

Abstract
With the trend in molecular epidemiology towards both genome-wide association studies and complex modelling, the need for large sample sizes to detect small effects and to allow for the estimation of many parameters within a model continues to increase. Unfortunately, most methods of association analysis have been restricted to either a family-based or a case-control design, resulting in the lack of synthesis of data from multiple studies. Transmission disequilibrium-type methods for detecting linkage disequilibrium from family data were developed as an effective way of preventing the detection of association due to population stratification. Because these methods condition on parental genotype, however, they have precluded the joint analysis of family and case-control data, although methods for case-control data may not protect against population stratification and do not allow for familial correlations. We present here an extension of a family-based association analysis method for continuous traits that will simultaneously test for, and if necessary control for, population stratification. We further extend this method to analyse binary traits (and therefore family and case-control data together) and accurately to estimate genetic effects in the population, even when using an ascertained family sample. Finally, we present the power of this binary extension for both family-only and joint family and case-control data, and demonstrate the accuracy of the association parameter and variance components in an ascertained family sample.
Keywords: ascertainment correction, family-based association, linkage disequilibrium
Introduction
For much of the past three decades, linkage analysis has been the primary tool for the initial exploration of complex diseases believed to have an underlying genetic aetiology and has resulted in many large cohorts of family data with DNA samples available. Unfortunately, however, the ability of linkage analysis to localise potentially segregating susceptibility or protective genotypes has been limited to, at best, regions of 5-10 centimorgans (cM) in length and, at worst, 20 cM in length [1]. This limitation has led to a rise in popularity of methods for detecting allelic (or gametic) association in candidate genes, in candidate linkage regions or genome-wide. This allelic association, coupled with linkage, allows for much more precise localisation of regions housing disease genes because, if it is due to linkage disequilibrium (LD), it will span a much shorter distance within the genome than is usually found by linkage analysis. With this rise in association studies, there has been a trend toward the collection of unrelated case-control samples, often with the abandonment of large family studies. Certainly, these samples are much easier to obtain than are family samples, but they also have certain limitations, even within the context of recent genome-wide association successes [2,3]. Further, allelic association can be due to factors other than LD (which we define as the combination of allelic association and linkage) or pleiotropy (a marker allele itself being involved in the aetiology of the disease) [4].
Population stratification, which exists when multiple strata within a given sample differ with respect to either the underlying trait distribution or the marker genotype distribution (and which leads to spurious association when it occurs with respect to both), is a commonly cited cause of false-positive findings in case-control association studies (eg Knowler et al. [5] ) and the most likely cause in genetic epidemiological studies. This threat of increased type I error rate has led to the development of many methods that guard against the effects of population stratification. The first two general classes use unlinked loci and can both be subsumed under the term 'genomic control': (1) test for population stratification using unlinked regions of the genome; (2) allow for the population stratification, as estimated from unlinked regions of the genome when performing an analysis of allelic association. The third general class guards against population stratification by using non-transmitted alleles as controls (ie a case-control design perfectly matched for ethnicity by appropriately using family data). While these methods are effective in controlling for population stratification, they each have their limitations with respect to power, efficiency and flexibility.
The limitations of genomic control methods [6-8] are the requirement of having genotypes at many loci unlinked to the disease allele. In the context of a genome-wide association scan, the choice of the best regions to use as a 'control' is difficult, as there is no guarantee that the markers being used are indeed unlinked to a disease gene. Applying this method to a candidate gene study suffers from the same limitation, but also requires significant additional cost and labour to type enough (and how many is enough?) additional loci.
Transmission disequilibrium tests (TDTs) - as they were termed by Spielman et al. [9] and are commonly referred to - comprise, in general, a unique study design (rather than a single statistical test) that protects against the effects of population stratification by comparing the frequencies of alleles (haplotypes or genotypes) transmitted from parents to their affected children with the frequencies of non-transmitted alleles to these same children. These tests of allelic association condition on, at least, parental genotypes and offspring disease phenotypes. Many TDT-type designs have been suggested since first proposed by Rubinstein et al., [10] including extensions for multiple siblings, missing parents and extended pedigrees - to name but a few (see Table S1 (Table 5)). All of these extensions, however, retain conditioning on part of the data available and therefore share the following limitations: (1) conditional tests are sensitive to sampling strategy, leading to very low power under several conditions; [11] and (2) missing parental data, transmissions from homozygous parents - or from heterozygous parents to heterozygous children - are non-informative, which results in a dramatic reduction of effective sample size and therefore of power, particularly when analysing single nucleotide polymorphism (SNP) data. This may also lead to an increased type I error rate if care is not taken to include the transmissions from two similarly heterozygous parents when the child is heterozygous [12]. Further, as for all tests of allelic association, the power of TDT-type designs rapidly decreases if the marker is not the disease locus and/or if the marker and disease allele frequencies differ [13-15].
Table S1.
Summary of TDT-type methods and their respective features
| Incorporation of: | ||||||||
|---|---|---|---|---|---|---|---|---|
| Method/Reference | Missing parents | Multiple alleles | Parental phenotypes | Quantitative traits | Extended pedigrees | Different family structures | Multiple markers | Covariates |
| Curtis (1997)S1 | * | * | ||||||
| S-TDT (Spielman and Ewens, 1998)S2 | * | * | * | |||||
| DAT (Boehnke and Langefeld, 1998)S3 | * | * | ||||||
| SDT (Horvath and Laird, 1998)S4 | * | * | * | |||||
| NFS (Whittemore and Tu, 2000)S5 | * | * | * | * | * | |||
| TRANSMIT (Clayton, 1999)S6 | * | * | * | * | ||||
| RC-TDT (Knapp, 1999)S7 | * | |||||||
| 1-TDT (Sun et al., 1999)S8 | * | * | ||||||
| Martin et al. (1997)S9 | * | * | * | |||||
| George et al. (1999)S10 | * | * | * | * | * | |||
| P-TDT (Abecasis et al., 2000)S11 | * | * | * | * | * | |||
| Bickeboller and Clerget-Darpoux(1995)S12 | * | |||||||
| Spielman and Ewens (1996)S13 | * | |||||||
| Purcell et al. (2005)S14 | * | * | * | |||||
| TDT(max) (Morris, 1997)S15 | * | |||||||
| Lazzeroni and Lange (1998)S16 | * | * | ||||||
| Monks and Kaplan(2000)S17 | * | * | * | * | ||||
| Xiong et al. (1998)S18 | * | * | ||||||
| Fan and Jung(2002)S19 | * | * | * | * | ||||
| TDT(Q1) - TDT(Q5) (Allison, 1997)S20, | * | * | ||||||
| Rabinowitz (1997)S21 | * | * | * | |||||
| Allison et al. (1999)S22 | * | * | * | |||||
| Sun et al. (2000)S23 | * | * | * | * | ||||
| Schaid and Rowlands (2000)S24 | * | * | * | |||||
| Waldman et al. (1999)S25 | * | * | ||||||
| Sinsheimer et al. (2000)S26 | * | * | * | * | * | |||
| Kistner and Weinberg (2004)S27 | * | * | * | |||||
| QTDT (Abecasis et al., 2000)S28 | * | * | * | * | ||||
| Zhu and Elston(2001)S29 | * | * | * | * | ||||
| PDT (Martin, 2000)S30 | * | * | ||||||
| Goring and Terwilliger(2000)S31 | * | * | ||||||
| Clayton and Jones(1999)S32 | * | * | ||||||
| ETDT (Sham and Curtis, 1995)S33 | * | |||||||
| TDT-EX (Cleves et al., 1997)S34 | * | * | ||||||
| Fulker (1999)S35 | * | * | * | |||||
| Fan et al. (2002)S36 | * | * | * | |||||
Table S1 References
S1. Curtis D. (1997), 'Use of siblings as controls in case-control association studies', Ann. Hum. Genet. vol. 61, No. 4, July, pp. 319-333.
S2. Spielman R.S. and Ewens W.J. (1998), 'A sibship test for linkage in the presence of association: The sib transmission/disequilibrium test', Am. J. Hum. Genet. vol. 62, No. 2, February, pp. 450-458.
S3. Boehnke M. and Langefeld C.D. (1998), 'Genetic association mapping based on discordant sib pairs: The discordant-alleles test', Am. J. Hum. Gene. vol. 62, No. 4, April, pp. 950-961.
S4. Horvath S. and Laird N.M. (1998), 'A discordant-sibship test for disequilibrium and linkage: No need for parental data', Am. J. Hum. Genet. vol. 63, No. 6, December, pp. 1886-1897.
S5. Whittemore A.S. and Tu I.P. (2000), 'Detection of disease genes by use of family data. I. Likelihood-based theory', Am. J. Hum. Genet. vol. 66, No. 4, April, pp. 1328-1340.
S6. Clayton D. (1999), 'A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission', Am. J. Hum. Genet. vol. 65, No. 4, October, pp. 1170-1177.
S7. Knapp M. (1999), 'The transmission/disequilibrium test and parental genotype reconstruction: The reconstruction-combined transmission/disequilibrium test', Am. J. Hum. Genet. vol. 64, No. 3, March, pp. 861-870.
S8. Sun F., Flanders W., Yang Q. and Khoury M. (1999), 'Transmission disequilbrium test (TDT) when only one parent is available: The 1-TDT', Am. J. Epidemiol. vol. 150, pp. 97-104.
S9. Martin E.R., Kaplan N.L. and Weir B.S. (1997), 'Tests for linkage and association in nuclear families', Am. J. Hum. Genet. Vol 61, No. 2, August, pp. 439-448.
S10. George V., Tiwari H.K., Zhu X. and Elston R.C. (1999), 'A test of transmission/disequilibrium for quantitative traits in pedigree data, by multiple regression', Am. J. Hum. Genet. vol. 65, No. 1, July, pp. 236-245.
S11. Abecasis G.R., Cookson W.O. and Cardon L.R. (2000), 'Pedigree tests of transmission disequilibrium', Eur. J. Hum. Genet. vol. 8, No. 7, pp. 545-551.
S12. Bickeboller H. and Clerget-Darpoux F. (1995), 'Statistical properties of the allelic and genotypic transmission/disequilibrium test for multiallelic markers', Genet. Epidemiol. vol. 12, No. 6, pp. 865-870.
S13. Spielman R.S. and Ewens W.J. (1996), 'The TDT and other family-based tests for linkage disequilibrium and association', Am. J. Hum. Genet. vol. 59, No. 5, November, pp. 983-989.
S14. Purcell S., Sham P.C. and Daly M.J. (2005), 'Parental phenotypes in family-based association analysis', Am. J. Hum. Genet. vol. 76, No. 2, pp. 249-259.
S15. Morris A.P., Curnow R.N. and Whittaker J.C. (1997), 'Randomization tests of disease-marker associations', Ann. Hum. Genet. vol. 61, No. 1, January, pp. 49-60.
S16. Lazzeroni L.C. and Lange K. (1998), 'A conditional inference framework for extending the transmission/disequilibrium test', Hum. Hered. vol. 48, No. 2, March, pp. 67-81.
S17. Monks S.A. and Kaplan N.L. (2000), 'Removing the sampling restrictions from family-based tests of association for a quantitative-trait locus', Am. J. Hum. Genet. vol. 66, No. 2, February, pp. 576-592.
S18. Xiong M.M., Krushkal J. and Boerwinkle E. (1998), 'TDT statistics for mapping quantitative trait loci', Ann. Hum. Genet. vol. 62, No. 5, September, pp. 431-452.
S19. Fan R. and Jung J. (2002), 'Association studies of QTL for multi-allele markers by mixed models', Hum. Hered. vol. 54, No. 3, pp. 132-150.
S20. Allison D.B. (1997), 'Transmission-disequilibrium tests for quantitative traits', Am. J. Hum. Genet. vol. 60, No. 3, March, pp. 676-690.
S21. Rabinowitz D. (1997), 'A transmission disequilibrium test for quantitative trait loci', Hum. Hered. vol. 47, No. 6, November, pp. 342-350.
S22. Allison D.B. Neale M.C., Zannolli R., Schork N.J. et al. (1999), 'The robustness of the likelihood-ratio test in a variance-component quantitative-trait loci-mapping procedure', Am. J. Hum. Genet. vol. 65, No. 2, August, pp. 531-544.
S23. Sun F., Flanders W., Yang Q. and Zhao H. (2000), 'Transmission/disequilibrium tests for quantitative traits', Ann. Hum. Genet. vol. 64, pp. 555-565.
S24. Schaid D.J. and Rowland C.M. (2000), 'Robust transmission regression models for linkage and association', Genet. Epidemiol. vol. 19, Suppl. 1. pp. S78-S84.
S25. Waldman I.D., Robinson B.F. and Rowe D.C. (1999), 'A logistic regression based extension of the TDT for continuous and categorical traits', Ann. Hum. Genet. vol. 63, No. 4, July, pp. 329-340.
S26. Sinsheimer J.S., Blangero J. and Lange K. (2000), 'Gamete-competition models', Am. J. Hum. Gene. vol. 66, No. 3, March, pp. 1168-1172.
S27. Kistner E.O. and Weinberg C.R. (2004), 'Method for using complete and incomplete trios to identify genes related to a quantitative trait', Genet. Epidemiol. vol. 27, No. 1, July, pp. 33-42.
S28. Abecasis G.R., Cardon L.R. and Cookson W.O. (2000), 'A general test of association for quantitative traits in nuclear families', Am. J. Hum. Genet. vol. 66, pp. 279-292.
S29. Zhu X. and Elston R.C. (2001), 'Transmission/disequilibrium tests for quantitative traits', Genet. Epidemiol. vol. 20, No. 1, January, pp. 57-74.
S30. Martin E.R., Monks S.A., Warren L.L. and Kaplan N.L. (2000), 'A test for linkage and association in general pedigrees: The pedigree disequilibrium test', Am. J. Hum. Genet. vol. 67, No. 1, pp. 146-154.
S31. Goring H.H. and Terwilliger J.D. (2000), 'Linkage analysis in the presence of errors IV: Joint pseudomarker analysis of linkage and/or linkage disequilibrium on a mixture of pedigrees and singletons when the mode of inheritance cannot be accurately specified', Am. J. Hum. Genet. vol. 66, No. 14, pp. 1310-1327.
S32. Clayton D. and Jones H. (1999), 'Transmission/disequilibrium tests for extended marker haplotypes', Am. J. Hum. Genet. vol. 65, No. 4, October, pp. 1161-1169.
S33. Sham P.C. and Curtis D. (1995), 'An extended transmission/disequilbrium test (TDT) for multiallele marker loci', An. Hum. Genet. vol. 59, Nos. 53/323, p. 336.
S34. Cleves M.A., Olson J.M. and Jacobs K.B. (1997), 'Exact transmission disequilibrium tests with multiallelic markers', Genet. Epidemiol. vol. 14, No. 4, pp. 337-347.
S35. Fulker D.W., Sham P.C. and Hewitt J.K. (1999), 'Combined linkage and association sib-pair analysis for quantitative traits', Am. J. Hum. Genet. vol. 64, pp. 259-267.
S36. Fan R., Floros J. and Xiong M. (2002), 'Models and tests of linkage and association studies of quantitative trait locus for multi-allele marker loci', Hum. Hered. vol. 53, No. 3, pp. 130-145.
Novel methodological approaches for the analysis of LD in family data include a class of variance component approaches and what are termed family-based association tests (FBATs). Fulker first proposed a test for both between-family association (or, more appropriately, 'among-family', as we typically expect more than two families), which models the phenotypic means given the marker locus genotypes, and within-family association (linkage) by using identity-by-descent status in modelling the sib-pair variance-covariance structure [16]. It was shown that the within-family component provides an estimate of the additive genetic effect unaffected by population stratification. Sham et al. [17] extended this method to incorporate larger sibships, dominance variance and multi-allelic markers. It was further extended to sibships with or without parental genotypes, and to multi-generational pedigree data by Abecasis et al. [18] FBAT is a unified approach to family-based tests of association that 'compares tests for association to their conditional distributions given the minimal sufficient statistics under the null hypothesis for the genetic model, sampling plan and population admixture', [19] in two steps: (1) building a test statistic that is sensitive to the co-variation of the trait and marker; and (2) finding the distribution of the test statistic under the null hypothesis. Broadly speaking, the test statistic is the 'covariance between a function of the genotype and a function of the trait', [20] the dependent variable being the offspring genotype. While the first step gives great flexibility in the choice of test statistic, the second is designed to ensure correct type I error rates (ie validity), regardless of population admixture, genetic model or ascertainment scheme [21]. These approaches are broad, in that they can handle different genetic models, different family structures (including extended pedigrees) and disease phenotypes (qualitative or quantitative, single or multiple). As with the original TDT, however, only heterozygous parents are informative in this framework; non-family data cannot be included and, in the case of FBAT, even if one does have a random sample, the effect size of the allele of interest is not estimated. This can lead to a dramatic loss of effective sample size and therefore potential power and/or precision when compared with an unconditional method such as that presented here and demonstrated in our previous work [22]. Other methods more robust to these particular limitations have been recently proposed for assessing quantitative traits in family-based samples [23] and binary traits in case-control samples, including related individuals [24,25]. Neither of these methods, however, includes an ascertainment correction (central to pooling family and case-control samples), nor do they estimate family or cluster effects. Further, the former does not allow for the inclusion of case-control data and the latter does not allow for the inclusion of covariates.
Based on the limitations of the existing strategies for testing LD, we present an alternative two-stage family-based association test in which we combine attributes of two existing methods, first to test whether population stratification is present and then appropriately test for and estimate the effect of, LD of a marker to a continuous trait. We further offer extensions of this method that can be applied to binary traits and hence allow an analysis of case-control data together with family data. We illustrate the power of this method for various sample sizes and structures, specifically for joint family and population-based samples that cannot be analysed by existing methods. We also extend this method to allow for the accurate estimation of association parameters and residual variance components from ascertained family data, and demonstrate, via simulation, that this method is effective in controlling ascertainment bias.
Methods
Continuous traits
The framework on which our approach is built was first described by George and Elston [26] and Elston et al. [27] in the special context of a randomly selected family sample with a measurable, quantitative trait of interest. For any individual i, with continuous trait (or, as we will later discuss, liability) yi, jth covariate values cji and a genotype indicator zi, we can construct a regression model of the form:
| (1) |
in which the number of A1 alleles, along with other covariates, is a predictor of phenotype. In this model, zi is coded such that the allelic effect of substituting A2 for A1 is . The random components include pi, a random polygenic effect, fi and , random nuclear family effects, mi, a random marital effect, si, a random sibship effect and εi, a random residual individual effect. In addition, the generalised power transformation (h), [28] applied to both the trait and its predictors, when simultaneously estimated under a model that assumes normality of the residuals, helps assure both linearity and normality, thus making the model robust to non-independence (as can be the case in large pedigrees). There are two random nuclear family effects fi and in model (1) because each individual is potentially a member of two different nuclear families, one in which we include the individual's parents and siblings and one in which we include the individual's spouse and children. All the random effects in the model are assumed to be mutually independent and, after the transformation, normally distributed with zero means and variances and such that: for families with more than two generations, and for families with only two generations. It is important to note that the total variance V[h(yi)] is made the same for all individuals by adjusting the residual variance separately for each person (see Elston et al. [27] for details). This model has recently been further extended to allow for each person to have more than two nuclear family effects, as can occur when there are half-sibships in the data, and other kinds of common environmental cluster effects.
As currently implemented in the S.A.G.E. program ASSOC, the likelihood is maximised numerically over all parameters, and standard errors are determined by numerical double differentiation of the log likelihood. Also, p-values, based on the likelihood ratio or a Wald test, can be calculated for the transformation parameters, any of the variance components and any regression coefficients. They are two-sided for all transformation parameters and regression coefficients, and one-sided for all variance components.
This method is meant to follow existing evidence of linkage because it does not control for population stratification. With the growing popularity of genome-wide and candidate gene association studies, however, there are likely to be many instances in which linkage is not known a priori. For this reason, we suggest - rather than automatically resorting to cumbersome genomic control methods or a less powerful TDT-type design - using a two-stage procedure to (1) test for a stratification effect and then (2) test for allelic association. If there is no stratification, then the association resulting from model (1) can be interpreted as LD effects. If there is stratification, then one can use the same regression model framework to perform a test like those mentioned above (TDT and FBAT) that conditions on parental genotype.
To test for stratification, we use the same regression model outlined above, but with the addition of transmitted and non-transmitted allele indicators (x1i and x2i) defined as:
Thus, the regression equation (3) for the trait value yi is now defined as
| (2) |
where ηi is the random effect comprising all of the familial, sibling, marital, polygenic and individual specific errors outlined above. George et al. [29] gave details of how the indicator variable x1i is constructed to form a TDT-type test by substituting it for zi in regression model (1). We point out that, because it includes components of a TDT-type test, it requires family data. The variable x2i is formed analogously for the other allele of an SNP. In the case of a multiallelic marker, all the other alleles can be pooled into a single allele for this purpose. To test for a stratification effect, we first test the null hypothesis that the genotypic effect is half the difference of the transmitted allele effects; that is, . If we do not reject this null hypothesis at some liberal significance level such as p = 0.2, we infer that there is no evidence of stratification, set β2 = β1 = 0 and estimate the allele A1 effect by . If there is any evidence of stratification, we set δ = 0 and estimate the allele A1 effect by β2 - β1. Thus, once either β2 = β1 or δ is set to 0, as appropriate, we return to a framework in which we simultaneously estimate the effect of allele A1, the residual variance components and one or more transformation parameters. We can use asymptotic results to obtain confidence intervals for all parameter estimates in the usual way, and the method can be extended to estimate genotype effects rather than allele effects. While other approaches like the principal component approach proposed by Zhu et al. [30] work well within this regression framework and are potentially more informative when many SNPs are available, this new approach is a viable option, even if only one or a few SNPs are typed (ie in the case of a candidate gene study).
Extension to binary traits
The generalised modulus power transformation mentioned above is fairly effective in inducing approximate normality, but does, of course, assume a continuous trait distribution. In many cases, continuous traits are not available to characterise complex diseases and only the presence or absence of disease is available. Therefore, we propose the following algorithmic extension of Zhu et al. [31] Let
| (3) |
where is the binary trait of interest, 1 represents affected individuals and 0 represents unaffected, and represents an initial estimate of . Our aim here is to define a new trait yi that, if μi were its expected value, would be approximately normally distributed with mean 0 and variance 1. We use the values of yi defined by equations (2) and (3) as the dependent variable in a simple generalised linear regression model of the form
| (4) |
We have shown that the iterative maximisation procedure currently implemented in our software (ASSOC) is quite robust to these initial estimates, regardless of family size or misspecified analysis model [32]. We do note, however, that the ease of maximisation and the accuracy of estimates depend on both the sample size and the number of parameters estimated. In general, we recommend at least 20 observations per parameter estimated to ensure accuracy (which can be assessed based on standard errors we provide).
Because the likelihood that is maximised by this process is perhaps not a true likelihood (it is a pseudo-likelihood, in that the estimates of the variance components may be based on incorrect model assumptions), the variance-covariance matrix of the estimators obtained by double differentiation of the likelihood may not equal the true variance matrix, even asymptotically. We may therefore estimate the variance-covariance matrix using the robust sandwich estimator, [33]
| (5) |
where is the estimated Fisher information matrix, which we need not assume is correctly specified, is the estimated outer product gradient and is the estimated outer product gradient expressed as
| (6) |
where, for the the kth pedigree, is a diagonal matrix with elements μik(1 - μik), yk is the vector of trait values for the kth pedigree, μk is the vector of means specific to the kth pedigree and Dk, with transpose , is the matrix of first order partial derivatives of μk with respect to β obtained assuming that the covariates are fixed:
| (7) |
In matrix (7), Nk is the number of persons in the kth pedigree, p is the total number of regression coefficients in equation (4), including the intercept, and βj represents any one of them.
Combining case-control and family data
One of the benefits of the regression framework outlined above is the flexibility to include families of any size or structure. This is vital, given, as mentioned above, that the magnitude of the effects associated with any given gene for a common complex disease is likely to be small. Certainly, provided we are only interested in hypothesis testing (we will discuss parameter estimation later), unmatched case-control data can be easily included as single person pedigrees with only an individual-specific variance. In this framework, however, matched case-control data can also be included by simply specifying the matched pairs as members of the same cluster (a cluster, of course, being a special case of a pedigree). We include in the model a cluster-specific variance component , such that and then adjust the residual variance so as to keep the total variance the same for all individuals. This approach does not limit the case-control cluster size or composition, as does conditional logistic regression.
Correcting for ascertainment
The underlying assumption of the method outlined above is that the sampling units (families, individuals, case-control clusters) represent a random sample from the same population. This is often not the case - particularly when families were sampled for a linkage study - and cannot be the case for case-control samples. The sample association and variance component estimates are thus not representative of the population values. We therefore present an ascertainment correction specifically for family data (and briefly address an extension to case-control data in the discussion).
Let the proband sampling frame (PSF) comprise those individuals who, regardless of phenotype, could have allowed the family to be ascertained by reason of being in the catchment area (the area from which the sample was collected). Then, let the ascertainment corrected natural log (ln) likelihood be:
| (8) |
where L(P) is the final likelihood, L(PAll) is the like-lihood for the whole sample on the assumption of random sampling of families and L(PPSF) is the like-lihood for the family members in the PSF, similarly on the assumption of random sampling. (For single ascertainment, only the probands are included in the PSF). Maximising this likelihood (8) leads to consistent estimators of all the parameters [34].
Power calculations for family data
To assess the power and type I error of our association analysis method as extended to binary traits, as well as to verify the accuracy of both the association parameters and the residual variance components for ascertained data, we simulated 2,000 replicates of samples of 1,000 individuals comprising either 200 nuclear families (two founders, three offspring) or 125 extended pedigrees (three founders, one of whom is a 'marry-in': three generations; one sibship of size 3 in generation 2; one sibship of size 2 in generation 3). A continuous liability was created according to the following linear model:
| (9) |
where i represents the ith individual; ai is the genotypic effect assigned based on an individual's major genotype defined as:
| (10) |
where q is the allele frequency and is locus-specific heritability, which we varied to have values 0 (the null hypothesis), 0.0025, 0.0125, 0.025, 0.0375, 0.05 and 0.0625; γ is the coefficient (set to 0.25) of the polygenic effect (or 'polygenotype') pi, generated from a N(0,1) distribution for founders and for non-founders derived as (polygenotype of the mother + polygenotype of the father) + a randomly generated value from a ; δj is the coefficient for the jth environmental effect which, in our examples were familial (F), sibling (S) and/or marital (M) (set to 0 when not included in the model and to 0.25 otherwise); dji is the environ-mental factor value assigned to all individuals within the same familial cluster and distributed N(0,1) across such clusters, ε (set to 0.5) is the coefficient of the random effect; and ei is generated separately for each individual from a N(0,1) distribution. The liability yi was then transformed to a binary phenotype. First, a standardised liability was created as:
| (11) |
where yi is the continuous liability created as defined above and is a mixture of three normal distributions with means equal to the genotypic effects of the three genotypes and a common variance (specific to the variance component model as shown in Table 1), is the sample mean, n is the total sample size, and is the sample variance. This transformation resulted in three distributions for the A1A1, A1A2, and A2A2 genotypes, with means (a - (q 2 a- (1 - q)2 a)), (0 - (q 2 a - (1 - q)2 a)), and (- a - (q 2 a - (1 - q)2 a)), respectively, the whole mixture distribution having a variance of 1. Then, an individual was classified as affected if zi >x, unaffected otherwise. For all simulations, x was fixed at 1.28, corresponding to a disease prevalence of approximately 0.1. Thus, A1 is the 'risk' allele.
Table 1.
Total variance of the non-major gene component of the continuous liability underlying the binary trait and the proportion of that variance represented by each variance component for each model
| Simulated proportion of variance for each variance component | ||||||
|---|---|---|---|---|---|---|
| Model Name | Total variance | Polygenic | Familial | Sibling | Marital | Random |
| P | 0.3125 | 0.200 | - - - - - - | - - - - - - | - - - - - - | 0.800 |
| FP | 0.3750 | 0.167 | 0.167 | - - - - - - | - - - - - - | 0.667 |
| SP | 0.3750 | 0.167 | - - - - - - | 0.167 | - - - - - - | 0.667 |
| MP | 0.3750 | 0.167 | - - - - - - | - - - - - - | 0.167 | 0.667 |
| SMP | 0.4375 | 0.143 | - - - - - - | 0.143 | 0.143 | 0.571 |
F = familial effect; M = marital effect; P = polygenic effect; S = sibling effect.
Creation of a random sample was achieved by simply collecting individuals (and thus their entire pedigree) from the simulated population in the order in which they were encountered until the desired sample size (1,000 individuals) was met. All replicates were analysed using both the simulated correlation model and an 'incorrect' correlation model. For example, if data were simulated to have both a familial and polygenic effect, they were analysed under a model (denoted as FP) including both effects and one (denoted P) that included only a polygenic effect. Names for all the models investigated are enumerated in Table 1.
Type I error was calculated as the number of replicates simulated under the null hypothesis meeting a recommended cut-off point for genome-wide association studies by the Wellcome Trust of α = 5 × 10-7 [35]. Power was calculated as the number of replicates meeting the same criterion but simulated under the alternate hypothesis.
Sample size estimation for joint family and case-control data
In addition to the simulations outlined above, in order to demonstrate the usefulness of this method for the joint analysis of family and population or case-control data, we analytically estimated, for a combination of unrelated individuals (50 per cent cases, 50 per cent controls), nuclear families and/or extended pedigrees, the number of individuals required to detect a given effect size at a fixed type I error rate and power.
For these calculations, we classified families according to the number of founders and non-founders (where unrelated individuals were simply one-founder pedigrees). Suppose there are ni families of the ith type, each with nfi founders and nnfi non-founders. Let yjm be the trait value underlying liability for the mth individual in the jth family with polymorphic marker value gji, and let αjm be a row vector whose elements are the intercept and effect of other covariates. Let β be the regression coefficient on the polymorphic marker and define Zjm = Yjm - αjm1 = β(gjm - E(gjm)), where 1 is a column vector of unities. The three genotypes of the marker are assumed to have the values (-2,1,1), (-1, -1,2) and (-1, 0, 1) for dominant, recessive and additive modes of inheritance, respectively. Then, letting , = the inverse of the variance-covariance matrix for the ith-type family and assuming multivariate normality, the log likelihood for the ith-type family is , giving the maximum likelihood estimator
This is an extension of Nick et al., [36] who gave approximate results for exactly two founders and a dominant mode of inheritance, and assumes the quantitative trait locus and marker variants are in perfect LD. We derived more generally for nfi founders, for both additive and dominant inheritance, as well as for relative pair specific correlations. We also allowed for incomplete LD by applying a 1/(0.8)-fold factor (equivalent to r2 = 0.8, D' ≈ 0.9).
For these calculations, we made some simplifying but conservative assumptions. First, we assumed that founder pairs have a correlation of 0 and that parent-offspring correlations (ρpo) and sib-sib correlations (ρss) correspond to a residual heritability of 2 ρpo = 2 ρss and that grandparent-grandchild pairs have a residual correlation of ρgg corresponding to a residual heritability of 4 ρgg. We further assumed, for simplicity, that all persons with the same genotype at the disease locus have the same disease risk and that LD between the locus and the closest SNP, assuming the same allele frequencies at the two loci, is given by r2 . Finally, we imposed the type I error recommended for genome-wide association studies by the Wellcome Trust of α = 5 × 10-7, [35] and assumed a fixed power equivalent to a sample of 500 cases and 500 controls (0.92 for an additive model and 0.86 for a dominant model), and a locus-specific heritability of - see equation (11) - of 0.05. We did this for samples of nuclear families only, extended pedigrees only and mixtures of both, for various sample sizes, and then, demonstrated the approximate linearity of the trend in sample size needed to detect the same effect given a fixed power and type I error.
Accuracy of association and variance component estimates
In addition to generating random family samples (RAND), we also generated a sample of singly ascertained families (ASC) by assigning each family a probability of entering the sample based on the number of affected members in the family: P(family enters sample) = Na/N, where Na is the number of affected members in the family and N is the number of family members. Each simulation output file was parsed and, if a family had an affected member, the above probability was calculated and, based on the appropriate Bernoulli distribution, the family was either ascertained or not until the desired sample size was met.
The accuracy of the locus-specific association parameter (β) and the polygenic and familial variance components were assessed after the appropriate analysis (ie with ascertainment correction if ASC and without if RAND) using the empirically found mean square error (MSE) averaged over 100 replicates, each comprising 1,000 individuals from either 200 nuclear families or 125 extended pedigrees. In all cases, the root mean square error (rMSE) compared with the simulated value (Tables 2-4) is reported. As mentioned, the accuracy of estimates in ascertained case-control samples was not addressed in this study, but is discussed below.
Table 2.
Accuracy of the association parameter as ln odds of being affected given two copies of the disease allele versus one copy for a sample size of 1,000 individuals
| Nuclear | Extended | ||||
|---|---|---|---|---|---|
| Model* | RAND | ASC | RAND | ASC | |
| FP-FP | Est | 2.529 | 2.479 | 2.511 | 2.561 |
| rMSE | 0.1709 | 0.1210 | 0.1530 | 0.2030 | |
| FP-P | Est | 2.537 | 2.509 | 2.517 | 2.524 |
| rMSE | 0.1789 | 0.1510 | 0.1591 | 0.1661 | |
| P-FP | Est | 2.780 | 2.655 | 2.763 | 2.722 |
| rMSE | 0.1775 | 0.0529 | 0.1603 | 0.1196 | |
| P-P | Est | 2.780 | 2.648 | 2.768 | 2.724 |
| rMSE | 0.1775 | 0.0458 | 0.1655 | 0.1212 | |
* Model indicates the variance components that were simulated followed by those included in the analysis model (F = familial and P = polygenic); Est is the average estimate across all replicates of that model; rMSE is the square root of the mean square error; ASC represents the analysis of an ascertained sample using ascertainment correction and RAND represents the analysis of a random sample without any such correction.
Table 4.
Accuracy of variance components as proportions of the total variance, N = 1,000
| Nuclear | Extended | |||||
|---|---|---|---|---|---|---|
| Parameter | Model | RAND | ASC | RAND | ASC | |
| Marital | MP-MP | Est | 0.079 | 0068 | 0.0775 | 0.0696 |
| rMSE | 0.0877 | 0.0990 | 0.0894 | 0.0693 | ||
| SMP-SMP | Est | 0.1743 | 0.0636 | 0.1291 | 0.0555 | |
| rMSE | 0.0316 | 0.0781 | 0.0141 | 0.0866 | ||
| Sibling | SP-SP | Est | 0.0574 | 0.0669 | 0.0549 | 0.0713 |
| rMSE | 0.1095 | 0.1000 | 0.1122 | 0.0959 | ||
| SMP-SMP | Est | 0.0549 | 0.0554 | 0.057 | 0.0579 | |
| rMSE | 0.0883 | 0.1118 | 0.0860 | 0.1090 | ||
| Polygenic | FP-FP | Est | 0.0896 | 0.0604 | 0.1711 | 0.1388 |
| rMSE | 0.0775 | 0.1068 | 0.0000 | 0.0283 | ||
| MP-MP | Est | 0.0741 | 0.0655 | 0.0775 | 0.0643 | |
| rMSE | 0.0927 | 0.1015 | 0.0894 | 0.1030 | ||
| P-P | Est | 0.063 | 0.0805 | 0.0602 | 0.0755 | |
| rMSE | 0.1039 | 0.1196 | 0.1068 | 0.1249 | ||
| SMP-SMP | Est | 0.2169 | 0.0617 | 0.1759 | 0.0559 | |
| rMSE | 0.0742 | 0.0800 | 0.0332 | 0.0860 | ||
| SP-SP | Est | 0.0962 | 0.0723 | 0.0782 | 0.0603 | |
| rMSE | 0.0707 | 0.0949 | 0.0889 | 0.1068 | ||
| Familial | FP-FP | Est | 0.1133 | 0.0122 | 0.033 | 0.0139 |
| rMSE | 0.0539 | 0.3521 | 0.1342 | 0.1530 | ||
| P-FP | Est | 0.0198 | 0.0032 | 0.048 | 0.0102 | |
| rMSE | 0.0200 | 0.0032 | 0.0480 | 0.0100 | ||
* Model indicates the variance components that were simulated followed by those included in the analysis model (F = familial, M = marital, S = sibling and P = polygenic); Est is the average estimate across all replicates of that model; rMSE is the square root of the mean square error; ASC represents the analysis of an ascertained sample using ascertainment correction and RAND represents the analysis of a random sample without any such correction.
Results
Type I error and power in family data
Under both additive and dominant models, the association method we present for detecting diallelic trait loci has stable type I error rates of less than 0.05 (mean = 0.0452) for the RAND sample of both the nuclear families and extended pedigrees.
The ASC sample had slightly higher type I error rates for the nuclear family sample (0.0523) but not for the extended pedigrees (0.0427). The power reached 100 per cent at a total heritability of 0.25 for both the additive and dominant models in both the nuclear family sample and the extended pedigrees, and there was virtually no power to detect a heritability of 0.01. The power curves for the RAND and ASC samples were virtually identical, so for the sake of space only the ASC curves are presented. The nuclear family sample (200 families, 1,000 individuals), out-performed the extended pedigree sample (125 families, 1,000 individuals) under both models. Further, there was a steep decline in power between heritabilities of 0.15 and 0.10 (Figure 1).
Figure 1.
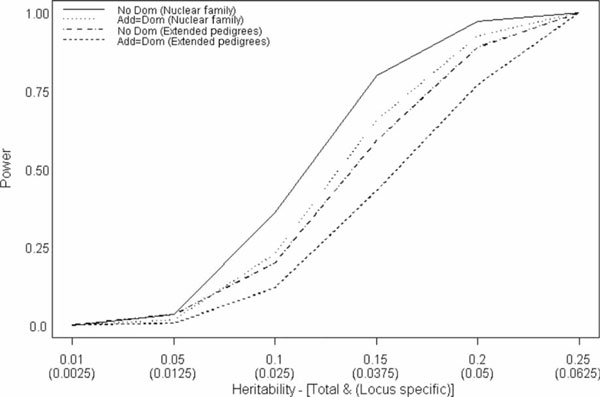
Power to detect association by both total and locus-specific heritability for nuclear families (nuc fam) under an additive model (No Dom) and a model with 50 per cent additive and 50 per cent dominance variance (Add = Dom).
Sample size estimation in joint family and case-control data
To demonstrate the usefulness of family data in association analysis, as well as the usefulness of combining samples from both linkage (family-based) and association (typically case-control) studies, we evaluated the number of unrelated individuals that need to be added to an existing family sample to be able to detect the same effect size as in a population-based sample of 1,000 unrelated cases and 1,000 controls. Beginning with a collection of 125 extended pedigrees or 200 nuclear families (samples that are quite prevalent), one can decrease the number of unrelated samples needed to detect a given effect size by at minimum close to 40 per cent and at maximum more than 50 per cent (Figure 2).
Figure 2.
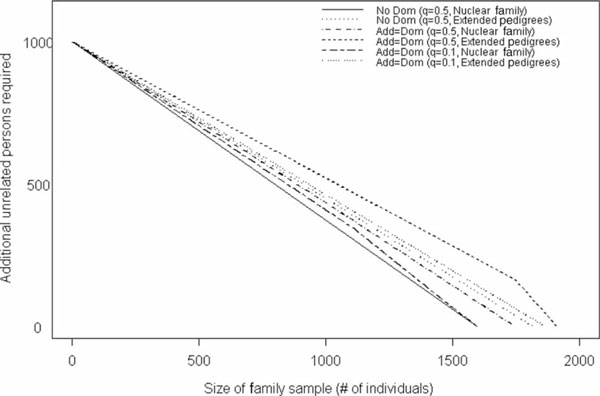
Number of unrelated case-control samples needed, in addition to a fixed sample of either nuclear or extended pedigrees, to achieve a power of 92 per cent under an additive model (No Dom) and 86 per cent under a model with 50 per cent additive and 50 per cent dominance variance (Add = Dom). Values were generated for fixed sample sizes of both nuclear families and extended pedigrees, as well as for allele frequencies of both 0.5 and 0.1.
Note that the equivalence of samples is shown, assuming (1) a common minor allele frequency (q = 0.5), for which the family data are not as informative as are the case-control data, and (2) an allele frequency under which the family sample is fairly informative (q = 0.1). As expected, the nuclear family sample (assuming an additive model with q = 0.5) requires the fewest additional unrelated individuals to detect a given effect, and the extended pedigree sample (assuming a dominant model with q = 0.5) requires the most additional unrelated individuals. The extended pedigree sample, under a dominant model with q = 0.5 or 0.1, performed similarly, as did the nuclear family sample under a dominant model with q = 0.5 or 0.1. The extended pedigree and nuclear family samples (assuming an additive model) require approximately the same number of additional unrelated persons to detect the given effect size.
In addition to providing the number of additional individuals necessary to detect a fixed effect size given a sample of nuclear or extended pedigrees, we further provide this information given a sample comprising varying proportions of nuclear and extended pedigrees. We found that, for the additive and dominant models, regardless of the allele frequency, the samples that contained 30 per cent extended pedigrees and 70 per cent nuclear families (30:70) required the fewest additional unrelated individuals (of the three mixtures examined) to attain the same power. For the model in which dominance and additive variance were equal (Add = Dom) with allele frequency 0.5, the sample with equal frequency of nuclear and extended pedigrees (50:50) and the sample that is 70 per cent extended and 30 per cent nuclear (70:30) require similar sample sizes except in the extreme cases where there are very few to no unrelated individuals (Figure 3). The results for the Add = Dom model with q = 0.1 are similar to those for the model with q = 0.5, except that the divergence of the 50:50 and 70:30 samples is not as large as in the previous case (Figure 4). The additive model indicates the least difference in the three sample types (Figure 5); for example, a sample of 140 nuclear families and 38 extended pedigrees requires 500 additional individuals to achieve the same power and type I error as 100 nuclear families, 63 extended pedigrees and 625 additional unrelated persons.
Figure 3.
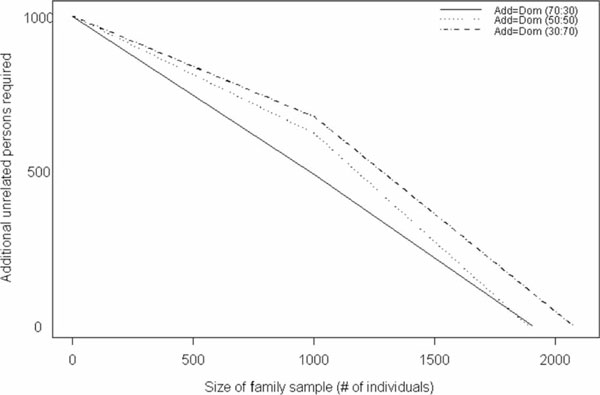
Number of unrelated case-control samples needed, in addition to a fixed, mixed sample of nuclear and extended pedigrees, to achieve a power of 86 per cent under a model with 50 per cent additive and 50 per cent dominance variance (Add = Dom), assuming an allele frequency of 0.5. Values were generated for samples that comprised 30 per cent nuclear families and 70 per cent extended pedigrees, 50 per cent and 50 per cent, and 30 per cent and 70 per cent, respectively.
Figure 4.
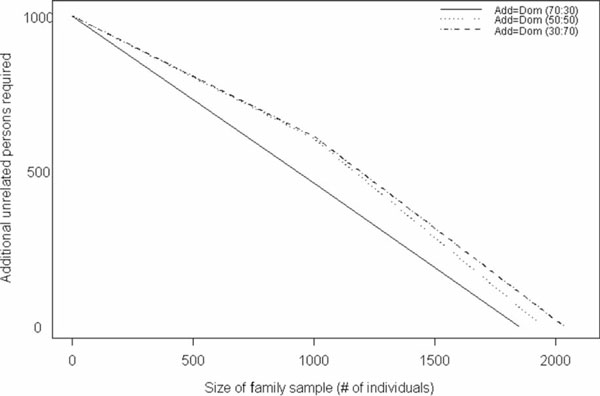
Number of unrelated case-control samples needed, in addition to a fixed, mixed sample of nuclear and extended pedigrees, to achieve a power of 86 per cent under a model with 50 per cent additive and 50 per cent dominance variance (Add = Dom), assuming an allele frequency of 0.1. Values were generated for samples that comprised 30 per cent nuclear families and 70 per cent extended pedigrees, 50 per cent and 50 per cent, and 30 per cent and 70 per cent, respectively.
Figure 5.
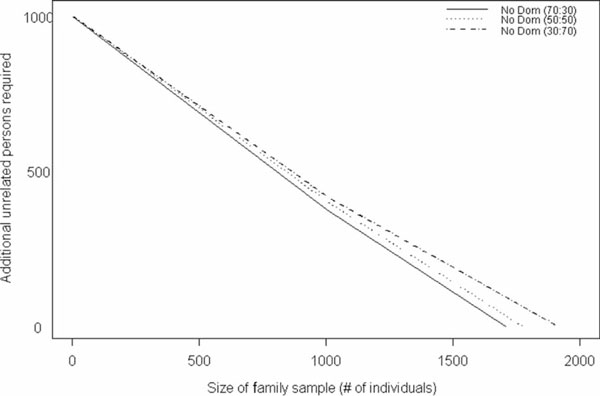
Number of unrelated case-control samples needed, in addition to a fixed, mixed sample of nuclear and extended pedigrees, to achieve a power of 92 per cent under as additive model (No Dom), assuming an allele frequency of 0.1. Values were generated for samples that comprised 30 per cent nuclear families and 70 per cent extended pedigrees, 50 per cent and 50 per cent, and 30 per cent and 70 per cent, respectively.
Estimation using family samples
Accuracy of the association parameter
The estimates of the association parameter (expressed as the ln odds of two copies of the disease allele versus one copy) were, on average, 2.615 for the nuclear family sample and 2.636 for the extended family sample - not too dissimilar to the simulated value of 2.48. The RAND and ASC samples had similar averages of 2.648 and 2.603, respectively. Note that we purposely generated the data under a (probit) model different from the (logit) model used to analyse the data, to illustrate the robustness of the analysis model, and that the accuracy of the ascertainment correction is seen in the small difference in parameter estimates between the RAND and ASC samples. The average estimate for the ascertained extended families (2.633) was overestimated by a factor of 1.06, a slightly larger deviation from the simulated value than seen in the nuclear family samples, which had an average of 2.573 - only 1.03 times the simulated value (and the closest to it). The rMSE averaged over all models was 0.210 and all estimates were within a factor of 1.15 of the simulated value (Table 2).
Results were similar for estimates comparing the odds of two disease susceptibility alleles to no susceptibility alleles, on average 5.251 - again, not too dissimilar to the simulated value of 5.616. The RAND samples had an average of 5.296 and the ASC samples almost the same (5.206). Nuclear (NUC) families had the estimate 5.230 and extended families 5.273. In these cases, the random nuclear family samples were the closest to the simulated value. The average rMSE was 0.351 and all estimates were within a factor of 0.88 of the simulated value (Table 3). Notice that for these comparisons the effect was always under- rather than overestimated, whereas in the previous comparisons they were overestimates.
Table 3.
Accuracy of the association parameter as ln odds of being affected given two copies of the disease allele versus no copies for a sample size of 1,000 individuals
| Nuclear | Extended | ||||
|---|---|---|---|---|---|
| Model* | RAND | ASC | RAND | ASC | |
| FP-FP | Est | 5.058 | 4.958 | 5.022 | 5.122 |
| rMSE | 0.2936 | 0.3936 | 0.3295 | 0.2296 | |
| FP-P | Est | 5.074 | 5.018 | 5.034 | 5.048 |
| rMSE | 0.2777 | 0.3336 | 0.3176 | 0.3036 | |
| P-FP | Est | 5.560 | 5.310 | 5.526 | 5.444 |
| rMSE | 0.1010 | 0.5696 | 0.3536 | 0.4357 | |
| P-P | Est | 5.560 | 5.296 | 5.536 | 5.448 |
| rMSE | 0.3195 | 0.5836 | 0.3437 | 0.4316 | |
* Model indicates the variance components that were simulated followed by those included in the analysis model (F = familial and P = polygenic); Est is the average estimate across all replicates of that model; rMSE is the square root of the mean square error; ASC represents the analysis of an ascertained sample using ascertainment correction and RAND represents the analysis of a random sample without any such correction.
Accuracy of the variance components
Overall, the rMSEs were, as might be expected, smaller for the RAND samples than for the ASC samples. When comparing the estimated values with the simulated proportions of variance (Table 4), the estimates from the RAND and ASC samples yielded good estimates of the true simulated population values for the polygenic and familial components, but the sibling and marital components were often over- or underestimated in the ASC sample, depending on both model and family structure. Specifically, sibling (S) and marital (M) components were consistently underestimated in the SMP-SMP scenarios and overestimated in all other scenarios.
The accuracy of the variance component estimates were affected by the sampling scheme, as expected. The RAND samples resulted in estimates closest to the simulated population values, but ASC samples yielded estimates reasonably reflective of the population values as well.
Discussion
The prediction of the future of genetic studies of complex disease is ever changing, but what remains true is that we must have methods of analysis that are both powerful and flexible. Whether searching for common genes with small effect or rare genes with large effect, we shall need large samples that are likely to come only from combining family, population-based and case-control data and we must have methods that analyse these combinations. In fact, the use of family samples was recently high-lighted by Visscher et al., [2] showing that including related individuals results in only a small loss of power but large gains in terms of quality control, flexibility of tests to be performed and ability to control for population stratification. Our results support these assertions and we further recommend that association methods must account for environmental covariates (which are certain to play a role in complex diseases) and must not be restricted by, but rather be effective in controlling for, population stratification. These tools will be powerful in aiding both genome-wide association and candidate gene studies.
We have present here a method to test and estimate the association between an allele or genotype and a continuous or binary trait, as well as approaches to combining family and case-control data that are powerful as well as robust to ascertainment. We also present a two-stage procedure to determine the need for a test that is robust to stratification. A purist would argue that a two-stage approach could affect type I error rate. The important thing to note, however, is that this decision should be made on the basis of the significance, not the magnitude, of the difference in the two estimates of marker effect, β2 - β1 versus , because a study whose sample size is powered to detect a small effect will automatically be powered to detect the small biases that stratification could induce.
We further present a method for correcting for ascertainment and accurately estimating association parameters, as well as variance components, even in ascertained family data. Two things should be pointed out, however. First, we examined only single ascertainment, when a more complex scheme is used to collect families such that most of the sample is in the PSF and/or the PSF is undefined, the estimates for the association parameter and the variance components will reflect only the effect in the sample. Note, however, that the test for association is still valid and it is only the parameter estimates that are affected. Secondly, when combining data from a case-control sample and an ascertained family sample, for the parameter estimates from this method to be reflective of the population from which the samples were drawn, certain assumptions must be met: (1) the cases in the population-based data should have been phenol-typed in a manner similar to the cases in the family data; (2) there must be appropriate correction for ascertainment; and (3) the non-cases or 'controls', although matched, should apart from this also be a random sample - if they are a completely random sample from the same population, it is possible to estimate a relative risk, while if they are a random sample of those showing absence of the phenotype of interest, only an odds ratio can be estimated. If the phenotype is sufficiently rare such that choosing controls based on absence of the trait of interest is essentially the same as random sampling, then the relative risk and odds ratio will be essentially the same. Because this is not the case for common complex diseases, we suggest and will investigate further in future studies, two other ways of combining case-control and family data for accurate estimation: (1) express the likelihood for the case-control data in terms of odds ratios, which are functions of the parameters in the pedigree likelyhood, and constrain the maximum likelihood for them such that the marginal probability of disease, given a set of regressors, is finite; [37] and (2) multiply the likelihood by a factor that summarises any information we have about the prevalence of disease independent of the sample data. This factor would be expressed as μR (1 - μ)N - R, where μ is the prevalence of the disease - expressed as a function of the parameters in the full likelihood at particular values of the covariates in the model - and R reflects our external information about the number of affected persons in a population of size N. For example, if we have an estimate of μ, and its standard error (s.e.), we can estimate reasonable values for N and R by noting , and hence and . It is known that constraining likelihood maximisation so that the estimated disease prevalence is equal to its true prevalence can be equivalent to a correction for single ascertainment [38]. These two options offer simple solutions for 'non-traditional' samples and will be examined in future work.
The general method described in this paper, which is currently being implemented in the program package S.A.G.E., is more flexible than other TDT-type methods and more efficient (in the practical sense) than genomic control methods. Further, we have shown the power of this method for binary traits in various types of family, population-based and combined samples at a constant type I error rate and, while we concede that a population-based sample could sometimes detect a smaller effect size than the respective family-based samples, as mentioned earlier, these scenarios assume the same degree of heterogeneity and sporadic cases in all samples after correction for ascertainment. We know that this is not likely to be the case, as family samples are designed to decrease greatly the number of sporadic cases and, at least to some extent, reduce the amount of heterogeneity in the sample in a manner that makes appropriate ascertainment correction difficult. Further, for most complex phenotypes, family samples of at least the size examined here (and usually much larger) already exist and, as shown in Figures 2 and 3, can drastically reduce the number of population-based samples needed to detect even very small effects. Other benefits of family data, such as increased ability to assess the effects of shared environment and parent-of-origin effects, to detect errors and many others are beyond the scope of this paper, but must also be considered. Finally, while having to correct for ascertainment is one of the reasons often cited for using population-based versus family data, we have demonstrated that, in principle, our method can be used to estimate fairly accurately the effect size of a given allele of interest for a given population, even if using an ascertained sample. For situations where most of the sample is in the PSF (and hence likelihood (8) contains little information), or the PSF is ill-defined, we suggest constraining the likelihood to give an accurate estimate of the disease prevalence. Future investigation will determine the accuracy of estimates obtained in this manner.
Acknowledgements
This work was supported in part by a US Public Health Service resource grant (RR03655) from the National Center for Research Resources, research grant (GM28356) from the National Institute of General Medical Sciences, Cancer Center support grant (P30CAD43703) and Transdisciplinary Research in Energetic and Cancer grant (U54CA116867), both from the National Cancer Institute, and training grant (HL07567) from the National Heart, Lung and Blood Institute, as well as from the Swiss National Foundation for Science (PROSPER: 3200BO-111362/1 and 3233BO-111361/1). Some of the results of this paper were obtained by using the program package S.A.G.E., which is supported by a US Public Health Service Resource grant (RR03655) from the NCRR.
References
- Cordell HJ. 'Sample size requirements to control for stochastic variation in magnitude and location of allele-sharing linkage statistics in affected sibling pairs'. Ann Hum Genet. 2001;65:491–502. doi: 10.1046/j.1469-1809.2001.6550491.x. [DOI] [PubMed] [Google Scholar]
- Visscher PM, Andrew T, Nyholt DR. 'Genome-wide association studies of quantitative traits with related individuals: Little (power) lost but much to be gained'. Eur J Hum Genet. 2008;16:387–390. doi: 10.1038/sj.ejhg.5201990. [DOI] [PubMed] [Google Scholar]
- Altshuler D, Clark AG. 'Genetics. Harvesting medical information from the human family tree'. Science. 2005;307:1052–1053. doi: 10.1126/science.1109682. [DOI] [PubMed] [Google Scholar]
- Elston RC. 'Linkage and association to genetic markers'. Exp Clin Immunogenet. 1995;12:129–140. doi: 10.1159/000424866. [DOI] [PubMed] [Google Scholar]
- Knowler WC, Williams RC, Pettitt DJ, Steinberg AG. 'Gm3;5,13,14 and type 2 diabetes mellitus: An association in American Indians with genetic admixture'. Am J Hum Genet. 1988;43:520–526. [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Rosenberg NA. 'Use of unlinked genetic markers to detect population stratification in association studies'. Am J Hum Genet. 1999;65:220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorroochurn P, Heiman GA, Hodge SE, Greenberg DA. 'Centralizing the non-central chi-square: A new method to control for population stratification in genetic case-control association studies'. Genet Epidemiol. 2006;30:277–289. doi: 10.1002/gepi.20143. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K. 'Genomic control for association studies'. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ. 'Transmission test for linkage disequilibriumml: The insulin gene region and insulin-dependent diabetes mellitus (IDDM)'. Am J Hum Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- Rubinstein P, Walker M, Carpenter C, Carrier C. et al. 'Genetics of HLA disease associations: The use of the haplotype relative risk (HRR) and the 'haplo-delta' (Dh) estimates in juvenile diabetes from three racial groups'. Hum Immunol. 1981;3:384. [Google Scholar]
- Abecasis GR, Cardon LR, Cookson WO, Sham PC. et al. 'Association analysis in a variance components framework'. Genet Epidemiol. 2001;21(Suppl 1):S341–S346. doi: 10.1002/gepi.2001.21.s1.s341. [DOI] [PubMed] [Google Scholar]
- Curtis D, Sham PC. 'A note on the application of the transmission disequilibrium test when a parent is missing'. Am J Hum Genet. 1995;56:811–812. [PMC free article] [PubMed] [Google Scholar]
- Abel L, Muller-Myhsok B. 'Maximum-likelihood expression of the transmission/disequilibrium test and power considerations'. Am J Hum Genet. 1998;63:664–667. doi: 10.1086/301975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu IP, Whittemore AS. 'Power of association and linakge tests when the disease alleles are unobserved'. Am J Hum Genet. 1999;64:641–649. doi: 10.1086/302253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Myhsok B, Abel L. 'Genetic analysis of complex diseases'. Science. 1997;275:1328–1329. [PubMed] [Google Scholar]
- Fulker DW, Cherny SS, Sham PC, Hewitt JK. 'Combined linkage and association sib-pair analysis for quantitative traits'. Am J Hum Genet. 1999;64:259–267. doi: 10.1086/302193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham PC, Cherny SS, Purcell S, Hewitt JK. 'Power of linkage versus association analysis of quantitative traits, by use of variance-components models, for sibship data'. Am J Hum Genet. 2000;66:1616–1630. doi: 10.1086/302891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Cardon LR, Cookson WO. 'A general test of association for quantitative traits in nuclear families'. Am J Hum Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz D, Laird N. 'A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information'. Hum Hered. 2000;50:211–223. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. 'Implementing a unified approach to family-based tests of association'. Genet Epidemiol. 2000;19(Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Horvath S, Xu X, Laird NM. 'The family based association test method: Strategies for studying general genotype - phenotype associations. Eur J Hum Genet. 2001;9:301–306. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- Gray-McGuire C, Song Y, Sinha R, Won S, 'Comparison of family based association methods and designs for genome-wide association scans'. Proceedings of the Genetic Analysis Workshop 15. 2006. pp. 14–18.http://www.geneticepi.org
- Chen WM, Abecasis GR. 'Family-based association tests for genomewide association scans'. Am J Hum Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton T, McPeek MS. 'Case-control association testing with related individuals: A more powerful quasi-likelihood score test'. Am J Hum Genet. 2007;81:321–337. doi: 10.1086/519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgain C, Hoffjan S, Nicolae R, Newman D. et al. 'Novel case-control test in a founder population identifies P-selectin as an atopy-susceptibility locus'. Am J Hum Genet. 2003;73:612–626. doi: 10.1086/378208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George VT, Elston RC. 'Testing the association between polymorphic markers and quantitative traits in pedigrees'. Genet Epidemiol. 1987;4:193–201. doi: 10.1002/gepi.1370040304. [DOI] [PubMed] [Google Scholar]
- Elston RC, George VT, Severtson F. 'The Elston- Stewart algorithm for continuous genotypes and environmental factors'. Hum Hered. 1992;42:16–27. doi: 10.1159/000154043. [DOI] [PubMed] [Google Scholar]
- George V, Elston RC. 'Generalized modulus power transformation'. Commun Stat Theory Methods. 1988;17:2933–2952. doi: 10.1080/03610928808829781. [DOI] [Google Scholar]
- George V, Tiwari HK, Zhu X, Elston RC. 'A test of transmission/disequilibrium for quantitative traits in pedigree data, by multiple regression'. Am J Hum Genet. 1999;65:236–245. doi: 10.1086/302444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Li S, Cooper RS, Elston RC. 'A unified association analysis approach for family and unrelated samples correcting for stratification'. Am J Hum Genet. 2008;82:352–365. doi: 10.1016/j.ajhg.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Elston RC, Bielefeld RA. 'Testing disease marker association in pedigree data'. Proceedings of the Annual Meeting of the American Statistical Association. 1997. pp. 34–43.
- Gray-McGuire C. 'Assessment of a variance component method for binary phenotype data: Model misspecification and effects of ascertainment'. Case Western Reserve University, Cleveland, OH, USA; 2004. (Thesis) [Google Scholar]
- Gourieroux C, Monfort A. 'Pseudo maximum likelihood methods'. Handbook of Statistics. 1993;11:335–362. [Google Scholar]
- Ginsburg E, Malkin I, Elston RC. 'Sampling correction in linkage analysis'. Genet Epidemiol. 2004;27:87–96. doi: 10.1002/gepi.20008. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control C. 'Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls'. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick TG, George V, Elston RC, Wilson AF. 'Statistical validity for testing associations between genetic markers and quantitative traits in family data'. Genet Epidemiol. 1995;12:145–161. doi: 10.1002/gepi.1370120204. [DOI] [PubMed] [Google Scholar]
- Prentice RL, Pyke R. 'Logistic disease incidence models and case-control studies'. Biometrika. 1979;66:403–411. doi: 10.1093/biomet/66.3.403. [DOI] [Google Scholar]
- Burton PR. 'Comment on "Ascertainment adjustment in complex diseases"'. Genet Epidemiol. 2002;23:214–218. doi: 10.1002/gepi.10199. [DOI] [PubMed] [Google Scholar]