doi: 10.1006/excr.2000.5043. [DOI] [PubMed] [Google Scholar]
- Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci USA. 2003;100(4):1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83(2):171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol. 2000;150(4):797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberg T, Geiger B, Citi S, Bershadsky AD. Effect of protein kinase inhibitor H-7 on the contractility, integrity, and membrane anchorage of the microfilament system. Cell Motil Cytoskeleton. 1994;29(4):321–338. doi: 10.1002/cm.970290405. [DOI] [PubMed] [Google Scholar]
- Wang HB, Dembo M, Hanks SK, Wang Y. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci USA. 2001;98(20):11295–11300. doi: 10.1073/pnas.201201198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260(5111):1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6(2):154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- Wolfenson H, Lubelski A, Regev T, Klafter J, Henis YI, Geiger B. A role for the juxtamembrane cytoplasm in the molecular dynamics of focal adhesions. PLoS ONE. 2009;4(1):e4304. doi: 10.1371/journal.pone.0004304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthylake RA, Burridge K. RhoA and ROCK promote migration by limiting membrane protrusions. J Biol Chem. 2003;278(15):13578–13584. doi: 10.1074/jbc.M211584200. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163(3):583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998;125(2):327–337. doi: 10.1242/dev.125.2.327. [DOI] [PubMed] [Google Scholar]
- Yanase M, Ikeda H, Ogata I, Matsui A, Noiri E, Tomiya T, Arai M, Inoue Y, Tejima K, Nagashima K, Nishikawa T, Shibata M, Ikebe M, Rojkind M, Fujiwara K. Functional diversity between Rho-kinase- and MLCK-mediated cytoskeletal actions in a myofibroblast-like hepatic stellate cell line. Biochem Biophys Res Commun. 2003;305(2):223–228. doi: 10.1016/s0006-291x(03)00726-5. [DOI] [PubMed] [Google Scholar]
- Zhu C, Bao G, Wang N. Cell mechanics: mechanical response, cell adhesion, and molecular deformation. Annu Rev Biomed Eng. 2000;2:189–226. doi: 10.1146/annurev.bioeng.2.1.189. [DOI] [PubMed] [Google Scholar]