

Abstract
Oligomers of acylated lysines (OAKs) are synthetic mimics of host defense peptides (HDPs) with promising antimicrobial properties. Here we challenged the OAK concept for its ability to generate both systemically efficient and economically viable lead compounds for fighting multidrug-resistant bacteria. We describe the design and characterization of a miniature OAK composed of only 3 lysyls and 2 acyls (designated C12(ω7)K-β12) that preferentially targets gram-positive species by a bacteriostatic mode of action. To gain insight into the mechanism of action, we examined the interaction of OAK with various potential targets, including phospholipid bilayers, using surface plasmon resonance, and Langmuir monolayers, using insertion assays, epifluorescence microscopy, and grazing incidence X-ray diffraction, in a complementary manner. Collectively, the data support the notion that C12(ω7)K-β12 damages the plasma-membrane architecture similarly to HDPs, that is, following a near-classic 2-step interaction including high-affinity electrostatic adhesion and a subsequent shallow insertion that was limited to the phospholipid head group region. Notably, preliminary acute toxicity and efficacy studies performed with mouse models of infection have consolidated the potential of OAK for treating bacterial infections, including systemic treatments of methicillin-resistant Staphylococcus aureus. Such simple yet robust chemicals might be useful for various antibacterial applications while circumventing potential adverse effects associated with cytolytic compounds.—Sarig, H., Livne, L., Held-Kuznetsov, V., Zaknoon, F., Ivankin, A., Gidalevitz, D., Mor, A. A miniature mimic of host defense peptides with systemic antibacterial efficacy.
Keywords: antimicrobial peptide, chemical mimicry, drug design, drug resistance, mechanism of action
Host defense peptides (HDPs) are ubiquitous cationic amphiphiles known to exert broad-spectrum cytotoxic activities via multiple mechanisms, including plasma-membrane disruption, inhibition of enzymatic activities, and biosynthesis (1,2,3,4). Such a multitarget mode of action might significantly overcome various drug resistance mechanisms, thereby rendering HDPs attractive in a variety of antimicrobial applications (5). However, various drawbacks, such as limited bioavailability, optional toxicity, and high manufacturing costs, hamper this potential, particularly toward systemic therapies.
The peptidomimetic approach has emerged in recent years as a powerful means for overcoming the inherent limitations of peptides’ physical characteristics (6, 7). Typical peptidomimetic strategies attempted to mimic the peptide primary structure using amide bond isosteres or backbone modifications by chain extension or heteroatom incorporation through the use of either β-amino acids (8, 9), fluorinated amino acids (10), peptoids (11, 12), arylamides (13), or acylated lysines (14). Nonpeptide mimics were also reported, namely, based on a steroid scaffold (15), phenylene ethynylene (16), polymethacrylate (17), or polynorbornene backbones (18). Although these mimics address the issue of susceptibility to proteolysis and in some cases exhibit interesting in vitro antimicrobial properties, most present one or more severe problems pertaining to toxicity, limited potency, and/or high production costs, while the in vivo properties of most of these mimics remain unknown.
Oligomers of acylated lysines (OAKs) are among the simpler HDP-mimetic designs, where the two essential characteristics for antimicrobial activity, hydrophobicity and charge, are represented by tandem repeats of an amide-linked amino fatty acid and a cationic amino acid (19). This design was shown to overcome limitations of conventional peptides with respect to in vivo efficacy and toxicity (20). Representative OAKs were shown to exert potent bactericidal activities by distinct sequence-specific mechanisms that either damaged the cytoplasmic membrane or directly inhibited DNA expression (20). Here we report that the OAK concept can generate bacteriostatic compounds that selectively target gram-positive bacteria, indicating that the minimal requirements for potent yet selective activity are embedded in a specific arrangement of 3 lysines and 2 fatty acids.
MATERIALS AND METHODS
Synthesis
The OAKs were synthesized by the solid-phase method (21) by applying the 9-fluorenylmethyloxy carbonyl active-ester chemistry (model 433A peptide synthesizer; Applied Biosystems, Foster City, CA, USA) essentially as described previously (14). The crude compounds were purified to chromatographic homogeneity in the range of >95% by reverse-phase high-performance liquid chromatography (HPLC) equipped with a mass spectrometer (MS) (Alliance-ZQ; Waters, Milford, MA, USA).
Cytotoxicity assays
Fresh human blood was collected from one volunteer into a heparinated test tube and washed 3 times in PBS. Next, 200-μl suspensions (20% hematocrit) were added to 200 μl of PBS alone (as blank) or PBS containing either OAKs (in serial 2-fold dilutions) or 0.2% Triton X-100 (for 100% hemolysis). After 15 min incubation (37°C under shaking), reaction was stopped by addition of 400 μl of cold PBS, followed by centrifugation (10,000 g, 2 min), and absorbance of the supernatant was read (350 nm).
Cytotoxicity to primary fibroblasts was assessed by the XTT [2,3-(bis-2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5- carboxanilide inner salt] assay after 24 h culture. Primary human foreskin fibroblasts were grown in DMEM supplemented with 10% fetal calf serum, 1% nonessential amino acids, 100 μM β-mercaptoethanol (Life Technologies, Gaithersburg, MD, USA), 2 mM l-glutamine, and 25 U/ml penicillin-streptomycin.
Organization in solution
OAKs at an initial concentration of 160 μg/ml were submitted to serial 2-fold dilutions in PBS and incubated for 2 h at room temperature, and light scattering of each dilution was evaluated by holding both the excitation and the emission at 400 nm (1 nm slits).
Bacteria
All bacteria were cultured in LB medium (10 g/L tryptone, 5 g/L yeast extract, and 5 g/L NaCl, pH 7.4).
Gram-positive bacteria
Staphylococcus aureus, American Type Culture Collection (ATCC; Manassas, VA, USA) strains 25923, 29213, and methicillin-resistant S. aureus (MRSA) 43300; methicillin-sensitive S. aureus (MSSA) clinical isolated strains 15873, U-17309, 15877, 16001, and 15885; MRSA clinical isolated strains 15903, 15819, 15852, 15918, and U-17314; Listeria seeligeri, ATCC 35967; Listeria grayi, ATCC 19120; Listeria ivanovii, ATCC 19119; Listeria innocua, ATCC 33090; Listeria welshineri, ATCC 35897; Streptococcus agalactiae, ATCC 13813; Bacillus polymyxa, ATCC 842; and Bacillus cereus, ATCC 11778.
Gram-negative bacteria
Escherichia coli, ATCC strains 35218, 25922, and 43894; clinical isolated strains 16223, 16328, 16359, and 14182; Pseudomonas aeruginosa, ATCC strains 9027 and 27853; Salmonella typhimurium, ATCC 14028; and Salmonella choleraesuis, ATCC 7308.
Antibacterial assays
The minimal inhibitory concentrations (MICs) were determined by microdilution assay, in sterilized 96-well plates (final volume of 200 μl), as follows. Bacteria were grown overnight in LB and diluted 10,000-fold in growth medium. Next, 100 μl of LB-containing bacteria (2–4×105 CFU/ml) was added to 100 μl of culture medium containing the test compound (C12(ω7)K-β12 and antibiotics in serial 2-fold dilutions). Inhibition of proliferation was determined by optical density measurements (620 nm) after incubation overnight at 37°C.
Bactericidal kinetics were assessed as follows. Bacteria were grown overnight in LB and diluted 100-fold in growth medium. Next, 100 μl of LB containing bacteria was added to 900 μl of the growth medium containing the test compounds at final concentrations equal to 4 times the MIC value and incubated at 37°C under shaking. At the specified time points, aliquots were diluted (serial 10-fold dilutions in saline) and plated on LB agar for CFU count, using the drop plate method (three 20-μl drops onto LB agar plates). CFUs were counted after an overnight incubation at 37°C. Reported results are from 2 independent experiments.
For the cytoplasmic membrane permeability assay based on ATP reaction, S. aureus (ATCC 29213) were grown overnight in LB (37°C, under shaking), diluted (1000-fold in the grown medium), and treated with either C12(ω7)K-β12 or control peptide (4×MIC each). Extracellular ATP was directly measured in bacterial medium at the specified time points following the producer recommendations (Cell Titer-Glow Luminecnt microbial cell viability assay; G7570; Promega, Madison, WI, USA).
Ethidium bromide (EtBr) uptake was measured as follows: cells were grown overnight in LB and resuspended in PBS containing 0.5% glucose. After incubation of 10 min at 37°C, samples were placed in a 96-well plate containing EtBr (final concentration, 1 μg/ml), and then either C12(ω7)K-β12 (4×MIC) or Triton (0.01%) was added. Fluorescence was recorded by a BioTeK Synergy HT Microplate Reader (excitation, 530 nm; emission, 645 nm; BioTek Instruments, Winooski, VT, USA).
Resistance studies were assessed by selective pressure as follows. MICs were determined against S. aureus ATCC 29213 as described above. After the initial MIC experiment, MICs were determined anew daily, for 15 d, as follows: for each compound tested, bacteria from half the MIC were used for MIC determination in the subsequent generation. In parallel, MIC evolution during these subcultures was compared concomitantly with each new generation, using bacteria harvested from control wells (wells cultured without antimicrobial agent from the previous generation). The relative MIC was calculated for each experiment from the ratio of MIC obtained for a given subculture to that obtained for first-time exposure.
The DNA binding assay was performed by incubation of OAKs (25 μM) with pure pUC19 (150 ng) for 30 min at 37°C, followed by incubation with DNase that opens the plasmid in one point (BamHI) according to the manufacturer’s procedure (1 h at 37°C; New England Biolabs, Beverly, MA, USA). The plasmids and marker (λHindIII) were run in 1% agarose gel for 40 min.
Whole-blood assay
Fresh human blood was collected from one volunteer into a heparinated test tube. Next, 270 μl blood was immediately infected by addition of 24 μl suspension of S. aureus CI 15903 (106 CFU/ml) that were grown overnight in LB and diluted in saline (0.9% NaCl). Inoculation was immediately followed with addition of 5.8 μl OAK solution (1 mg/ml) in distilled water (final concentration of 20 μg/ml, i.e., 4 multiples of the MIC value). After 12 h incubation at 37°C under shaking, samples were diluted (serial 10-fold dilutions in saline) and plated on LB agar for CFU count. Reported results are from 2 independent experiments.
Surface plasmon resonance (SPR)
Binding properties to phospholipid membranes were determined using the BIAcore 2000 optical biosensor system (Biacore Life Science, Uppsala, Sweden) using small unilamellar vesicles composed of 1-palmitoyl 2-oleoyl-sn-glycero-3-phosphoglycerol:1-phosphatidylethanol-amine (POPG:PE, 1:4), and 1-palmitoyl-2-oleoyl-sn-glycero-3phospho glycerol/cardiolipin (POPG:CL, 1.4:1) prepared as described previously (22). Experimental procedures for the SPR assay, analysis of binding kinetics, and calculation of the resulting affinity constants were performed as described (22). The OAK concentrations used were selected to be close to the affinity constant for optimal accuracy.
Insertion assays
To quantify the propensity of OAKs to incorporate into membrane mimics, insertion experiments were carried out as described previously (23). Langmuir monolayers composed of 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol/1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPG:DPPE, 1/4), and 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG) (Avanti Polar Lipids, Alabaster, AL, USA) were used to model inner gram-negative and gram-positive membranes, respectively. Saturated lipids represent up to 50 mol% of membrane lipids, and it is essential to understand their contribution to OAK-membrane interaction. Therefore, saturated analogs of PG and PE lipids were used in the monolayer experiments, which also allowed us to take full advantage of epifluorescence microscopy and grazing incidence X-ray diffraction (GIXD) techniques. Throughout the insertion experiments, surface pressure of the monolayers was kept at 30 mN/m, which is equivalent to the packing density of the cell membrane. The experiments were carried out on Dublecco’s phosphate buffered saline without calcium and magnesium (D-PBS) (Invitrogen, Carlsbad, CA, USA) at 30 ± 0.2°C.
Epifluorescence microscopy
Evolution of the membrane mimics morphology was monitored concurrently with insertion experiments using epifluorescence microscopy as described previously (24). Lipid-linked Texas red-DHPE fluorescence probe (0.5 mol%; Molecular Probes, Eugene, OR, USA) was incorporated into the phospholipid monolayers. Excitation between 530 and 590 nm and emission between 610 and 690 nm was gained through usage of a HYQ Texas Red filter cube.
X-ray experiments
GIXD was employed to study evolution of the in-plane order of the monolayers at subatomic scale upon OAK insertion (25). Depth and orientation of the OAK insertion were determined using the specular X-ray reflectivity (XR) technique (25). Liquid surface X-ray-scattering experiments were performed at the 15-ID (ChemMatCARS synchrotron) beam line at the Advanced Phtoon Source (APS), Argonne National Laboratory (Argonne, IL, USA), as described previously (24, 26). The synchrotron radiation beam was monochromated to a wavelength λ = 1.24 Å. Analysis of collected XR data was approached with so-called slab model refinement using RFIT2000 software (Oleg Konovalov, European Synchrotron Radiation Facility, Grenoble, France) (24, 26).
In vivo studies
All animal studies were performed using male ICR mice (20±2 g). Procedures, care, and handling of animals were approved by the Technion Animal Care and Use Committee.
Toxicity
Acute toxicity was examined after intravenous (i.v.), intraperitoneal (i.p.), and subcutaneous (s.c.) injections of C12(ω7)K-β12 (0.15, 0.5, and 0.15 ml PBS, respectively) at the specified doses. Animals were directly inspected for adverse effects for 4 h, and mortality was monitored for 6 d thereafter.
Efficacy
The peritonitis-sepsis model was investigated as described previously (14) using i.p. injection of S. aureus (1×108 CFU/mice in 0.5 ml PBS containing 5% porcine mucin) and i.p. treatment (0.5 ml C12(ω7)K-β12 in PBS) 1 h post-inoculation. Mice were monitored for survival for 6 d post-treatment. In the thigh infection model, mice were inoculated intramuscularly (1.4–1.5×106 CFU of S. aureus ATCC 29213) and treated subcutaneously 2 h post-inoculation. Alternatively, single or double doses were administered immediately and 6 h post-inoculation. All mice in this model were sacrificed 24 h post-infection, and the infected thigh muscle was dissected, homogenized, and plated for CFU enumeration.
Pharmacokinetics
Blood concentrations of C12(ω7)K-β12 were determined by LC-MS using calibrated standard curves as described previously (14). The OAK was administered in an s.c. injection (0.15 ml PBS) to male ICR 4-wk-old mice. Doses of 100, 200, or 400 μg/mouse (corresponding to ∼5, 10, and 20 mg/kg body weight, respectively) were administrated to 2 mice/data point. At the specified time points, mice were euthanized by CO2 asphyxiation, and blood samples were collected from the portal vein. For analysis, 0.2 ml whole blood from each mouse was added to 1 ml of extraction buffer (acetonitrile:formic acid 9:1, w/w, fortified with 50 mM ammonium formiate) and shaken for 30 min, then centrifuged for 2 min at 14,000 g. Extract supernatants (150 μl) were diluted 2-fold in water containing 0.1% TFA and analyzed by LC-MS. For quantitative calibration, the identical procedure was performed to establish standard curves in mouse blood.
RESULTS
Design rationale
The relationships existing between hydrophobicity, aggregation in solution, and selective antimicrobial properties have been investigated for numerous HDPs (27,28,29) and OAKs (30, 31). Allegedly, excess hydrophobicity and the consequent self-assembly in aqueous media are responsible for poor antibacterial performance as well as potential toxicity, as reflected by hemolysis. To address this issue, we recently attempted the manipulation of a hydrophobic OAK, palmitoyl-KKc12K (C16K-β12) (31), by substitution of the N-terminal acyl with its unsaturated counterpart (C16(ω7)K-β12), hypothesizing that the double bond should interfere with self-assembly. Although this has indeed significantly altered the OAK’s supramolecular organization and improved its antibacterial performance, hemolytic activity of C16(ω7)K-β12 remained nonetheless high enough to compromise its potential use in therapeutics. Therefore, building on these findings, we used here a shorter acyl. Presumably, the anticipated interference caused by the double bond should be more efficient in lauroyl (C12) because of the reduced hydrophobic forces at play.
The resulting compound, C12(ω7)K-β12 (molecular structure is shown in Fig. 1A), displayed reduced self-assembly and hemolysis but maintained potent, albeit selective, antibacterial activity. Figure 1B depicts the dose-dependent light-scattering plots of C12(ω7)K-β12 and of its saturated analog. The critical aggregation concentrations (CACs) indicated that C12(ω7)K-β12 had a significantly lower tendency for aggregation than C12K-β12 (CAC ∼ 40 and 8 μg/ml, respectively). The considerably higher light-scattering intensities (above CAC values) also reflected the higher tendency of C12K-β12 to form larger aggregates. Reversed-phase HPLC analysis of these compounds confirmed that the acyl substitution was accompanied with reduced hydrophobicity, as reflected by their elution times, which corresponded to 50 and 54% acetonitrile eluent, respectively. As anticipated, disassembly correlated with reduced toxicity to mammalian cells, as assessed with erythrocytes (Fig. 1C) and with primary fibroblasts (Fig. 1D). Interestingly, C12(ω7)K-β12 displayed potent growth inhibitory activity preferentially against gram-positive species: MIC against 90% of tested strains was ≤5 μg/ml (≤6.2 μM), while gram-negative bacteria were generally less susceptible (Table 1). For comparison, widely used antibiotics such as oxacillin, piperacillin, and penicillin G that were assessed under identical conditions against 4 strains of S. aureus have yielded the following MIC values: 1, 16, and 4 μg/ml (ATCC 43300); 4, 16, and 16 μg/ml (clinical isolate 15819); 128, 32, and 8 μg/ml (clinical isolate 15877); and 128, 128, and 32 μg/ml (clinical isolate 15852), respectively.
Figure 1.

Structures and in vitro biophysical properties. A) Molecular structure of C12K-β12 and C12(ω7)K-β12 (MW 781 g/mol); difference is highlighted by dashed circles. B) Dose-dependent self-assembly in PBS after 2 h incubation at room temperature. CAC was evaluated by extrapolating the curve to the intercept with the x axis. C) Hemoglobin leakage from human erythrocytes (10% hematocrit) in PBS as determined after 15 min incubation under shaking at 37°C and compared with 0.2% Triton X-100. D) XTT cytotoxicity assay using primary fibroblasts after 24 h culture. Solid circles, C12K-β12; open circles, C12(ω7)K-β12. Error bars = sd calculated from ≥2 independent experiments performed in duplicate. Lack of error bars indicates consistency.
TABLE 1.
MIC values of C12(ω7)K-β12 determined against 33 strains
| Bacteria tested | Strains (n) | MIC range (μg/ml) |
|---|---|---|
| Gram-positive | ||
| Staphylococcus aureus | 14 | 2.5–5 |
| Listeria sp. | 5 | 2.5–5 |
| Streptococcus agalactiae | 1 | 5 |
| Bacillus sp. | 2 | 5–10 |
| Gram-negative | ||
| Escherichia coli | 7 | ≥40 |
| Pseudomonas aeruginosa | 2 | 10 |
| Salmonella sp. | 2 | 40 |
Mechanistic studies
To investigate the mechanism of action, we first asked how growth inhibition was related to bacterial viability. At concentrations representing up to 4 multiples of the MIC value (4×MIC), C12(ω7)K-β12 exerted a bacteriostatic effect in that the OAK managed to reduce the CFU count by 1 log unit at most, within 3 h of exposure. Figure 2A shows the inability of a representative strain of S. aureus (ATCC 29213) to proliferate in the presence of the OAK as opposed to the rapid bactericidal activity of a control antimicrobial peptide, the dermaseptin derivative S4(1–16). Similar results were obtained with two additional S. aureus strains, CI 15903 and ATCC 25913 (data not shown).
Figure 2.

Mechanistic studies. A) Viability of a representative strain (S. aureus ATCC 29213 in LB) on treatment with 4×MIC of C12(ω7)K-β12 (asterisks) or dermaseptin S4(1–16) (rectangles) and untreated control (circles). B) Extracellular ATP levels in treated S. aureus suspension, with C12(ω7)K-β12 or dermaseptin (4×MIC each). Symbols are as in panel A. C) Mean EtBr accumulation in S. aureus suspended in PBS in the absence and presence of OAK at 4×MIC (2 independent experiments, P<0.05). Asterisks, OAK; asterisk; circles, EtBr alone (untreated control); rectangles, 0.01% Triton X100 (positive control). D) MIC evolution after successive exposures of S. aureus to subinhibitory concentrations of the specified antibacterial agent: oxacillin (circles), ciprofloxacin (rectangles), and C12(ω7)K-β12 (asterisks). Relative MIC is the normalized ratio of MIC obtained for a given subculture to the MIC obtained for first-time exposure. E) Representative binding curves for various concentrations (3.125, 6.25, 12.5, 25, and 50 μM) of C12(ω7)K-β12 in PBS using a model phospholipid membrane (POPG:PE, 1:4). Higher resonance intensities correspond to higher OAK concentrations. F) Representative agarose gel runs of bacterial plasmid pUC19 in its native state (lane 2) and after 1 h exposure to BamHI (lane 3). Lane 1: molecular weight marker (λHindIII); lanes 4 and 5: pUC19 after preincubation (30 min) with C12(ω7)K-β12 and C12K-5α8 (25 μM each), respectively, followed by exposure to BamHI.
Bacterial viability in the treated cultures was also determined based on ATP concentration as an indicator of metabolically active cells. Figure 2B reflects the extracellular ATP levels in treated bacterial suspensions (4×MIC). The control antimicrobial peptide, known for its membrane-disruptive properties (20), has induced an intensive and nearly immediate ATP leakage, unlike C12(ω7)K-β12, which induced a slight luminescence signal, providing evidence for a slow and discrete ATP leakage.
The nonabrupt permeabilization of OAK-treated bacteria was evidenced by another experiment that monitored intracellular accumulation of EtBr, known for its slow spontaneous translocation across the plasma membrane and interaction with nucleic acids (32). As shown in Fig. 2C, the slow and limited EtBr uptake was enhanced in OAK-treated bacteria.
In addition, we assessed emergence of resistance following multiple exposures of staphylococci to sub-MIC concentrations of C12(ω7)K-β12 or antibiotics (oxacillin and ciprofloxacin). Figure 2D shows that over time bacteria have developed resistance to both antibiotics (as reflected by 4- and 8-fold increase of the relative MIC, respectively) but not to the OAK, indicating that despite its ultrashort sequence and its bacteriostatic mode of action, C12(ω7)K-β12 has retained the property of long OAKs, shown to be quite indifferent to drug-resistance mechanisms (14).
Collectively, these findings raised the possibility that C12(ω7)K-β12 exerted a bacteriostatic effect by a mechanism implicating mild (nonabrupt) damage to the plasma membrane. In fact, the data are quite in agreement with recent work suggesting that the OAK induced a slow and partial membrane depolarization of S. aureus (33). To verify this hypothesis, we investigated OAK binding properties with respect to phospholipid membranes as well as to nucleic acids, also believed to represent potential targets of OAKs (6) and HDPs in general (34).
Membrane-binding properties were assessed using a variety of complementary technologies. Figure 2E portrays a representative SPR trace (dose-dependent association/dissociation curves) using 5 concentrations of C12(ω7)K-β12 in PBS. The resulting apparent affinity constants (determined using the 2-stage binding model) (22) demonstrated a high binding affinity (Kapp 1.7×107/M) to a bilayer of POPG:CL (which is often used as a model for S. aureus plasma membrane) (35, 36) and significantly lower affinity to POPG:PE, the mimic of E. coli cytoplasmic membrane (Kapp 5.2×103/M). These findings correlated well with the cytotoxicity results, suggesting that the observed selectivity of C12(ω7)K-β12 could be at least partially linked to its preferential interaction with the highly anionic staphylococcal membrane phospholipids.
In addition to bilayers, the monolayer of phospholipids may be used to investigate the membrane in a fluid environment (23). In this approach, Langmuir monolayers mimic the external leaflet of the cell or bacterial membrane, while the OAK is introduced into the subphase of a Langmuir trough to comprise the extracellular fluid and thus mimic the approach of a peptide toward the cell surface (23, 26). Changes in membrane structure resulting from its interaction with peptides allow a possible mechanism of peptide action to be suggested (37, 38). Thus, further details about the mechanism of action of the OAK were gained from examining its interactions with Langmuir monolayers, using constant-pressure insertion assays, epifluorescence microscopy, and liquid surface XR and GIXD in a complementary manner.
As followed from changes in mean lipid area at constant surface pressure (Fig. 3A), C12(ω7)K-β12 readily altered the DPPG monolayer (∼56% increase in area per lipid molecule), whereas the mixed monolayer DPPG:DPPE (1:4) mainly remained intact (∼4% area increase). Incorporation of the OAK into the monolayer structure was accompanied by evolution of the membrane in-plane morphology. Epifluorescence microscopy illustrates that C12(ω7)K-β12 is incapable of inducing noticeable morphological changes in DPPG:DPPE (1:4) monolayer (Fig. 3B). In contrast, in-plane order of the DPPG monolayer undergoes significant evolution on insertion of the OAK (Fig. 3C). Dark domains, which represent the liquid-ordered phase of the DPPG monolayer, gradually changed their shape and decreased in size until total disappearance. Simultaneously, an increase in the bright portion of the monolayer, which is associated with the liquid-disordered lipid phase, was observed. In other words, C12(ω7)K-β12 commences membrane disruption from incorporation into the DPPG liquid-disordered phase followed by disorganization of the liquid-ordered phase through insertion into its boundary.
Figure 3.

Constant-pressure insertion and epifluorescence microscopy. A) Insertion isotherms of DPPG and DPPG:DPPE (1:4) monolayers showing percentage change in area per molecule after injection of C12(ω7)K-β12. B) Epifluorescence images of DPPG:DPPE (1:4) monolayer before and after C12(ω7)K-β12 injection. C) Epifluorescence images of DPPG monolayer before and after C12(ω7)K-β12 injection. Lipid-linked Texas Red-DHPE fluorescence probe (0.5 mol%) was incorporated into the phospholipid monolayers for EFM experiments. Because of steric hindrance, the dye partitions into the liquid-disordered phase, rendering it bright and the liquid-ordered phase dark.
This membrane-disruptive behavior of the OAK against the DPPG monolayer was corroborated by diffraction data (Fig. 4D), where the lipid order disappears completely on C12(ω7)K-β12 insertion. GIXD has also provided molecular-level details on the effect of the OAK on the mixed monolayer. Three-dimensional Bragg peak diffraction patterns of the DPPG:DPPE (1:4) monolayer (Fig. 4A, C) imply that phospholipid acyl chains pack into an oblique unit cell with parameters a = 4.9 Å, b = 4.875 Å, and γ = 118.8°. Coherence length L of the order constitutes 112 Å in {0,1}, 139 Å in {1,0}, and 453 Å in {1,1} + {1,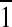 } directions. Figure 4B, C implies that C12(ω7)K-β12 does not alter the packing character of the acyl chains, inducing only minor changes in unit cell parameters a = 4.88 Å, b = 4.87 Å, and γ = 118.9°. However, the OAK reduces the coherence length of the order in {0,1} direction to 94 Å, in {1,0} to 123 Å, and most significantly in {1,1} + {1,
} directions. Figure 4B, C implies that C12(ω7)K-β12 does not alter the packing character of the acyl chains, inducing only minor changes in unit cell parameters a = 4.88 Å, b = 4.87 Å, and γ = 118.9°. However, the OAK reduces the coherence length of the order in {0,1} direction to 94 Å, in {1,0} to 123 Å, and most significantly in {1,1} + {1,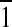 } to 111 Å. Although {1,1} + {1,
} to 111 Å. Although {1,1} + {1,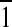 } reflection originates from both DPPG and DPPE acyl chains, 3-D Bragg peak diffraction pattern of the monolayer registered after C12(ω7)K-β12 insertion is a fingerprint of DPPE, and therefore, C12(ω7)K-β12 interacts preferentially with anionic DPPG phospholipids.
} reflection originates from both DPPG and DPPE acyl chains, 3-D Bragg peak diffraction pattern of the monolayer registered after C12(ω7)K-β12 insertion is a fingerprint of DPPE, and therefore, C12(ω7)K-β12 interacts preferentially with anionic DPPG phospholipids.
Figure 4.

Grazing incidence X-ray diffraction. A, B) Three-dimensional plots of Bragg peaks (qXY) against Bragg rods (qZ) as a function of intensity of the DPPG:DPPE (1:4) monolayer at 30 mN/m before (A) and after (B) C12(ω7)K-β12 injection, respectively. C, D) Two-dimensional plots of DPPG:DPPE (1:4) and DPPG monolayer GIXD Bragg peaks (qXY) integrated over qZ as a function of intensity.
Further evidence in support of the notion that the dominant mode of OAK action on the bacterial membrane is electrostatic rather than hydrophobic interaction follows from XR data. To fit the XR profile of the DPPG monolayer, a 2-slab model was sufficient (Fig. 5A). The upper slab was ascribed to phospholipid acyl chains, and the bottom one to head groups. Thickness of the acyl-chain slab is 18.5 Å, and its electron density is 0.327 e−/Å3; the head-group slab returned values of 5.9 Å and 0.498 e−/Å3. When the OAK was introduced into the monolayer, a minimum of 3 slabs were required to fit the experimental data. The upper slab contained only phospholipid acyl chains and now was 9.8 Å thick and had an electron density of 0.304 e−/Å3. The second upper slab was characterized by a thickness of 8.3 Å and electron density of 0.437 e−/Å3 and corresponded to phospholipid head groups and cationic lysine chains. The bottom slab was mainly represented with the OAK acyl backbone and was 9.7 Å thick and had an electron density of 0.371 e−/Å3. The decrease in thickness of the monolayer suggests that the OAK may utilize the membrane thinning effect as part of the mechanism for cell permeabilization. The DPPG:DPPE (1:4) monolayer was also characterized by 2 slabs with thicknesses of 17.2 and 8.9 Å and electron densities of 0.318 and 0.430 e−/Å3 for acyl-chain and head-group slabs, respectively. When C12(ω7)K-β12 was incorporated, trends similar to those in DPPG were observed. Thickness and electron density of acyl-chain slabs decreased to 16.2 Å and 0.309 e−/Å3, indicating no presence of the OAK in acyl chains. On the other hand, the head-group slab was now described by parameters of 9.6 Å and 0.437 e−/Å3, which suggests that in the mixed monolayer, lysine chains also insert into DPPG. Model refinement returned no third slab for the OAK acyl backbone, which, in fact, can be explained by limited incorporation of the OAK into DPPG:DPPE (1:4) monolayer (∼4%). Accordingly, discrepancy between a slab of the OAK acyl backbones and buffer is within experimental error. Figure 5B shows a cartoon representation of C12(ω7)K-β12 mode of interaction with a mixed monolayer.
Figure 5.

X-ray reflectivity. A) X-ray specular reflectivity data (symbols) and corresponding fits (lines) of DPPG and DPPG:DPPE (1:4) monolayers before and after C12(ω7)K-β12 injection normalized by Fresnel reflectivity plotted against scattering vector qz. For clarity, data have been offset vertically. B) Cartoon representation of the mechanism of C12(ω7)K-β12 and bacterial membrane interactions. Synchrotron radiation beam was monochromated to a wavelength λ = 1.24 Å. XR data were analyzed with both slab model and model-independent refinement.
Collectively, the studies using monolayers, which correlated well with the SPR data using bilayers, reinforced the view of plasma membrane acting as a likely target and further provided support to the mechanistic experiments, pointing to the shallow insertion of C12(ω7)K-β12 as a potential instigator of membrane destabilization.
We also verified the option of the OAK targeting bacterial DNA, by assessing the ability of OAK to protect plasmidial DNA (pUC19) from digestion by the DNase BamHI. When run in a 1% agarose gel, pUC19 displayed one band, which on treatment with BamHI runs as a higher-molecular-weight band (Fig. 2F). As shown in Fig. 2F, preincubation of pUC19 with C12(ω7)K-β12 (lane 4) did not inhibit the DNase action, unlike the positive control experiment using another OAK, C12K-5α8 (lane 5), known for its ability to bind DNA and thereby prevent the DNase action (20). The inability of C12(ω7)K-β12 to similarly prevent BamHI action on the plasmid argues against a mode of action involving DNA interactions. Moreover, the apparent high binding affinity of OAK to the membrane also argues against an effective OAK translocation across the plasma membrane and interactions with additional cytoplasmic targets, thereby suggesting that the inward progression of C12(ω7)K-β12 is likely to be stopped at the plasma membrane level, the preferred target of many HDPs.
In vivo studies
The ability of C12(ω7)K-β12 to affect bacterial proliferation in vivo was assessed using two mouse models of infection. The peritonitis-sepsis model was used at first to assess efficacy in protecting mice from a lethal challenge under conditions previously used to test activity of different OAKs against gram-negative bacteria (14, 20). In this model, ICR mice were inoculated intraperitoneally with a virulent clinical isolate of S. aureus (1×108 CFU/mouse) and treated by a single i.p. dose 1 h postinoculation and monitored for survival for 6 d thereafter. As shown in Fig. 6A, under conditions that killed all mice within 24 h, the OAK increased the survival rate by 30 and 90% at the respective doses of 2 and 5 mg/kg, a dose range that previously proved the efficacy of various OAKs and HDPs (39,40,41).
Figure 6.

In vivo studies in mice. A) Efficacy of C12(ω7)K-β12 in protecting mice from a lethal challenge using the peritonitis-sepsis model. ICR mice were inoculated IP with 1 × 108 CFU of S. aureus (CI 15903) and treated by i.p. injection 1 h postinoculation. Number of mice per group is specified in brackets. Data are values at 6 d postinoculation. B, C) Ability of C12(ω7)K-β12 to affect bacterial viability systemically using the thigh infection model. Mice were inoculated intramuscularly with 1.4–1.5 × 106 CFU of S. aureus (ATCC 29213) and treated 2 h after (B) or immediately after inoculation (C). Vanco, vancomycin. Unless specified otherwise, treatments were s.c. Horizontal bars indicate mean values. D) Acute toxicity of C12(ω7)K-β12 at 6 d after various routes of administrations. n = 2 mice/group for highest doses; 6 mice/group for all lower doses. Triangles, i.v. route; rectangles, i.p. route; circles, s.c. route. E) Blood concentration of C12(ω7)K-β12 as determined after extraction and analysis by LC-MS for single-dose administrations of 5, 10, or 20 mg/kg (open, shaded, and solid circles, respectively). Data are means ± sd from 2 mice/data point. F) Inhibition of S. aureus (CI 15903) after 12 h incubation with C12(ω7)K-β12 (20 μg/ml) in 90% human whole blood. Horizontal dashed line indicates initial inoculum.
To assess systemic effectiveness, we used the thigh infection model, where mice were inoculated intramuscularly with a nonlethal dose (1.4–1.5×106 CFU) of S. aureus (ATCC 29213) and treated 2 h after inoculation. To determine efficacy, mice were sacrificed 24 h postinfection, and the thigh was homogenized and plated for CFU enumeration. As shown in Fig. 6B, after a single-dose s.c. administration, C12(ω7)K-β12 has significantly (P<0.0001) reduced bacterial load, similarly to the well-known antibiotic vancomycin. Notably, comparable efficacies were observed on investigating either alternative routes of OAK administration, including i.v. and i.p. (Fig. 6B) or high OAK doses administered in either a single or double dose of 20 mg/kg (Fig. 6C).
Acute toxicity experiments revealed the maximal tolerated dose (MTD) of C12(ω7)K-β12 for each route of administration (Fig. 6D). The MTDs for i.v. and i.p. routes were 5 and 10 mg/kg, respectively, whereas the s.c. route was well tolerated at least up to 20 mg/kg. No signs of toxicity were detected for all doses lower than MTD, after up to 6 d observation.
To investigate the reason for the efficacy observed, we performed preliminary pharmacokinetic studies by monitoring the blood levels of C12(ω7)K-β12. The dose of 20 mg/kg was chosen as the maximal dose, representing the highest nontoxic dose in s.c. administration. As shown in Fig. 6E, after s.c. administration, the OAK rapidly entered circulation, remained stable for ≥1 h (5, 10 mg/kg), or even displayed increased concentration (20 mg/kg). Two hours postadministration, the OAK concentration was similar (∼30 μg/ml). A supportive experiment was conducted in vitro, determining S. aureus viability in human whole blood, after incubation with C12(ω7)K-β12. As shown in Fig. 6F, the OAK displayed antibacterial activity when bacteria were treated with 20 μg/ml (4×MIC), but not with 10 μg/ml. The data suggest that despite exposure to a complex environment such as blood, some of the OAK remains available for treating the bacteria.
DISCUSSION
The data presented provide several novel mechanistic and functional aspects of an OAK derivative with potential for development as a new anti-infective agent, an endeavor crucially needed in light of the ever-increasing resistance to current therapies (42, 43). From the design point of view, this study presents an approach for amending flaws of potentially interesting short OAKs and possibly of related compounds as well. Thus, the fact that C12(ω7)K-β12 was essentially in a nonaggregative state at biologically relevant concentrations agreed with previous findings that linked hydrophobicity-based self-assembly and hemolytic activity. Namely, as observed previously with the pair C16(ω7)K-β12 and C16K-β12 (31), the present comparison between C12(ω7)K-β12 and C12K-β12 demonstrated again that the N-terminal unsaturated acyl residue was responsible for significant alteration of biophysical properties while retaining antibacterial activity. Self-assembly of C12(ω7)K-β12 is likely to be inhibited owing to a dual effect imposed by the single double bond: reduced hydrophobicity and wobbly packing, as demonstrated in the former case (31). These findings therefore suggest a strategy for amending/improving a variety of related acyl-antibiotics (44) and lipopeptides in general (45,46,47).
Another interesting outcome of this study pertains to the mode of action. Although the mechanism of action of HDPs has been intensely investigated for >2 decades (48, 49), various aspects remain poorly understood. Thus, using a plethora of mechanisms, many HDPs are known to induce rapid cell death, while relatively few are bacteriostatic (50); however, the molecular basis for selecting one mechanism over another is fundamentally unknown. A complicating factor is the exceedingly high number of variables in the peptide characteristics and the consequent hurdles set by the insights provided through SAR studies. Therefore, simplified yet robust HDP-mimics might be useful in shedding new light into the mechanisms of action, as recently illustrated using two closely analogous OAKs (20, 35). Furthermore, unlike previously characterized OAKs that were typically bactericidal, C12(ω7)K-β12 exerted a bacteriostatic effect that evidently did not involve either the abrupt disruption of the plasma membrane or the direct inhibition of DNA functions. The smaller molecular size, per se, may not represent a major reason necessarily because various OAKs (31) and additional comparable compounds (44, 46, 51) clearly exert a bactericidal activity. Based on the data presented, we propose that the molecular basis for the observed bacteriostatic effect is linked to the shallow insertion of C12(ω7)K-β12 within plasma membrane anionic phospholipids. A limited insertion might explain the inefficiency in disrupting the membrane abruptly. The OAK interactions with the plasma membrane are also likely to nonspecifically alter processes linked to membrane functions that depend on fluidity and charge distribution, as proposed previously (52, 53). Thus, by acting as a membrane “modifier” C12(ω7)K-β12 may, slowly but substantially, affect the barrier function, as reflected by the slow augmented entry of EtBr or the slow leakage of ATP. This effect is expected to be more pronounced for gram-positive bacteria because of the lack of an additional permeability barrier, the outer membrane of gram-negative bacteria. Therefore, although additional experiments are needed toward better understanding of the mode of action of these peptides, the present findings reinforce the notion that synthetic OAKs can reflect the global complexity of the prevailing modes of action of HDPs and hence that the OAK concept might help address the vast challenges facing a better understanding of HDPs mechanisms of action.
The data also demonstrated in vivo activity of C12(ω7)K-β12 against gram-positive bacteria such as S. aureus. The staphylococci are important pathogenic bacteria responsible for a variety of diseases (54). For instance, they are the most common cause of hospital-acquired infections, while antibiotic-resistant strains (e.g., MRSA) have become endemic in hospitals in most countries, causing major public health issues. The cyclic lipopeptide antibiotic daptomycin exhibits rapid bactericidal activity against S. aureus by a calcium-dependent membrane-depolarization mechanism (44). In 2003, the FDA approved its use for the treatment of skin and soft-tissue infections caused by gram-positive bacteria; resistance was reported shortly thereafter (55). There is thus a clear need for alternative therapeutic agents. In that sense, this study established C12(ω7)K-β12 as a potentially interesting compound for treating staphylococcal infections. Notably, while 2 different OAK sequences have previously shown in vivo efficacy in a peritonitis-sepsis model (20), a model that is often feared to reflect local treatment because both drug and pathogen are introduced in the peritoneal cavity, the present study demonstrates the unambiguous efficacy of an OAK administrated by systemic routes. Future studies could shed light onto the question of to what extent the host immune system is implicated in the potency observed for this HDP-mimic.
The development of small/inexpensive chemicals with systemic efficacy that moreover are unaffected by common mechanisms of chemoresistance would be a major advance in treatment of infectious diseases. Interestingly, short arylamide foldamers were recently reported to exhibit bactericidal activities. The most potent compound reduced staphylococcal load on systemic administration of 1–10 mg/kg with MTD at 20 mg/kg (51). Also, several lipopeptides were introduced in recent years but whose in vivo properties are unknown: the extremely short sequence palmitoyl-lysyl-lysyl-NH2 was found active in vitro against a large panel of gram-positive bacteria (46). Another acylated diastereoisomeric tetrapeptide (45) was reported to display interesting antibacterial activities in vitro with fast bactericidal kinetics, which is typical for cytolytic agents that abruptly disrupt the cytoplasmic membrane. In comparison, C12(ω7)K-β12 displayed distinct properties in terms of either selectivity, mode of action, and/or in vivo potency. It is noteworthy, nevertheless, that these examples seem to delineate the rough minimal requirements for membrane-active properties, while their common denominators raise intriguing questions as to the importance of molecular rigidity in selectivity—beyond the established factors of hydrophobicity, charge, and amphipathic organization. It is likely, therefore, that comparative studies of these tiny chemicals might help in designing improved antimicrobials.
In summary, this study established C12(ω7)K-β12 as the smallest OAK that combines antibacterial potency with low toxicity. Such simple yet robust chemicals whose attributes could include high safety and low production costs might be useful for various antibacterial applications while circumventing potential adverse effects associated with lytic compounds.
Acknowledgments
This research was supported by Israel Science Foundation grant 283/08 (A.M.) and U.S. National Institutes of Health grant R01 AI073892 (D.G.). The authors are indebted to Binhua Lin and Mati Meron for their help with X-ray measurements at APS. The OAK sequences described here have been submitted as part of a U.S. patent application.
References
- Brogden K A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- Dhople V, Krukemeyer A, Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim Biophys Acta. 2006;1758:1499–1512. doi: 10.1016/j.bbamem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Gottler L M, Ramamoorthy A. Structure, membrane orientation, mechanism, and function of pexiganan—a highly potent antimicrobial peptide designed from magainin. Biochim Biophys Acta. 2009;1788:1680–1686. doi: 10.1016/j.bbamem.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- Hancock R E, Sahl H G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- Rotem S, Mor A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim Biophys Acta. 2008;1788:1582–1592. doi: 10.1016/j.bbamem.2008.10.020. [DOI] [PubMed] [Google Scholar]
- Scott R W, DeGrado W F, Tew G N. De novo designed synthetic mimics of antimicrobial peptides. Curr Opin Biotechnol. 2008;19:620–627. doi: 10.1016/j.copbio.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter E A, Wang X, Lee H S, Weisblum B, Gellman S H. Non-haemolytic beta-amino-acid oligomers. Nature. 2000;404:565. doi: 10.1038/35007145. [DOI] [PubMed] [Google Scholar]
- Liu D, DeGrado W F. De novo design, synthesis, and characterization of antimicrobial beta-peptides. J Am Chem Soc. 2001;123:7553–7559. doi: 10.1021/ja0107475. [DOI] [PubMed] [Google Scholar]
- Gottler L M, Lee H Y, Shelburne C E, Ramamoorthy A, Marsh E N. Using fluorous amino acids to modulate the biological activity of an antimicrobial peptide. ChemBioChem. 2008;9:370–373. doi: 10.1002/cbic.200700643. [DOI] [PubMed] [Google Scholar]
- Patch J A, Barron A E. Helical peptoid mimics of magainin-2 amide. J Am Chem Soc. 2003;125:12092–12093. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- Chongsiriwatana N P, Patch J A, Czyzewski A M, Dohm M T, Ivankin A, Gidalevitz D, Zuckermann R N, Barron A E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci U S A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew G N, Liu D, Chen B, Doerksen R J, Kaplan J, Carroll P J, Klein M L, DeGrado W F. De novo design of biomimetic antimicrobial polymers. Proc Natl Acad Sci U S A. 2002;99:5110–5114. doi: 10.1073/pnas.082046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzishevsky I S, Rotem S, Bourdetsky D, Navon-Venezia S, Carmeli Y, Mor A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat Biotechnol. 2007;25:657–659. doi: 10.1038/nbt1309. [DOI] [PubMed] [Google Scholar]
- Savage P B, Li C, Taotafa U, Ding B, Guan Q. Antibacterial properties of cationic steroid antibiotics. FEMS Microbiol Lett. 2002;217:1–7. doi: 10.1111/j.1574-6968.2002.tb11448.x. [DOI] [PubMed] [Google Scholar]
- Ishitsuka Y, Arnt L, Majewski J, Frey S, Ratajczek M, Kjaer K, Tew G N, Lee K Y. Amphiphilic poly(phenyleneethynylene)s can mimic antimicrobial peptide membrane disordering effect by membrane insertion. J Am Chem Soc. 2006;128:13123–13129. doi: 10.1021/ja061186q. [DOI] [PubMed] [Google Scholar]
- Kuroda K, DeGrado W F. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J Am Chem Soc. 2005;127:4128–4129. doi: 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]
- Ilker M F, Nusslein K, Tew G N, Coughlin E B. Tuning the hemolytic and antibacterial activities of amphiphilic polynorbornene derivatives. J Am Chem Soc. 2004;126:15870–15875. doi: 10.1021/ja045664d. [DOI] [PubMed] [Google Scholar]
- Radzishevsky I, Krugliak M, Ginsburg H, Mor A. Antiplasmodial activity of lauryl-lysine oligomers. Antimicrob Agents Chemother. 2007;51:1753–1759. doi: 10.1128/AAC.01288-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem S, Radzishevsky I S, Bourdetsky D, Navon-Venezia S, Carmeli Y, Mor A. Analogous oligo-acyl-lysines with distinct antibacterial mechanisms. FASEB J. 2008;22:2652–2661. doi: 10.1096/fj.07-105015. [DOI] [PubMed] [Google Scholar]
- Fields G B, Noble R L. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- Gaidukov L, Fish A, Mor A. Analysis of membrane-binding properties of dermaseptin analogues: relationships between binding and cytotoxicity. Biochemistry. 2003;42:12866–12874. doi: 10.1021/bi034514x. [DOI] [PubMed] [Google Scholar]
- Maget-Dana R. The monolayer technique: a potent tool for studying the interfacial properties of antimicrobial and membrane-lytic peptides and their interactions with lipid membranes. Biochim Biophys Acta. 1999;1462:109–140. doi: 10.1016/s0005-2736(99)00203-5. [DOI] [PubMed] [Google Scholar]
- Brockman H. Lipid monolayers: why use half a membrane to characterize protein-membrane interactions? Curr Opin Struct Biol. 1999;9:438–443. doi: 10.1016/S0959-440X(99)80061-X. [DOI] [PubMed] [Google Scholar]
- Berge B, Konovalov O, Lajzerowicz J, Renault A, Rieu J P, Vallade M, Als-Nielsen J, Grubel G, Legrand J F. Melting of short 1-alcohol monolayers on water: thermodynamics and x-ray scattering studies. Phys Rev Lett. 1994;73:1652–1655. doi: 10.1103/PhysRevLett.73.1652. [DOI] [PubMed] [Google Scholar]
- Neville F, Ishitsuka Y, Hodges C S, Waring A J, Lehrer R I, Lee K Y C, Gidalevitz D. Protegrin interaction with lipid monolayers: grazing incidence X-ray diffraction and X-ray reflectivity study. Soft Matters. 2008;4:1665–1674. doi: 10.1039/b718295c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadpour M M, Barkley M D. Self-assembly of designed antimicrobial peptides in solution and micelles. Biochemistry. 1997;36:9540–9549. doi: 10.1021/bi961644f. [DOI] [PubMed] [Google Scholar]
- Avrahami D, Oren Z, Shai Y. Effect of multiple aliphatic amino acids substitutions on the structure, function, and mode of action of diastereomeric membrane active peptides. Biochemistry. 2001;40:12591–12603. doi: 10.1021/bi0105330. [DOI] [PubMed] [Google Scholar]
- Rotem S, Radzishevsky I, Mor A. Physicochemical properties that enhance discriminative antibacterial activity of short dermaseptin derivatives. Antimicrob Agents Chemother. 2006;50:2666–2672. doi: 10.1128/AAC.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzishevsky I S, Kovachi T, Porat Y, Ziserman L, Zaknoon F, Danino D, Mor A. Structure-activity relationships of antibacterial acyl-lysine oligomers. Chem Biol. 2008;15:354–362. doi: 10.1016/j.chembiol.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Sarig H, Rotem S, Ziserman L, Danino D, Mor A. Impact of self-assembly properties on antibacterial activity of short acyl-lysine oligomers. Antimicrob Agents Chemother. 2008;52:4308–4314. doi: 10.1128/AAC.00656-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midoux P, Mayer R, Monsigny M. Membrane permeabilization by alpha-helical peptides: a flow cytometry study. Biochim Biophys Acta. 1995;1239:249–256. doi: 10.1016/0005-2736(95)00163-w. [DOI] [PubMed] [Google Scholar]
- Epand R F, Sarig H, Mor A, Epand R M. Cell-wall interactions and the selective bacteriostatic activity of a miniature oligo-acyl-lysyl. Biophys J. 2009;97:2250–2257. doi: 10.1016/j.bpj.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- Epand R M, Rotem S, Mor A, Berno B, Epand R F. Bacterial membranes as predictors of antimicrobial potency. J Am Chem Soc. 2008;130:14346–14352. doi: 10.1021/ja8062327. [DOI] [PubMed] [Google Scholar]
- Lohner K, Prenner E J. Differential scanning calorimetry and X-ray diffraction studies of the specificity of the interaction of antimicrobial peptides with membrane-mimetic systems. Biochim Biophys Acta. 1999;1462:141–156. doi: 10.1016/s0005-2736(99)00204-7. [DOI] [PubMed] [Google Scholar]
- Gidalevitz D, Ishitsuka Y, Muresan A S, Konovalov O, Waring A J, Lehrer R I, Lee K Y. Interaction of antimicrobial peptide protegrin with biomembranes. Proc Natl Acad Sci U S A. 2003;100:6302–6307. doi: 10.1073/pnas.0934731100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville F, Cahuzac M, Konovalov O, Ishitsuka Y, Lee K Y, Kuzmenko I, Kale G M, Gidalevitz D. Lipid headgroup discrimination by antimicrobial peptide LL-37: insight into mechanism of action. Biophys J. 2006;90:1275–1287. doi: 10.1529/biophysj.105.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navon-Venezia S, Feder R, Gaidukov L, Carmeli Y, Mor A. Antibacterial properties of dermaseptin S4 derivatives with in vivo activity. Antimicrob Agents Chemother. 2002;46:689–694. doi: 10.1128/AAC.46.3.689-694.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mygind P H, Fischer R L, Schnorr K M, Hansen M T, Sonksen C P, Ludvigsen S, Raventos D, Buskov S, Christensen B, De M L, Taboureau O, Yaver D, Elvig-Jorgensen S G, Sorensen M V, Christensen B E, Kjaerulff S, Frimodt-Moller N, Lehrer R I, Zasloff M, Kristensen H H. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature. 2005;437:975–980. doi: 10.1038/nature04051. [DOI] [PubMed] [Google Scholar]
- Cherkasov A, Hilpert K, Jenssen H, Fjell C D, Waldbrook M, Mullaly S C, Volkmer R, Hancock R E. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem Biol. 2009;4:65–74. doi: 10.1021/cb800240j. [DOI] [PubMed] [Google Scholar]
- Levy S B, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- Van B F, Mingeot-Leclercq M P, Struelens M J, Tulkens P M. The bacterial envelope as a target for novel anti-MRSA antibiotics. Trends Pharmacol Sci. 2008;29:124–134. doi: 10.1016/j.tips.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Steenbergen J N, Alder J, Thorne G M, Tally F P. Daptomycin: a lipopeptide antibiotic for the treatment of serious gram-positive infections. J Antimicrob Chemother. 2005;55:283–288. doi: 10.1093/jac/dkh546. [DOI] [PubMed] [Google Scholar]
- Makovitzki A, Avrahami D, Shai Y. Ultrashort antibacterial and antifungal lipopeptides. Proc Natl Acad Sci U S A. 2006;103:15997–16002. doi: 10.1073/pnas.0606129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamysz W, Silvestri C, Cirioni O, Giacometti A, Licci A, Della V A, Okroj M, Scalise G. In vitro activities of the lipopeptides palmitoyl (Pal)-Lys-Lys-NH(2) and Pal-Lys-Lys alone and in combination with antimicrobial agents against multiresistant gram-positive cocci. Antimicrob Agents Chemother. 2007;51:354–358. doi: 10.1128/AAC.00344-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thennarasu S, Lee D K, Tan A, Prasad K U, Ramamoorthy A. Antimicrobial activity and membrane selective interactions of a synthetic lipopeptide MSI-843. Biochim Biophys Acta. 2005;1711:49–58. doi: 10.1016/j.bbamem.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman M R, Yount N Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- Hale J D, Hancock R E. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev Anti-Infect Ther. 2007;5:951–959. doi: 10.1586/14787210.5.6.951. [DOI] [PubMed] [Google Scholar]
- Epand R F, Epand R M, Formaggio F, Crisma M, Wu H, Lehrer R I, Toniolo C. Analogs of the antimicrobial peptide trichogin having opposite membrane properties. Eur J Biochem. 2001;268:703–712. doi: 10.1046/j.1432-1327.2001.01922.x. [DOI] [PubMed] [Google Scholar]
- Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott R W, Winkler J D, DeGrado W F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc Natl Acad Sci U S A. 2009;106:6968–6973. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand R M, Epand R F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim Biophys Acta. 2009;1788:289–294. doi: 10.1016/j.bbamem.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Jean-François F, Castano S, Desbat B, Odaert B, Roux M, Metz-Boutigue M H, Dufourc E J. Aggregation of cateslytin beta-sheets on negatively charged lipids promotes rigid membrane domains: a new mode of action for antimicrobial peptides? Biochemistry. 2008;47:6394–6402. doi: 10.1021/bi800448h. [DOI] [PubMed] [Google Scholar]
- Zetola N, Francis J S, Nuermberger E L, Bishai W R. Community-acquired meticillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect Dis. 2005;5:275–286. doi: 10.1016/S1473-3099(05)70112-2. [DOI] [PubMed] [Google Scholar]
- Mangili A, Bica I, Snydman D R, Hamer D H. Daptomycin resistant, methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis. 2005;40:1058–1060. doi: 10.1086/428616. [DOI] [PubMed] [Google Scholar]