

Abstract
Recent studies have suggested that schizophrenia is associated with alterations in the synaptic connectivity involving cytoskeletal proteins. The microtubule-associated protein STOP (Stable Tubule Only Polypeptide) plays a key-role in neuronal architecture and synaptic plasticity and it has been demonstrated that STOP gene deletion in mice leads to a phenotype mimicking aspects of positive and negative symptoms and cognitive deficits classically observed in schizophrenic patients. In STOP-null mice, behavioral defects are associated with synaptic plasticity abnormalities including defects in long-term potentiation. In these mice, long-term administration of typical antipsychotics has been shown to partially alleviate behavioral defects but, as in humans, such a treatment was poorly active on deficits related to negative symptoms and cognitive impairments. Here, we assessed the effects of risperidone and clozapine, two atypical antipsychotics, on STOP-null mice behavior and synaptic plasticity. Long-term administration of either drug results in alleviation of behavioral alterations mimicking some negative symptoms and partial amelioration of some cognitive defects in STOP-null mice. Interestingly, clozapine treatment also improves synaptic plasticity of the STOP- null animals by restoring long-term potentiation in the hippocampus. All together, the pharmacological reactivity of STOP-null mice to antipsychotics evokes the pharmacological response of humans to such drugs. Totally, our study suggests that STOP-null mice may provide a useful preclinical model to evaluate pharmacological properties of antipsychotic drugs.
Keywords: Animals; Antipsychotic Agents; pharmacology; Behavior, Animal; drug effects; Clozapine; pharmacology; Drug Evaluation, Preclinical; methods; Hippocampus; drug effects; metabolism; Male; Mice; Mice, Knockout; Microtubule-Associated Proteins; genetics; Neuronal Plasticity; drug effects; Risperidone; pharmacology
Keywords: schizophrenia, STOP-null mice, cognitive defects, social investigation, antipsychotics
Introduction
Schizophrenia is a chronic disease characterized by positive, negative or mood symptoms and cognitive deficits. Common negative symptoms include lack of motivation, anhedonia and social withdrawal. Cognitive dysfunctions encompass affected attention and perception, problem-solving, short- and long-term memory. Schizophrenia symptoms are sensitive to antipsychotics (APs) which can be classified as follows: typical APs known to be mainly efficient in improving positive symptoms (Ereshefsky et al. 1990; Mueser and McGurk 2004), atypical AP drugs (e.g., risperidone and clozapine) known to partially act on negative symptoms and cognitive function (Meltzer 2004). Atypical agents have been found to increase cortical dopamine and acetylcholine release, and have some effects on the glutamatergic system not shared by the typical agents (Coyle and Tsai 2004). Effects on neuronal survival and plasticity, together with decreased neurotoxicity, might also contribute to their clinical advantage over typical AP drugs (Meltzer 2004).
However, there is consensus that none of the currently available APs completely alleviate the core symptoms and the debilitating cognitive dysfunction associated with schizophrenia (Peuskens et al. 2005; Sergi et al. 2007). Monitoring reactivity of the symptoms to AP treatment in a preclinical model exhibiting positive, negative and cognitive disorders may help developing drugs that better target each kind of symptoms and could ultimately lead to better outcome and quality of life for patients.
At the physiopathological level, several lines of evidence suggest that schizophrenia is associated with alterations of neuronal architecture, particularly in the synaptic connectivity (Frankle et al. 2003; Mirnics et al. 2001; Owen et al. 2005a; Owen et al. 2005b). Recently, the protein disrupted-in-schizophrenia1 (DISC1) has been demonstrated to bind to microtubules; its truncation in schizophrenic patients contributes to impair intraneuronal transport, neurite cytoarchitecture, and/or neuronal migration (Duan et al. 2007; Mackie et al. 2007; Matsuzaki and Tohyama 2007; Shinoda et al. 2007; Taya et al. 2007). Dysbindin, another protein linked to schizophrenia is also able to bind and regulate microtubules (Talbot et al. 2006). Thus, cytoskeletal alterations in general and more specifically abnormal microtubule dynamics may contribute to connectivity disorders observed in schizophrenia (Blackwood et al. 2007; Camargo et al. 2008; Chiba et al. 2006; Clapcote et al. 2007; Hikida et al. 2007; Ishizuka et al. 2006).
Consistent with this hypothesis, disruption of the MAP6 gene (Denarier et al. 1998) encoding a microtubule-associated protein called STOP (Stable Tubule Only Polypeptide) in mice generated a set of defects similar to those of schizophrenia disorders (Andrieux et al. 2002). Correlatively, the MAP6 gene - the human homologous of STOP gene - is localized on chromosome 11q14, a region highly associated with schizoid personality disorders (Lewis et al. 2003). Interestingly, an association between single nucleotide polymorphism markers of the MAP6 gene and schizophrenia has been found, together with a dysregulation of the isoform2 of MAP6 transcripts in the frontal cortex of schizophrenic patients (Shimizu et al. 2006).
The STOP-null mice, at the behavioral level, have been shown to display behavioral abnormalities thought to simulate some aspects of positive symptoms classically observed in schizophrenic patients (Andrieux et al. 2002; Brun et al. 2005). These mice exhibit increased basal locomotor activity during the dark phase of the light/dark cycle associated with purposeless and disorganized activity (Andrieux et al. 2002; Brun et al. 2005). Additionally, STOP-null mice display a hyper-sensitive locomotor reaction to acutely stressful situations, such as a single saline injection or exposure to novelty and an increased locomotor stimulatory effect of psychotomimetic drugs such as amphetamine (Brun et al. 2005). Some of these locomotor activity defects are present in juvenile mice, whereas others appear only in adulthood mimicking thus the life-long evolution of the pathology (Begou et al. 2007). Regarding negative or cognitive abilities, STOP-null mice exhibit strong defects such as social withdrawal with dramatically reduced aggressive encounters (Andrieux et al. 2002), deficits in prepulse inhibition (Andrieux et al. 2002; Bouvrais-Veret et al. 2007; Fradley et al. 2005; Powell et al. 2007) and recognition memory (Powell et al. 2007), spatial and social learning impairments (Bégou et al. 2008). STOP-null mice also exhibit hyper-reactivity of dopamine release (Brun et al. 2005) associated with an hypoglutamatergic phenotype (Andrieux et al. 2002; Brenner et al. 2007), in accordance with the dopamine/glutamate neurotransmission imbalance hypothesis in schizophrenia. Hence, STOP-null mice exhibit both short and long term synaptic plasticity defects in the hippocampus (Andrieux et al. 2002) associated with depleted glutamatergic vesicle pools. Moreover, levels of mRNAs encoding synaptic proteins, such as synaptophysin, growth-associated protein-43 and spinophilin are decreased in specific hippocampic sub-regions (Eastwood et al. 2007).
Consistently with clinical data, long-term treatment with typical APs, such as haloperidol, has been shown to suppress hyperlocomotion disorders in STOP-null mice (Brun et al. 2005) as well as poorly effective on negative and cognitive defects (Bégou et al. 2008). At the biological level, typical AP treatment has been shown to reverse short term plasticity defects and to be ineffective on long term plasticity (Andrieux et al. 2002). Here, we tested the effect of atypical APs, namely clozapine and risperidone, administrated chronically, on several impaired behaviors exhibited by STOP-null mice. We found that atypical AP administration in STOP-null mice resulted in noticeable amelioration of the negative symptoms and in a partial alleviation of the cognitive defects. Correlatively, we showed that clozapine treatment was able to restore hippocampal long term synaptic plasticity in STOP-null mice.
Materials and methods
Animals
Male STOP-null mice (STOP−/−) and their wild-type (WT) littermates (STOP +/+) were generated as previously described (Andrieux et al. 2002). Eight mice were housed per cage in a temperature-controlled environment (22 ± 1°C) under a 12:12 light/dark cycle (light from 6:00 AM to 6:00 PM), with ad libitum access to familiar food (Irradiated Global 18% Protein Diet, Harlan Teklad) and water. All animals used in this study arose from the same colony (BALBc/129 Sv mixed background). Experiments were performed in accordance with French and European Community guidelines for the care and use of laboratory animals. All efforts were made to minimize the number of animals and their suffering (86/609/EEC-Council Directive of November, 24th 1986).
Drugs and long-term treatments
Drugs used for long-term treatments in this study were either (i) risperidone purchased from Janssen-Cilag Ltd (Risperdal®, Beerse, Belgium), dissolved in drinking water or (ii) clozapine provided by Tocris Bioscience (Bristol, United Kingdom), resuspended with a few drops of HCl before its volume was completed with NaCl (NaCl 0,9% sterile, Physiologica) and injected daily intra-peritoneally. We carefully estimated water consumption of WT and STOP-null mice in the presence or in the absence of risperidone and found no differences between genotypes or treatments (average value for all groups: 15% of the body weight/day). Risperidone concentrations in the drinking water were calculated to obtain the desired delivered doses (0.1mg/kg/day and 0.3 mg/kg/day).
When mice reached 3 months, they were submitted to a long-term treatment with either antipsychotic drug or vehicle during 4 weeks. The treatment was then pursued during a 3 to 4-weeks behavioral testing period.
Behavioral tests
Drug-free and long-term APs-treated animals (4- to 5-month-old, 25 to 30 g) were investigated during the light phase, in a random order for comparison between genotypes and treatments. Experimenter was blind to genotype during testing. In all experiments, animals were allowed to habituate to the testing room 1 h before the test. The usual sequence of tests was as follows: Y-maze test, social investigation test, marble burying test, forced swim test and electrophysiological recordings (see below). All the devices used for testing were extensively washed with water between two animals to avoid olfactive cues. The luminosity of the testing room was controlled and fixed around 30–35 Lux.
Y-maze Test
Spontaneous alternation behavior and exploratory activity were recorded in a Y-maze. The apparatus was made of three equal arms (dimensions: 20 cm long, 8 cm wide, 15 cm high) made of white polyvinyl chloride. The starting arm and the two arms forming the Y were radiating at an angle of 120° from each other. Each mouse was placed in the starting arm of the maze and its position recorded during a 5-min session of free roaming by a video tracking system placed above the maze (Smart®, Panlab, Harvard Apparatus). The total distance traveled across the maze was automatically recorded. The sequence and number of arm entries were manually counted from videotrack recordings. It was considered that a mouse had entered an arm when all four paws were positioned in the arm runway. A spontaneous alternation was defined as entries into all three arms on 3 consecutive choices. The alternation score (%) for each mouse was defined as the ratio of the actual number of alternations to the total possible number of alternations over the testing period: % Alternation = 100 × [ (Number of alternations)/(Total arm entries-2) ]. Mice that completed less than eight arm entries within 5 min were excluded from further analysis (Sakaguchi et al. 2006).
Social investigation test
The social behavior of STOP and WT mice was assessed towards an encaged WT NMRI male mouse (Janvier, France) used as intruder. Without any former isolation, a tested mouse was placed in an open field (50 cm long, 50 cm wide, 20 cm high) containing a wire mesh cylinder in a corner (diameter: 12 cm; height: 14 cm; mesh dimensions: 6 mm) and covered with a lid. After habituation of the animal to its new environment for 5 min, the intruder was introduced in the cylinder and the number of contacts and contact duration were measured during a 10-min session. A contact was defined as a direct sniffing and full use of vibrissae performed by the tested animal towards the intruder.
Marble burying test
The procedure for marble burying was adapted with minor modifications from that described by (Millan et al. 2001). Briefly, mice were individually placed in transparent polysulfone cages (dimensions: 36 X 21 X 14 cm) containing a 4-cm layer of sawdust and 12 glass marbles (diameter: 1.6 cm) evenly spaced over the cage (3 rows of 4 marbles). In order to prevent escape, the cage was covered with a filtering lid. Thirty minutes later, the animals were removed from the cages and the number of marbles buried more than two-thirds in the sawdust was scored.
Forced Swim Test
Studies were carried out on mice according to the slightly modified method of Porsolt and co-workers (Porsolt et al. 1977). Animals were placed individually into polypropylene beakers (height 20 cm, diameter 16 cm) containing 2.5 liters of water (12-cm high) maintained at 20–22°C. The animals were left in the beaker for 6 min. After the first 2 min, total duration of immobility was measured by a well-trained experimenter during a 4-min session. Visually, the mouse was judged to be immobile when it remained floating passively, performing slow motion to keep its head above the water.
Electrophysiological recordings
For preparation of hippocampal slices, adult mice (5 month-old) were deeply anesthetized with ketamine. Brain slices (300 μm thick) were cut in a cold “cutting” artificial cerebrospinal fluid (ACSF) of the following composition (in mM): NaCl (0), KCl (2.5), MgCl2 (7), CaCl2 (0.5), D-Glucose (10), NaHCO3 (26), NaH2PO4 (1.25), Sucrose (230). Slices were then submerged in a waiting chamber in “recording” ACSF equilibrated with 95% O2 and 5% CO2 of the following composition (in mM): NaCl (124), KCl (5), MgCl2 (2), CaCl2 (2), D-Glucose (11), NaHCO3 (26), NaH2PO4 (1.25) and maintained at room temperature for at least one hour before recording. Recordings were done using microelectrode array (MEA) technology allowing stimulation and recording of bioelectrical activity from different areas of the slice. For recordings, slices were placed within the recording chamber of a MEA probe (64×64 planar microelectrodes for Med64 apparatus, Panasonic) with perfusion of “recording” ACSF equilibrated with 95% O2 and 5% CO2 and maintained at 29–30°C. Stimulation was applied on Schaeffer collaterals every 30 sec during baseline (10 min) and after tetanus (TB, 100 Hz, 1 sec) (60 min) and recordings of field EPSPs were made within Ammon’s Horn 1. Normalized field EPSP amplitudes were calculated and only slices showing i) less than 10% variation during the first 10 min of recording before tetanus, ii) more than 10% variation directly following the tetanus and iii) less than 20% variation during the post-tetanus recording were kept for further analysis. Electrophysiological data were plotted as a function of time and comparisons were performed between pooled data recorded 30 to 40 minutes after occurrence of the LTP protocol as previously described (Andrieux et al. 2002).
Statistical analysis
All statistical analyses were performed using Prism 4.0 (GraphPad Software, La Jolla, CA) and data were represented as boxed medians with 25th and 75th percentiles and error bars corresponding to the lowest and the highest data points. For all tests, results were compared between WT and KO placebo groups first, using unpaired non-parametric Mann- Whitney tests. To assay APs effect, comparisons were then separately performed for each genotype (WT: placebo, dose 1, dose 2 and KO: placebo, dose 1, dose2) using non-parametric Kruskals-Wallis ANOVA tests followed by Dunns’ post-hoc comparisons.
Results
We monitored the effect of long-term treatments with atypical antipsychotics (APs) on STOP-null mice, using several behavioral tasks. Mice of WT and STOP KO genotypes were submitted to a chronic treatment with risperidone, clozapine or vehicle during 4 weeks. The treatment was then pursued during the 3 to 4-weeks behavioral testing period. Since risperidone was administrated in the drinking water while clozapine was injected daily, placebo groups can not be directly compared and each treated group will be compared to the corresponding placebo.
Social investigation test
Social investigation has been addressed by measuring the number of contacts and contact duration between an immobilized intruder mouse (male NMRI) and a freely-moving mouse to be tested. Results are presented in Fig. 1. In drug free animals, as compared to WT, STOP-null mice exhibited a reduction of social investigation affecting both the number of contacts with the intruder [9.5 vs 20 for KO and WT placebo mice in risperidone groups (p = 0.006) and 4.5 vs 12.5 for KO and WT placebo mice in clozapine groups (p = 0.200)] and the time spent in social investigation [9 vs 29 sec for KO and WT placebo mice in risperidone groups (p = 0.006) and 6.5 vs 30.5 sec for KO and WT placebo mice in clozapine groups (p = 0.029)].
Fig. 1.
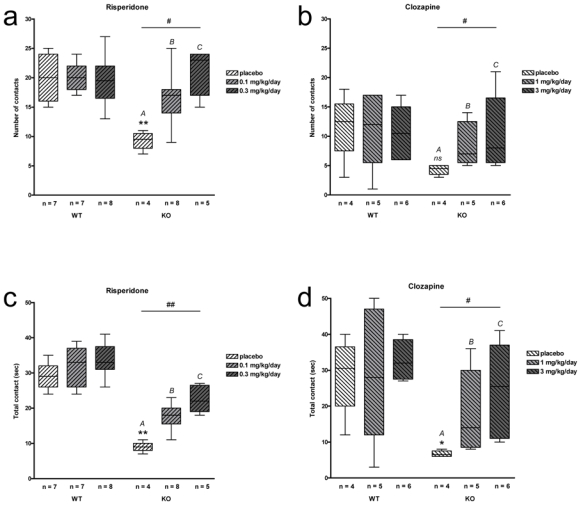
Social investigation test. Number (a and b) and total duration (c and d) of contacts with the intruder in placebo and long-term risperidone (a and c) or clozapine (b and d) -treated WT and STOP-null mice. STOP-null mice treated with placebo present a social withdrawal when compared with their WT littermates [ns, *, **: p>0.05, p<0.05, p<0.01, respectively, KO vs WT placebo]. Chronic treatment with risperidone or clozapine significantly improved social behaviour in STOP-null mice [#, ##: p<0.05, p<0.01, respectively, between KO groups, with A different from C, p<0.01for risperidone (a and c) and p<0.05 for clozapine (b and d) long-term treatments]. Note that clozapine and risperidone are not administrated the same way (injection and drinking water, respectively) preventing direct comparisons between the two groups
We then assessed social behavior of STOP-null mice after long-term administration of risperidone or clozapine.
In WT mice, risperidone treatment did not affect the number of contacts [20 (placebo), 20 (0.1mg/kg/day), 19.5 (0.3mg/kg/day); p = 0.890] or time spent in investigation [29 sec (placebo), 33 sec (0.1mg/kg/day), 33 sec (0.3mg/kg/day); p = 0.316]. STOP-null mice groups displayed significant differences in number of contacts [9.5 (placebo), 17 (0.1mg/kg/day), 23 (0.3mg/kg/day); p = 0.012] and time spent in investigation [9 sec (placebo), 18 sec (0.1mg/kg/day), 22 (0.3 mg/kg/day); p = 0.005]. In both sets of observations, post-hoc analyses show significant improvement of STOP-null mice treated with risperidone 0.3 mg/kg/day as compared to placebo (p < 0.01).
In WT mice, clozapine treatment did not affect the number of contacts [12.5 (placebo), 12 (1mg/kg/day), 10.5 (3mg/kg/day); p = 0.924] or time spent in investigation [30.5 sec (placebo), 28 sec (1mg/kg/day), 32 sec (3mg/kg/day); p = 0.909]. STOP-null mice groups also displayed significant differences in number of contacts [4.5 (placebo), 7 (1mg/kg/day), 8 (3 mg/kg/day); p = 0.030] and time spent in investigation [6.5 sec (placebo), 14 sec (1mg/kg/day), 25.5 sec (3 mg/kg/day); p = 0.015]. Again, post-hoc analyses show significant improvement of STOP-null mice treated with clozapine 3 mg/kg/day as compared to placebo (p < 0.05).
Altogether these results indicate that long term treatment with APs risperidone or clozapine alleviate social behavioral impairment in STOP-null mice.
Marble burying test
In rodents, digging of bedding and burying of objects considered as dangerous for the animal are natural tendencies that do not depend on the nature of their environment (laboratory or nature). The marble burying assay has been predominantly used for the screening of anxiolytic compounds (Nicolas et al. 2006; Young et al. 2006).
In the marble burying test, each mouse was introduced in a cage containing 12 marbles and left free to roam for 30 minutes; the number of buried marbles was then counted. Results are presented in Fig. 2.
Fig. 2.
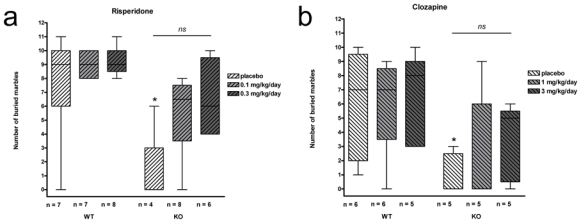
Marble burying test. Number of buried marbles in placebo and long-term risperidone (a) or clozapine (b) -treated WT and STOP-null mice. STOP-null mice treated with placebo bury fewer marbles when compared with their WT littermates [*: p<0.05, KO vs WT placebo]. Chronic treatment of STOP-null mice with risperidone or clozapine enhanced the number of buried marbles in a non significant [ns] way
In drug free animals, as compared to WT, STOP-null mice buried less marbles [0 vs 9 for KO and WT placebo mice in risperidone groups (p =0.042) and 0 vs 7 for KO and WT placebo mice in clozapine groups (p = 0.017)].
The ability to bury marbles was then investigated in risperidone or clozapine long-term-treated animals.
In WT mice, risperidone treatment did not affect the number of buried marbles [9 (placebo), 9 (0.1mg/kg/day), 9 (0.3mg/kg/day); p = 0.887] while STOP-null mice groups showed nearly significant increases in number of buried marbles [0 (placebo), 6.5 (0.1mg/kg/day), 6 (0.3mg/kg/day); p = 0.084].
In WT mice, clozapine treatment did not affect the number of buried marbles [7 (placebo), 7 (0.1mg/kg/day), 8 (0.3mg/kg/day); p = 0.920] while STOP-null mice groups showed non-significant increases in number of buried marbles [0 (placebo), 0 (0.1mg/kg/day), 5 (0.3mg/kg/day); p = 0.329].
These results indicate that although both long-term treatments increased STOP-null mice ability to bury marbles, none did so to a significant level.
Forced swim test
Resignation/despair was assessed in a forced swim test. Mice were placed in a glass beaker containing water for 6 minutes and immobility was measured during the last 4 minutes. Results are presented in Fig. 3.
Fig. 3.

Forced swim test. Total time of immobility in placebo and long-term risperidone (a) or clozapine (b) -treated WT and STOP-null mice. Immobility time was longer in STOP-null mice treated with placebo compared with their WT littermates [ns, *: p>0.05, p<0.05, respectively, KO vs WT placebo]. Administration of a chronic treatment with either risperidone or clozapine produced a significant decrease in immobility time in STOP-null mice [#, ##: p<0.05, p<0.01, respectively, between KO groups, with A different from C, p<0.01 for risperidone (a) and p<0.05 for clozapine (b) long-term treatments]
In drug free animals, as compared to WT, STOP-null mice stayed immobile longer [127.5 vs 73.5 sec for KO and WT placebo mice in risperidone groups (p =0.114) and 95.5 vs 49.5 sec for KO and WT placebo mice in clozapine groups (p = 0.019)].
We then assessed resignation of STOP-null mice after long-term administration of risperidone or clozapine.
Total immobility of WT animals was not modified by risperidone treatment [73.5 sec (placebo), 73 sec (0.1mg/kg/day), 68.5 sec (0.3mg/kg/day); p = 0.362]. STOP-null mice groups displayed significant differences in the time of immobility [127.5 sec (placebo), 93.5 sec (0.1mg/kg/day) and 70.5 sec (0.3mg/kg/day); p = 0.005]. Post-hoc analysis showed significant improvement of STOP-null mice treated with risperidone 0.3 mg/kg/day as compared to placebo (p < 0.01).
Total immobility of WT animals was not modified by clozapine treatment [49.5 sec (placebo), 68 sec (1mg/kg/day), 60 sec (3mg/kg/day); p = 0.551]. STOP-null mice groups displayed significant differences in the time of immobility [95.5 sec (placebo), 88 ± 7.19 sec (0.1mg/kg/day), 52 sec (0.3mg/kg/day); p = 0.028]. Post-hoc analyzes showed significant improvement of STOP-null mice treated with risperidone 0.3 mg/kg/day as compared to placebo (p < 0.05).
Altogether, these results indicate increased resignation in STOP-null mice which is ameliorated by AP long-term treatments.
Y-maze test
Spatial working memory was assessed by testing the spontaneous alternation performance of mice in a Y-maze during a 5-min session of free roaming. Exploratory activity was estimated by the total distance traveled across the maze. The alternation score for each mouse was defined as the ratio of the actual number of alternations to the total possible number of alternations. Results are presented in Fig. 4.
Fig. 4.
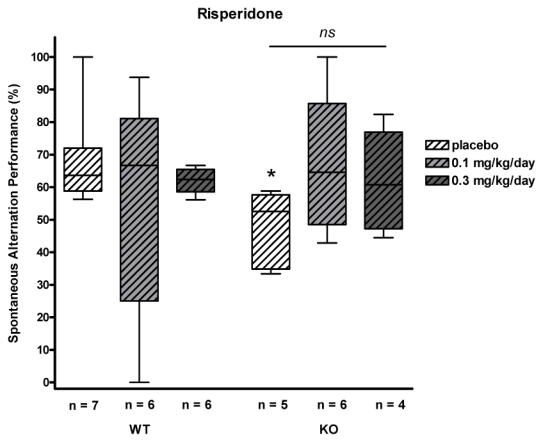
Spontaneous alternation task (Y-Maze) in placebo and long-term risperidone-treated WT and STOP-null mice. STOP-null mice treated with placebo present an impairment of spatial working memory when compared to their WT littermates [*: p<0.05, KO vs WT placebo]. Chronic treatment with risperidone slightly improved STOP-null mice performance in the Y maze although not significantly [ns]
In drug-free animals, as compared to WT, STOP-null mice displayed the same exploratory activity measured from the total distance traveled (data not shown) but displayed significantly less spontaneous alternation performance [52.5 vs 63.6 % for KO and WT placebo mice in risperidone groups (p =0.010)].
Risperidone treated or untreated STOP-null mice only showed slight differences in spontaneous alternation [52.5 % (placebo), 64.6 % (0.1mg/kg/day), 60.7 % (0.3mg/kg/day); p =0.219]. These results indicate impairment of working memory in STOP-null mice and a limited beneficial effect of risperidone long-term treatment.
Hippocampal Long-Term Potentiation
STOP-null mice have been shown to display severe defects in long-term synaptic plasticity with highly reduced Long-Term Potentiation (LTP) in the CA1 region of the hippocampus (Andrieux et al. 2002). Here, we studied the effect of chronic treatment with risperidone or clozapine on LTP. In the clozapine group (daily injection of either clozapine or vehicle), STOP-null mice exhibited reduced LTP as compared to wild type [127.9% vs 165.5% (normalized field EPSP amplitudes measured between 30 and 40 minutes post-tetanus) for KO and WT placebo mice (p =0.006)] (Fig. 5b). Synaptic potentiation was obviously improved by clozapine treatments at early time points and persisted for at least 1 hour (Fig. 5a). Quantitative analysis showed that after clozapine treatment, STOP-null mice groups exhibited significant differences in LTP values measured between 30 and 40 minutes post-tetanus [127.9% (placebo), 136.3% (3mg/kg/day), 171.1% (10 mg/kg/day); normalized field EPSP amplitudes, p =0.027] (Fig. 5b). Post-hoc analysis showed significant improvement of STOP-null mice LTP with clozapine 10 mg/kg/day as compared to placebo (p < 0.05). Risperidone treatment did not alleviate LTP defects of STOP-null mice (data not shown).
Fig. 5.
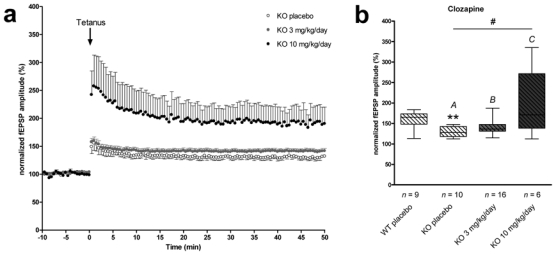
Electrophysiological recordings of LTP. (a) Normalized field EPSP amplitude recorded during 10 min before and 60 min after tetanus (100 Hz, 1 sec) for placebo (n = 10 slices), clozapine 3 mg/kg/day (n = 16 slices) and clozapine 10mg/kg/day (n = 6 slices) STOP null mice. (b) Normalized field EPSP amplitudes between 30 and 40 min after tetanus. In wild type mice, LTP values were similar in all studied groups (untreated, placebo and clozapine-treated) and the value for WT placebo group is shown for comparisons; STOP-null mice treated with placebo present an impairment of LTP when compared to their WT littermates [**: p<0.01, KO vs WT placebo]. Only the dose of 10 mg/kg/day of clozapine significantly improved LTP of chronically-treated mice [#: p<0.05 between KO groups, with A different from C, p<0.05]
Discussion
The goal of the present work was to assay the sensitivity to a long-term atypical AP treatment, namely clozapine and risperidone, on behavioural and synaptic impairments exhibited by STOP-null mice. The present study extended the behavioural phenotype of STOP-null mice revealing that STOP (MAP6) gene deletion in mice leads to multiple behavioural abnormalities relevant to negative symptoms and cognitive impairments of schizophrenia.
In schizophrenia, negative symptoms are close to most of depression symptoms (Barnes et al. 1989). Social interactions measured in rodents are directly comparable to social interaction measured in humans. It has been previously described that STOP-null mice show severe social withdrawal both in aggressive and non-aggressive components (Andrieux et al.2002; Bégou et al. 2008). In this study, using a new paradigm, we found a strong decrease in number of contacts and time spent sniffing in STOP-null mice, indicating a marked social deficit. Interestingly, both clozapine and risperidone long-term treatments strongly improved social interactions in STOP-null mice whereas typical AP long-term treatments have been shown to be less active (Bégou et al. 2008).
The behavioural despair of the animal, placed in an unusual and inescapable environment (Porsolt et al. 1978; Porsolt et al. 1977) and revealed through increased immobility in the forced swim test, could be notably due to a loss of motivation (Noda et al. 1997; Noda et al. 1995) and so relevant to the negative symptomatology in animal models of schizophrenia. In this study, STOP-null mice spent more time floating with minimum movements than WT mice. This altered behaviour of STOP-null mice may potentially be the consequence of an intrinsic lack of motivation as already suspected in a previous study (Bégou et al. 2008). Comparable deficits have been observed in transgenic mice expressing dominant negative copies of DISC1, another genetic model for schizophrenia (Hikida et al. 2007). Chronic treatments with either clozapine or risperidone alleviate behavioural despair in STOP-null mice, potentially by improving their motivational ability.
Whereas marble burying is more closely related to impulsive behaviour than anxious states, this test has been predominantly used as a screening model of detection of new anxiolytic compounds (Nicolas et al. 2006; Young et al. 2006). STOP-null mice bury a substantially lower number of marbles than WT mice. Such behaviour could rely on low anxious state or low motivation for physical environment. Low anxious state is also observed in the elevated plus maze test, as STOP-null mice spent more time in the open arms than WT animals (D. D. unpublished observations) and low motivation for physical environment has already been suggested from STOP-null mice unable to perform object recognition tests (Andrieux et al. 2002). In any case, long-term treatment with either clozapine or risperidone increased the number of buried marbles, but never to significant levels. Better suited tests for anxiety versus motivation should be used in the future.
It has been previously demonstrated that STOP-null mice present cognitive deficits related to the episodic (Andrieux et al. 2002), social, spatial and working memories (Bégou et al. 2008). Here, we actually show that chronic treatment with risperidone does not restore impairment in spatial short-term working memory of STOP-null mice in the spontaneous alternation task.
Altogether these results clearly indicate that STOP-null mice exhibit strong deficits related to negative symptoms and cognitive impairments of schizophrenia. Atypical APs significantly improve deficits related to negative symptoms in STOP-null mice while typical APs only slightly alleviate them (Bégou et al. 2008). In parallel, atypical APs display a slight benefit effect on the cognitive impairments in STOP-null mice, known to be resistant to typical treatment with haloperidol (Bégou et al. 2008).
Typical versus atypical APs sensitivity of STOP-null mice are in accordance with clinical data showing that clozapine and risperidone have a trend to a positive action on negative impairments equivalent or superior to the typical APs, particularly versus haloperidol (Leucht et al. 2009; Volavka et al. 2002). The poor effect of atypical APs treatment on cognitive functions in STOP-null mice is compatible with the effects of atypical APs in humans. Typical APs have been shown to be deleterious on cognitive abilities in humans whereas atypical APs do not induce these defects. In total, we detected the same pharmacological reactivity of typical/atypical APs on STOP-null mice behaviour. The beneficial effect of atypical APs on negative and cognitive impairments over typical drugs is thought to be linked to their pharmacological profile, atypical APs exhibiting higher affinity for 5-HT-2 receptors than typical APs (Duncan et al. 1999).
A growing body of evidence involves glutamatergic transmission in the aetiology of schizophrenia and drugs directly targeting glutamatergic receptors have been shown to exhibit antipsychotic properties (Patil et al. 2007). Accordingly, STOP-null mice display abnormal glutamatergic neurotransmission revealed by impaired long-term potentiation (Andrieux et al. 2002). Some beneficial effects of clozapine are thought to occur through glutamatergic transmission (Coyle and Tsai 2004; Frankle et al. 2003) and interestingly, long-term treatment of STOP-null mice with clozapine restored LTP whereas risperidone did not. These results indicate that LTP measurements in STOP-null mice may discriminate between APs with or without glutamatergic profiles.
A translational preclinical model of human psychiatric disorder is all the more relevant that it respects construct, face and predictive validity (Ellenbroek and Cools 1990; Miczek and de Wit 2008). As outlined in the introduction STOP-null mouse model fulfils the construct and face validity: it is constructed from the neurodevelopmental hypothesis of schizophrenia, with synaptic impairment. Indeed, an association between single nucleotide polymorphism markers of the MAP6 gene and schizophrenia has been found (Shimizu et al. 2006) as well as a deregulation of STOP expression inprefrontal cortex of schizophrenic patients (Martins-de-Souza et al. 2009). STOP-null mice have been shown to display behavioral abnormalities thought to simulate aspects of positive symptoms classically observed in schizophrenic patients. The present data, added to all previous ones, give a comprehensive phenotype of STOP null mice according to the behavioral defects related to the core symptoms of schizophrenia, namely negative symptoms and cognitive deficits, which are known to be resistant to current treatments. The pharmacological results of the present study referring to the possibility to mimic the benefit effects of APs in humans are relevant for the predictive validity of STOP-null mice. However, knowing that current atypical AP treatment in humans displayed limited effects, the predictive validity of STOP null mice need to be further supported by studies showing behavioral alterations resistant to APs. Altogether, we suggest that STOP null mice could be a very useful tool to develop AP drugs by monitoring pharmacological effects both at the behavioural and biological levels.
Acknowledgments
We thank C. Dumont and D. Proietto for technical help.
Abbreviations
- APs
Antipsychotics
- DISC1
Disrupted-In-Schizophrenia 1
- MAP6
Microtubule-Associated Protein 6
- PPI
Pre-Pulse Inhibition
- STOP
Stable Tubule Only Polypeptide
- WT
Wild-Type
- LTP
Long Term Potentiation
References
- Andrieux A, Salin PA, Vernet M, Kujala P, Baratier J, Gory-Fauré S, Bosc C, Pointu H, Proietto D, Schweitzer A, Denarier E, Klumperman J, Job D. The suppression of brain cold-stable microtubules in mice induces synaptic defects associated with neuroleptic-sensitive behavioral disorders. Genes Dev. 2002;16:2350–64. doi: 10.1101/gad.223302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes TR, Curson DA, Liddle PF, Patel M. The nature and prevalence of depression in chronic schizophrenic in-patients. Br J Psychiatry. 1989;154:486–91. doi: 10.1192/bjp.154.4.486. [DOI] [PubMed] [Google Scholar]
- Begou M, Brun P, Bertrand JB, Job D, Schweitzer A, D’Amato T, Saoud M, Andrieux A, Suaud-Chagny MF. Post-pubertal emergence of alterations in locomotor activity in stop null mice. Synapse. 2007;61:689–697. doi: 10.1002/syn.20409. [DOI] [PubMed] [Google Scholar]
- Bégou M, Volle J, Bertrand JB, Brun P, Job D, Schweitzer A, Saoud M, D’Amato T, Andrieux A, Suaud-Chagny MF. The stop null mice model for schizophrenia displays cognitive and social deficits partly alleviated by neuroleptics. Neuroscience. 2008;157:29–39. doi: 10.1016/j.neuroscience.2008.07.080. [DOI] [PubMed] [Google Scholar]
- Blackwood DH, Pickard BJ, Thomson PA, Evans KL, Porteous DJ, Muir WJ. Are some genetic risk factors common to schizophrenia, bipolar disorder and depression? Evidence from DISC1, GRIK4 and NRG1. Neurotox Res. 2007;11:73–83. doi: 10.1007/BF03033484. [DOI] [PubMed] [Google Scholar]
- Bouvrais-Veret C, Weiss S, Andrieux A, Schweitzer A, McIntosh JM, Job D, Giros B, Martres MP. Sustained increase of alpha7 nicotinic receptors and choline-induced improvement of learning deficit in STOP knock-out mice. Neuropharmacology. 2007;52:1691–700. doi: 10.1016/j.neuropharm.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Brenner E, Sonnewald U, Schweitzer A, Andrieux A, Nehlig A. Hypoglutamatergic activity in the STOP knockout mouse: A potential model for chronic untreated schizophrenia. J Neurosci Res. 2007 doi: 10.1002/jnr.21200. [DOI] [PubMed] [Google Scholar]
- Brun P, Bégou M, Andrieux A, Mouly-Badina L, Clerget M, Schweitzer A, Scarna H, Renaud B, Job D, Suaud-Chagny MF. Dopaminergic transmission in STOP null mice. J Neurochem. 2005;94:63–73. doi: 10.1111/j.1471-4159.2005.03166.x. [DOI] [PubMed] [Google Scholar]
- Camargo LM, Wang Q, Brandon NJ. What can we learn from the disrupted in schizophrenia 1 interactome: lessons for target identification and disease biology? Novartis Found Symp. 2008;289:208–16. doi: 10.1002/9780470751251.ch17. discussion 216–21,238–40. [DOI] [PubMed] [Google Scholar]
- Chiba S, Hashimoto R, Hattori S, Yohda M, Lipska B, Weinberger DR, Kunugi H. Effect of antipsychotic drugson DISC1 and dysbindin expression in mouse frontal cortex and hippocampus. J Neural Transm. 2006 doi: 10.1007/s00702-005-0414-1. [DOI] [PubMed] [Google Scholar]
- Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, Lerch JP, Trimble K, Uchiyama M, Sakuraba Y, Kaneda H, Shiroishi T, Houslay MD, Henkelman RM, Sled JG, Gondo Y, Porteous DJ, Roder JC. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G. NMDA receptor function, neuroplasticity, and the pathophysiology of schizophrenia. Int Rev Neurobiol. 2004;59:491–515. doi: 10.1016/S0074-7742(04)59019-0. [DOI] [PubMed] [Google Scholar]
- Denarier E, Aguezzoul M, Jolly C, Vourc’h C, Roure A, Andrieux A, Bosc C, Job D. Genomic structure and chromosomal mapping of the mouse STOP gene (Mtap6) Biochem Biophys Res Commun. 1998;243:791–6. doi: 10.1006/bbrc.1998.8179. [DOI] [PubMed] [Google Scholar]
- Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, Liu XB, Yang CH, Jordan JD, Ma DK, Liu CY, Ganesan S, Cheng HJ, Ming GL, Lu B, Song H. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–58. doi: 10.1016/j.cell.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Zorn S, Lieberman JA. Mechanisms of typical and atypical antipsychotic drug action in relation to dopamine and NMDA receptor hypofunction hypotheses of schizophrenia. Mol Psychiatry. 1999;4:418–28. doi: 10.1038/sj.mp.4000581. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Lyon L, George L, Andrieux A, Job D, Harrison PJ. Altered expression of synaptic protein mRNAs in STOP (MAP6) mutant mice. J Psychopharmacol. 2007;21:635–44. doi: 10.1177/0269881106068825. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Cools AR. Animal models with construct validity for schizophrenia. Behav Pharmacol. 1990;1:469–490. [PubMed] [Google Scholar]
- Ereshefsky L, Tran-Johnson TK, Watanabe MD. Pathophysiologic basis for schizophrenia and the efficacy of antipsychotics. Clin Pharm. 1990;9:682–707. [PubMed] [Google Scholar]
- Fradley RL, O’Meara GF, Newman RJ, Andrieux A, Job D, Reynolds DS. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behav Brain Res. 2005;163:257–64. doi: 10.1016/j.bbr.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Frankle WG, Lerma J, Laruelle M. The synaptic hypothesis of schizophrenia. Neuron. 2003;39:205–16. doi: 10.1016/s0896-6273(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, Wu D, Xue R, Andrade M, Tankou S, Mori S, Gallagher M, Ishizuka K, Pletnikov M, Kida S, Sawa A. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka K, Paek M, Kamiya A, Sawa A. A review of Disrupted-In-Schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol Psychiatry. 2006;59:1189–97. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373:31–41. doi: 10.1016/S0140-6736(08)61764-X. [DOI] [PubMed] [Google Scholar]
- Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, Brzustowicz LM, Kaufmann CA, Garver DL, Gurling HM, Lindholm E, Coon H, Moises HW, Byerley W, Shaw SH, Mesen A, Sherrington R, O’Neill FA, Walsh D, Kendler KS, Ekelund J, Paunio T, Lonnqvist J, Peltonen L, O’Donovan MC, Owen MJ, Wildenauer DB, Maier W, Nestadt G, Blouin JL, Antonarakis SE, Mowry BJ, Silverman JM, Crowe RR, Cloninger CR, Tsuang MT, Malaspina D, Harkavy-Friedman JM, Svrakic DM, Bassett AS, Holcomb J, Kalsi G, McQuillin A, Brynjolfson J, Sigmundsson T, Petursson H, Jazin E, Zoega T, Helgason T. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie S, Millar JK, Porteous DJ. Role of DISC1 in neural development and schizophrenia. Curr Opin Neurobiol. 2007;17:95–102. doi: 10.1016/j.conb.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Martins-de-Souza D, Gattaz WF, Schmitt A, Rewerts C, Maccarrone G, Dias-Neto E, Turck CW. Prefrontal cortex shotgun proteome analysis reveals altered calcium homeostasis and immune system imbalance in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2009;259:151–63. doi: 10.1007/s00406-008-0847-2. [DOI] [PubMed] [Google Scholar]
- Matsuzaki S, Tohyama M. Molecular mechanism of schizophrenia with reference to disrupted-in-schizophrenia 1(DISC1) Neurochem Int. 2007;51:165–72. doi: 10.1016/j.neuint.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. What’s atypical about atypical antipsychotic drugs? Curr Opin Pharmacol. 2004;4:53–7. doi: 10.1016/j.coph.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Miczek KA, de Wit H. Challenges for translational psychopharmacology research--some basic principles. Psychopharmacology (Berl) 2008;199:291–301. doi: 10.1007/s00213-008-1198-4. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Dekeyne A, Papp M, La Rochelle CD, MacSweeny C, Peglion JL, Brocco M. S33005, a novel ligand at both serotonin and norepinephrine transporters: II. Behavioral profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J Pharmacol Exp Ther. 2001;298:581–91. [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Lewis DA, Levitt P. Analysis of complex brain disorders with gene expression microarrays: schizophrenia as a disease of the synapse. Trends Neurosci. 2001;24:479–86. doi: 10.1016/s0166-2236(00)01862-2. [DOI] [PubMed] [Google Scholar]
- Mueser KT, McGurk SR. Schizophrenia. Lancet. 2004;363:2063–72. doi: 10.1016/S0140-6736(04)16458-1. [DOI] [PubMed] [Google Scholar]
- Nicolas LB, Kolb Y, Prinssen EP. A combined marble burying-locomotor activity test in mice: a practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur J Pharmacol. 2006;547:106–15. doi: 10.1016/j.ejphar.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Noda Y, Mamiya T, Furukawa H, Nabeshima T. Effects of antidepressants on phencyclidine-induced enhancement of immobility in a forced swimming test in mice. Eur J Pharmacol. 1997;324:135–40. doi: 10.1016/s0014-2999(97)00067-8. [DOI] [PubMed] [Google Scholar]
- Noda Y, Yamada K, Furukawa H, Nabeshima T. Enhancement of immobility in a forced swimming test by subacute or repeated treatment with phencyclidine: a new model of schizophrenia. Br J Pharmacol. 1995;116:2531–7. doi: 10.1111/j.1476-5381.1995.tb15106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MJ, Craddock N, O’Donovan MC. Schizophrenia: genes at last? Trends Genet. 2005a;21:518–25. doi: 10.1016/j.tig.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Owen MJ, O’Donovan MC, Harrison PJ. Schizophrenia: a genetic disorder of the synapse? BMJ. 2005b;330:158–9. doi: 10.1136/bmj.330.7484.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–7. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Peuskens J, Demily C, Thibaut F. Treatment of cognitive dysfunction in schizophrenia. Clin Ther. 2005;27(Suppl A):S25–37. doi: 10.1016/j.clinthera.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Anton G, Blavet N, Jalfre M. Behavioural despair in rats: a new model sensitive to antidepressant treatments. Eur J Pharmacol. 1978;47:379–91. doi: 10.1016/0014-2999(78)90118-8. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–36. [PubMed] [Google Scholar]
- Powell KJ, Hori SE, Leslie R, Andrieux A, Schellinck H, Thorne M, Robertson GS. Cognitive impairments in the STOP null mouse model of schizophrenia. Behav Neurosci. 2007;121:826–35. doi: 10.1037/0735-7044.121.5.826. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Koseki M, Wakamatsu M, Matsumura E. Effects of systemic administration of beta-casomorphin-5 on learning and memory in mice. Eur J Pharmacol. 2006;530:81–7. doi: 10.1016/j.ejphar.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Sergi MJ, Rassovsky Y, Widmark C, Reist C, Erhart S, Braff DL, Marder SR, Green MF. Social cognition in schizophrenia: relationships with neurocognition and negative symptoms. Schizophr Res. 2007;90:316–24. doi: 10.1016/j.schres.2006.09.028. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Iwayama Y, Yamada K, Toyota T, Minabe Y, Nakamura K, Nakajima M, Hattori E, Mori N, Osumi N, Yoshikawa T. Genetic and expression analyses of the STOP (MAP6) gene in schizophrenia. Schizophr Res. 2006;84:244–52. doi: 10.1016/j.schres.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Shinoda T, Taya S, Tsuboi D, Hikita T, Matsuzawa R, Kuroda S, Iwamatsu A, Kaibuchi K. DISC1 regulates neurotrophin-induced axon elongation via interaction with Grb2. J Neurosci. 2007;27:4–14. doi: 10.1523/JNEUROSCI.3825-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Cho DS, Ong WY, Benson MA, Han LY, Kazi HA, Kamins J, Hahn CG, Blake DJ, Arnold SE. Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum Mol Genet. 2006;15:3041–54. doi: 10.1093/hmg/ddl246. [DOI] [PubMed] [Google Scholar]
- Taya S, Shinoda T, Tsuboi D, Asaki J, Nagai K, Hikita T, Kuroda S, Kuroda K, Shimizu M, Hirotsune S, Iwamatsu A, Kaibuchi K. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J Neurosci. 2007;27:15–26. doi: 10.1523/JNEUROSCI.3826-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volavka J, Czobor P, Sheitman B, Lindenmayer JP, Citrome L, McEvoy JP, Cooper TB, Chakos M, Lieberman JA. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry. 2002;159:255–62. doi: 10.1176/appi.ajp.159.2.255. [DOI] [PubMed] [Google Scholar]
- Young R, Batkai S, Dukat M, Glennon RA. TDIQ (5,6,7,8-tetrahydro-1,3-dioxolo[4,5-g]isoquinoline) exhibits anxiolytic-like activity in a marble-burying assay in mice. Pharmacol Biochem Behav. 2006;84:62–73. doi: 10.1016/j.pbb.2006.04.006. [DOI] [PubMed] [Google Scholar]