

Abstract
Neonatal necrotizing enterocolitis is a devastating inflammatory bowel disease of premature infants. The pathogenesis remains incompletely understood and there is no specific treatment. Efforts are ongoing to understand aspects of intestinal immaturity which contribute to susceptibility to this disease. This review focuses on bacterial colonization patterns, intestinal barrier function, and inflammatory responses of immature enterocytes leading to a unique vulnerability of the preterm gut. In addition the possible therapeutic potential of factors in human milk and probiotic bacteria is discussed.
Keywords: Neonatal necrotizing enterocolitis, premature infant, probiotic, human milk, platelet activating factor, nuclear factor kappaB
INTRODUCTION
Premature infants, defined as infants born at less than 37 weeks gestational age, account for over 12% of deliveries in the United States [1]. These infants have many acute problems, including fluid and electrolyte imbalance, impaired oxygenation associated with lung immaturity, and increased risk of sepsis all due to the premature assumption of roles expected to be performed by maternal circulation and the protective intra-uterine environment of fetal life. However, there is evidence that many of the long-term problems that account for much of the morbidity of prematurity are inflammatory in nature. The cytokine balance for preterm infants appears skewed toward inflammation [2]. Exposure to increased inflammatory cytokines comes from both maternal and infant sources, and risk for inflammatory conditions such as chronic lung disease, periventricular leukomalacia, cerebral palsy and necrotizing enterocolitis may be increased by maternal chorioamnionitis, postnatal sepsis, and intensive care unit interventions which increase the production of pro-inflammatory cytokines in preterm infants known to have lower levels of anti-inflammatory cytokines such as IL-10 [3–5]. This review will focus on the disease necrotizing enterocolitis (NEC) as an inflammatory disorder of the preterm infant.
NECROTIZING ENTEROCOLITIS
NEC is an inflammatory bowel necrosis that primarily afflicts premature infants after the initiation of enteral feeds. NEC affects about 10% of premature infants <1500grams, or 1–5% of all neonatal intensive care unit admissions. It characteristically strikes between 7 and 14 days of life, although, increasingly NEC has been documented several weeks after birth, particularly in very low birth weight infants [6–8]. Susceptibility to NEC is inversely related to gestational age [9]. Thus, more immature infants are more likely to get this disease and are at risk for a longer window of time suggesting that degree of immaturity plays an important role. While full term infants can develop a similar clinical picture, these infants generally have specific underlying risk factors for intestinal perfusion compromise such as birth asphyxia, polycythemia, exchange transfusion, intrauterine growth restriction, or cyanotic congenital heart disease. In these patients NEC often presents within the first days of life. Thus the pathophysiology in full term infants may be quite different from that in premature infants and will not be the focus of this review. There does not appear to be a sex or race predilection.
NEC presents with variable symptoms which are often non-specific signs of gastrointestinal dysfunction. These signs include abdominal distension, feeding intolerance, gastric aspirates, bilious vomiting and hematochezia; with sudden progression to pneumoperitoneum and/or systemic shock and rapid death in severe cases. The pathognomonic feature is pneumatosis intestinalis on abdominal radiograph which represents air tracking within the bowel wall as a result of bacterial fermentation of intraluminal substrates.
NEC classically affects the terminal ileum. The histopathologic landmark is coagulation necrosis indicating prior ischemia, with a combination of ischemic necrosis, acute and chronic inflammation, bacterial overgrowth, and tissue repair suggesting that NEC is an evolving process rather than an acute event [10, 11]. This disease can have focal, segmental, and non-continuous lesions. Inflammation can be limited to the mucosa and submucosa of the intestine, or progress to transmural involvement in the most severe cases. In addition, lesions may include submucosal or subserosal collections of gas which is what is seen as pneumatosis on abdominal radiograph.
Although numerous studies have been done on animals and humans and many theories have been proposed, the cellular basis of this disease remains poorly understood. Treatment is currently merely supportive and includes full bowel rest with immediate cessation of enteral feeds, institution of nasogastric suction, fluid resuscitation, close monitoring of acid/base and electrolyte balance, broad spectrum parenteral antibiotics, parenteral nutrition and frequent abdominal radiographs to document progression to pneumoperitoneum. Surgery is indicated for pneumoperitoneum, peritonitis, and intestinal obstruction. However, current treatment modalities are often inadequate because of the rapid progression of NEC from its initial diagnosis. In particular, the outcomes for these infants are very concerning. There is a high mortality rate recently documented at 16–42% depending on birth-weight, along with risk of intestinal stricture, short-gut syndrome, and the complications of difficulty providing adequate nutrition and parenteral nutrition-induced cholestasis [9]. Furthermore, studies have shown that NEC, particularly NEC requiring surgery, is an independent risk factor for poor neurodevelopmental outcome [12].
The primary risk factors for NEC are prematurity, bacterial colonization, hypoxia/altered intestinal blood flow, and enteral feeding [13]. Prematurity with its associated immature GI host and blood flow regulation results in mucosal injury. Combined with enteral feeds and bacterial colonization, inflammatory mediators are released leading to a propagated inflammatory response with both pro and anti inflammatory influences. If counter-regulatory responses to these inflammatory events are insufficient, pathologic changes to gut mucosa occur. In the preterm infant the balance appears to favor pro inflammatory influences resulting in NEC.
SUSCEPTIBILITY OF THE IMMATURE INTESTINE
It has been hypothesized that the injury in NEC begins with a breach in the intestinal mucosal barrier leading to bacterial translocation across the epithelium, and exacerbation of the inflammatory cascade, resulting in the clinical signs of NEC. Thus to understand the intestinal inflammation progression, this review will focus on bacterial colonization patterns in preterm infants, means by which bacteria breach the intestinal barrier, and the inflammatory response of the immature enterocyte to these bacteria.
Bacterial Colonization and the Preterm Gut
As opposed to the adult intestinal microbiota which is comprised of more than 1013 microorganisms, the newborn gut is sterile at birth. Furthermore, colonization of the blank slate of the preterm intestine is influenced by iatrogenic manipulations in the neonatal intensive care unit (NICU). This includes a hospital environment; frequent use of broad spectrum antibiotics, opioids, and H2 blockers; and instrumentation with endotracheal tubes, feeding tubes, and suctioning tubes. Resulting altered microbial flora may have significant implications for development of the immature preterm gut and susceptibility to NEC.
Bacteria are believed to be important in the pathogenesis of NEC; however no specific pathogen has been identified. Previous studies have been limited by the inability of conventional microbiological cultivation techniques to thoroughly characterize the human gastrointestinal microbiota. It has been reported that 80% of the human colonic microbiota are not detected by conventional culture methods [14]. Molecular profiling of microbiomes are now possible by sequencing the highly conserved 16S small subunit bacterial ribosomal RNA gene (rRNA), allowing identification of previously undetectable microbes [15]. This approach therefore provides a more complete picture of the composition of human intestinal microbiota.
Sequencing of the intestinal microbiome of infants with and without NEC has demonstrated that sequences from NEC patients cluster separately from sequences from control patients, even between genetically identical twin pairs [16]. A specific pathogen has still not been identified, rather, the microbial community structure in NEC patients is distinct based on a significant decrease in diversity of microbial species with an increase in Proteobacteria dominance compared to other preterm infants, and a specific bloom of a single genus of Proteobacteria to >50% of the overall bacterial composition [16]. This data suggests that while bacteria are important in the pathogenesis of NEC, NEC does not appear to be an infection in classic sense. A specific organism is not causal, but rather a trigger for a rapid inflammatory cascade in the preterm gut, leading to signs and symptoms of disease. There are 2 sides to this interaction – the microbe and the host.
Host Defense in the Preterm Infant
The preterm gut is essentially a fetal gut, expecting conditions of the intrauterine environment. This includes absence of bacteria. Preterm infants may not yet be prepared for bacterial interaction when initially colonized and fed, potentially placing them at higher risk for NEC.
The mature intestine has many physical barriers to bacteria including peristalsis, gastric acid, proteolytic enzymes, intestinal mucus, cell surface glycoconjugates, and tight junctions between intestinal epithelial cells. These are designed to limit bacteria to the gut lumen and prevent attachment and translocation across the intestinal epithelium. Animal studies have shown that pathogenic organisms adhere to and translocate across the intestine to a greater extent in immature vs. mature animals Fig. (1). Abnormal peristaltic activity in these infants may increase bacterial adherence, allowing for bacterial overgrowth that could increase endotoxin exposure and predispose the infant to NEC [17–19]. Cell surface glycoconjugates which serve as adhesion sites for a variety of microbes, have a different pattern of carbohydrate residues in the immature compared to the adult intestine, which may result in increased pathogenic colonization in preterm infants [20–22]. Furthermore, it is known that intestinal mucus, which protects against bacterial and toxin invasion, is different in developing animals and perhaps in premature infants in terms of carbohydrate composition, density and possibly in inclusion of secretory immunoglobulin [23].
Fig. 1.
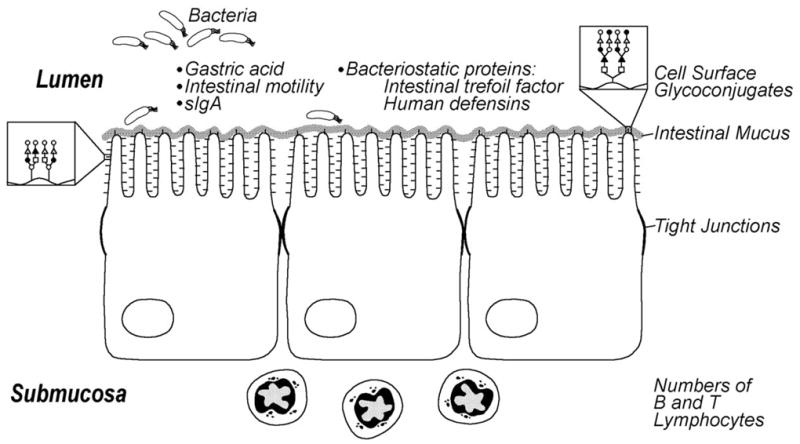
Aspects of immature intestinal host defense in the premature infant that may contribute to susceptibility to necrotizing enterocolitis. Reproduced with permission [146].
In addition, there is immaturity of the functional barrier that limits growth of bacteria that breach the physical barrier. This functional barrier is comprised of the immunologic host defense and various biochemical factors. It is known that numbers of intestinal B and T lymphocytes are decreased in neonates and do not approach adult levels until 3–4 weeks of life. Newborns also have reduced levels of secretory IgA in salivary samples, presumably reflecting decreased activity in the intestine [24–26]. Additionally, the premature infant has lower gastric acid production compared to older children, and immature proteolytic enzyme activity may lead to incomplete breakdown of toxins [24]. Finally, key proteins secreted from the intestinal epithelium such as intestinal trefoil factor are developmentally regulated and deficient in premature infants [27–29] while human defensins (or cryptidins) synthesized and secreted from paneth cells to protect against bacterial translocation are also altered in premature infants and those with NEC [30, 31]. All of these issues increase the likelihood that bacteria will interact with the immature enterocyte. A premature infant has a potentially more pathogenic bacterial balance and an immature gut defense to shield against bacteria, which collectively increases the risk for NEC.
Intestinal Barrier Disruption
A unique characteristic of the gastrointestinal system is its lining by a single epithelial layer in continuous contact with intestinal bacterial flora. This layer is a key host defense mechanism critical for confining pathogenic bacteria to the intestinal lumen, but must also allow passage of nutrients. Preterm infants have increased intestinal permeability, perhaps to allow expected passage of important macromolecules from amniotic fluid or breast milk [32]. However, this same increased permeability could lead to increased bacterial translocation. The inflammatory response of immature intestinal epithelial cells can be triggered by either commensal or pathogenic bacteria [33]. Disruption of the intestinal epithelial barrier increases this interaction and is thought to be an early event in the pathogenic cascade of NEC.
A breach in the mucosal barrier could be caused by disruption of the tight junctions between epithelial cells. The intestinal barrier normally consists of intestinal epithelial cells (IEC) connected by a system of both intracellular (zonula occludin proteins ZO-1,ZO-2, and ZO-3) and membrane-spanning (occludin, junction adhesion molecule (JAM) and the claudin family) proteins [34]. The epithelial tight junction (TJ) forms a selectively permeable barrier which allows fluids and solutes to cross while maintaining a protective barrier against other contents of the intestinal lumen. Occludin and claudin-3 have specifically shown to be altered in animal models of NEC [35].
Another means of barrier disruption is destruction of the cells themselves. One means of cell destruction is apoptosis, or programmed cell death. Apoptosis is a process of removing damaged cells characterized by cell shrinkage, chromatin condensation, and DNA fragmentation [36]. Caspase proteases are executors of apoptotic cell death. Caspase activation regulates endonucleases, resulting in cleavage of internucleosomal DNA and cell death [37]). In addition, there are caspase independent pathways in which proteases other than caspases initiate chromatin cleavage [38]. While apoptosis is a normal aspect of enterocyte turnover, accelerated apoptosis may lead to a breach in the critical intestinal mucosal barrier. Studies in animal models have demonstrated that apoptosis precedes necrosis in NEC, and that inhibition of apoptosis can decrease incidence of disease [39].
Apoptosis can be triggered by both intrinsic and extrinsic pathways [40]. In the intrinsic pathway, cell stress leads to binding of cytoplasmic proapoptotic proteins such as Bcl-2 family members BAX and BID to the mitochondria. This results in collapse of the mitochondrial potential. Cytochrome C is then released from mitochondria and binds to apoptotic protease activating factor 1 resulting in formation of the apoptosome. Exposure of the caspase recruitment domain recruits caspase -9 resulting in ultimate activation of caspase 3 and DNA fragmentation resulting in cell death. In the extrinsic pathway, binding of Fas ligand or tumor necrosis factor (TNF) to its receptor results in the recruitment of Fas associated death domain [41] proteins. Subsequent recruitment of caspase 8 results in the formation of the death inducing signal complex (DISC). This complex can directly activate caspase 3 in a mitochondria independent fashion, or act through BID to induce mitochondrial release of cytochrome C. Apoptosis can also result from DNA fragmentation induced by sustained calcium increases which act through activation of a Ca2+/Mg2+ endonuclease or intracellular acidosis which can enhance activation of caspases and activate of pH sensitive endonucleases [42, 43].
Platelet activating factor (PAF) is a phospholipid inter- and intra-cellular mediator which has been implicated in the pathology of inflammatory bowel disease and shown to induce intestinal epithelial cell apoptosis. However, the mechanism of PAF induced intestinal injury is incompletely understood. It is known that tissue and/or serum PAF levels are elevated in patients with Crohn’s disease, ulcerative colitis, and NEC and levels appear to correlate with disease severity [44–47]. In animal models of NEC, PAF receptor blockade or administration of the PAF-degrading enzyme PAF acetylhydrolase reduced the incidence of experimental NEC [48]. Moreover, in human studies of NEC, PAF has been shown to rise several days before the onset of clinical symptoms in some patients suggesting that this is a critical, and possibly initiating, factor in the development of the disease [49]. PAF levels have been shown to be increased by both hypoxia and enteral feeds, some of the specific risk factors implicated in NEC [50, 51].
Regulation begins at the receptor level where PAF acts through binding to a seven transmembrane domain G protein coupled receptor. Both high and low affinity PAF receptors have been identified [52]. Multiple signaling cascades are then linked to the PAF receptor. Depending on the route of administration and the animal model used, PAF can induce a variety of effects including platelet aggregation, hypotension, increased vascular permeability, vasoconstriction, intestinal ischemia, neutrophil recruitment, and production of reactive oxygen species [53]. In the intestine it is thought that PAF, once activated, initiates production of other inflammatory mediators such as TNFα, prostaglandins, thromboxane, and complement that then lead to the clinical signs and symptoms of inflammatory bowel disease [54–56]. However, several lines of evidence suggest that the pathologic consequences of elevated PAF levels in the intestine are not only the result of the initiation of an inflammatory cascade, but that PAF itself has direct effects on intestinal epithelial cells.
In isolated IEC lines and an animal model of NEC, PAF has been shown to induce apoptosis via three mechanisms [39, 57]. PAF induces translocation of BAX to mitochondria resulting in loss of mitochondrial membrane potential and caspase activation, an effect blocked by the anti-apoptotic protein Bcl2 [57]. PAF also inhibits the phosphatidylinositol 3-kinase/protein kinase B Akt signaling pathway [58]. Lastly, PAF has been shown to induce intracellular acidosis via activation of the Ca2+ dependent Cl− channel ClC-3 resulting in apoptosis of IEC [59].
Studies of intestinal tissue of preterm human infants with NEC have noted additional markers to give clues about the mechanism of altered barrier function. These studies have demonstrated upregulation of inducible nitric oxide which correlated with the degree of intestinal injury and number of apoptotic nuclei, Local intestinal production of nitric oxide (NO) induced by cytokines leads to intestinal apoptosis, potentially exacerbating intestinal injury [60]. In addition increased levels of high mobility group box 1 (HMGB1) protein have been found, which is released by macrophages and necrotic cells and is associated with increased barrier dysfunction and bacterial translocation [61].
Intestinal Inflammation and NEC
The necrosis which follows apoptosis results from progression of the triggered inflammatory cascade. IL-8 is a chemokine that stimulates migration of neutrophils from intravascular to interstitial sites and can directly activate neutrophils and regulate the expression of neutrophil adhesion molecules [62–64]. Thus by recruiting and activating immune cells, IL-8 may play an important role in inflammation. Previous studies have shown that concentrations of serum IL-8 were significantly elevated in severe cases of NEC from its onset through the first 24 hours [65]. Surgical specimens of intestine from infants with acute NEC show upregulation of IL-8 mRNA throughout the serosa, muscularis, and intestinal epithelium compared to those with other inflammatory conditions or those without disease [66]. Studies also demonstrate increased serum levels of the pro-inflammatory cytokines IL-6 and TNFα in infants with NEC [51, 67, 68].
Preterm infants not only have altered bacterial colonization and immature host defense, but also an immature response to bacteria. It is potentially an important developmental step for enterocytes to decrease inflammatory responsiveness in order to prevent immune defense mechanisms against normal flora. Preterm infants may not have completed this maturation when initially fed and colonized by bacteria. Evidence suggests that the premature neonate may be predisposed to intestinal inflammation. Studies have demonstrated that compared to adult IEC, human fetal IEC have an exaggerated production of IL-8 in response to both pathogenic and commensal bacteria as well as to endogenous inflammatory mediators such as TNFα and IL-1γ [69, 70].
Toll-like receptors (TLR) are a highly conserved family of pathogen associated molecular pattern (PAMP) receptors which recognize bacterial components. TLR4 specifically recognizes the bacterial cell wall component lipopolysaccharide (LPS). Interestingly, TLR4 expression decreases after birth in the intestines of healthy mother fed rat pups, but increases in the intestinal epithelium when pups are exposed to stresses common to preterm infants such as formula feeding and asphyxia [71]. Furthermore, animal models of NEC have demonstrated that TLR4 mutant mice were protected from NEC [71].
TLR activation, as well as TNFα and IL-1β can activate nuclear factor kappaB (NF-κB) proteins which lead to transcription of a wide variety of genes important in inflammatory and immune responses Fig. (2). In its resting state NF-κB dimers are bound in the cytoplasm to inhibitory κB (IκB) proteins [72]. Cell stimulation can trigger signaling pathways leading to phosphorylation, ubiquitination, and ultimately degradation of IκB by the 26S proteasome [73, 74]. NF-κB, thus liberated, moves to the nucleus where it activates gene transcription. Interestingly, NF-κB binding sites are located on the inhibitory IκBα promoter. Thus an elegant autoregulatory feedback loop exists in which NF-κB activation leads to IκBα synthesis to terminate the NF-κB response [75, 76]. However, in vitro and in vivo studies have shown that immature enterocytes have increased NF-κB activity associated with decreased baseline expression of IκB isoforms [33]. In addition, inflammatory stimuli trigger increased phosphorylation, ubiquitination, leading to increased degradation of IκBα and prolonged NF-κB activation in immature enterocytes compared to adult enterocytes [33, 77]. Differences in IκB levels appear to be key as increasing expression of IκBα by transfection in immature intestinal epithelial cells attenuated transcriptional up-regulation of inflammatory cytokines, and suggest that developmentally-regulated differences in IκB could contribute to the differential responsiveness of immature and mature enterocytes to inflammatory stimuli [33]. A critical balance between NF-κB activation and inhibition is important to allow appropriate defense against pathogens yet avoid host damaging inflammatory responses to commensal flora.
Fig. 2.
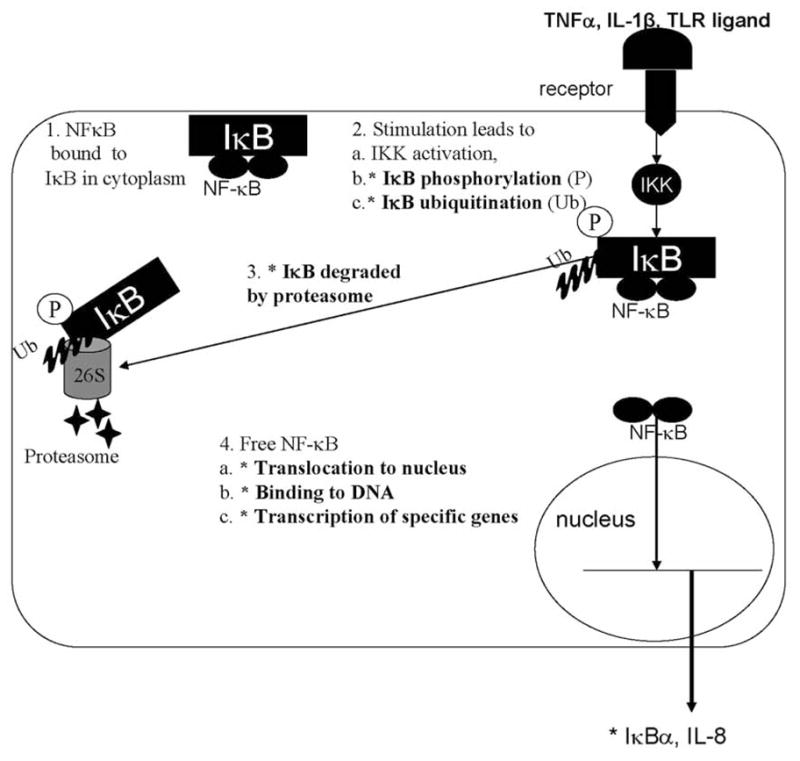
Nuclear factor kappa B signaling cascade. Asterisks denote steps with differential response to inflammatory stimuli in immature and mature intestinal epithelial cells.
POTENTIAL THERAPEUTIC OPTIONS FOR LIMITING INFLAMMATION IN NEC
NEC is a multi-factorial disease, leading to intestinal inflammation resulting in the signs and symptoms of disease. Many studies have investigated potential therapeutic agents to specifically limit the inflammatory cascade. Single studies in animal models of NEC have demonstrated the ability of various compounds to inhibit intestinal inflammatory injury when given prior to intestinal insult. These have included compounds to decrease NF-κB activation such as the NEMO-binding domain (NBD) peptide that selectively inhibits the critical upstream IκB kinase [78] within the NF-κB pathway and the peroxisome proliferation receptor γ (PPAR-γ) agonist 15d-PGJ2 [79, 80]. PPARγ has been shown to directly associate with the nuclear NF-κB p65 protein, promoting its export from the nucleus, thus limiting its duration of action [81, 82]. The macrophage deactivator semapimod [61], decreased incidence of NEC potentially via limitation of production of pro-inflammatory mediators such as HMGB-1. Pentoxyphylline which inhibits the effects of the pro-inflammatory cytokine TNFα has also been shown to decrease the incidence of NEC [83]. While these studies add insight to mechanism of intestinal injury, therapeutic potential is unclear. These compounds, which inhibit fundamental pathways important in many tissues besides the intestine, raise concerns for unwanted side effects or collateral damage in the developing gut. Furthermore, studies have noted efficacy with administration prior to intestinal insult, which may be difficult to determine in the clinical setting of a disease without a discrete point of injury.
As previously described, factors contributing to the development of intestinal inflammation include a pathogenic bacterial colonization balance, disruption of the intestinal barrier, and immature regulation of NF-κB signaling. Targeting these deficiencies may allow prevention of disease or limitation of injury prior to onset of clinical symptoms. Two areas of promise are human milk and probiotics.
Human milk has been shown to be protective against NEC. A clinical study by Lucas and Cole demonstrated a 10–20 fold reduced incidence of NEC in human milk fed preterm infants compared to formula fed infants [84]. Recent studies suggest a dose-related association between human milk feeding and decreased incidence of NEC [85]. Many of the same factors present in amniotic fluid are present in human breast milk, and vary in amount with changing gestational age of the infant. Human milk may provide necessary factors for the preterm infant in the extra-uterine environment in the same way that amniotic fluid provides for the fetus in the intra-uterine environment [86].
Probiotics are living microorganisms in food and dietary supplements which have beneficial health effects beyond their inherent nutritive value. Probiotics are most commonly Lactobacillus or Bifidobacterium strains, which are the dominant species in the intestinal microbiota of breast fed infants. Since bacteria are necessary for maturation of the intestine and appropriate containment of inflammatory responses. It may be important to influence bacterial colonization. Health may not hinge on the presence of bad vs. good bacteria but the creation of optimal bacterial communities versus pathogenic bacterial communities. Thus for a premature infant it has been suggested that optimizing the bacterial flora can enhance intestinal maturation and decrease the incidence of NEC [87]. Probiotics have been shown to be beneficial in treating inflammatory bowel diseases, rotavirus and antibiotic associated diarrhea, and C. difficile colitis [88–91].
Four recent clinical studies have suggested a beneficial effect of probiotics on the incidence of necrotizing enterocolitis. Hoyos et al. administered Infloran Berna7 containing a mixture of Lactobacillus acidophilus and Bifidobacterium infantis to all newborns admitted to the neonatal intensive care unit at Hospital Simon Bolivar in Colombia and compared incidence of NEC in this cohort to the incidence of NEC in historical controls from the previous year [92]. This study found a significantly lower incidence of NEC – 2.9% in the probiotic treated infants vs. 6.6 % in historical controls, and no adverse events. Lin et al. also evaluated the efficacy of the probiotic product Infloran in a prospective, masked randomized control trial of 367 infants <1500gm at China Medical University Hospital in Taiwan and documented decreased incidence of NEC (≥ Bell stage 2) from 5.3 to 1.1% (P = 0.04) with no reported adverse effects [93]. This trial was recently repeated as a prospective, blinded, randomized, multi-center controlled trial at 7 neonatal intensive care units in Taiwan and enrolled 434 infants <1500gm [94]. Patients received Infloran with a new formulation containing a combination of L. Acidophilus 109 cfu and Bifidobacterium bifidum 109 cfu and incidence of NEC (≥ Bell stage 2) was again decreased from 6.5% to 1.8% in the probiotic treatment group with no reported adverse events. Last, Binnun et al. conducted a trial at the Shaare Zedek Medical Center in Israel. 145 infants <1500gm were enrolled in a prospective, randomized, double blind study of ABC Dophilus containing B. infantis, Streptococcus thermophilus, and Bifidobacterium bifidus [95]. This study demonstrated a significant decrease in incidence of NEC (≥ Bell stage 2) from 14% to 1% (P=0.013) with no adverse effects.
Both human milk and probiotics have potential to influence many of the features of NEC pathogenesis including bacterial colonization and translocation, intestinal barrier dysfunction, and the intestinal inflammatory response.
Optimizing Bacterial Colonization
Many factors contribute to the acquisition of intestinal flora including mode of delivery, gestational age, exposure to antibiotics in either the mother or the infants, and importantly, feeding. In breast fed infants, Bifidobacterium is a primary organism with Lactobacillus and Streptococcus as minor components. In formula-fed infants, similar amounts of Bacteroides and Bifidobacterium are found with minor components of the more pathogenic species Staphylococcus, Escherichia coli, and Clostridia [96–100]. As previously noted, oligosaccharides on the surface of intestinal epithelial cells function as binding sites for bacteria, and have different carbohydrate residue patterns in immature vs. mature intestine, potentially contributing to a different adherent bacterial profile in preterm infants. Human milk also contains free and conjugated oligosaccharides known as glycans, which protect breast fed infants against infection by functioning as soluble receptors to inhibit pathogens from adhering to the gastrointestinal mucosa [101]. Human milk contains many other factors which may influence bacterial colonization including secretory IgA which can compensate for the decreased intestinal IgA in immature gut and the antimicrobial factors lysozyme and lactoferrin. [102–104]. Human milk has even been described as “Bifidogenic” - a reference to a complex interaction of proteins and growth factors which appear to function as prebiotics to promote the specific growth of beneficial Bifidobacterium species [105].
Probiotics may directly influence bacterial colonization patterns. Necessary conditions of care of the preterm infant such as frequent use of broad spectrum antibiotics, instrumentation, and formula feeding may predispose to altered bacterial balance. It has been suggested that probiotic treatment may improve this balance. Multiple studies have shown that different organisms have unique properties, and even different strains of the same organism may behave differently [106–108]. Probiotics may influence bacterial colonization patterns via the production of ammonia, hydrogen peroxide, and bacteriocins which directly inhibit the growth of pathogenic bacteria [109–114].
Strengthening of the Intestinal Barrier
Disruption of the intestinal barrier is a known aspect of NEC thought to lead to bacterial translocation, thus increasing inflammatory stimuli and furthering intestinal injury. Both disruption of the tight junctions between IEC as well as destruction of the cells themselves via direct oxidant injury, apoptosis, or necrosis have been linked to disruption of this key host defense mechanism. Several factors in breast milk and probiotics bacteria have demonstrated specific ability to improve barrier function.
Epidermal growth factor (EGF) is a heat and acid stable protein found in both amniotic fluid and breast milk in amounts that vary with gestational age [115]. It has been shown to have many trophic effects on the intestine. EGF receptors are present throughout the gastrointestinal tract on the basolateral membrane, which may be more accessible in the preterm intestine with increased permeability [116, 117]. It promotes intestinal growth, stimulates intestinal repair, and protects against intestinal epithelial cell apoptosis [118, 119]. EGF levels are decreased in the saliva and serum of patients with NEC versus nondiseased age matched controls. Animal studies have shown a decreased incidence and severity of NEC in a rat model treated with EGF associated with normalization of occludin and claudin 3 tight junction proteins [120, 121]. There has also been a case report of an infant with enterocolitis improving after treatment with intravenous EGF along with reports of improved intestinal repair measured in rectal biopsy samples of preterm infants with NEC receiving recombinant EGF [122, 115]. Heparin-binding EGF is another growth factor in the EGF family also found in both breast milk and amniotic fluid and upregulated in response to tissue damage. It has been shown to protect intestinal barrier function, protect against proinflammatory cytokine induced apoptosis, and induce intestinal barrier restitution following injury. In addition it has specifically been shown to decrease the incidence of NEC in animal models [123, 124].
Erythropoietin (Epo) is a glycoprotein originally described as a hormone important for red blood cell production. However, it is also present in both breast milk and amniotic fluid. Epo receptors have been demonstrated on human fetal and postnatal intestine. Enterally administered Epo appears to have limited systemic absorption, but rather acts locally [125]. It has been shown to increase small bowel length and villus surface area and decrease TNFα induced IEC apoptosis [126]. Interestingly a retrospective study of preterm infants showed a decreased incidence of NEC in infants given Epo compared to infants who did not receive Epo [127].
Glutamine is found in increasing amounts throughout lactation in human milk. It has been shown to maintain intestinal mucosal integrity, and glutamine deprivation has been shown in in vitro models to increase bacterial translocation. Studies have further specifically found decreases in the tight junction proteins claudin-1, occludin, and ZO-1 under conditions of glutamine deprivation [128].
Probiotics can also improve intestinal barrier function. The immature gut is “leaky” compared to mature IEC as demonstrated by permeability studies. One means of evaluating barrier function is measuring transepithelial resistance (TER). Studies have demonstrated that when intestinal cells are treated with commensal bacteria such as E. coli, Streptococcus thermophilus, Lactobacillus, and Bifidobacterium, TER increases, while treatment with pathogenic Salmonella decreases TER [129]. The specific combination of live Streptococcus thermophilus and Lactobacillus acidophilus has been shown in cell culture models to improve TER, associated with maintenance of tight junction protein phosphorylation [130]. Utilizing a model of directly measuring intestinal permeability to dextran or mannitol in the presence of different bacteria, it was similarly found that colonization with E. coli, Klebsiella, or Streptococcus viridans increased permeability while colonization with Lactobacillus brevis decreased permeability [131]. This has clinical relevance as a clinical study in infants demonstrated that when intestinal flora was manipulated to increase levels of anaerobic bacteria including Bifidobacterium, there were lower levels of serum endotoxin, suggesting decreased translocation of endotoxin producing organisms [132]. Furthermore, Ussing chamber experiments of transport of intact proteins, have demonstrated that Lactobacillus GG can specifically protect against increased intestinal permeability in suckling rats [133]. L. plantarum has also been shown to protect against E. coli induced intestinal permeability [134].
Modulation of the Inflammatory Response
Lastly, human milk and probiotics contain factors that can be viewed as anti inflammatory. Human milk specifically contains soluble TNF receptor, IL-1 receptor antagonist, and the PAF degrading enzyme acetylhydrolase [135]. In addition transforming growth factor beta and Epo in human milk have been shown to dampen the exaggerated IL-8 production of immature enterocytes exposed to TNFα or IL-1β [69].
Some probiotics also possess anti-inflammatory properties and specifically inhibit activity of NF-κB [136–138]. The previously cited clinical studies of probiotics and NEC were not sufficiently powered to fully assess the safety of administration of live bacteria to preterm infants. Thus, despite potential biologic plausibility for the use of probiotics in the treatment of NEC, safety is a primary concern. Live probiotic bacteria have the potential to become pathogenic when host defenses are compromised [139–142]. Studies reporting increased infant mortality in probiotic-treated animal models have raised questions as to the prudence of using live bacteria in relatively immunocompromised premature infants [143].
While some protective effects of probiotics require direct bacterial-epithelial cell-to-cell contact with live bacteria, other studies have shown that beneficial effects can be conferred by synthesized bioactive factors. Thus research is progressing to investigate the possibility of using secreted bacterial products to confer protection without the risk of live organisms. Conditioned media from probiotic organisms is the broth used to grow the probiotic bacteria subsequently filtered to remove all bacteria to result in a complex formulation of natural bioactive components synthesized and secreted by the probiotics in a solution that is bacteria-free. Particularly relevant to the premature infant with increased activation of NF-κB dependent inflammatory cytokine production, the combination of probiotic organisms called VSL#3 containing a mixture of Streptococcus, Lactobacillus and Bifidobacterium can decrease NF-κB activation by inhibition of the proteasome, preventing degradation of the inhibitor IκB [136]. Recent studies have demonstrated that conditioned media from Lactobacillus plantarum alone is similarly able to directly inhibit the chymotrypsin-like activity of the 26S proteasome [144]. Preserving IκB may be specifically important as this is a point of developmental differentiation between immature and mature IEC [33]. Other studies have shown that bacterial CpG-DNA can activate TLR 9 signaling in intestinal epithelial cells and dampen the LPS-mediated TLR 4 inflammatory signaling which has been shown to be important in NEC. Thus activation of TLR9 by bacterial DNA may be another means by which probiotics can offer protection, and suggests another potential bacterial product that could be harnessed separate from live bacteria as a therapeutic option [71, 145]. However, further research is required to determine which probiotic organisms are optimal and means of effecting the greatest benefit while minimizing harm.
CONCLUSIONS
Much work has been done in cell culture and animal models regarding possible mechanisms of injury which must now be translated to clinical utility. Future research must be focused on identifying means to predict which preterm infants are at greatest risk for this disease, an early point in the inflammatory cascade at which intervention can be beneficial prior to clinical symptoms, as well as means to appropriately mature host defense and inflammatory responses of the preterm infant gut in order to prevent this disease. Unique characteristics of the preterm gut predispose to an exaggerated inflammatory response. These include limited diversity and Proteobacteria predominance of the intestinal microbiota, immature intestinal host defense, and immature NF-κB regulation leading to exaggerated production of inflammatory cytokines. Identifying means of predicting which infants are at highest risk for this disease by microbial profiling would allow early intervention, perhaps utilizing probiotic agents to improve the microbial profile.
NEC is a multifactorial disease and likely requires a multifactorial intervention. A combination of human milk and probiotic factors targeting areas of susceptibility of the immature gut may be necessary. Breast milk feeding should be encouraged for all preterm infants. In addition much work remains to understand the beneficial aspects of human milk, as well as beneficial probiotic organisms for this patient population. Human studies to translate bench research into therapeutic options are still required. However, factors in human milk as well as factors secreted by probiotic bacteria hold great promise as means to mature responses, strengthen intestinal barrier function, as well as modulate inflammation to improve the health of preterm infants.
Acknowledgments
E. Claud has received funding from NIH HD043839, HD059123, AT004044, and the March of Dimes Basil O’Connor Starter Scholar Research Award.
ABBREVIATIONS
- NEC
Necrotizing enterocolitis
- NICU
Neonatal intensive care unit
- IEC
Intestinal epithelial cell
- JAM
Junction adhesion molecule
- ZO
Zonula occludin
- TJ
Tight junction
- TNF
Tumor necrosis factor
- DISC
Death inducing signal complex
- PAF
Platelet activating factor
- NO
Nitric oxide
- HMGB1
High mobility group box 1
- TLR
Toll-like receptor
- PAMP
Pathogen associated molecular pattern
- LPS
Lipopolysaccharide
- NF-κB
Nuclear factor kappaB
- IκB
Inhibitor of kappB
- NBD
NEMO binding domain
- PPARγ
Peroxisome proliferation receptor gamma
- EGF
Epidermal growth factor
- Epo
Erythropoietin
- TER
Transepithelial resistance
References
- 1.Martin JA, Kung HC, Mathews TJ, Hoyert DL, Strobino DM, Guyer B, Sutton SR. Annual summary of vital statistics: 2006. Pediatrics. 2008;121(4):788–801. doi: 10.1542/peds.2007-3753. [DOI] [PubMed] [Google Scholar]
- 2.Van Marter LJ, Dammann O, Allred EN, Leviton A, Pagano M, Moore M, Martin C. Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr. 2002;140(2):171–176. doi: 10.1067/mpd.2002.121381. [DOI] [PubMed] [Google Scholar]
- 3.Blahnik MJ, Ramanathan R, Riley CR, Minoo P. Lipopolysaccharide-induced tumor necrosis factor-alpha and IL-10 production by lung macrophages from preterm and term neonates. Pediatr Res. 2001;50(6):726–731. doi: 10.1203/00006450-200112000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Chheda S, Palkowetz KH, Garofalo R, Rassin DK, Goldman AS. Decreased interleukin-10 production by neonatal monocytes and T cells: relationship to decreased production and expression of tumor necrosis factor-alpha and its receptors. Pediatr Res. 1996;40(3):475–483. doi: 10.1203/00006450-199609000-00018. [DOI] [PubMed] [Google Scholar]
- 5.Jones CA, Cayabyab RG, Kwong KY, Stotts C, Wong B, Hamdan H, Minoo P, de Lemos RA. Undetectable interleukin (IL)-10 and persistent IL-8 expression early in hyaline membrane disease: a possible developmental basis for the predisposition to chronic lung inflammation in preterm newborns. Pediatr Res. 1996;39(6):966–975. doi: 10.1203/00006450-199606000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosloske AM. Epidemiology of necrotizing enterocolitis. Acta Paediatr Suppl. 1994;396:2–7. doi: 10.1111/j.1651-2227.1994.tb13232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacKendrick W, Caplan M. Necrotizing enterocolitis. New thoughts about pathogenesis and potential treatments. Pediatr Clin North Am. 1993;40(5):1047–1059. doi: 10.1016/s0031-3955(16)38622-9. [DOI] [PubMed] [Google Scholar]
- 8.Rayyis SF, Ambalavanan N, Wright L, Carlo WA. Randomized trial of “slow”; versus “fast”; feed advancements on the incidence of necrotizing enterocolitis in very low birth weight infants. J Pediatr. 1999;134(3):293–297. doi: 10.1016/s0022-3476(99)70452-x. [DOI] [PubMed] [Google Scholar]
- 9.Fitzgibbons SC, Ching Y, Yu D, Carpenter J, Kenny M, Weldon C, Lillehei C, Valim C, Horbar JD, Jaksic T. Mortality of necrotizing enterocolitis expressed by birth weight categories. J Pediatr Surg. 2009;44(6):1072–1075. doi: 10.1016/j.jpedsurg.2009.02.013. discussion 1075–1076. [DOI] [PubMed] [Google Scholar]
- 10.Ballance WA, Dahms BB, Shenker N, Kliegman RM. Pathology of neonatal necrotizing enterocolitis: a ten-year experience. J Pediatr. 1990;117(1 Pt 2):S6–13. doi: 10.1016/s0022-3476(05)81124-2. [DOI] [PubMed] [Google Scholar]
- 11.Faix RG, Adams JT. Neonatal necrotizing enterocolitis: current concepts and controversies. Adv Pediatr Infect Dis. 1994;9:1–36. [PubMed] [Google Scholar]
- 12.Hintz SR, Kendrick DE, Stoll BJ, Vohr BR, Fanaroff AA, Donovan EF, Poole WK, Blakely ML, Wright L, Higgins R. Neurodevelopmental and growth outcomes of extremely low birth weight infants after necrotizing enterocolitis. Pediatrics. 2005;115(3):696–703. doi: 10.1542/peds.2004-0569. [DOI] [PubMed] [Google Scholar]
- 13.Claud EC, Walker WA. Hypothesis: inappropriate colonization of the premature intestine can cause neonatal necrotizing enterocolitis. FASEB J. 2001;15(8):1398–1403. doi: 10.1096/fj.00-0833hyp. [DOI] [PubMed] [Google Scholar]
- 14.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Relman DA. The search for unrecognized pathogens. Science. 1999;284(5418):1308–1310. doi: 10.1126/science.284.5418.1308. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Hoenig JD, Malin KJ, Qamar S, Petrof EO, Sun J, Antonopoulos DA, Chang EB, Claud EC. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009 doi: 10.1038/ismej.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berseth CL. Gestational evolution of small intestine motility in preterm and term infants. J Pediatr. 1989;115(4):646–651. doi: 10.1016/s0022-3476(89)80302-6. [DOI] [PubMed] [Google Scholar]
- 18.Berseth CL. Neonatal small intestinal motility: motor responses to feeding in term and preterm infants. J Pediatr. 1990;117(5):777–782. doi: 10.1016/s0022-3476(05)83343-8. [DOI] [PubMed] [Google Scholar]
- 19.Berseth CL. Gut motility and the pathogenesis of necrotizing enterocolitis. Clin Perinatol. 1994;21(2):263–270. [PubMed] [Google Scholar]
- 20.Chu SH, Walker WA. Developmental changes in the activities of sialyl- and fucosyltransferases in rat small intestine. Biochim Biophys Acta. 1986;883(3):496–500. doi: 10.1016/0304-4165(86)90289-8. [DOI] [PubMed] [Google Scholar]
- 21.Dai D, Nanthakumar NN, Savidge TC, Newburg DS, Walker WA. Region-specific ontogeny of alpha-2,6-sialyltransferase during normal and cortisone-induced maturation in mouse intestine. Am J Physiol Gastrointest Liver Physiol. 2002;282(3):G480–490. doi: 10.1152/ajpgi.00531.2000. [DOI] [PubMed] [Google Scholar]
- 22.Dai D, Nanthkumar NN, Newburg DS, Walker WA. Role of oligosaccharides and glycoconjugates in intestinal host defense. J Pediatr Gastroenterol Nutr. 2000;30(Suppl 2):S23–33. [PubMed] [Google Scholar]
- 23.Snyder JD, Walker WA. Structure and function of intestinal mucin: developmental aspects. Int Arch Allergy Appl Immunol. 1987;82(3–4):351–356. doi: 10.1159/000234225. [DOI] [PubMed] [Google Scholar]
- 24.Udall JN., Jr Gastrointestinal host defense and necrotizing enterocolitis. J Pediatr. 1990;117(1 Pt 2):S33–43. doi: 10.1016/S0022-3476(05)81128-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eibl MM, Wolf HM, Furnkranz H, Rosenkranz A. Prevention of necrotizing enterocolitis in low-birth-weight infants by IgA-IgG feeding. N Engl J Med. 1988;319(1):1–7. doi: 10.1056/NEJM198807073190101. [DOI] [PubMed] [Google Scholar]
- 26.Roberts SA, Freed DL. Neonatal IgA secretion enhanced by breast feeding. Lancet. 1977;2(8048):1131. doi: 10.1016/s0140-6736(77)90576-1. [DOI] [PubMed] [Google Scholar]
- 27.Tan XD, Hsueh W, Chang H, Wei KR, Gonzalez-Crussi F. Characterization of a putative receptor for intestinal trefoil factor in rat small intestine: identification by in situ binding and ligand blotting. Biochem Biophys Res Commun. 1997;237(3):673–677. doi: 10.1006/bbrc.1997.7144. [DOI] [PubMed] [Google Scholar]
- 28.Sands BE, Podolsky DK. The trefoil peptide family. Annu Rev Physiol. 1996;58:253–273. doi: 10.1146/annurev.ph.58.030196.001345. [DOI] [PubMed] [Google Scholar]
- 29.Lin J, Holzman IR, Jiang P, Babyatsky MW. Expression of intestinal trefoil factor in developing rat intestine. Biol Neonate. 1999;76(2):92–97. doi: 10.1159/000014146. [DOI] [PubMed] [Google Scholar]
- 30.Ouellette AJ. Paneth cells and innate immunity in the crypt micro-environment. Gastroenterology. 1997;113(5):1779–1784. doi: 10.1053/gast.1997.v113.pm9352884. [DOI] [PubMed] [Google Scholar]
- 31.Salzman NH, Polin RA, Harris MC, Ruchelli E, Hebra A, Zirin-Butler S, Jawad A, Martin Porter E, Bevins CL. Enteric defensin expression in necrotizing enterocolitis. Pediatr Res. 1998;44(1):20–26. doi: 10.1203/00006450-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Rouwet EV, Heineman E, Buurman WA, ter Riet G, Ramsay G, Blanco CE. Intestinal permeability and carrier-mediated monosaccharide absorption in preterm neonates during the early postnatal period. Pediatr Res. 2002;51(1):64–70. doi: 10.1203/00006450-200201000-00012. [DOI] [PubMed] [Google Scholar]
- 33.Claud EC, Lu L, Anton PM, Savidge T, Walker WA, Cherayil BJ. Developmentally regulated IkappaB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc Natl Acad Sci USA. 2004;101(19):7404–7408. doi: 10.1073/pnas.0401710101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nusrat A, Turner JR, Madara JL. Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol. 2000;279(5):G851–857. doi: 10.1152/ajpgi.2000.279.5.G851. [DOI] [PubMed] [Google Scholar]
- 35.Clark JA, Doelle SM, Halpern MD, Saunders TA, Holubec H, Dvorak K, Boitano SA, Dvorak B. Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am J Physiol Gastrointest Liver Physiol. 2006;291(5):G938–949. doi: 10.1152/ajpgi.00090.2006. [DOI] [PubMed] [Google Scholar]
- 36.Metcalfe A, Streuli C. Epithelial apoptosis. Bioessays. 1997;19(8):711–720. doi: 10.1002/bies.950190812. [DOI] [PubMed] [Google Scholar]
- 37.McConkey DJ, Orrenius S. Signal transduction pathways in apoptosis. Stem Cells. 1996;14(6):619–631. doi: 10.1002/stem.140619. [DOI] [PubMed] [Google Scholar]
- 38.Torriglia A, Perani P, Brossas JY, Altairac S, Zeggai S, Martin E, Treton J, Courtois Y, Counis MF. A caspase-independent cell clearance program. The LEI/L-DNase II pathway. Ann NY Acad Sci. 2000;926:192–203. doi: 10.1111/j.1749-6632.2000.tb05612.x. [DOI] [PubMed] [Google Scholar]
- 39.Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res. 2004;55(4):622–629. doi: 10.1203/01.PDR.0000113463.70435.74. [DOI] [PubMed] [Google Scholar]
- 40.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 41.Lane JS, Todd KE, Lewis MP, Gloor B, Ashley SW, Reber HA, McFadden DW, Chandler CF. Interleukin-10 reduces the systemic inflammatory response in a murine model of intestinal ischemia/reperfusion. Surgery. 1997;122(2):288–294. doi: 10.1016/s0039-6060(97)90020-9. [DOI] [PubMed] [Google Scholar]
- 42.Barry MA, Eastman A. Endonuclease activation during apoptosis: the role of cytosolic Ca2+ and pH. Biochem Biophys Res Commun. 1992;186(2):782–789. doi: 10.1016/0006-291x(92)90814-2. [DOI] [PubMed] [Google Scholar]
- 43.Park HJ, Lyons JC, Ohtsubo T, Song CW. Acidic environment causes apoptosis by increasing caspase activity. Br J Cancer. 1999;80(12):1892–1897. doi: 10.1038/sj.bjc.6690617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rachmilewitz D, Eliakim R, Simon P, Ligumsky M, Karmeli F. Cytokines and platelet-activating factor in human inflamed colonic mucosa. Agents Actions. 1992:C32–36. [PubMed] [Google Scholar]
- 45.Sobhani I, Hochlaf S, Denizot Y, Vissuzaine C, Rene E, Benveniste J, Lewin MM, Mignon M. Raised concentrations of platelet activating factor in colonic mucosa of Crohn’s disease patients. Gut. 1992;33(9):1220–1225. doi: 10.1136/gut.33.9.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Denizot Y, Chaussade S, Nathan N, Colombel JF, Bossant MJ, Cherouki N, Benveniste J, Couturier D. PAF-acether and acetylhydrolase in stool of patients with Crohn’s disease. Dig Dis Sci. 1992;37(3):432–437. doi: 10.1007/BF01307739. [DOI] [PubMed] [Google Scholar]
- 47.Kald B, Smedh K, Olaison G, Sjodahl R, Tagesson C. Platelet-activating factor acetylhydrolase activity in intestinal mucosa and plasma of patients with Crohn’s disease. Digestion. 1996;57(6):472–477. doi: 10.1159/000201376. [DOI] [PubMed] [Google Scholar]
- 48.Caplan MS, Hedlund E, Adler L, Lickerman M, Hsueh W. The platelet-activating factor receptor antagonist WEB 2170 prevents neonatal necrotizing enterocolitis in rats. J Pediatr Gastroenterol Nutr. 1997;24(3):296–301. doi: 10.1097/00005176-199703000-00012. [DOI] [PubMed] [Google Scholar]
- 49.Rabinowitz SS, Dzakpasu P, Piecuch S, Leblanc P, Valencia G, Kornecki E. Platelet-activating factor in infants at risk for necrotizing enterocolitis. J Pediatr. 2001;138(1):81–86. doi: 10.1067/mpd.2001.110132. [DOI] [PubMed] [Google Scholar]
- 50.Amer MD, Hedlund E, Rochester J, Caplan MS. Platelet-activating factor concentration in the stool of human newborns: effects of enteral feeding and neonatal necrotizing enterocolitis. Biol Neonate. 2004;85(3):159–166. doi: 10.1159/000075306. [DOI] [PubMed] [Google Scholar]
- 51.Caplan MS, Sun XM, Hseuh W, Hageman JR. Role of platelet activating factor and tumor necrosis factor-alpha in neonatal necrotizing enterocolitis. J Pediatr. 1990;116(6):960–964. doi: 10.1016/s0022-3476(05)80661-4. [DOI] [PubMed] [Google Scholar]
- 52.Hwang S. Specific receptors of platelet-activating factor, receptor heterogeneity, and signal transduction mechanisms. J Lipid Mediat. 1990;2(3–4):123–158. [PubMed] [Google Scholar]
- 53.Hsueh W, Gonzalez-Crussi F. Ischemic bowel necrosis induced by platelet-activating factor: an experimental model. Methods Achiev Exp Pathol. 1988;13:208–239. [PubMed] [Google Scholar]
- 54.Caplan MS, Kelly A, Hsueh W. Endotoxin and hypoxia-induced intestinal necrosis in rats: the role of platelet activating factor. Pediatr Res. 1992;31(5):428–434. doi: 10.1203/00006450-199205000-00002. [DOI] [PubMed] [Google Scholar]
- 55.Hsueh W, Gonzalez-Crussi F, Arroyave JL, Anderson RC, Lee ML, Houlihan WJ. Platelet activating factor-induced ischemic bowel necrosis: the effect of PAF antagonists. Eur J Pharmacol. 1986;123(1):79–83. doi: 10.1016/0014-2999(86)90690-4. [DOI] [PubMed] [Google Scholar]
- 56.Wallace JL, Steel G, Whittle BJ, Lagente V, Vargaftig B. Evidence for platelet-activating factor as a mediator of endotoxin-induced gastrointestinal damage in the rat:effects of three platelet-activating factor antagonists. Gastroenterology. 1987;93(4):765–773. doi: 10.1016/0016-5085(87)90438-0. [DOI] [PubMed] [Google Scholar]
- 57.Lu J, Caplan MS, Saraf AP, Li D, Adler L, Liu X, Jilling T. Platelet-activating factor-induced apoptosis is blocked by Bcl-2 in rat intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;286(2):G340–350. doi: 10.1152/ajpgi.00182.2003. [DOI] [PubMed] [Google Scholar]
- 58.Lu J, Caplan MS, Li D, Jilling T. Polyunsaturated fatty acids block platelet-activating factor-induced phosphatidylinositol 3 kinase/Akt-mediated apoptosis in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008;294(5):G1181–1190. doi: 10.1152/ajpgi.00343.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Claud EC, Lu J, Wang XQ, Abe M, Petrof EO, Sun J, Nelson DJ, Marks J, Jilling T. Platelet-activating factor-induced chloride channel activation is associated with intracellular acidosis and apoptosis of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008;294(5):G1191–1200. doi: 10.1152/ajpgi.00318.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ford H, Watkins S, Reblock K, Rowe M. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg. 1997;32(2):275–282. doi: 10.1016/s0022-3468(97)90194-9. [DOI] [PubMed] [Google Scholar]
- 61.Zamora R, Grishin A, Wong C, Boyle P, Wang J, Hackam D, Upperman JS, Tracey KJ, Ford HR. High-mobility group box 1 protein is an inflammatory mediator in necrotizing enterocolitis: protective effect of the macrophage deactivator semapimod. Am J Physiol Gastrointest Liver Physiol. 2005;289(4):G643–652. doi: 10.1152/ajpgi.00067.2005. [DOI] [PubMed] [Google Scholar]
- 62.Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84(4):1045–1049. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Djeu JY, Matsushima K, Oppenheim JJ, Shiotsuki K, Blanchard DK. Functional activation of human neutrophils by recombinant monocyte-derived neutrophil chemotactic factor/IL-8. J Immunol. 1990;144(6):2205–2210. [PubMed] [Google Scholar]
- 64.Huber AR, Kunkel SL, Todd RF, 3rd, Weiss SJ. Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science. 1991;254(5028):99–102. doi: 10.1126/science.1718038. [DOI] [PubMed] [Google Scholar]
- 65.Edelson MB, Bagwell CE, Rozycki HJ. Circulating pro- and counterinflammatory cytokine levels and severity in necrotizing enterocolitis. Pediatrics. 1999;103(4 Pt 1):766–771. doi: 10.1542/peds.103.4.766. [DOI] [PubMed] [Google Scholar]
- 66.Nadler EP, Stanford A, Zhang XR, Schall LC, Alber SM, Watkins SC, Ford HR. Intestinal cytokine gene expression in infants with acute necrotizing enterocolitis: interleukin-11 mRNA expression inversely correlates with extent of disease. J Pediatr Surg. 2001;36(8):1122–1129. doi: 10.1053/jpsu.2001.25726. [DOI] [PubMed] [Google Scholar]
- 67.Morecroft JA, Spitz L, Hamilton PA, Holmes SJ. Plasma cytokine levels in necrotizing enterocolitis. Acta Paediatr Suppl. 1994;396:18–20. doi: 10.1111/j.1651-2227.1994.tb13235.x. [DOI] [PubMed] [Google Scholar]
- 68.Morecroft JA, Spitz L, Hamilton PA, Holmes SJ. Plasma interleukin-6 and tumour necrosis factor levels as predictors of disease severity and outcome in necrotizing enterocolitis. J Pediatr Surg. 1994;29(6):798–800. doi: 10.1016/0022-3468(94)90374-3. [DOI] [PubMed] [Google Scholar]
- 69.Claud EC, Savidge T, Walker WA. Modulation of human intestinal epithelial cell IL-8 secretion by human milk factors. Pediatr Res. 2003;53(3):419–425. doi: 10.1203/01.PDR.0000050141.73528.AD. [DOI] [PubMed] [Google Scholar]
- 70.Nanthakumar NN, Fusunyan RD, Sanderson I, Walker WA. Inflammation in the developing human intestine: A possible patho-physiologic contribution to necrotizing enterocolitis. Proc Natl Acad Sci USA. 2000;97(11):6043–6048. doi: 10.1073/pnas.97.11.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jilling T, Simon D, Lu Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol. 2006;177(5):3273–3282. doi: 10.4049/jimmunol.177.5.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 73.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388(6642):548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 74.Roff M, Thompson J, Rodriguez MS, Jacque JM, Baleux F, Arenzana-Seisdedos F, Hay RT. Role of IkappaBalpha ubiquitination in signal-induced activation of NFkappaB in vivo. J Biol Chem. 1996;271(13):7844–7850. doi: 10.1074/jbc.271.13.7844. [DOI] [PubMed] [Google Scholar]
- 75.Chiao PJ, Miyamoto S, Verma IM. Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci USA. 1994;91(1):28–32. doi: 10.1073/pnas.91.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259(5103):1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 77.Claud EC, Zhang X, Petrof EO, Sun J. Developmentally regulated tumor necrosis factor-alpha induced nuclear factor-kappaB activation in intestinal epithelium. Am J Physiol Gastrointest Liver Physiol. 2007;292(5):G1411–1419. doi: 10.1152/ajpgi.00557.2006. [DOI] [PubMed] [Google Scholar]
- 78.Kassinen A, Krogius-Kurikka L, Makivuokko H, Rinttila T, Paulin L, Corander J, Malinen E, Apajalahti J, Palva A. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology. 2007;133(1):24–33. doi: 10.1053/j.gastro.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 79.Baregamian N, Mourot JM, Ballard AR, Evers BM, Chung DH. PPAR-gamma agonist protects against intestinal injury during necrotizing enterocolitis. Biochem Biophys Res Commun. 2009;379(2):423–427. doi: 10.1016/j.bbrc.2008.11.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Plaen IG, Liu SX, Tian R, Neequaye I, May MJ, Han XB, Hsueh W, Jilling T, Lu J, Caplan MS. Inhibition of nuclear factor-kappaB ameliorates bowel injury and prolongs survival in a neonatal rat model of necrotizing enterocolitis. Pediatr Res. 2007;61(6):716–721. doi: 10.1203/pdr.0b013e3180534219. [DOI] [PubMed] [Google Scholar]
- 81.Shibolet O, Podolsky DK. TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1469–1473. doi: 10.1152/ajpgi.00531.2006. [DOI] [PubMed] [Google Scholar]
- 82.Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. 2004;5(1):104–112. doi: 10.1038/ni1018. [DOI] [PubMed] [Google Scholar]
- 83.Travadi J, Patole S, Charles A, Dvorak B, Doherty D, Simmer K. Pentoxifylline reduces the incidence and severity of necrotizing enterocolitis in a neonatal rat model. Pediatr Res. 2006;60(2):185–189. doi: 10.1203/01.pdr.0000228325.24945.ac. [DOI] [PubMed] [Google Scholar]
- 84.Lucas A, Cole TJ. Breast milk and neonatal necrotising enterocolitis. Lancet. 1990;336(8730):1519–1523. doi: 10.1016/0140-6736(90)93304-8. [DOI] [PubMed] [Google Scholar]
- 85.Meinzen-Derr J, Poindexter B, Wrage L, Morrow AL, Stoll B, Donovan EF. Role of human milk in extremely low birth weight infants’ risk of necrotizing enterocolitis or death. J Perinatol. 2009;29(1):57–62. doi: 10.1038/jp.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wagner CL. Amniotic fluid and human milk: a continuum of effect? J Pediatr Gastroenterol Nutr. 2002;34(5):513–514. doi: 10.1097/00005176-200205000-00007. [DOI] [PubMed] [Google Scholar]
- 87.Lawrence G, Bates J, Gaul A. Pathogenesis of neonatal necrotising enterocolitis. Lancet. 1982;1(8264):137–139. doi: 10.1016/s0140-6736(82)90383-x. [DOI] [PubMed] [Google Scholar]
- 88.Arvola T, Laiho K, Torkkeli S, Mykkanen H, Salminen S, Maunula L, Isolauri E. Prophylactic Lactobacillus GG reduces antibiotic-associated diarrhea in children with respiratory infections: a randomized study. Pediatrics. 1999;104(5):e64. doi: 10.1542/peds.104.5.e64. [DOI] [PubMed] [Google Scholar]
- 89.Gionchetti P, Rizzello F, Venturi A, Brigidi P, Matteuzzi D, Bazzocchi G, Poggioli G, Miglioli M, Campieri M. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119(2):305–309. doi: 10.1053/gast.2000.9370. [DOI] [PubMed] [Google Scholar]
- 90.Gionchetti P, Rizzello F, Venturi A, Campieri M. Probiotics in infective diarrhoea and inflammatory bowel diseases. J Gastroenterol Hepatol. 2000;15(5):489–493. doi: 10.1046/j.1440-1746.2000.02162.x. [DOI] [PubMed] [Google Scholar]
- 91.Surawicz CM. Probiotics, antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in humans. Best Pract Res Clin Gastroenterol. 2003;17(5):775–783. doi: 10.1016/s1521-6918(03)00054-4. [DOI] [PubMed] [Google Scholar]
- 92.Hoyos AB. Reduced incidence of necrotizing enterocolitis associated with enteral administration of Lactobacillus acidophilus and Bifidobacterium infantis to neonates in an intensive care unit. Int J Infect Dis. 1999;3(4):197–202. doi: 10.1016/s1201-9712(99)90024-3. [DOI] [PubMed] [Google Scholar]
- 93.Lin HC, Su BH, Chen AC, Lin TW, Tsai CH, Yeh TF, Oh W. Oral probiotics reduce the incidence and severity of necrotizing enterocolitis in very low birth weight infants. Pediatrics. 2005;115(1):1–4. doi: 10.1542/peds.2004-1463. [DOI] [PubMed] [Google Scholar]
- 94.Lin HC, Hsu CH, Chen HL, Chung MY, Hsu JF, Lien RI, Tsao LY, Chen CH, Su BH. Oral probiotics prevent necrotizing enterocolitis in very low birth weight preterm infants: a multicenter, randomized, controlled trial. Pediatrics. 2008;122(4):693–700. doi: 10.1542/peds.2007-3007. [DOI] [PubMed] [Google Scholar]
- 95.Bin-Nun A, Bromiker R, Wilschanski M, Kaplan M, Rudensky B, Caplan M, Hammerman C. Oral probiotics prevent necrotizing enterocolitis in very low birth weight neonates. J Pediatr. 2005;147(2):192–196. doi: 10.1016/j.jpeds.2005.03.054. [DOI] [PubMed] [Google Scholar]
- 96.Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, Welling GW. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr. 2000;30(1):61–67. doi: 10.1097/00005176-200001000-00019. [DOI] [PubMed] [Google Scholar]
- 97.Gewolb IH, Schwalbe RS, Taciak VL, Harrison TS, Panigrahi P. Stool microflora in extremely low birthweight infants. Arch Dis Child Fetal Neonatal Ed. 1999;80(3):F167–173. doi: 10.1136/fn.80.3.f167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rubaltelli FF, Biadaioli R, Pecile P, Nicoletti P. Intestinal flora in breast- and bottle-fed infants. J Perinat Med. 1998;26(3):186–191. doi: 10.1515/jpme.1998.26.3.186. [DOI] [PubMed] [Google Scholar]
- 99.Tomkins AM, Bradley AK, Oswald S, Drasar BS. Diet and the faecal microflora of infants, children and adults in rural Nigeria and urban U.K. J Hyg (Lond) 1981;86(3):285–293. doi: 10.1017/s0022172400069035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wold AE, Adlerberth I. Breast feeding and the intestinal micro-flora of the infant--implications for protection against infectious diseases. Adv Exp Med Biol. 2000;478:77–93. doi: 10.1007/0-306-46830-1_7. [DOI] [PubMed] [Google Scholar]
- 101.Morrow AL, Ruiz-Palacios GM, Jiang X, Newburg DS. Human-milk glycans that inhibit pathogen binding protect breast-feeding infants against infectious diarrhea. J Nutr. 2005;135(5):1304–1307. doi: 10.1093/jn/135.5.1304. [DOI] [PubMed] [Google Scholar]
- 102.Newburg DS. Oligosaccharides in human milk and bacterial colonization. J Pediatr Gastroenterol Nutr. 2000;30(Suppl 2):S8–17. [PubMed] [Google Scholar]
- 103.Goldman AS, Garza C, Nichols BL, Goldblum RM. Immunologic factors in human milk during the first year of lactation. J Pediatr. 1982;100(4):563–567. doi: 10.1016/s0022-3476(82)80753-1. [DOI] [PubMed] [Google Scholar]
- 104.Goldman AS, Goldblum RM, Hanson LA. Anti-inflammatory systems in human milk. Adv Exp Med Biol. 1990;262:69–76. doi: 10.1007/978-1-4613-0553-8_6. [DOI] [PubMed] [Google Scholar]
- 105.Coppa GV, Zampini L, Galeazzi T, Gabrielli O. Prebiotics in human milk: a review. Dig Liver Dis. 2006;38(Suppl 2):S291–294. doi: 10.1016/S1590-8658(07)60013-9. [DOI] [PubMed] [Google Scholar]
- 106.Christensen HR, Frokiaer H, Pestka JJ. Lactobacilli differentially modulate expression of cytokines and maturation surface markers in murine dendritic cells. J Immunol. 2002;168(1):171–178. doi: 10.4049/jimmunol.168.1.171. [DOI] [PubMed] [Google Scholar]
- 107.Yan F, Polk DB. Probiotic bacterium prevents cytokine-induced apoptosis in intestinal epithelial cells. J Biol Chem. 2002;277(52):50959–50965. doi: 10.1074/jbc.M207050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291(5505):881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 109.Annuk H, Shchepetova J, Kullisaar T, Songisepp E, Zilmer M, Mikelsaar M. Characterization of intestinal lactobacilli as putative probiotic candidates. J Appl Microbiol. 2003;94(3):403–412. doi: 10.1046/j.1365-2672.2003.01847.x. [DOI] [PubMed] [Google Scholar]
- 110.Braude AI, Siemienski JS. The influence of bacteriocins on resistance to infection by gram-negative bacteria. II. Colicin action, transfer of colicinogeny, and transfer of antibiotic resistance in urinary infections. J Clin Invest. 1968;47(8):1763–1773. doi: 10.1172/JCI105866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cleveland J, Montville TJ, Nes IF, Chikindas ML. Bacteriocins: safe, natural antimicrobials for food preservation. Int J Food Microbiol. 2001;71(1):1–20. doi: 10.1016/s0168-1605(01)00560-8. [DOI] [PubMed] [Google Scholar]
- 112.Kullisaar T, Zilmer M, Mikelsaar M, Vihalemm T, Annuk H, Kairane C, Kilk A. Two antioxidative lactobacilli strains as promising probiotics. Int J Food Microbiol. 2002;72(3):215–224. doi: 10.1016/s0168-1605(01)00674-2. [DOI] [PubMed] [Google Scholar]
- 113.Ocana VS, Pesce de Ruiz Holgado AA, Nader-Macias ME. Selection of vaginal H2O2-generating Lactobacillus species for probiotic use. Curr Microbiol. 1999;38(5):279–284. doi: 10.1007/pl00006802. [DOI] [PubMed] [Google Scholar]
- 114.Paraje MG, Albesa I, Eraso AJ. Conservation in probiotic preparations of Lactobacillus with inhibitory capacity on other species. New Microbiol. 2000;23(4):423–431. [PubMed] [Google Scholar]
- 115.Sullivan PB, Lewindon PJ, Cheng C, Lenehan PF, Kuo BS, Haskins JR, Goodlad RA, Wright NA, de la Iglesia FA. Intestinal mucosa remodeling by recombinant human epidermal growth factor(1–48) in neonates with severe necrotizing enterocolitis. J Pediatr Surg. 2007;42(3):462–469. doi: 10.1016/j.jpedsurg.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 116.Fagbemi AO, Wright N, Lakhoo K, Edwards AD. Immunoreactive epidermal growth factor receptors are present in gastrointestinal epithelial cells of preterm infants with necrotising enterocolitis. Early Hum Dev. 2001;65(1):1–9. doi: 10.1016/s0378-3782(01)00164-5. [DOI] [PubMed] [Google Scholar]
- 117.Menard D, Pothier P. Radioautographic localization of epidermal growth factor receptors in human fetal gut. Gastroenterology. 1991;101(3):640–649. doi: 10.1016/0016-5085(91)90520-u. [DOI] [PubMed] [Google Scholar]
- 118.Swaniker F, Guo W, Fonkalsrud EW, Diamond J. The effect of epidermal growth factor on mucosal function after ileal resection. J Surg Res. 1995;58(6):565–569. doi: 10.1006/jsre.1995.1089. [DOI] [PubMed] [Google Scholar]
- 119.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376(6538):337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 120.Shin CE, Falcone RA, Jr, Stuart L, Erwin CR, Warner BW. Diminished epidermal growth factor levels in infants with necrotizing enterocolitis. J Pediatr Surg. 2000;35(2):173–176. doi: 10.1016/s0022-3468(00)90005-8. discussion 177. [DOI] [PubMed] [Google Scholar]
- 121.Dvorak B, Halpern MD, Holubec H, Williams CS, McWilliam DL, Dominguez JA, Stepankova R, Payne CM, McCuskey RS. Epidermal growth factor reduces the development of necrotizing enterocolitis in a neonatal rat model. Am J Physiol Gastroin-test Liver Physiol. 2002;282(1):G156–164. doi: 10.1152/ajpgi.00196.2001. [DOI] [PubMed] [Google Scholar]
- 122.Sullivan PB, Brueton MJ, Tabara ZB, Goodlad RA, Lee CY, Wright NA. Epidermal growth factor in necrotising enteritis. Lancet. 1991;338(8758):53–54. doi: 10.1016/0140-6736(91)90042-n. [DOI] [PubMed] [Google Scholar]
- 123.Feng J, El-Assal ON, Besner GE. Heparin-binding epidermal growth factor-like growth factor decreases the incidence of necrotizing enterocolitis in neonatal rats. J Pediatr Surg. 2006;41(1):144–149. doi: 10.1016/j.jpedsurg.2005.10.018. discussion 144-149. [DOI] [PubMed] [Google Scholar]
- 124.Yu X, Radulescu A, Zorko N, Besner GE. Heparin-binding EGF-like growth factor increases intestinal microvascular blood flow in necrotizing enterocolitis. Gastroenterology. 2009;137(1):221–230. doi: 10.1053/j.gastro.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Juul SE, Ledbetter DJ, Joyce AE, Dame C, Christensen RD, Zhao Y, DeMarco V. Erythropoietin acts as a trophic factor in neonatal rat intestine. Gut. 2001;49(2):182–189. doi: 10.1136/gut.49.2.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Juul SE, Joyce AE, Zhao Y, Ledbetter DJ. Why is erythro-poietin present in human milk? Studies of erythropoietin receptors on enterocytes of human and rat neonates. Pediatr Res. 1999;46(3):263–268. doi: 10.1203/00006450-199909000-00003. [DOI] [PubMed] [Google Scholar]
- 127.Ledbetter DJ, Juul SE. Erythropoietin and the incidence of necrotizing enterocolitis in infants with very low birth weight. J Pediatr Surg. 2000;35(2):178–181. doi: 10.1016/s0022-3468(00)90006-x. discussion 182. [DOI] [PubMed] [Google Scholar]
- 128.Li N, Lewis P, Samuelson D, Liboni K, Neu J. Glutamine regulates Caco-2 cell tight junction proteins. Am J Physiol Gastroin-test Liver Physiol. 2004;287(3):G726–733. doi: 10.1152/ajpgi.00012.2004. [DOI] [PubMed] [Google Scholar]
- 129.Otte JM, Podolsky DK. Functional modulation of enterocytes by gram-positive and gram-negative microorganisms. Am J Physiol Gastrointest Liver Physiol. 2004;286(4):G613–626. doi: 10.1152/ajpgi.00341.2003. [DOI] [PubMed] [Google Scholar]
- 130.Resta-Lenert S, Barrett KE. Live probiotics protect intestinal epithelial cells from the effects of infection with enteroinvasive Escherichia coli (EIEC) Gut. 2003;52(7):988–997. doi: 10.1136/gut.52.7.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Garcia-Lafuente A, Antolin M, Guarner F, Crespo E, Malagelada JR. Modulation of colonic barrier function by the composition of the commensal flora in the rat. Gut. 2001;48(4):503–507. doi: 10.1136/gut.48.4.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Urao M, Fujimoto T, Lane GJ, Seo G, Miyano T. Does probiotics administration decrease serum endotoxin levels in infants? J Pediatr Surg. 1999;34(2):273–276. doi: 10.1016/s0022-3468(99)90189-6. [DOI] [PubMed] [Google Scholar]
- 133.Isolauri E, Majamaa H, Arvola T, Rantala I, Virtanen E, Arvilommi H. Lactobacillus casei strain GG reverses increased intestinal permeability induced by cow milk in suckling rats. Gastroenterology. 1993;105(6):1643–1650. doi: 10.1016/0016-5085(93)91059-q. [DOI] [PubMed] [Google Scholar]
- 134.Mangell P, Nejdfors P, Wang M, Ahrne S, Westrom B, Thorlacius H, Jeppsson B. Lactobacillus plantarum 299v inhibits Escherichia coli-induced intestinal permeability. Dig Dis Sci. 2002;47(3):511–516. doi: 10.1023/a:1017947531536. [DOI] [PubMed] [Google Scholar]
- 135.Moya FR, Eguchi H, Zhao B, Furukawa M, Sfeir J, Osorio M, Ogawa Y, Johnston JM. Platelet-activating factor acetylhydrolase in term and preterm human milk: a preliminary report. J Pediatr Gastroenterol Nutr. 1994;19(2):236–239. doi: 10.1097/00005176-199408000-00015. [DOI] [PubMed] [Google Scholar]
- 136.Petrof EO, Kojima K, Ropeleski MJ, Musch MW, Tao Y, De Simone C, Chang EB. Probiotics inhibit nuclear factor-kappaB and induce heat shock proteins in colonic epithelial cells through proteasome inhibition. Gastroenterology. 2004;127(5):1474–1487. doi: 10.1053/j.gastro.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 137.Lammers KM, Helwig U, Swennen E, Rizzello F, Venturi A, Caramelli E, Kamm MA, Brigidi P, Gionchetti P, Campieri M. Effect of probiotic strains on interleukin 8 production by HT29/19A cells. Am J Gastroenterol. 2002;97(5):1182–1186. doi: 10.1111/j.1572-0241.2002.05693.x. [DOI] [PubMed] [Google Scholar]
- 138.Jijon H, Backer J, Diaz H, Yeung H, Thiel D, McKaigney C, De Simone C, Madsen K. DNA from probiotic bacteria modulates murine and human epithelial and immune function. Gastroenterology. 2004;126(5):1358–1373. doi: 10.1053/j.gastro.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 139.Antony SJ. Lactobacillemia: an emerging cause of infection in both the immunocompromised and the immunocompetent host. J Natl Med Assoc. 2000;92(2):83–86. [PMC free article] [PubMed] [Google Scholar]
- 140.Horwitch CA, Furseth HA, Larson AM, Jones TL, Olliffe JF, Spach DH. Lactobacillemia in three patients with AIDS. Clin Infect Dis. 1995;21(6):1460–1462. doi: 10.1093/clinids/21.6.1460. [DOI] [PubMed] [Google Scholar]
- 141.Schlegel L, Lemerle S, Geslin P. Lactobacillus species as opportunistic pathogens in immunocompromised patients. Eur J Clin Microbiol Infect Dis. 1998;17(12):887–888. doi: 10.1007/s100960050216. [DOI] [PubMed] [Google Scholar]
- 142.De Groote MA, Frank DN, Dowell E, Glode MP, Pace NR. Lactobacillus rhamnosus GG bacteremia associated with probiotic use in a child with short gut syndrome. Pediatr Infect Dis J. 2005;24(3):278–280. doi: 10.1097/01.inf.0000154588.79356.e6. [DOI] [PubMed] [Google Scholar]
- 143.Wagner RD, Warner T, Roberts L, Farmer J, Balish E. Colonization of congenitally immunodeficient mice with probiotic bacteria. Infect Immun. 1997;65(8):3345–3351. doi: 10.1128/iai.65.8.3345-3351.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Petrof EO, Claud EC, Sun J, Abramova T, Guo Y, Waypa TS, He SM, Nakagawa Y, Chang EB. Bacteria-free solution derived from lactobacillus plantarum inhibits multiple NF-kappaB pathways and inhibits proteasome function. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.20930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gribar SC, Sodhi CP, Richardson WM, Anand RJ, Gittes GK, Branca MF, Jakub A, Shi XH, Shah S, Ozolek JA, Hackam DA. Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J Immunol. 2009;182(1):636–646. doi: 10.4049/jimmunol.182.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Claud EC, Caplan M. Necrotizing Enterocolitis. In: Walker G, Kleinman S, Shneider S, editors. Pediatric Gastrointestinal Disease Pathophysiology, Diagnosis, Management. 4. BC Decker; Canada: 2004. pp. 871–879. [Google Scholar]