

Abstract
Perturbations in the regulation of the core cell cycle machinery are frequently observed in human cancers. Cyclin D1 which functions as a mitogenic sensor and allosteric activator of CDK4/6, is one of the more frequently altered cell cycle regulators in cancers. Cyclin D1 is frequently overexpressed in cancers and its overexpression can be attributed to many factors including increased transcription, translation, and protein stability. Although cyclin D1 overexpression is clearly implicated in the affected cancers, overexpression of cyclin D1 is not sufficient to drive oncogenic transformation. Rather, emerging evidence suggests that nuclear retention of cyclin D1 resulting from altered nuclear trafficking and proteolysis is critical for the manifestation of its oncogenicity. This review provides a brief overview of current data documenting various mechanisms underlying aberrant cyclin D1 regulation in human cancers and their impact on neoplastic transformation.
Work during the past two decades has provided a detailed molecular understanding of processes that drive cell cycle division. Early work resulted in the identification of the individual cyclin-dependent kinases that are activated during distinct cell cycle phases and contributed to our understanding of how phosphorylation of downstream targets in turn contributes to regulated cell cycle transitions. The recent generation of mice wherein individual cyclins or their cognate CDKs have been eliminated from the mouse germline has revealed the potential for significant redundancy with regard to CDK function and substrate regulation.
While our fundamental understanding of the normal physiological functions of the cyclin-dependent kinase has increased dramatically, the potential pathological functions/activities remain to be established. This is particularly evident when one considers the potential contribution of G1 cyclins and CDKs to neoplastic transformation and growth. Regulation of G1 to S-phase progression is disrupted in nearly all cancers, due to its intimate function in the integration of growth factor signals with the cell cycle network. Of the G1 regulatory molecules, the cyclin D1/CDK4 complex is deregulated at a high frequency and is thought to contribute directly to neoplastic transformation and growth. Recent efforts have provided insights into the precise mechanisms whereby the cyclin D1-dependent kinase promotes tumorigenic growth and are discussed below.
D-cyclins: Mediators of mitogenic signals to core cell cycle machinery
Cyclins function as allosteric regulatory subunits for the cyclin-dependent kinases (CDKs). While the CDK components are generally expressed throughout cell division cycles, cyclins accumulate periodically at which point they associate with their cognate CDK to form active holoenzymes; through phosphorylation of distinct substrates, they trigger transition to distinct cell cycle phases. D-type cyclins (D1, D2, and D3) are G1-specific cyclins that associate with CDK4 or CDK6, and promote restriction point progression during G1 phase (Sherr, 1993). Unlike other cyclins that are periodically synthesized during cell cycle progression, expression and accumulation of D-cyclins are strongly dependent on extracellular mitogenic cues. Due to this property of D-cyclins, they are regarded as mitogenic sensors that relay signals from the extracellular environment to the core cell cycle machinery (Sherr and Roberts, 1999).
Our understanding of mitogen-dependent cyclin D1 regulation is more advanced than that for cyclin D2 and D3. Cyclin D1 expression and accumulation are induced by growth factors and occur at multiple levels including increased transcription, translation, and protein stability. Regulation mediated primarily through Ras-signaling pathways (Musgrove, 2006). Upon cell exposure to mitogenic stimuli, activated receptor tyrosine kinase triggers a sequential signaling cascade involving Ras-Raf-MEK (MAPK kinase)-ERK (extracellular signal-regulated kinase), resulting in transcriptional up-regulation of cyclin D1 (Albanese et al., 1995; Cheng et al., 1998; Weber et al., 1997). The increase in transcription is accompanied by enhanced translation and reduced proteolysis, which is triggered through other distinct Ras-mediated pathway involving PI3K (phosphatidylinositol-3-kinase) and Akt, further potentiating the induction of cyclin D1. Increased cyclin D1 translation is mediated by the PI3K-Akt-mTOR (mammalian target of rapamycin)-S6K1 (S6 kinase 1) cascade (Koziczak and Hynes, 2004; Muise-Helmericks et al., 1998), while the pathway that directs decreased protein degradation involves PI3K-Akt-GSK3β (glycogen synthase kinase-3β) (Diehl et al., 1998). The Akt-mediated phosphorylation of GSK3β decreases GSK3β catalytic activity, which inhibits nuclear export and cytoplasmic degradation of cyclin D1 resulting in nuclear accumulation of cyclin D1 and its associated CDK activity during G1 phase (Diehl et al., 1998).
Following its induction by mitogenic growth factors, newly synthesized cyclin D1 associates with CDK4; the predominant function of the D1/CDK4 enzyme involves the phosphorylation-dependent inactivation of the retinoblastoma protein, RB. RB phosphorylation is necessary for the activation of a gene expression network that regulates entry and progression through S-phase. The release of E2F family transcription factors from RB-dependent repression is both necessary and sufficient for the G1/S transition (Weinberg, 1995). In addition, cyclin D1 plays another important role for G1 phase transition, which is a kinase-independent function by sequestering CDK inhibitors such as p27 Kip1 and p21Cip1 for efficient activation of CDK2-containing complexes (Polyak et al., 1994; Sherr and Roberts, 1999). Interestingly, the binding of Cip/Kip inhibitors was shown to stabilize the association between cyclin D1 and CDK4/6 (Cheng et al., 1999; LaBaer et al., 1997). Given that cyclin D1 plays a crucial role in mitogen-driven cell cycle regulation, it may not be surprising to detect frequent dysregulation of cyclin D1 homeostasis in human malignancies.
D-cyclin Overexpression in Human Cancer
Molecular analysis of human tumors has revealed that cyclin D1 overexpression prevails over that of cyclin D2 and D3. While it is unclear why cyclin D1 expression is frequently altered in human cancers, as opposed to the other D-type cyclins, its overexpression is implicated as driving feature in various types of cancer, including mantle cell lymphoma (MCL) (Jares et al., 2007), non-small cell lung cancer (Jin et al., 2001), and carcinomas of breast (Barnes and Gillett, 1998), head and neck (Bartkova et al., 1995), and esophagus (Shamma et al., 2000). Cyclin D1 overexpression in human tumors is driven by multiple mechanisms comprising genomic alterations, post-transcriptional regulation, and post-translational protein stabilization.
Genomic Alterations: chromosomal translocations, gene amplification, and polymorphisms
Genomic alterations leading to aberrant cyclin D1 expression have been the subject of intensive investigation in a variety of tumors because of their predicted role for dysregulation of cyclin D1 abundance. The observed alterations range from a single nucleotide polymorphism to massive chromosomal translocations and gene amplification. Chromosomal translocation is a common genetic mechanism of pathogenesis of B-cell lymphoma (Kuppers, 2005). In particular, over 90 % of MCL, a mature B-cell malignancy representing 5–10 % of all non-Hodgkin’s lymphomas, is genetically characterized by t(11;14)(q13;q32) translocation that juxtaposes the cyclin D1-encoding gene (CCND1) to the immunoglobulin (Ig) heavy chain gene (Bertoni et al., 2006). As a consequence of this translocation, cyclin D1 is constitutively expressed under the control of an active Ig locus in B-lymphocytes where cyclin D1 is not expressed in normal conditions. While cyclin D1 is most frequently implicated in human cancers, chromosomal alterations that encompass the cyclin D3 locus have also been reported. The chromosomal translocation t(6;14)(p21.1;q32.3) is a rare but recurrent event in multiple myeloma (Bergsagel and Kuehl, 2001). In addition to multiple myeloma, the t(6;14)(p21.1;q32.3) translocation and an accompanying overexpression of cyclin D3 has been observed in several histologic subtypes of mature B-cell malignancies. With regard to diffuse large B-cell lymphoma (DLBCL) cases (Sonoki et al., 2001), cyclin D3 overexpression is noted at a frequency approaching 10 %.
Gene amplifications that encompass the cyclin D1 locus have also been observed in a number of cancers. Cyclin D1 overexpression resulting from amplification of 11q13 has been reported in various tumor types such as head and neck carcinoma (Izzo et al., 1998), pituitary tumor (Hibberts et al., 1999), esophageal squamous cell carcinoma (Shinozaki et al., 1996), and breast cancer (Buckley et al., 1993; Gillett et al., 1994). In addition to these massive chromosomal alterations that are accompanied by cyclin D1 overexpression, small genetic changes such as single nucleotide polymorphisms may also contribute to increased cyclin D1 by producing a specific isoform that is an alternative splice variant of cyclin D1. Among more than 100 polymorphisms identified in cyclin D1, G/A870 polymorphism has gained the most attention due to its potential contribution to cyclin D1 alternative splicing. The G/A polymorphism occurring at nucleotide position 870 is thought to hinder normal splicing at the exon 4/5 boundary, allowing for read-through into intron 4 and resulting in a truncated cyclin D1 (Betticher et al., 1995). This splice variant of cyclin D1 (cyclin D1b) can form an active complex with Cdk4 (Lu et al., 2003; Solomon et al., 2003) but lacks the C-terminal threonine residue whose phosphorylation is critical for nuclear export and subsequent cytoplasmic degradation of cyclin D1 (Diehl et al., 1997). This structural difference of the cyclin D1b renders the protein refractory to nuclear export through the cell cycle, which increases its oncogenic potency (Alt et al., 2000; Lu et al., 2003).
Post-transcriptional Mechanisms: a target of microRNA action
MicroRNAs (miRNAs) are small non-coding RNAs that regulate a variety of cellular processes including apoptosis, differentiation, and proliferation. Recently, it has been shown that truncations in 3′ untranslated region (UTR) of CCND1 mRNA in MCL cell lines eliminate miR-16-1 binding sites in the 3′ UTR, resulting in escape of the CCND1 transcripts from miR-16-1-mediated down-regulation (Chen et al., 2008). This provides another potential mechanism contributing to cyclin D1 overexpression in MCL. The mechanisms that lead to the observed 3′UTR truncations remain to be determined.
Post-translational Mechanism: perturbed nuclear export and proteolysis
Cell cycle progression requires orderly recruitment of distinct cyclin/CDK activities driven by timely synthesis and destruction of specific cyclins. As a key regulator of G1 reentry and progression, cyclin D1 turnover is subject to a tight regulation by a defined mechanism involving GSK3β-mediated phosphorylation of Thr286 (Diehl et al., 1998; Diehl et al., 1997), nuclear export by CRM1 (Benzeno and Diehl, 2004), and subsequent ubiquitylation by SCFFbx4/αB-crystallin ligase targeting the cyclin D1 to the 26S proteasome (Lin et al., 2006) (Figure 1A). Alterations in cyclin D1 turnover can lead to aberrant accumulation of cyclin D1 independent of changes in cyclin D1 transcription and translation. Given the relatively low frequency of genetic alterations that directly involve the cyclin D1 locus in breast and esophagus cancers (10 %) while cyclin D1 overexpression occurs at a frequency approaching 50 % (Barnes and Gillett, 1998; Bartkova et al., 1994; Shinozaki et al., 1996), it seems logical to predict frequent alterations in pathways that regulate cyclin D1 protein degradation. Consistent with this notion, mutations targeting the nuclear export or proteolysis of cyclin D1 have been well documented in human cancers while mutations directed at the Thr286 seem to be rare. For example, several mutations that impair phosphorylation-dependent nuclear export of cyclin D1 have been identified in esophageal carcinoma and esophageal cancer-derived cell lines (Benzeno et al., 2006). Similarly, cyclin D1 gene in endometrial cancer also possesses mutations or deletions that are expected to affect the phosphorylation of Thr286 and CRM1 binding (Moreno-Bueno et al., 2003). In addition to these mutations, we recently discovered that esophageal tumors have a number of mutations targeting the E3 ligase for cyclin D1 proteolysis so that the tumors can maintain high levels of cyclin D1 due to the decreased proteolysis efficiency (Barbash et al., 2008). These mutations occur in Fbx4 that is a substrate specificity factor for cyclin D1 in the SCF ligase, and affect the ligase activity by impairing its dimerization or interaction with other E3 ligase component. Along with the identification of cyclin D1b splice variant in human cancer, mutations targeting cyclin D1 nuclear export and proteolysis suggest that perturbed protein turnover and consequent accumulation of nuclear cyclin D1 may be more prevailing in cancers than previously thought.
Figure 1. Cyclin D1 regulation in normal versus cancer cells.
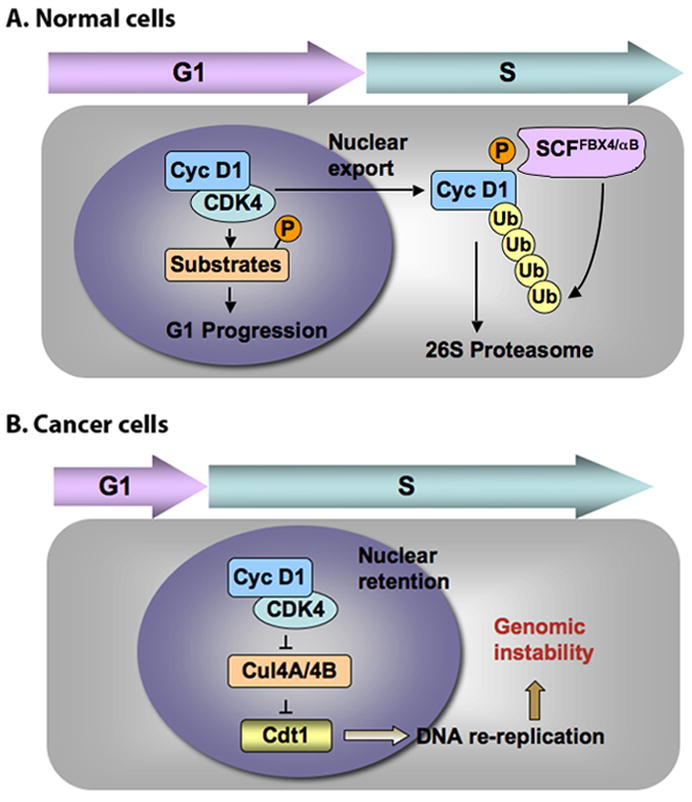
A: In normal cells, following G1 progression, cyclin D1 exits the nucleus via a distinct mechanism that involves GSK3β-mediated phosphorylation of Thr286 and CRM1-dependent nuclear export. Once in cytoplasm, phosphorylated cyclin D1 is recognized and ubiquitylated by SCFFbx4/αB-crystallin ligase, targeting the cyclin D1 to the 26S proteasome for degradation. B: In cancer cells, constitutive nuclear accumulation of active cyclin D1/CDK4 complexes can be achieved through one of several mechanisms: 1) mutations in cyclin D1 that prevent recognition by Crm1 (Benzeno et al., 2006; Moreno-Bueno et al., 2003); 2) mutation of Fbx4 which blocks ubiquitin-dependent degradation (Barbash et al., 2008); 3) alternative splicing of the cyclin D1 transcript which removes sequences that direct nuclear export and Fbx4-dependent destruction (Lu et al., 2003). The accumulation of active nuclear cyclin D1/CDK4 complex during S-phase stabilizes Cdt1 by transcriptionally inhibiting Cul4A and Cul4B, promoting DNA re-replication during S phase and consequently inducing genomic stability (Aggarwal et al., 2007).
Aberrant nuclear accumulation of the Cyclin D1/CDK4 kinase and neoplastic transformation
The initial cloning of the cyclin D1 cDNA as a component of a chromosomal translocation from a parathyroid adenoma (Motokura et al., 1991) immediately suggested the propensity of cyclin D1 to function as a potential initiating factor in neoplastic growth. Such implications were supported when a second group revealed that cyclin D1 was synonymous with the bcl-1 oncogene present in the 11;14 translocation present in nearly 100% of mantle cell lymphomas (Motokura et al., 1991). Additional data revealing that cyclin D1-expression was acutely regulated by mitogen-dependent growth factors was also consistent with this notion (Sherr and Roberts, 1999). Strikingly however, in vitro assays that evaluated the potential of cyclin D1 to function as a bona fide oncogene presented a paradox. Unlike ras and myc oncogenes, which readily transformed murine fibroblasts, neither cyclin D1, D2, nor D3 showed such potential, but rather could only potentiate Ras-dependent transformation in vitro (Quelle et al., 1993). Results from transgenic animals were equally unrevealing. Transgenic expression of wild type cyclin D1 in murine lymphocytes failed to elicit any appreciable phenotype unless mice also expressed a c-myc transgene (Bodrug et al., 1994; Lovec et al., 1994). Similar results were obtained in a transgenic mouse model of oral-esophageal cancer wherein cyclin D1 overexpression resulted in invasive oral-esophageal cancer only in combination with p53 deletion (Opitz et al., 2002). The only hint of oncogenic activity came from mice programmed to overexpress cyclin D1 under the control of mouse mammary tumor virus (MMTV) promoter in mammary epithelial cells. These MMTV-D1 mice succumb to adenocarcinomas after a long latency and with incomplete penetrance (Wang et al., 1994).
One interpretation of the above data is that cyclin D1 does not possess overt oncogenic activity. However, given the large amount of data demonstrating its overexpression in numerous cancers this seems unlikely. Thus a second and more likely possibility seems that cyclin D1 overexpression per se is not sufficient to drive neoplastic growth. If so, the latter would suggest that cells maintain additional regulatory mechanisms that maintain tight control over cyclin D1 activity thereby preventing manifestation of its neoplastic potential. Insight into this possibility stemmed from early observations that cyclin D1/CDK4 complexes shuttled between nuclear and cytoplasmic compartments during cell cycle progression (Alt et al., 2000; Diehl et al., 1998). This work revealed that the rate of nuclear import effectively maintains cyclin D1/CDK4 complexes in the nucleus during G1 phase; however, as cells enter S-phase, phosphorylation of cyclin D1 dramatically increased its rate of nuclear export (Diehl et al., 1997). Nuclear export coupled with ubiquitin-dependent destruction of cyclin D1 in the cytoplasm during S-phase effectively limits access of the cyclin D1/CDK4 kinase to nuclear substrates during S-phase.
Experimental models support the idea that mutations that interfere with nuclear exclusion of cyclin D1/CDK4 during S-phase trigger neoplastic conversion. Indeed overexpression of a mutant cyclin D1 (D1T286A) that is defective in phosphorylation-mediated nuclear export and subsequent proteolysis, induces cell transformation in cell culture assays and triggers B-cell lymphoma in a mouse model of mantle cell lymphoma (Alt et al., 2000; Gladden et al., 2006). Furthermore, transgenic mice that over-express the identical mutant cyclin D1 driven by MMTV promoter (MMTV-D1T286A) developed mammary adenocarcinoma with a shorter latency relative to mice over-expressing the wild-type cyclin D1 (MMTV-D1) (Lin et al., 2008). These observations suggest that subcellular localization and stabilization of cyclin D1 through its altered nuclear trafficking and proteolysis may exert more profound effects on tumorigenesis than its overexpression itself.
The experimental data described above stem from use of cyclin D1 mutants that are refractory to both CRM1-dependent nuclear export and Fbx4- and ubiquitin-dependent turnover. While such mutations have been observed in human cancers, they appear at low frequency (<3%). Mutations that inactivate Fbx4 and thereby stabilize cyclin D1 occur at a much higher frequency (~15 %), suggesting that loss of this E3 ligase is sufficient for the manifestation of the neoplastic potential of cyclin D1 (Barbash et al., 2008). While no knockout mouse for Fbx4 is currently available to directly examine this possibility, in vitro studies are consistent with this notion. ShRNA-dependent reduction of Fbx4 not only resulted stabilized cyclin D1 but also constitutively nuclear cyclin D1/CDK4 and spontaneous transformation, a phenotype similar to expression of cyclin D1T286A (Barbash et al., 2008). This would suggest that the Fbx4 E3 ligase functions as a critical off switch that prevents inappropriate nuclear accumulation of the D1/CDK4 complex and thereby prevents cell transformation.
Genomic instability in tumors harboring a nuclear cyclin D1/CDK4 kinase
Despite the obvious involvement of cyclin D1 in many human cancers, detailed mechanisms underlying cyclin D1-induced tumorigenesis are not well understood. To date, there seem to be at least two major mechanisms of cyclin D1-mediated tumorigenesis attributed to its association with CDKs. The first mechanism is based on the ability of cyclin D1 to increase CDK activity and consequently promote continuous proliferation. This hypothesis is supported by the observations that overexpression of p16 INK4a or p15INK4b suppresses Ras-induced transformation in vitro (Malumbres et al., 2000; Serrano et al., 1995). Moreover, expression of p16 INK4a blocks ErbB2-induced mammary tumor development in mice (Yang et al., 2004). The cyclin E “knockin” mice to the cyclin D1 locus were shown to develop Neu-driven mammary tumors like their wild-type counterparts (Geng et al., 1999). This indicates that cyclin E expression can compensate for the absence of cyclin D1, and also points to the role of cyclin D1 in cell cycle progression/proliferation during Neu-induced tumorigenesis.
In the above model, cyclin D1-dependent kinase functions largely through its canonical role as an integrator of mitogen-dependent signaling as opposed to directly contributing to early neoplastic conversion. As such, the assumption is that cellular controls of cyclin D1 localization and degradation are intact. In tumors where this latter mechanism is lost, recent investigation suggests that the cyclin D1/CDK4 kinase is likely to participate more directly in the neoplastic process. Recently, we provided the first experimental evidence linking the constitutively nuclear cyclin D1/CDK4 complex with induction of genomic instability in mouse models and human cancer-derived cells (Aggarwal et al., 2007). Nuclear accumulation of catalytically active mutant cyclin D1/CDK4 complex stabilizes Cdt1, an origin-licensing factor that is normally degraded during S phase to prevent reloading of the replicative MCM helicase. As a result, stabilized Cdt1 continually prime DNA re-replication during S phase and induces genomic instability characterized by aneuploidy. The perturbed proteolysis of Cdt1 in nuclear cyclin D1-overexpressing cells resulted from transcriptional repression of Cul4A and Cul4B that are component of E3 ligase for Cdt1 proteolysis (Figure 1B). Although the precise mechanism of this transcriptional regulation remains to be uncovered, the kinase-inactive mutant of CDK4 was shown to abrogate the nuclear cyclin D1-induced Cdt1 stabilization by relieving the transcriptional repression of Cul4A and Cul4B, suggesting that nuclear cyclin D1 induces DNA re-replication during S phase through its associated CDK4 activity. Consistent with the role of nuclear cyclin D1/CDK4 complex in Cdt1 stabilization, down-regulation of cyclin D1 E3 ligase was also shown to stabilize Cdt1 through the accumulation of nuclear cyclin D1 by its disrupted proteolysis (Barbash et al., 2008). All these observations suggest that nuclear retention of active cyclin D1/CDK complex may be a critical determinant to elicit the oncogenicity of cyclin D1 in addition to its prevalent overexpression in cancers. Then, the next logical question would be to elucidate the detailed functions of nuclear cyclin D1/CDK complex in the context of neoplastic transformation. Given the requirement of CDK activity to induce genomic instability, identification of specific substrates targeted by the nuclear cyclin D1/CDK activity would be of a great interest to address this question.
In addition to its critical role as a regulator of CDK4/6 activity, a growing body of work has suggested CDK-independent function for cyclin D1. Several groups have observed interactions between cyclin D1 and steroid hormone receptors. Current data suggest that direct interaction between cyclin D1 and estrogen (or androgen) receptor can promote association of the nuclear receptor with its coactivator(s) even in the absence of the hormone ligands (Knudsen et al., 1999; Petre et al., 2002; Roy and Thompson, 2006; Zwijsen et al., 1997). Thus cyclin D1 can induce nuclear receptor activity independent of soluble ligand, participating in gene expression program as a transcriptional regulator in these hormone-responsive tissues (Coqueret, 2002). This CDK-independent, nuclear receptor-agonistic activity of cyclin D1 may contribute to its oncogenicity in certain types of cancers including breast cancer. A potential kinase-dependent role for cyclin D1 in transcriptional repression of Cul4A and Cul4B by nuclear cyclin D1 was recently described (Aggarwal et al., 2007). It is currently unknown whether cyclin D1 plays a direct transcriptional role or it involves additional effector(s) to mediate the transcriptional regulation. The relative contribution of this noncanonical function for cyclin D1 relative to its role as a CDK4 activator remains to be determined.
Targeting cyclin D/CDK activity as a therapeutic modality
The oncogenic capacity of cyclin D1 explored in various cancer models and human cancers seems to present cyclin D1 and its associated CDK activity as potential therapeutic targets. In fact, CDK4 turned out to be a promising therapeutic target in MCL cells. Treatment of MCL cells that express predominantly cyclin D1a isoform (full-length cyclin D1) with a small molecule CDK4/6 inhibitor PD0332991 efficiently suppresses RB phosphorylation and cell proliferation at nanomolar concentrations (Marzec et al., 2006). While there are no inhibitors that target cyclin D1 directly so far, alternative therapeutic strategies targeting cyclin D1 turnover may be postulated, such as modulation of GSK3β activity especially when nuclear retention of cyclin D1 is considered to be a causative factor for initial oncogenic events. Because GSK3β was shown to increase cyclin D1 proteolysis by generating degradable cyclin D1 substrates for ubiquitin ligase and also augmenting the E3 ligase activity by promoting dimerization of the ligase (Barbash et al., 2008), GSK3β appears to be an attractive target if selective modulation of its activity can be achieved for cyclin D1-overexpressing cells.
Would such an approach only be useful in the subset of tumors harboring mutations in cyclin D1 or Fbx4? This is unlikely to be the case. Experimental evidence is also available demonstrating that cyclin D1 serves as an effector during tumorigenesis. This has been most elegantly demonstrated in mouse models of mammary carcinoma where cyclin D1 deficient mice were found to be resistant to mammary tumors induced by either the neu or ras oncogenes (Yu et al., 2001). Strikingly, the D1−/− mice were not resistant to the oncogenic effects of myc or wnt demonstrating that the coupling of oncogenic pathways to the cell cycle machinery is highly specific. Additional work using mice wherein endogenous CDK4 was deleted via homologous recombination also reported resistance to mammary tumorigenesis (Reddy et al., 2005; Yu et al., 2006). This set of experiments provides the most striking evidence that cyclin D1-dependent kinase activity is necessary for neoplastic growth and could represent point of therapeutic intervention.
Acknowledgments
This work was supported by NIH CA93237: JAD is a Leukemia Lymphoma Society Scholar.
References
- Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, Goradia A, Wasik MA, Klein-Szanto AJ, Rustgi AK, Bassing CH, Diehl JA. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007;21(22):2908–2922. doi: 10.1101/gad.1586007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270(40):23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14(24):3102–3114. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash O, Zamfirova P, Lin DI, Chen X, Yang K, Nakagawa H, Lu F, Rustgi AK, Diehl JA. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell. 2008;14(1):68–78. doi: 10.1016/j.ccr.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DM, Gillett CE. Cyclin D1 in breast cancer. Breast Cancer Res Treat. 1998;52(1–3):1–15. doi: 10.1023/a:1006103831990. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer. 1994;57(3):353–361. doi: 10.1002/ijc.2910570311. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas J, Muller H, Strauss M, Gusterson B, Bartek J. Abnormal patterns of D-type cyclin expression and G1 regulation in human head and neck cancer. Cancer Res. 1995;55(4):949–956. [PubMed] [Google Scholar]
- Benzeno S, Diehl JA. C-terminal sequences direct cyclin D1-CRM1 binding. J Biol Chem. 2004;279(53):56061–56066. doi: 10.1074/jbc.M411910200. [DOI] [PubMed] [Google Scholar]
- Benzeno S, Lu F, Guo M, Barbash O, Zhang F, Herman JG, Klein PS, Rustgi A, Diehl JA. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene. 2006;25(47):6291–6303. doi: 10.1038/sj.onc.1209644. [DOI] [PubMed] [Google Scholar]
- Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene. 2001;20(40):5611–5622. doi: 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- Bertoni F, Rinaldi A, Zucca E, Cavalli F. Update on the molecular biology of mantle cell lymphoma. Hematol Oncol. 2006;24(1):22–27. doi: 10.1002/hon.767. [DOI] [PubMed] [Google Scholar]
- Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD, Heighway J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene. 1995;11(5):1005–1011. [PubMed] [Google Scholar]
- Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW, Adams JM. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. 1994;13(9):2124–2130. doi: 10.1002/j.1460-2075.1994.tb06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA, Sutherland RL. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8(8):2127–2133. [PubMed] [Google Scholar]
- Chen RW, Bemis LT, Amato CM, Myint H, Tran H, Birks DK, Eckhardt SG, Robinson WA. Truncation in CCND1 mRNA alters miR-16-1 regulation in mantle cell lymphoma. Blood. 2008;112(3):822–829. doi: 10.1182/blood-2008-03-142182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18(6):1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci U S A. 1998;95(3):1091–1096. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coqueret O. Linking cyclins to transcriptional control. Gene. 2002;299(1–2):35–55. doi: 10.1016/s0378-1119(02)01055-7. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11(8):957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97(6):767–777. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, Barnes D, Peters G. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 1994;54(7):1812–1817. [PubMed] [Google Scholar]
- Gladden AB, Woolery R, Aggarwal P, Wasik MA, Diehl JA. Expression of constitutively nuclear cyclin D1 in murine lymphocytes induces B-cell lymphoma. Oncogene. 2006;25(7):998–1007. doi: 10.1038/sj.onc.1209147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibberts NA, Simpson DJ, Bicknell JE, Broome JC, Hoban PR, Clayton RN, Farrell WE. Analysis of cyclin D1 (CCND1) allelic imbalance and overexpression in sporadic human pituitary tumors. Clin Cancer Res. 1999;5(8):2133–2139. [PubMed] [Google Scholar]
- Izzo JG, Papadimitrakopoulou VA, Li XQ, Ibarguen H, Lee JS, Ro JY, El-Naggar A, Hong WK, Hittelman WN. Dysregulated cyclin D1 expression early in head and neck tumorigenesis: in vivo evidence for an association with subsequent gene amplification. Oncogene. 1998;17(18):2313–2322. doi: 10.1038/sj.onc.1202153. [DOI] [PubMed] [Google Scholar]
- Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750–762. doi: 10.1038/nrc2230. [DOI] [PubMed] [Google Scholar]
- Jin M, Inoue S, Umemura T, Moriya J, Arakawa M, Nagashima K, Kato H. Cyclin D1, p16 and retinoblastoma gene product expression as a predictor for prognosis in non-small cell lung cancer at stages I and II. Lung Cancer. 2001;34(2):207–218. doi: 10.1016/s0169-5002(01)00225-2. [DOI] [PubMed] [Google Scholar]
- Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res. 1999;59(10):2297–2301. [PubMed] [Google Scholar]
- Koziczak M, Hynes NE. Cooperation between fibroblast growth factor receptor-4 and ErbB2 in regulation of cyclin D1 translation. J Biol Chem. 2004;279(48):50004–50011. doi: 10.1074/jbc.M404252200. [DOI] [PubMed] [Google Scholar]
- Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5(4):251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11(7):847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(Fbx4-alphaB crystallin) complex. Mol Cell. 2006;24(3):355–366. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene. 2008;27(9):1231–1242. doi: 10.1038/sj.onc.1210738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovec H, Grzeschiczek A, Kowalski MB, Moroy T. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J. 1994;13(15):3487–3495. doi: 10.1002/j.1460-2075.1994.tb06655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003;63(21):7056–7061. [PubMed] [Google Scholar]
- Malumbres M, Perez De Castro I, Hernandez MI, Jimenez M, Corral T, Pellicer A. Cellular response to oncogenic ras involves induction of the Cdk4 and Cdk6 inhibitor p15(INK4b) Mol Cell Biol. 2000;20(8):2915–2925. doi: 10.1128/mcb.20.8.2915-2925.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec M, Kasprzycka M, Lai R, Gladden AB, Wlodarski P, Tomczak E, Nowell P, Deprimo SE, Sadis S, Eck S, Schuster SJ, Diehl JA, Wasik MA. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood. 2006;108(5):1744–1750. doi: 10.1182/blood-2006-04-016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Bueno G, Rodriguez-Perales S, Sanchez-Estevez C, Hardisson D, Sarrio D, Prat J, Cigudosa JC, Matias-Guiu X, Palacios J. Cyclin D1 gene (CCND1) mutations in endometrial cancer. Oncogene. 2003;22(38):6115–6118. doi: 10.1038/sj.onc.1206868. [DOI] [PubMed] [Google Scholar]
- Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350(6318):512–515. doi: 10.1038/350512a0. [DOI] [PubMed] [Google Scholar]
- Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem. 1998;273(45):29864–29872. doi: 10.1074/jbc.273.45.29864. [DOI] [PubMed] [Google Scholar]
- Musgrove EA. Cyclins: roles in mitogenic signaling and oncogenic transformation. Growth Factors. 2006;24(1):13–19. doi: 10.1080/08977190500361812. [DOI] [PubMed] [Google Scholar]
- Opitz OG, Harada H, Suliman Y, Rhoades B, Sharpless NE, Kent R, Kopelovich L, Nakagawa H, Rustgi AK. A mouse model of human oral-esophageal cancer. J Clin Invest. 2002;110(6):761–769. doi: 10.1172/JCI15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petre CE, Wetherill YB, Danielsen M, Knudsen KE. Cyclin D1: mechanism and consequence of androgen receptor co-repressor activity. J Biol Chem. 2002;277(3):2207–2215. doi: 10.1074/jbc.M106399200. [DOI] [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8(1):9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Ashmun RA, Shurtleff SA, Kato JY, Bar-Sagi D, Roussel MF, Sherr CJ. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993;7(8):1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- Reddy HK, Mettus RV, Rane SG, Grana X, Litvin J, Reddy EP. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65(22):10174–10178. doi: 10.1158/0008-5472.CAN-05-2639. [DOI] [PubMed] [Google Scholar]
- Roy PG, Thompson AM. Cyclin D1 and breast cancer. Breast. 2006;15(6):718–727. doi: 10.1016/j.breast.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Serrano M, Gomez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995;267:249–252. doi: 10.1126/science.7809631. [DOI] [PubMed] [Google Scholar]
- Shamma A, Doki Y, Shiozaki H, Tsujinaka T, Yamamoto M, Inoue M, Yano M, Monden M. Cyclin D1 overexpression in esophageal dysplasia: a possible biomarker for carcinogenesis of esophageal squamous cell carcinoma. Int J Oncol. 2000;16(2):261–266. doi: 10.3892/ijo.16.2.261. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73(6):1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Shinozaki H, Ozawa S, Ando N, Tsuruta H, Terada M, Ueda M, Kitajima M. Cyclin D1 amplification as a new predictive classification for squamous cell carcinoma of the esophagus, adding gene information. Clin Cancer Res. 1996;2(7):1155–1161. [PubMed] [Google Scholar]
- Solomon DA, Wang Y, Fox SR, Lambeck TC, Giesting S, Lan Z, Senderowicz AM, Conti CJ, Knudsen ES. Cyclin D1 splice variants. Differential effects on localization, RB phosphorylation, and cellular transformation. J Biol Chem. 2003;278(32):30339–30347. doi: 10.1074/jbc.M303969200. [DOI] [PubMed] [Google Scholar]
- Sonoki T, Harder L, Horsman DE, Karran L, Taniguchi I, Willis TG, Gesk S, Steinemann D, Zucca E, Schlegelberger B, Sole F, Mungall AJ, Gascoyne RD, Siebert R, Dyer MJ. Cyclin D3 is a target gene of t(6;14)(p21.1;q32.3) of mature B-cell malignancies. Blood. 2001;98(9):2837–2844. doi: 10.1182/blood.v98.9.2837. [DOI] [PubMed] [Google Scholar]
- Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369(6482):669–671. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J. 1997;326 ( Pt 1):61–68. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Yang C, Ionescu-Tiba V, Burns K, Gadd M, Zukerberg L, Louis DN, Sgroi D, Schmidt EV. The role of the cyclin D1-dependent kinases in ErbB2-mediated breast cancer. Am J Pathol. 2004;164(3):1031–1038. doi: 10.1016/S0002-9440(10)63190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411(6841):1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, Sicinski P. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9(1):23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88(3):405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]