

Abstract
Hexavalent chromium (Cr(VI)) compounds are known human carcinogens associated with the incidence of lung cancer. Although a direct correlation between Cr(VI) exposure and lung cancer has been established, several studies aimed at generating animal models for Cr(VI) have yielded inconsistent data that do not affirmatively support findings from epidemiologic studies. Because the lack of a good animal model has hindered the identification of molecular mechanisms involved in Cr(VI) exposure, we developed an in vitro model that facilitates mechanistic studies of Cr(VI)-induced carcinogenesis. We report here that long-term exposure to Cr(VI) leads to the malignant transformation of nontumorigenic human lung epithelial cells. Cr(VI)-transformed cells exhibited loss of contact inhibition, colony formation, and increased rates of cell invasion, migration, and proliferation, as compared with passage-matched control cells. Cr(VI)-transformed cells evaded apoptosis by a mechanism involving S-nitrosylation and stabilization of Bcl-2 protein in a nitric oxide–dependent manner. This study establishes an important in vitro model that facilitates mechanistic studies of Cr(VI)-induced carcinogenesis, and elucidates a novel mechanism that causes apoptosis-resistant malignant transformation of nontumorigenic lung cells in response to a human carcinogen.
Keywords: nitric oxide, Bcl-2, malignant transformation, hexavalent chromium, apoptosis
CLINICAL RELEVANCE.
In the present study, a novel in vitro model was developed to facilitate the mechanistic studies of hexavalent chromium–induced lung carcinogenesis. Such an in vitro model may also be applied to other carcinogens that lack sufficient animal data, and will prove useful in facilitating their mechanistic studies. Using this model, we demonstrate an important mechanism that implicates nitric oxide, Bcl-2, and superoxide anion in the development of apoptosis-resistant malignant phenotype. This novel mechanism may also be critical in malignant transformation of normal cells under various conditions in response to other human carcinogens.
Lung cancer is one of the major causes of fatalities worldwide, with over 1 million deaths annually (1). In the United States, 160,440 deaths were attributed to cancer of the lung and bronchus in 2003 (1, 2). Hexavalent chromium (Cr(VI)) compounds have been classified as group I human carcinogens by the International Agency of Research in Cancer in 1990 (3). In the United States, Machle and Gregorius (4) reported the first epidemiologic evidence to demonstrate increased mortality due to lung cancer among chromium-exposed workers. Subsequently, several epidemiologic studies in the last few decades have confirmed that exposure to Cr(VI) compounds is associated with the induction of lung cancer in workers in various occupational settings (5–8). Cr(VI) compounds are also present in cigarette smoke, and an increased incidence of lung cancer has been reported in smokers with Cr(VI) exposure (3, 6, 9). Although several epidemiological studies demonstrated the carcinogenic potential of Cr(VI) compounds, in vivo studies have been inconsistent and unreliable. Studies in a number of animal models, including rat, mouse, guinea pig, and rabbit, have demonstrated no significant increase in lung tumors in Cr(VI)-treated animals versus untreated controls (9–13). The solubility of Cr(VI) compounds has been shown to be an important factor in determining the formation of tumors in animal models (12). The problems associated with the development of animal models to study Cr(VI)-induced carcinogenesis are poorly understood. In general, the intricate nature of the interaction of metals with the biological system has hindered the elucidation of mechanisms involved in metal carcinogenesis. Additionally, the lack of in vivo models has hindered efforts toward identifying the underlying mechanisms and delineation of the associated causative factors for cancer induced by Cr(VI). As a result, although Cr(VI) compounds have been extensively studied and associated with the induction of human lung cancer for more than a century (6), the mechanisms involved in Cr(VI)-induced carcinogenesis are still unclear.
To investigate the mechanisms involved in Cr(VI)-induced carcinogenesis, we developed an in vitro model by subjecting nontumorigenic human bronchial epithelial Beas-2B cells to long-term Cr(VI) exposure. Chronic exposure to Cr(VI) led to the malignant transformation of nontumorigenic Beas-2B cells. Furthermore, nitric oxide (NO)–mediated S-nitrosylation of Bcl-2 was observed to play a crucial role in apoptosis resistance and malignant transformation of human lung epithelial cells in response to Cr(VI) exposure. This novel in vitro model may also be applied to other carcinogens that lack sufficient animal data, and will prove useful in facilitating their mechanistic studies.
MATERIALS AND METHODS
Chemicals and Reagents
Sodium dichromate (Na2Cr2O7.H2O), NO inhibitor aminoguanidine (AG), diaminonaphthalene (DAN), mercury(II)chloride (HgCl2), and sodium hydroxide (NaOH) were obtained from Sigma-Aldrich (St. Louis, MO). NO donor dipropylenetriamine (DPTA) NONOate and the fluorogenic caspase substrates, LEHD–amino-4-methylcoumarin (AMC), were from Alexis Biochemicals (San Diego, CA). Dihydroethidium bromide (DHE), diaminofluorescein (DAF)-diacetate (DA), and the apoptosis dye, Hoechst 33,342, were purchased from Molecular Probes (Eugene, OR). Bcl-2 antibody, peroxidase-labeled secondary antibodies, anti-myc agarose beads, and protein A agarose beads were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for ubiquitin, S-nitrosocysteine, and β-actin were from Sigma-Aldrich, and the transfecting agent, Lipofectamine 2000, was from Invitrogen (Carlsbad, CA).
Cell Culture
Nontumorigenic human bronchial epithelial Beas-2B cells were obtained from American Type Culture Collection (Manassas, VA). The cells were cultured in Dulbecco's modified Eagle medium (Sigma-Aldrich) supplemented with 5% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in a 5% CO2 environment at 37°C.
Derivation of Cr(VI)-Transformed Cells
Beas-2B cells were continuously exposed to 5 μM Cr(VI) in culture in a 5% CO2 environment at 37°C. Cells were passaged weekly at preconfluent densities with a solution containing 0.05% trypsin and 0.5 mM EDTA (Invitrogen). Cr(VI)-exposed Beas-2B cells are designated as B-Cr cells so as to distinguish them from the parental Beas-2B cells. Parallel cultures grown in Cr(VI)-free medium provided passage-matched controls. After 24 weeks of exposure, the transformed cells were cultured in normal medium, and their tumorigenic potential was assessed by various experiments, described subsequently here.
Cyquant Cell Proliferation Assay
Beas-2B and B-Cr cells were plated in 96-well plates at a density of 2 × 104 cells/well in growth medium, and were incubated for various time points. After specific treatments, the incubating medium was changed with 50 μl of 1× Cyquant dye binding solution (Invitrogen) and incubated for 60 minutes at 37°C. The fluorescence intensity of each sample was measured at the excitation and emission wavelengths of 485 and 535 nm, respectively.
Cell Invasion and Migration Assays
In vitro invasion and migration was determined by a modified Boyden chamber assay with cell culture inserts with a polycarbonate filter coated with Matrigel (BD Biosciences, Franklin Lakes, NJ) for invasion, or control inserts for migration, in a 24-well format. Briefly, 3 × 104 cells (invasion) or 1.5 × 104 cells (migration) in 500 μl of serum-free medium, along with the specific test agents, were added to the inserts. The lower chambers were filled with normal growth medium containing 5% FBS. Chambers were incubated at 37°C in a 5% CO2 atmosphere for 24 hours for migration assay, and 48 hours for invasion assay. The noninvading or nonmigrating cells were removed from the insides of the inserts by cotton swabs. The cells that invaded the Matrigel or migrated to the underside of the coated membrane were fixed and stained with Diff-Quik fixative and stain (Dade Behring, Newark, DE). The inserts were examined under a light microscope (Leica DM IL, Wetzlar, Germany), and the number of cells that migrated or invaded through the inserts was scored.
Soft Agar Colony Formation Assay
Soft agar assay was performed as previously described with minor modifications (14). Beas-2B and B-Cr cells (3 × 104 cells for each cell type) were mixed with tissue culture medium containing 0.5% agar, resulting in a final agar concentration of 0.33%. Cell suspension (1.5 ml), along with the specific treatment, was immediately plated in 60-mm dishes coated with 7 ml of 0.5% agar in tissue culture medium. After 2 weeks, the average number of colonies per 3 × 104 cells was scored under a light microscope.
Apoptosis Assay
After specific treatments, apoptosis was determined by incubating cells with 10 μg/ml Hoechst 33342 nuclear stain for 30 minutes at 37°C and scoring the percentage of cells having intensely condensed chromatin and/or fragmented nuclei by fluorescence microscopy (Leica DM IL) with Leica software. From random fields, 1,000 nuclei were analyzed for each sample. The apoptotic index was calculated as apoptotic nuclei/total nuclei × 100 (%).
Caspase Assay
Caspase activity was determined by fluorometric assay with the enzyme substrate LEHD-AMC, which is specifically cleaved by caspase-9 at the Asp residue to release the fluorescent group, AMC. Cell extracts containing 20 μg of protein were incubated with 100 mM Hepes containing 10% sucrose, 10 mM dithiothreitol, 0.1% 3-[(3-cholamidiopropyl)-dimethylammonio]-1-propane sulfonate, and 50 μM caspase substrate in a total reaction volume of 0.25 ml. The reaction mixture was incubated for 60 minutes at 37°C, and quantified fluorometrically at the excitation and emission wavelengths of 380 and 460 nm, respectively, with an RF5301PC spectrofluorometer (Shimadzu, Kyoto, Japan).
Reactive Oxygen Species Detection
Intracellular superoxide anion (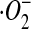 ) production was determined by spectrofluorometry with DHE as the fluorescent probe. After specific treatments, cells (1 × 106/ml) were incubated with the fluorescent probe (10 μM) for 30 minutes at 37°C. Cells were analyzed for DHE fluorescence at the excitation and emission wavelengths of 535 and 617 nm, respectively, with a FLUOstar OPTIMA plate reader (BMG Inc., Durham, NC).
) production was determined by spectrofluorometry with DHE as the fluorescent probe. After specific treatments, cells (1 × 106/ml) were incubated with the fluorescent probe (10 μM) for 30 minutes at 37°C. Cells were analyzed for DHE fluorescence at the excitation and emission wavelengths of 535 and 617 nm, respectively, with a FLUOstar OPTIMA plate reader (BMG Inc., Durham, NC).
NO Detection
Intracellular NO production was determined by spectrofluorometry with DAF-DA fluorescent probe. After specific treatments, cells (1 × 106/ml) were incubated with 10 μM DAF-DA for 30 minutes at 37°C. Cells were analyzed for DAF fluorescence at the excitation and emission wavelengths of 488 and 538 nm, respectively, with a FLUOstar OPTIMA plate reader.
Plasmids and Transfection
Beas-2B and B-Cr cells were seeded in 60-mm dishes until they reached 80% confluence. The cells were then transfected with myc-tagged wild-type (WT) Bcl-2 (WT Bcl-2), double-mutant (C158A/C229A) Bcl-2 (DM Bcl-2), or control (pcDNA3) plasmid by nucleofection (Amaxa GmbH, Köln, Germany), according to the manufacturer's protocol. Cells were pelleted and resuspended in 100 μl human cell nucleofector solution. Plasmid DNA (4 μg) was mixed with the cell suspension, transferred to a 2.0-mm electroporation cuvette, and nucleofected with a Nucleofector apparatus (Amaxa, GmbH). After electroporation, cells were immediately mixed with 500 μl prewarmed Nucleofector recovery medium (Hyclone, Logan, UT) and transferred into culture plates containing prewarmed complete medium. Cells were incubated at 37°C over a time period of 1 to 2 days until being analyzed. Transfection efficiency of the cells by this method was typically 70 to 80%.
Western Blotting
After specific treatments, cells were incubated in lysis buffer containing 20 mM Tris-HCl (pH 7.5), 1% Triton X-100, 150 mM NaCl, 10% glycerol, 1 mM sodium orthovanadate, 50 mM sodium formate, 100 mM PMSF, and a commercial protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN) for 20 minutes on ice. After insoluble debris was precipitated by centrifugation at 14,000 × g for 15 minutes at 4°C, the supernatants were collected and assayed for protein content with bicinchoninic acid assay kit (Pierce Biotechnology, Rockford, IL). Equal amount of proteins per sample (15 μg) were resolved on 10% SDS-PAGE and transferred onto 0.45-μm nitrocellulose membranes. The transferred membranes were blocked for 1 hour in 5% nonfat dry milk in Tris-buffered saline and Tween (25 mM Tris-HCl [pH 7.4], 125 mM NaCl, 0.05% Tween-20) and incubated with appropriate primary antibodies, followed by horseradish peroxidase–conjugated isotype-specific secondary antibodies. The immune complexes were detected by chemiluminescence (Supersignal West Pico; Pierce Biotechnology), and quantified by imaging densitometry with UN-SCAN-IT automated digitizing software (Silk Scientific, Orem, UT). Mean densitometry data from independent experiments were normalized to the control.
Immunoprecipitation
After treatments, cells were washed with ice-cold PBS and lysed in lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 10% glycerol, 0.2% NP-40, 100 mM PMSF, and a commercial protease inhibitor mixture) at 4°C for 20 minutes. After centrifugation at 14,000 × g for 15 minutes at 4°C, the supernatants were collected and the protein content was determined by bicinchoninic acid protein assay. Cleared lysates were normalized, and 60 μg proteins were incubated with 8 μl of anti-myc agarose bead (Santa Cruz Biotechnology) diluted with 12 μl protein A agarose for 4 hours at room temperature. The immune complexes were washed three times with 500 μl lysis buffer, resuspended in 2× Laemmli sample buffer, and boiled at 95°C for 5 minutes. The immune complexes were separated by 10% SDS-PAGE and analyzed by Western blotting, as described previously here.
Measurement of Bcl-2 S-Nitrosylation
S-nitrosylation of Bcl-2 was measured as previously described (15). In brief, cells were treated, harvested, lysed, and subjected to immunoprecipitation as described previously here. The immunoprecipitates were rinsed four times with lysis buffer and twice with PBS. The pellets were resuspended in 500 μl of PBS and incubated with HgCl2 (200 μM) and DAN (200 μM) for 0.5 hour in the dark at room temperature, followed by the addition of 1 M NaOH. Samples were then quantified with a fluorometer (FLUOstar OPTIMA) at the excitation and emission wavelengths of 375 and 450 nm, respectively.
Statistical Analysis
The data represent means (±SD) from three or more independent experiments. Statistical analysis was performed by Student's t test at a significance level of P less than 0.05.
RESULTS
Chronic Cr(VI) Exposure Leads to Malignant Transformation of Nontumorigenic Lung Epithelial Cells
Nontumorigenic Beas-2B cells were continuously exposed to a subcytotoxic concentration of Cr(VI) (5 μM) in culture, and passaged weekly. Phenotypic changes were observed in Cr(VI)-treated Beas-2B cells within 10–12 weeks of exposure. Cr(VI)-transformed cells began forming cell mounds, whereas mounding was not observed in passage-matched control cells, and colony formation was observed even in cells plated at subconfluent densities after 24 weeks of exposure to Cr(VI). Cell mounding (observed using Hoechst 33342 dye stain) indicated loss of contact inhibition, and was the first indication of the malignant transformation of nontumorigenic Beas-2B cells (Figure 1A). Soft agar colony formation assay was performed to confirm Cr(VI)-induced malignant transformation of lung epithelial cells. Passage-matched control Beas-2B cells and B-Cr cells were grown on agar plates so as to assess anchorage-independent growth. At 2 weeks after seeding, significant colony formation was observed in B-Cr cells, with a fourfold increase as compared with passage-matched control Beas-2B cells (Figures 1B and 1C). We further characterized the malignant potential of Cr(VI)-transformed cells by analyzing their proliferative, invasive, and migratory properties as compared with the passage-matched control cells in complete medium. B-Cr cells proliferated twice as fast as the control Beas-2B cells (Figure 1D), consistent with their malignant behavior. Furthermore, B-Cr cells demonstrated a roughly fourfold increase in invasion, and a nearly fivefold increase in migration rate as compared with the passage-matched control cells (Figures 1E and 1F).
Figure 1.

Hexavalent chromium (Cr(VI))–induced malignant transformation of human lung epithelial Beas-2B cells. (A) Cultured Beas-2B cells were continuously exposed to 5 μM Cr(VI) for 24 weeks. Fluorescence micrographs of passage-matched control Beas-2B and Cr(VI)-transformed, Cr(VI)-exposed Beas-2B (B-Cr) cells stained with Hoechst 33342 dye. (B) Beas-2B and B-Cr cells (3 × 104 cells) were seeded on 0.5% agar plates and incubated at 37°C in a 5% CO2 incubator. After 2 weeks, colonies were scored under a light microscope. (C) Representative micrographs of colonies formed by Beas-2B and B-Cr cells in soft agar are shown. (D) Beas-2B and B-Cr cells were plated in a 96-well plate at a density of 2 × 104 cells in growth medium. After 12, 24, and 48 hours, cells were incubated with 50 μl of 1× Cyquant dye binding solution, and cell proliferation was measured at the excitation and emission wavelengths of 485 and 535 nm, respectively. Beas-2B and B-Cr cells, at a density of 1.5 × 104 cells/ml or 3 × 104 cells/ml, were added to control inserts (migration) or inserts coated with Matrigel (invasion), and incubated for 24 and 48 hours, respectively. Cells were counted under a fluorescence microscope after staining, and the average number of cells was scored in each case. (E) Plots show relative invasion and migration of B-Cr cells as compared with passage-matched Beas-2B cells. (F) Representative micrographs of cells stained for invasion are shown. Values are means (±SD) (n = 4). *P < 0.05 versus passage-matched control cells.
Apoptosis and Caspase-9 Activity in Cr(VI)-Transformed Cells
To characterize the apoptotic response in our in vitro system, both Cr(VI)-transformed cells and passage-matched control cells were treated with different doses of Cr(VI) (0–50 μM), and apoptosis was determined after 12 hours by Hoechst 33342 assay. Figures 2A and 2B show that Cr(VI) treatment caused a dose-dependent increase in apoptosis in Beas-2B cells. In contrast, B-Cr cells demonstrated significantly reduced apoptosis. We confirmed the apoptotic response by measuring caspase-9 levels in both Beas-2B and B-Cr cells (Figure 2C). Both basal and stimulated levels of caspase-9 were significantly lower in B-Cr cells as compared with passage-matched Beas-2B cells.
Figure 2.

Apoptosis and caspase-9 levels in Beas-2B and B-Cr cells. (A) Subconfluent (90%) monolayers of Beas-2B and B-Cr cells were exposed to varying concentrations of Cr(VI) (0–50 μM) for 12 hours and analyzed for apoptosis by Hoechst 33342 assay. (B) Fluorescence micrographs of Beas-2B and B-Cr cells treated with Cr(VI) (0–50 μM) for 12 hours and stained with Hoechst 33,342 dye. Plots show relative apoptosis as compared with nontreated control. (C) Caspase-9 activity assay of Beas-2B and B-Cr cells treated with Cr(VI) (0–50 μM) for 12 hours. Cell lysates (20 μg protein) were prepared and analyzed for caspase-9 activity with specific fluorescent substrates LEHD-AMC. Plots show relative fluorescence intensity over nontreated control. Values are means (±SD) (n = 3). *P < 0.05 versus nontreated control.
Cellular NO Level Is Increased in Cr(VI)-Transformed Cells
Because NO has been implicated as an important mediator of carcinogenesis in a number of tumors, we assessed cellular NO levels in Cr(VI)-transformed cells and passage-matched control cells. Cr(VI)-induced NO production was significantly higher in the transformed B-Cr cells compared with control cells (Figure 3A), which was confirmed using NO modulators, such as AG (NO inhibitor) and DPTA NONOate (NO donor) (Figure 3B). To further demonstrate the role of NO in Cr(VI)-induced apoptosis and its resistance, we assessed the effect of NO modulators on apoptosis and caspase-9 levels induced by Cr(VI) in both Beas-2B and transformed B-Cr cells. Figures 3C and 3D show that treatment of the cells with NO donor DPTA NONOate significantly inhibited Cr(VI)-induced apoptosis and caspase-9 in both control and transformed cells, whereas NO inhibitor AG augmented this effect. The effect was more pronounced in B-Cr cells.
Figure 3.

Cellular nitric oxide (NO) levels in Beas-2B and B-Cr cells. (A) Spectrofluorometric measurement of diaminofluorescein (DAF) fluorescence in Beas-2B and B-Cr cells. Cells were treated with Cr(VI) (0–50 μM) for 1 hour. Plots show relative fluorescence intensity over nontreated control. (B) Beas-2B and B-Cr cells were pretreated for 0.5 hour with aminoguanidine (AG) (300 μM) or dipropylenetriamine (DPTA) NONOate (400 μM). The cells were then treated with Cr(VI) (20 μM) for 1 hour and analyzed for DAF fluorescence. Plots show relative fluorescence intensity over nontreated control. (C) Subconfluent (90%) monolayers of Beas-2B and B-Cr cells were pretreated with AG (300 μM) or DPTA NONOate (400 μM) for 1 hour. The cells were then treated with Cr(VI) (20 μM) for 12 hours and analyzed for apoptosis by Hoechst 33342 assay. Plots show relative apoptosis as compared with nontreated control. (D) Subconfluent (90%) monolayers of Beas-2B and B-Cr cells were either left untreated or pretreated with AG (300 μM) or DPTA NONOate (400 μM) for 1 hour, followed by Cr(VI) treatment (20 μM) for 12 hours. Caspase activity was measured, as described in Materials and Methods. Values are means (±SD) (n = 3). *P < 0.05 versus nontreated control or passage-matched control cells; #P < 0.05 versus Cr(VI)-treated control.
S-Nitrosylation of Bcl-2 Up-Regulates Its Expression Levels in Cr(VI)-Transformed Cells
NO-mediated S-nitrosylation has been shown to be important post-translational modification in regulating the action of a number of intracellular proteins in the apoptotic pathway, including c-FLIP and Bcl-2 (15–17). Because we observed elevated levels of NO in Cr(VI)-exposed transformed cells, we investigated whether Bcl-2 expression was modulated in B-Cr cells with Western blot analysis. Figure 4A shows that Bcl-2 is down-regulated in response to Cr(VI) stimulation in Beas-2B cells (lanes 1–3); however, sustained levels of Bcl-2 were observed in B-Cr cells (lanes 4–6). To ascertain that the stabilization of Bcl-2 occurred due to its NO-mediated S-nitrosyation, both Beas-2B and B-Cr cells were transiently transfected with myc–Bcl-2 plasmid, and analyzed for ubiquitination or S-nitrosylation. The results show a decrease in ubiquitinated Bcl-2 (Figure 4B) and an increase in S-nitrosylated Bcl-2 (Figures 4C and 4D) levels in B-Cr cells as compared with passage-matched control Beas-2B cells.
Figure 4.

Bcl-2 expression and ·O2− levels in Beas-2B and B-Cr cells. (A) Beas-2B and B-Cr cells were treated with Cr(VI) (10 and 20 μM) for 12 hours, and cell lysates were prepared and analyzed for Bcl-2 expression by Western blotting. Densitometry was performed to determine the relative Bcl-2 levels after reprobing the membrane with β-actin antibody. Beas-2B and B-Cr cells were transfected with myc-tagged WT Bcl-2 plasmid. Transfected cells were treated with Cr(VI) (10 and 20 μM) for 3 hours and immunoprecipitated with anti-myc antibody. Immunopellets were analyzed for (B) ubiquitin or (C) S-nitrocysteine by Western blotting. Densitometry was performed to determine the relative S-nitrocysteine levels after reprobing the membrane with anti–Bcl-2 antibody. (D) Similarly transfected and treated immunopellets were incubated with 200 μM HgCl2 and 200 μM diaminonaphthalene (DAN). NO released from S-nitrosylated Bcl-2 was quantified at 375 nm/450 nm. (E) Spectrofluorometric measurement of dihydroethidium bromide (DHE) fluorescence in Beas-2B and B-Cr cells. Cells were treated with Cr(VI) (0–50 μM) for 1 hour and analyzed for DHE fluorescence. Plots show relative fluorescence intensity over nontreated control. (F) Beas-2B and B-Cr cells were treated with Cr(VI) (20 μM) in the presence or absence of AG (300 μM) or DPTA NONOate (400 μM) for 1 hour and analyzed for DHE fluorescence. Plots show relative fluorescence intensity over nontreated control. Values are means (±SD) (n = 3). *P < 0.05 versus nontreated control; #P < 0.05 versus Cr(VI)-treated control.
We also investigated the role of reactive oxygen species in our model system, and observed significantly decreased ·O2− generation in B-Cr cells as compared with Beas-2B cells (Figure 4E). Furthermore, ·O2− level was significantly augmented in the presence of NO inhibitor, AG, and inhibited in the presence of NO donor, DPTA NONOate, in both transformed and passage-matched controls (Figure 4F), indicating an additional regulatory role for NO in mediating the effects of Cr(VI) exposure.
Effect of NO Modulators on Cr(VI)-Induced Malignant Transformation
We furthered assessed the importance of NO in contributing to the carcinogenic phenotype observed in Cr(VI)-exposed transformed cells with NO modulators. DPTA NONOate increased cell invasion, migration, proliferation, and colony formation in both B-Cr and Beas-2B cells, whereas AG decreased this effect; however, the effect of NO modulators was more pronounced in B-Cr cells as compared with normal Beas-2B cells (Figures 5A–5D).
Figure 5.

Effect of NO modulators on Cr(VI)-induced malignant transformation. (A and B) Beas-2B and B-Cr cells at a density of 1.5 × 104 cells/ml or 3 × 104 cells/ml with AG (300 μM) or DPTA NONOate (400 μM) were added to control inserts (migration) or inserts coated with Matrigel (invasion) and incubated for 24 and 48 hours, respectively. After staining, cells were counted under a fluorescence microscope, and the average number of cells was scored in each case. (C) Beas-2B and B-Cr cells with AG (300 μM) or DPTA NONOate (400 μM) were plated in a 96-well plate at a density of 2 × 104 cells in growth medium. After 24 hours, cells were incubated with 50 μl of 1× Cyquant dye binding solution, and cell proliferation was measured at 485 nm/535 nm. (D) Beas-2B and B-Cr cells (3 × 104 cells) with AG (300 μM) or DPTA NONOate (400 μM) were seeded on 0.5% agar plates and incubated at 37°C in a 5% CO2 incubator. After 2 weeks, colonies were scored under a light microscope. Values are means (±SD) (n = 3). *P < 0.05 versus nontreated control.
Effect of Nonnitrosylable Mutant of Bcl-2 on Apoptosis and Malignant Transformation
To confirm that NO-mediated S-nitrosylation of Bcl-2 may be an important mechanism in Cr(VI)-induced malignant transformation of cells, Beas-2B and B-Cr cells were transfected with WT Bcl-2, nonnitrosylable (C158A/C229A) double-mutant (DM) Bcl-2, or pcDNA3 (control) plasmid (Figure 6A), and their apoptosis response and malignant properties were examined by cell invasion, migration, proliferation, and colony formation assays. Ectopic expression of WT Bcl-2 significantly reduced apoptosis whereas DM Bcl-2 expression increased it as compared with control (pcDNA3) transfected cells (Figure 6B). Furthermore, ectopic expression of WT Bcl-2 increased cell invasion, migration, proliferation, and colony formation in both B-Cr and Beas-2B cells as compared with the control transfectant (Figures 6C–6F). In contrast, expression of DM Bcl-2 showed inhibitory effects on the malignant properties of the cells.
Figure 6.

Effect of wild-type and cysteine-mutated Bcl-2 on Cr(VI)-induced apoptosis and malignant transformation. (A) Beas-2B and B-Cr cells were transfected with WT Bcl-2, DM (C158A/C229A) Bcl-2, or control (pcDNA3) plasmid. Cell lysates were analyzed for Bcl-2 expression by Western blotting. β-Actin was used as a loading control. (B) Transfected cells were treated with Cr(VI) (20 μM) for 12 hours and analyzed for apoptosis by Hoechst 33342 assay. Plots show relative apoptosis as compared with nontreated control. (C) Transfected cells were plated in a 96-well plate at a density of 2 × 104 cells in growth medium. After 24 hours, cells were incubated with 50 μl of 1× Cyquant dye binding solution, and cell proliferation was measured at the excitation/emission wavelengths of 485 nm/535 nm. (D) Transfected cells (3 × 104 cells) were seeded on 0.5% agar plates and incubated at 37°C in a 5% CO2 incubator. After 2 weeks, colonies were scored under a light microscope. (E and F) Transfected cells at a density of 1.5 × 104 cells/ml or 3 × 104 cells/ml were added to control inserts (migration) or inserts coated with Matrigel (invasion) and incubated for 24 and 48 hours, respectively. After staining, cells were counted under a fluorescence microscope, and the average number of cells was scored in each case. Values are means (±SD) (n = 4). *P < 0.05 versus control transfectant.
DISCUSSION
Although Cr(VI) has been identified as a human carcinogen, in vivo studies have yielded negative or inconsistent results. To overcome this problem, we have developed an in vitro model by subjecting nontumorigenic human lung epithelial Beas-2B cells to long-term Cr(VI) exposure. Beas-2B cells have been widely used in the literature to define conditions under which various oncogenes and biological agents cause neoplastic transformation (18–20). In our study, Cr(VI) exposure led to the malignant transformation of nontumorigenic Beas-2B cells, as indicated by the loss of contact inhibition, colony formation, and increased rates of cell invasion, migration, and proliferation. Loss of contact inhibition is an exclusive property of cancer cells and an early indication of malignant transformation of nontumorigenic (21, 22). In our study, Cr(VI)-transformed B-Cr cells demonstrated extensive cell mounding, whereas passage-matched control Beas-2B cells formed a monolayer (Figure 1). We exposed cells to Cr(VI), and eventually selected cells that became resistant to Cr(VI)-induced cell death and kept proliferating, eventually forming cell mounds. Phenotypic changes were observed in Cr(VI)-transformed B-Cr cells that appeared small and circular as compared with elongated passage-matched control Beas-2B cells. Cr(VI)-transformed B-Cr cells also exhibited anchorage-independent growth and increased colony formation in soft agar, indicating the malignant potential of Cr(VI). Anchorage-independent growth has been widely correlated with tumorigenicity and invasiveness in several cancer cell types, including small-cell lung carcinoma (23). Interestingly, nontumorigenic human bronchial epithelial Beas-2B cells obtained from the autopsy of individuals without cancer also formed a small number of slow-growing colonies on soft agar. This could be due to the fact that Beas-2B cells are reported to possess mutated and dysfunctional p53 tumor suppressor gene (18, 24). Additionally, the American Type Culture Collection indicates that the Beas-2B cell line forms colonies in semisolid medium, but was nontumorigenic in immunosuppressed mice. Therefore, the colony formation observed in Beas-2B cells is due to the indigenous properties of the cell, and not because of their tumorigenic potential. Recent reports also indicate that, in early passage, Beas-2B are nontumorigenic, aneuploid cells, but undergo squamous differentiation in response to serum and transforming growth factor-β1 (25). However, because passage-control Beas-2B cells showed no phenotypic changes or malignant behavior, it can be concluded that B-Cr cells are Cr(VI)-transformed Beas-2B cells, and not Beas-2B cells that have undergone differentiation and show altered phenotype due to continued passaging.
We have previously shown that Cr(VI) induces apoptosis, mainly via the intrinsic mitochondrial pathway (15, 26). Cr(VI)-induced apoptosis and caspase-9 activity was significantly decreased in Cr(VI)-transformed cells as compared with passage-matched control, which indicated that continuous exposure of Cr(VI) may modify the cellular system such that the cells develop a mechanism to escape apoptosis. We have also reported that Cr(VI)-induced NO production regulates cellular events such that the cells become resistant to Cr(VI)-induced apoptosis (15). Therefore, it is plausible that NO-mediated regulation of Cr(VI)-induced apoptosis may be a key mechanism by which cells acquire apoptosis-resistant phenotype. NO is elevated in the apoptosis-resistant B-Cr cells as compared with passage-matched control cells. Furthermore, NO donor inhibited Cr(VI)-induced apoptosis and caspase-9 activation, whereas NO inhibitor augmented this effect (Figure 3). The effect of NO modulators on apoptosis and caspase-9 activation confirmed the antiapoptotic role of NO in response to Cr(VI) treatment, and suggested a key role of NO in apoptosis resistance and malignant transformation. This is supported by the evidence that NO inhibitor (AG) inhibited cell invasion, migration, proliferation, and colony formation of Cr(VI)-transformed cells, whereas NO donor (DPTA NONOate) promoted these effects (Figure 5). The effects of NO were nominal in Beas-2B cells, but strongly enhanced in Cr(VI)-transformed B-Cr cells.
Sensitivity to apoptosis depends on the expression levels of various apoptosis-regulatory proteins. A functional loss of proapoptotic proteins and/or increased expression of antiapoptotic protein can confer resistance to apoptotic stimuli (27, 28). The oncogenic potential of Bcl-2 is well established, with its overexpression reported in various cancers, including lung cancer (29–33). NO-mediated S-nitrosylation is an important post-translational modification that regulates the action of important intracellular proteins in the apoptotic pathway. S-nitrosylation has been shown to prevent apoptosis by the inactivation of the proapoptotic caspase family of proteins (34) or stabilization of antiapoptotic proteins, such as c-FLIP (17) and Bcl-2 (15). We have earlier reported that Bcl-2 is also involved in cell death resistance in response to Cr(VI) exposure. S-nitrosylation of Bcl-2 by Cr(VI)-induced NO inhibited its down-regulation and degradation via the ubiquitin–proteasomal degradation pathway (15). In this study, we found that Bcl-2 levels are sustained in B-Cr cells in response to Cr(VI) as opposed to passage-matched controls, primarily through increased S-nitrosylation of Bcl-2, which precludes its ubiquitin-dependent degradation. Elevated levels of NO in B-Cr cells induced increased nitrosylation of Bcl-2 as compared with Beas-2B cells, whereas levels of ubiquitinated Bcl-2 were decreased in transformed cells. In light of our previous work, this observation confirmed that NO-mediated S-nitrosylation of Bcl-2 prevents it ubiquitin proteasomal degradation and stabilizes the protein, thus inhibiting apoptosis (15). B-Cr cells transfected with a nonnitrosylable mutant (DM Bcl-2) demonstrated decreased migration, proliferation, and invasion as compared with B-Cr cells transfected with empty vector. This confirmed that NO-mediated S-nitrosylation of Bcl-2 is a critical event in the malignant transformation of nontumorigenic lung epithelial cells in response to Cr(VI) exposure, as cysteine-mutated Bcl-2 is incapable of undergoing S-nitrosylation. In our previous report, we showed that cells transfected with WT Bcl-2 plasmid underwent S-nitrosylation in response to Cr(VI) treatment, whereas DM Bcl-2–transfected cells, in which the cysteine residues at the 158 and 229 positions were replaced with alanines, were incapable of S-nitrosylation, and showed increased Bcl-2 ubiquitination (15). Furthermore, basal levels of ·O2− anions in B-Cr cells were also decreased as compared with passage-matched control cells, and were susceptible to NO modulators (Figure 4). We have previously demonstrated that induction of apoptosis by Cr(VI) is dependent on reactive oxygen species, particularly ·O2− generation (26). Therefore, it is plausible that the high basal levels of NO in B-Cr cells contribute to the decreased levels of ·O2− in our system, leading to increased Bcl-2 levels regulated by NO-mediated nitrosylation and stabilization. Overall, the data suggest that NO-mediated S-nitrosylation of Bcl-2 and down-regulation of ·O2− are critical events in the malignant transformation of nontumorigenic lung epithelial cells in response to Cr(VI) exposure.
The present study fortifies earlier epidemiological reports by providing evidence that human lung epithelial cells are susceptible to Cr(VI)-induced malignant transformation. We have established an important in vitro model in which the molecular and genetic events associated with Cr(VI)-induced carcinogenesis can be studied. This study also reveals an important mechanism that implicates ·O2−, NO, and Bcl-2 regulation in the development of an apoptosis-resistant, malignant phenotype. This novel mechanism may also be critical in malignant transformation of normal cells under various conditions in response to other human carcinogens, and warrants investigation in the future with similar in vitro approaches.
This work was supported by National Institutes of Health grant R01HL76340.
The findings and conclusions in this article are those of the authors, and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
Originally Published in Press as DOI: 10.1165/rcmb.2009-0094OC on June 25, 2009
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Jemal A, Clegg LX, Ward E, Ries LA, Wu X, Jamison PM, Wingo PA, Howe HL, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975–2001, with a special feature regarding survival. Cancer 2004;101:3–27. [DOI] [PubMed] [Google Scholar]
- 2.Azad N, Rojanasakul Y, Vallyathan V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev 2008;11:1–15. [DOI] [PubMed] [Google Scholar]
- 3.IARC. IARC monograph on the evaluation of carcinogenic risk to humans: chromium, nickel and welding. Lyon, France; IARC: 1990. pp. 49–445. [PMC free article] [PubMed]
- 4.Machle W, Gregorius F. Cancer of the respiratory system in the United States chromate-producing industry. Public Health Rep 1948;63:1114–1127. [PubMed] [Google Scholar]
- 5.De Flora S. Threshold mechanisms and site specificity in chromium(VI) carcinogenesis. Carcinogenesis 2000;21:533–541. [DOI] [PubMed] [Google Scholar]
- 6.Langard S. One hundred years of chromium and cancer: a review of epidemiological evidence and selected case reports. Am J Ind Med 1990;17:189–215. [DOI] [PubMed] [Google Scholar]
- 7.Langard S. Role of chemical species and exposure characteristics in cancer among persons occupationally exposed to chromium compounds. Scand J Work Environ Health 1993;19:81–89. [PubMed] [Google Scholar]
- 8.Simonato L, Fletcher AC, Andersen A, Anderson K, Becker N, Chang-Claude J, Ferro G, Gerin M, Gray CN, Hansen KS, et al. A historical prospective study of European stainless steel, mild steel, and shipyard welders. Br J Ind Med 1991;48:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balansky RM, D'Agostini F, Izzotti A, De Flora S. Less than additive interaction between cigarette smoke and chromium(VI) in inducing clastogenic damage in rodents. Carcinogenesis 2000;21:1677–1682. [DOI] [PubMed] [Google Scholar]
- 10.Mackenzie RD, Byerrum RU, Decker CF, Hoppert CA, Langham RF. Chronic toxicity studies. II. Hexavalent and trivalent chromium administered in drinking water to rats. AMA Arch Ind Health 1958;18:232–234. [PubMed] [Google Scholar]
- 11.Bucher J. NTP toxicity studies of sodium dichromate dihydrate (CAS no. 7789-12-0) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice. Toxic Rep Ser 2007;72:1–G4. [PubMed] [Google Scholar]
- 12.Holmes AL, Wise SS, Wise JP Sr. Carcinogenicity of hexavalent chromium. Indian J Med Res 2008;128:353–372. [PubMed] [Google Scholar]
- 13.Levy LS, Martin PA, Bidstrup PL. Investigation of the potential carcinogenicity of a range of chromium containing materials on rat lung. Br J Ind Med 1986;43:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark GJ, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods Enzymol 1995;255:395–412. [DOI] [PubMed] [Google Scholar]
- 15.Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation: a novel antiapoptotic mechanism that suppresses apoptosis. J Biol Chem 2006;281:34124–34134. [DOI] [PubMed] [Google Scholar]
- 16.Chanvorachote P, Nimmannit U, Wang L, Stehlik C, Lu B, Azad N, Rojanasakul Y. Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin–proteasome–mediated degradation of FLICE inhibitory protein. J Biol Chem 2005;280:42044–42050. [DOI] [PubMed] [Google Scholar]
- 17.Iyer AK, Azad N, Wang L, Rojanasakul Y. Role of S-nitrosylation in apoptosis resistance and carcinogenesis. Nitric Oxide 2008;19:146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerwin BI, Spillare E, Forrester K, Lehman TA, Kispert J, Welsh JA, Pfeifer AM, Lechner JF, Baker SJ, Vogelstein B, et al. Mutant p53 can induce tumorigenic conversion of human bronchial epithelial cells and reduce their responsiveness to a negative growth factor, transforming growth factor beta 1. Proc Natl Acad Sci USA 1992;89:2759–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khatlani TS, Wislez M, Sun M, Srinivas H, Iwanaga K, Ma L, Hanna AE, Liu D, Girard L, Kim YH, et al. c-Jun N-terminal kinase is activated in non–small-cell lung cancer and promotes neoplastic transformation in human bronchial epithelial cells. Oncogene 2007;26:2658–2666. [DOI] [PubMed] [Google Scholar]
- 20.Lehman TA, Reddel R, Peiifer AM, Spillare E, Kaighn ME, Weston A, Gerwin BI, Harris CC. Oncogenes and tumor-suppressor genes. Environ Health Perspect 1991;93:133–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velge P, Kaeffer B, Bottreau E, Van Langendonck N. The loss of contact inhibition and anchorage-dependent growth are key steps in the acquisition of Listeria monocytogenes susceptibility phenotype by non-phagocytic cells. Biol Cell 1995;85:55–66. [PubMed] [Google Scholar]
- 22.Abercrombie M. Contact inhibition in tissue culture. In Vitro 1970;6:128–142. [DOI] [PubMed] [Google Scholar]
- 23.Carney DN, Gazdar AF, Minna JD. Positive correlation between histological tumor involvement and generation of tumor cell colonies in agarose in specimens taken directly from patients with small-cell carcinoma of the lung. Cancer Res 1980;40:1820–1823. [PubMed] [Google Scholar]
- 24.Van Vleet TR, Watterson TL, Klein PJ, Coulombe RA Jr. Aflatoxin B1 alters the expression of p53 in cytochrome P450-expressing human lung cells. Toxicol Sci 2006;89:399–407. [DOI] [PubMed] [Google Scholar]
- 25.Ke Y, Reddel RR, Gerwin BI, Miyashita M, McMenamin M, Lechner JF, Harris CC. Human bronchial epithelial cells with integrated SV40 virus T antigen genes retain the ability to undergo squamous differentiation. Differentiation 1988;38:60–66. [DOI] [PubMed] [Google Scholar]
- 26.Azad N, Iyer AK, Manosroi A, Wang L, Rojanasakul Y. Superoxide-mediated proteasomal degradation of Bcl-2 determines cell susceptibility to Cr(VI)-induced apoptosis. Carcinogenesis 2008;29:1538–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol 1998;16:395–419. [DOI] [PubMed] [Google Scholar]
- 28.Hickman JA. Apoptosis and tumourigenesis. Curr Opin Genet Dev 2002;12:67–72. [DOI] [PubMed] [Google Scholar]
- 29.Buolamwini JK. Novel anticancer drug discovery. Curr Opin Chem Biol 1999;3:500–509. [DOI] [PubMed] [Google Scholar]
- 30.Jiang SX, Sato Y, Kuwao S, Kameya T. Expression of bcl-2 oncogene protein is prevalent in small cell lung carcinomas. J Pathol 1995;177:135–138. [DOI] [PubMed] [Google Scholar]
- 31.Osford SM, Dallman CL, Johnson PW, Ganesan A, Packham G. Current strategies to target the anti-apoptotic Bcl-2 protein in cancer cells. Curr Med Chem 2004;11:1031–1039. [DOI] [PubMed] [Google Scholar]
- 32.Ben-Ezra JM, Kornstein MJ, Grimes MM, Krystal G. Small cell carcinomas of the lung express the Bcl-2 protein. Am J Pathol 1994;145:1036–1040. [PMC free article] [PubMed] [Google Scholar]
- 33.Ikegaki N, Katsumata M, Minna J, Tsujimoto Y. Expression of bcl-2 in small cell lung carcinoma cells. Cancer Res 1994;54:6–8. [PubMed] [Google Scholar]
- 34.Mannick JB. Regulation of apoptosis by protein S-nitrosylation. Amino Acids 2007;32:523–526. [DOI] [PubMed] [Google Scholar]