

Abstract
Microdeletion 15q24 is an emerging syndrome recently described, mainly due to increased use of array-CGH. Clinical features associate mild to moderate developmental delay, typical facial characteristics (high forehead and frontal hairline, broad eyebrows, down-slanting palpebral features, long philtrum), hands (particularly proximal implanted thumbs) and genital anomalies (micropenis, hypospadias).
We report here four de novo cases having 2.5–6.1 Mb deletions involving 15q24: one 15q23q24.2 (patient 1) and three 15q24.1q24.2 deletions (patients 2, 3 and 4). We correlate phenotype to genotype according to molecular boundaries of these deletions. Since bilateral iris coloboma and severe ano-rectal malformation were only present in patient 1, we could link these anomalies to haploinsufficiency of 15q23 genes. Neither hypospadias nor micropenis were present in patient 3 bearing the smallest deletion, therefore we could define 500 kb 15q24.1 region linked to these anomalies.
Keywords: Child; Child, Preschool; Chromosome Breakage; Chromosome Deletion; Chromosomes, Human, Pair 15; genetics; Comparative Genomic Hybridization; Female; Genotype; Humans; Infant; Infant, Newborn; Male; Phenotype; Pregnancy
Keywords: 15q24 deletion, genotype-phenotype correlation, array-CGH
Introduction
Microdeletion of 15q24 is a recent emerging syndrome mainly identified due to widespread use of array-CGH widespread use. Although there is an important phenotypic variability, patients may share some common findings, and these deletions could potentially represent a clinically identifiable syndrome (Sharp and others 2007). Clinical features therefore associate mild to moderate developmental delay, distinctive facial characteristics (high forehead and frontal hairline, broad eyebrows, down-slanting palpebral features, long philtrum), hand anomalies (particularly proximal placement of thumbs), and genital malformations in males (micropenis, hypospadias) (Cushman and others 2005; Sharp and others 2007) (Klopocki and others 2008).
We report detailed studies of four patients with 15q24 deletions, having common features and compare these findings with 9 previously reported cases in the literature (Cushman and others 2005; Klopocki and others 2008; Masurel-Paulet and others 2009; Sharp and others 2007; Van Esch and others 2009).
Patients and Methods
Patient 1 is the first child of healthy non consanguineous parents, born after a previous early miscarriage. During pregnancy, intra uterine growth retardation (IUGR) was diagnosed at 26 weeks’ gestation (WG). Later on, ventricular septal defect was identified on the third trimester ultrasound, together with single umbilical artery. Chromosome analysis and 22q11.2 FISH performed on amniotic fluid were normal. He was born at 35 WG by caesarean section performed for maternal preeclampsia, with a birth weight of 1.900 kg (10–25th centile), a length of 43 cm (10–25th centile), and a head circumference of 30 cm (10–25th centile). At birth, several malformations were identified including an imperforate anus, a bilateral iris coloboma, bilateral single palmar creases, bilateral II–III toe syndactyly, hypoplastic 5th toes with absent 5th toe nails, and a micropenis with a coronal hypospadias and a left cryptorchidism. The cardiac malformation was confirmed. He was first referred to the genetic clinic at 4 months, having severe hypotonia and post natal growth retardation with a weight of -4SD, a height of -3DS and a HC of -3SD. Striking dysmorphic features were identified comprising of dysplastic asymmetric ears, large and high forehead, hypoplastic nasal bridge, small nose with anteverted nares, up-slanting palpebral fissures, bilateral epicanthus and a high palate (Fig. 1a). His skin appeared very dry. Later on, he underwent surgery for left inguinal hernia where nystagmus was noted.
Figure 1.
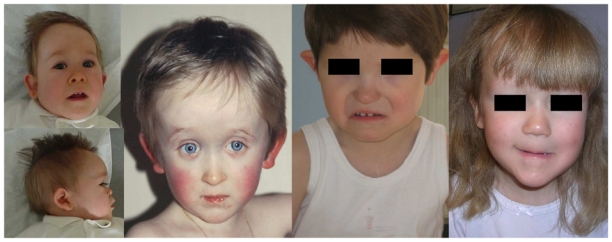
Facial characteristics of the 4 patients. A: Patient 1. B: Patient 2. C: Patient 3. D: Patient 4. Note high and large forehead on patients 1, 2 and 4.
Developmental delay occurred secondarily. Head control was acquired at 19 months. At 2 years, he was sitting by himself and his growth parameters were in the normal range with height -1SD, weight -1SD and HC -1SD. Unfortunately, he died at home by the age of 25 months due to unknown cause. Parents declined post-mortem examination. Several investigations were performed that includes, high resolution chromosome banding which was normal, along with renal scan, skeletal x-rays, electromyogram and nerve conduction velocities. Visual evoked potentials and 7-dehydrocholesterol were also normal except brain MRI that revealed cerebral atrophy and enlarged ventricles.
Patient 2 is the third child of healthy non consanguineous parents. He was born after a normal pregnancy, at 38 WG with normal growth parameters (weight 3.170 kg, height 53 cm and HC 33 cm). Hypospadias was noted at birth in association with hypotonia and short proximal implanted thumbs. Dysmorphic features comprising of high and large forehead with small mouth were observed (Fig. 1b). Developmental delay occurred later on, and walking independantly was achieved at 29 months. When referred to the genetic clinic at 3 years, growth parameters were normal. However, joint laxity was noted, but thumbs and facial features were still abnormal. High resolution chromosome banding was normal, along with renal and heart ultrasounds. However, skeletal x-rays revealed bilateral short first metacarpals, eye examination identified bilateral papilloedema and thick corpus callosum was noted on brain MRI.
Patient 3 is the second child born to healthy non consanguineous parents. During pregnancy, Fallot tetralogy was suspected and chromosomes including 22q11.2 by FISH analysis were observed to be normal on aminotic fluid. He was born at 39 WG, with normal growth parameters (weight 3.450 kg, height 49.5 cm and HC 35 cm). At birth, the cardiac anomaly was confirmed and he underwent several surgical interventions. Developmental delay was secondarily noted with head control achieved at 9 months, sitting at 15 months and walking at 3 years. Speech was also delayed (first words at 3 years). When referred to the genetic clinic at the age of 3 years, dysmorphic features were noted with asymmetric dysplastic ears, thin upper lip and widely-spaced teeth (Fig. 1c). His skin was dry. Eye examination revealed strabismus and hypermetropia. A bilateral sensorineural deafness was identified. High resolution chromosome banding was normal. Brain MRI revealed hypoplastic olfactory bulbs. Semi-circular canals were normal. CHD7 molecular analysis was normal that ruled out CHARGE syndrome.
Patient 4 is the first child of healthy non consanguineous parents. During pregnancy, intra uterine growth retardation was noted. She was born at 37 WG with a weight of 1.850 kg, height of 45.5 cm and HC of 31 cm. At birth, dysmorphic facial features were noted with large and high forehead, flat midface, thin lips, small mouth and posteriorly rotated ears. Developmental delay was noted, with sitting at 11 months, walking at 2½ years and first words at 3 years. When reviewed in the genetic clinic at the age of 7 years, growth parameters were normal. Dysmorphic features were still present (Fig. 1d) with adducted thumbs and genu valgum. Eye examination revealed hypermetropia. High resolution chromosome banding was normal together with metabolic investigations, brain MRI and molecular analyses that ruled out Prader-Willi syndrome and Steinert disease.
Array-CGH analysis
Genomic DNA was extracted from the four patients’ peripheral blood lymphocytes using the QIAamp DNA Blood Midi kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. DNA concentration was determined with NanoDrop ND-1000 spectrophotometer and software (NanoDrop Technologies, Berlin, Germany). Detection of gene copy number was performed by array-Comparative Genomic Hybridization (array-CGH) experiments following standard and manufacturer’s recommendations using 44.000 oligo probes approximately spaced at 40–100 kb intervals across the genome (Human Genome CGH microarray 44B kit, Agilent™). Male or female genomic DNA (Promega™) was used as reference in sex-match hybridizations which were analyzed with the CGH-analytics software v3.4 by applying Z-score segmentation algorithm with a window size of 10 points to identify chromosome aberrations. Analysis was performed with filter settings: 3-point filter and 0.2 of variation.
BACs and PACs DNAs were extracted by Macherey-Nagel kit (Nucleobond, 74579, Hoerdt, France) and labelled with Cy3-dUTP (Amersham Bioscience, PA53022, GEhealthcare, Europe Gmbh, Munich, Germany) by nick-translation (Nick Translation System, 18160-010, Invitrogen, San Diego, CA, USA). The Cy3-labelled DNA probe was precipitated and hybridized using standard procedures. Twenty metaphases were analyzed under fluorescence DMRB microscope (Leica) equipped with Metasystem Isis software (Altlussheim, Germany). The following academic BAC clone probes were used: on chromosome 15: RP11-375D9 (67.2 Mb), RP11-64K10 (68.7 Mb), RP11-141J4 (72.8 Mb), RP11-78M2 (74.5 Mb) (Sanger institute, UK).
Results
Array-CGH 44K Agilent™ showed 2.5–6.1 Mb deletions involving 15q24 (Fig. 2): one 15q23q24.2 (patient 1) and three 15q24.1q24.2 deletions (Table I).
Figure 2.
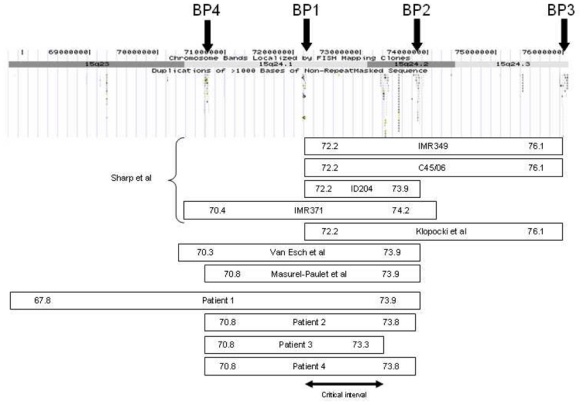
Schematic representation of 15q24 rearrangements published cases characterized by array-CGH with their respective breakpoints and recurrent breakpoints BP1, BP2 and BP3 (Sharp and others 2007). Our four cases are also represented and segmental duplications featured. Recurrent breakpoint 4 (BP4) is also represented (Masurel-Paulet and others 2009). Three of our patients’ deletions have BP4 as proximal breakpoint.
Table 1.
Phenotypic comparison between cases from (Cushman and others 2005; Klopocki and others 2008; Masurel-Paulet and others 2009; Sharp and others 2007; Van Esch and others 2009) and our 4 patients. HC: head circumference. ENT: Ear-Nose-Throat.
| Patient 1 15q23q24.2 deletion |
Patient 2 15q24.1q24.2 deletion |
Patient 3 15q24.1q24.2 deletion |
Patient 4 15q24.1q24.2 deletion |
(Cushman and others 2005) 3 cases (Sharp and others 2007) 4 cases (Klopocki and others 2008) 1 case (Van Esch and others 2009) 1 case (Masurel-Paulet and others 2009) 1 case |
|
|---|---|---|---|---|---|
| Array-CGH hg18 | 67,807,119–73,907,050 | 70,764,625–73,807,021 | 70,764,684–73,316,640 | 70,764,625–73,806,962 | available from (Klopocki and others 2008), (Sharp and others 2007), (Van Esch and others 2009) and (Masurel-Paulet and others 2009) |
| Birth parameters | 33WG weight: 1900g height: 43 cm HC: 30 cm |
38 WG weight: 3170g height: 53 cm HC: 33 cm |
39 WG weight: 3450g height: 49,5 cm HC: 35 cm |
37 WG weight: 1850g height: 45,5cm HC: 31cm |
|
| Sex, age at diagnosis | M, 2 years | M, 3 years | M, 7 years | F, 5 years | 9 M, 1 F 13 months to 33 years |
| Facial characteristics | large and high forehead hypoplastic nasal bridge small nose with anteverted nares up-slanting palpebral fissures bilateral epicanthus high palate |
large and high forehead small mouth |
thin upper lip widely-spaced teeth |
large and high forehead thin lips flat midface down-slanting palpebral fissures |
high forehead (7/7) broad medial eyebrows (6/7) hypertelorism (6/7) epicanthal folds (4/8) down-slanting palpebral fissures (6/7) long and smooth philtrum (7/7) full lower lip (5/6) broad nasal base (3/5) micrognathia (3/8) |
| Eye abnormalities | bilateral iris coloboma nystagmus |
papilloedema | strabismus phypermetropia |
hypermetropia | strabismus (5/7) nystagmus (1/5) |
| Ear abnormalities | dysplastic asymmetric ears | no | dysplastic asymmetric ears | posteriorly rotated ears | 5/8 |
| Limb abnormalities | bilateral single palmar creases bilateral II–III toe syndactyly hypoplastic 5th toes |
short and proximal placed thumbs | no | adducted thumbs genu valgum |
thumbs anomalies (4/8) other fingers anomalies (6/8) |
| Musculo-skeletal abnormalities | no | joint laxity | no | no | joint laxity (4/8) scoliosis (2/7) |
| Genital abnormalities | micropenis/coronal hypospadias left ectopic testis | hypospadias | no | no | hypospadias, micropenis and/or small scrotum (8/9) |
| Developmental delay | head control at 19 months sitting at 2 years |
walking at 29 months | sitting at 15 months walking at 3 years first words at 3 years |
sitting at 11 months walking at 2 years first words at 3 years |
mild to moderate mental retardation (10/10) |
| Microcephaly | no | no | no | no | 3/10 |
| Neurological findings | cerebral atrophy enlarged ventricles |
thick corpus callosum | hypoplastic olfactory bulbs | no | wide basal cisterna (1/5) dysplastic corpus callosum hypoplasia of the adenohypohysis and ectopic neurohypophysis (1/5) cysts in the corpus callosum (1/5) |
| Cardiovascular abnormalities | ventricular septal defect | no | Fallot tetralogy | no | pulmonary stenosis (1/4) |
| Recurrent infections | no | no | ENT | ENT (3/4) | |
| Deafness/hearing loss | no | bilateral sensorineural deafness |
deafness/hearing loss (2/5) | ||
| Skin | dry | normal | dry | normal | |
| Other | left inguinal hernia imperforate anus |
inguinal hernia (3/9) growth hormone deficiency (2/3) delayed puberty (2/2) |
FISH studies confirmed the 15q23q24.2 aberration elicited by array-CGH, since the RP11-64K10 and RP11-141J4 clones were deleted. The deletion was absent on parental DNA and appeared to be de novo in patient 1: 46, XY, ish del(15)(q23q24.2)(RP11-375D9+, RP11-64K10-, RP11-141J4-, RP11-78M2+)[25]dn. For the deleted 15q24.1q24.2 in patients 2, 3 and 4: RP11-141J4 clone was deleted and absence of deletion in the parents confirmed de novo origin.
Discussion
There are several reports of 15q deletions involving q24. However, to the best of our knowledge, only 7 published cases have been identified by array-CGH (Klopocki and others 2008; Masurel-Paulet and others 2009; Sharp and others 2007; Van Esch and others 2009). Taking together with our results we could define a 1.1 Mb region on 15q24.1q24.2 in which 23 genes are located. Among them, several are interesting in delineating the 15q24 deletion phenotype (table 2).
Table 2.
Genes potentially involved in the 15q24 phenotype.
| Gene | Expression | Function | Localization | Animal model |
|---|---|---|---|---|
| SIN3A (transcriptional co-repressor Sin3A) | Testis | Transcriptional regulatory protein, histone deacetylase interacting | 15q24.2 | No known KO |
| ODF3L1 (outer dense fiber of sperm tails 3-like 1) | Testis | Transport of keratins to microtubule-containing structures such as the manchette and axoneme during mammalian spermiogenesis | 15q24.2 | No known KO |
| CYP11A1 (cytochrome P450, family 11, subfamily A, polypeptide 1) | Adrenal glands | Localizes to the mitochondrial inner membrane and catalyzes the conversion of cholesterol to pregnenolone | 15q24.1 | No known KO |
| CCDC33 (coiled-coil domain containing 33) | Testis | C2 calcium-dependent membrane targeting, down-regulated in cryptorchidism | 15q24.1 | No known KO |
| STRA6 (stimulated by retinoic acid gene 6 homolog) | Eye | Receptor for retinol/retinol binding protein complexes. Removes the retinol from the complex and transports it across the cell membrane | 15q24.1 | No known KO Defects in this gene cause syndromic microphthalmia type 9 (MCOPS9). |
| UACA (uveal autoantigen with coiled-coil domains and ankyrin repeats) | Eye | Recruits and transports the apoptosome complex during stress-induced apoptosis | 15q23 | No known KO |
| PARP6 (poly (ADP-ribose) polymerase family, member 6) | Testis, eye | NAD+ ADP-ribosyltransferase activity | 15q23 | No known KO |
| CPLX3 (complexin 3) | Brain, eye | Positive regulator of neurotransmitter release in mouse hippocampal neurons, syntaxin binding | 15q24.1 | No known KO |
| SEMA7A (semaphorin 7A) | Brain | Mediate neuronal functions | 15q24.1 | No known KO Defects in this gene can result in loss of bone mineral density |
| HCN4 (hyperpolarization activated cyclic nucleotide-gated potassium channel 4) | Heart | Necessary for the cardiac pacemaking process | 15q24.1 | No known KO Mutations in this gene have been linked to sick sinus syndrome 2 |
Patient 1 is bearing the largest deletion and showed the most severe phenotype. Additional clinical characteristics may thus be correlated to the 3Mb additional 15q23 deletion between 67.8 and 70.8 Mb.
Some of the common facial characteristics previously described in 15q24 deletion syndrome were observed in our patients, mainly high forehead (3/4) and dysplastic ears (3/4) (Cushman and others 2005; Klopocki and others 2008; Masurel-Paulet and others 2009; Van Esch and others 2009). Musculo-skeletal anomalies such as limb anomalies, joint laxity and scoliosis have also been frequently observed in 15q24 deleted patients. Malformations of the extremities were present in 3/4 patients with proximal implanted thumbs in patients 2 and 4 and bilateral single palmar creases and II–III toe syndactyly in patient 1.
Genital abnormalities in males are frequent in 15q24 deletion (Klopocki and others 2008). Hypospadias was present in two of the male patients (1 and 2) and absent in patient 3 bearing the smallest deletion. Thus we hypothesized that genital anomalies may be linked to the 15q24.2 genes deleted in patients 1 and 2 but not in patient 3, i.e. between chr15:73,316,640 and chr15:73,807,021 bp. Ten genes are located within these 490 kb and among them SIN3A and ODF3L1 are significantly expressed in human testis and may potentially be involved. CYP11A1 is strongly expressed in adrenal gland and haploinsufficiency of this gene may also contribute to the genital abnormalities seen in 15q24 deleted patients (Klopocki and others 2008). Although CCDC33 is not deleted in patient 3, its strong expression in testis could be linked with genital anomalies in patients 1 and 2. Absence of genital anomalies in patient 3 may therefore be explained by incomplete penetrance, variable expressivity or other genes involvement.
Since eye abnormalities have been previously reported in the literature and also identified in our patients, we focused our attention on STRA6 which is involved in syndromic autosomal recessive microphthalmia type 9 (MCOPS9). Bilateral iris coloboma was present in patient 1. Ophthalmologic examination of two patients with 15q22q25 and 15q21q24 deletions who died at the age of 2 years with respiratory problems showed hypopigmentation and coloboma of the iris in one of them (Formiga and others 1988). Among the 15q23 genes deleted in patient 1, UACA is expressed in the eye anterior segment (Ohkura and others 2004) as well as PARP6 (Hassa and Hottiger 2008).
Patients 1, 2 and 3 showed different brain anomalies on MRI including hypoplastic olfactory bulbs in patient 3. CPLX3 is strongly expressed in the brain and SEMA7A also. Semaphorins are involved in axon guidance during neuronal development and particularly in olfactory bulbs, hippocampus, pontine nucleus and thalamocortical axon (Maruyama and others 2008; Miyazaki and others 1999).
HCN4 is strongly expressed in the heart and loss of function of HCN4 can be associated with sinus nodal dysfunction such as QT prolongation and polymorphic ventricular tachycardia which may lead to sudden unexpected death (Ueda and others 2004). Therefore, cardiac follow-up may be useful in 15q24 deleted patients. ECG had been performed in all 4 patients and was normal.
Array-CGH allowed in these 4 cases presenting with 15q24 microdeletion to compare their clinical features with previously reported cases, although it is still difficult to delineate specific diagnostic criteria for this rare microdeletion syndrome. We also suggest several candidate genes involvement in the neurological, genital and ophthalmological aspects of the phenotype.
Acknowledgments
The authors gratefully thank the parents for their cooperation.
Footnotes
Electronic database
UCSC Genome Browser on Human March 2006 Assembly - hg18: http://genome.ucsc.edu/cgi-bin/hgGateway?org=human.
EMBL-EBI microarray database: http://www.ebi.ac.uk/microarray-as/atlas/
References
- Cushman LJ, Torres-Martinez W, Cherry AM, Manning MA, Abdul-Rahman O, Anderson CE, Punnett HH, Thurston VC, Sweeney D, Vance GH. A report of three patients with an interstitial deletion of chromosome 15q24. Am J Med Genet Part A. 2005;137A:65–71. doi: 10.1002/ajmg.a.30836. [DOI] [PubMed] [Google Scholar]
- Formiga LD, Poenaru L, Couronne F, Flori E, Eibel JL, Deminatti MM, Savary JB, Lai JL, Gilgenkrantz S, Pierson M. Interstitial deletion of chromosome 15: two cases. Hum Genet. 1988;80:401–404. doi: 10.1007/BF00273663. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- Klopocki E, Graul-Neumann LM, Grieben U, Tonnies H, Ropers HH, Horn D, Mundlos S, Ullmann R. A further case of the recurrent 15q24 microdeletion syndrome, detected by array CGH. Eur J Pediatr. 2008;167:903–908. doi: 10.1007/s00431-007-0616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Matsuura M, Suzuki K, Yamamoto N. Cooperative activity of multiple upper layer proteins for thalamocortical axon growth. Dev Neurobiol. 2008;68:317–331. doi: 10.1002/dneu.20592. [DOI] [PubMed] [Google Scholar]
- Masurel-Paulet A, Callier P, Thauvin-Robinet C, Chouchane M, Mejean N, Marle N, Mosca AL, Salem DB, Giroud M, Guibaud L, et al. Multiple cysts of the corpus callosum and psychomotor delay in a patient with a 3.1 Mb 15q24.1q24.2 interstitial deletion identified by array-CGH. Am J Med Genet Part A. 2009;149A:1504–1510. doi: 10.1002/ajmg.a.32904. [DOI] [PubMed] [Google Scholar]
- Miyazaki N, Furuyama T, Sakai T, Fujioka S, Mori T, Ohoka Y, Takeda N, Kubo T, Inagaki S. Developmental localization of semaphorin H messenger RNA acting as a collapsing factor on sensory axons in the mouse brain. Neuroscience. 1999;93:401–408. doi: 10.1016/s0306-4522(99)00134-7. [DOI] [PubMed] [Google Scholar]
- Ohkura T, Taniguchi S, Yamada K, Nishio N, Okamura T, Yoshida A, Kamijou K, Fukata S, Kuma K, Inoue Y, et al. Detection of the novel autoantibody (anti-UACA antibody) in patients with Graves’ disease. Biochem Biophys Res Commun. 2004;321:432–440. doi: 10.1016/j.bbrc.2004.06.162. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Selzer RR, Veltman JA, Gimelli S, Gimelli G, Striano P, Coppola A, Regan R, Price SM, Knoers NV, et al. Characterization of a recurrent 15q24 microdeletion syndrome. Hum Mol Genet. 2007;16:567–572. doi: 10.1093/hmg/ddm016. [DOI] [PubMed] [Google Scholar]
- Ueda K, Nakamura K, Hayashi T, Inagaki N, Takahashi M, Arimura T, Morita H, Higashiuesato Y, Hirano Y, Yasunami M, et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem. 2004;279:27194–27198. doi: 10.1074/jbc.M311953200. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Backx L, Pijkels E, Fryns JP. Congenital diaphragmatic hernia is part of the new 15q24 microdeletion syndrome. Eur J Med Genet. 2009;52:153–156. doi: 10.1016/j.ejmg.2009.02.003. [DOI] [PubMed] [Google Scholar]