

Abstract
The transcription factor CREB is diminished in smooth muscle cells (SMCs) in remodeled, hypertensive pulmonary arteries (PAs) in animals exposed to chronic hypoxia. Forced depletion of CREB in PA SMCs stimulates their proliferation and migration in vitro. PDGF produced in the hypoxic PA wall promotes CREB proteasomal degradation in SMCs via PI3Kinase/Akt signaling, which promotes phosphorylation of CREB at two casein kinase 2 (CK2) sites. Here we tested whether thiazolidinediones, agents that inhibit hypoxia-induced PA remodeling, attenuate SMC CREB loss. We found that the thiazolidinedione rosiglitazone prevented PA remodeling and SMC CREB loss in rats exposed to chronic hypoxia. Likewise, the thiazolidinedione troglitazone blocked PA SMC proliferation and CREB depletion induced by PDGF in vitro. Thiazolidinediones did not repress Akt activation by hypoxia in vivo or by PDGF in vitro. However, PDGF induced CK2 α′ catalytic subunit expression and activity in PA SMCs, and depletion of CK2 α′ subunit prevented PDGF-stimulated CREB loss. Troglitazone inhibited PDGF-induced CK2 α′ subunit expression in vitro and rosiglitazone blocked induction of CK2 catalytic subunit expression by hypoxia in PA SMCs in vivo. We conclude that thiazolidine-diones prevent PA remodeling in part by suppressing upregulation of CK2 and loss of CREB in PA SMCs.
Keywords: thiazolidinedione, CREB, casein kinase 2, PDGF-BB, pulmonary artery, smooth muscle cell, pulmonary hypertension
Introduction
Hypoxia-induced pulmonary hypertension (PH) observed in chronic obstructive pulmonary diseases such as emphysema and chronic bronchitis, and sleep-related alveolar hypoventilation disorders (1) is a major cause of morbidity and mortality. There has been a doubling of PH and increased death rate from this set of diseases in the past two decades. Despite major advances in the treatment of severe cardiopulmonary conditions, PH remains a deadly disease that is largely unresponsive to current treatment regimens.
Hypoxia-induced PH is characterized by profound structural remodeling of the PA wall due in large part to the proliferation, migration, and hypertrophy of PA SMCs and adventitial fibroblasts. The most important changes involve the remodeling of thin-walled intermediate size arteries and small arterioles to thickened walled “resistance vessels” which are believed to have the greatest impact on pulmonary vascular resistance (2). Remodeling of these arteries is characterized by medial thickening as the result of SMC proliferation, hypertrophy, and extracellular matrix (ECM) deposition. In response to injury, normally quiescent SMCs switch to a proliferative and synthetic phenotype as demonstrated by an increase in DNA synthesis and production of ECM components (3).
Changes in SMC phenotype reflect the action of extracellular signals and intracellular signaling systems that regulate biochemical processes and the expression of genes that regulate growth. Hypoxia elicits changes in SMC phenotype via a variety of mechanisms including induction of Ca2+ influx (4, 5), and the production of reactive oxygen species by SMCs (6–10). Importantly, hypoxia, like other vascular insults, stimulates the expression of growth factors including PDGF-BB and IGF-1 (11, 12), VEGF (12), TGF-β, endothelin-1 (11, 12), and thrombos-pondin-1 (12) that modulate SMC phenotype.
These remodeling pathways are normally restrained in healthy arteries due to the low level of growth factor and cytokine production in healthy arterial walls. In addition, vasodilatory agents such as prostacyclins and NO exert antiproliferative effects on SMCs by increasing intracellular levels of cyclic nucleotides (13), which stimulate the activity of protein kinases A and G (PKA and PKG). There is now substantial evidence that cAMP/PKA signaling acts as a molecular gate to block MAP kinase induced proliferation in response to mitogens like PDGF (14, 15). Cyclic AMP signaling in SMCs has also been shown to decrease the expression of cyclin D1 and Cdk2 (16), and increase the expression of antiproliferative molecules like p53 and p21 (17).
The transcription factor CREB is a target for cAMP/PKA signaling and the primary regulator of gene transcription in response to elevated cAMP levels. Therefore, we hypothesized that CREB might regulate PA SMC phenotype. In testing this hypothesis we found that phosphorylated (P-CREB) and total CREB levels were reduced in SMCs treated with PDGF-BB or exposed to hypoxia, but elevated in quiescent cells (18). These data were confirmed in studies of lung and PA tissue samples from adult rats and neonatal cows raised under hypoxic conditions to produce PH. Total CREB content was reduced in actin positive SMCs surrounding remodeled, hypertensive vessels. Subsequent studies revealed that ectopic expression of wild type and constitutively active CREB isoforms in isolated PA SMCs inhibited their proliferation under basal conditions or in response to PDGF-BB (18). These CREB isoforms also attenuated PDGF-stimulated migration, but dominant negative CREBs enhanced PDGF-stimulated migration (18).
In more recent studies we found that siRNA-mediated depletion of CREB inhibited the expression of several SMC markers including SM-myosin, calponin, and fibronectin (19). Cyclin D1 expression, and DNA synthesis were upregulated by CREB depletion. CREB siRNA also increased intracellular elastin production and deposition of extracellular elastin fibers, a hallmark of dedifferentiated or immature SMCs. The changes induced by loss of CREB are consistent with the changes in SMC phenotype noted in PH and other vascular pathologies including decreased expression of SMC markers, elevated SMC cell cycle entry and DNA synthesis, and enhanced extracellular matrix deposition. These data implicate CREB in the regulation of SMC phenotypic regulation, and link CREB to PA remodeling in hypoxia-induced PH.
In a separate series of experiments we investigated whether thiazolidinediones (TZDs), agonists of the transcription factor peroxisome proliferator-activated receptor γ (PPARγ), would prevent or reverse PA remodeling induced by chronic hypoxia. These studies were based on the ability of TZDs to prevent arterial remodeling and vasoconstriction in the systemic vasculature (20–26). We reported that PA remodeling was reduced in rats exposed to hypoxia and simultaneously treated with the TZD rosiglitazone (ROSI). ROSI treatment also blocked muscularization of distal pulmonary arterioles, and reversed remodeling and neomuscularization in lungs of animals previously exposed to chronic hypoxia.
Here we test the hypothesis that TZDs suppress PA remodeling and PA SMC phenotypic modulation, in part, by preventing the loss of CREB induced by hypoxia or PDGF. The data demonstrate that ROSI prevents CREB depletion and PA remodeling in rats exposed to chronic hypoxia. Likewise, ROSI blocks CREB loss and PA SMC proliferation induced with PDGF-BB in culture. Loss of CREB in response to PDGF is due to increased expression of CK2 catalytic subunit, which is increased in the PA media of animals exposed to chronic hypoxia. ROSI prevents upregulation of CK2 catalytic subunit and thereby prevents CREB depletion by hypoxia or PDGF.
Methods
Materials
Recombinant human PDGF-BB was purchased from Bachem (King of Prussia, PA) and LY294002 was obtained from Cayman Chemicals (Ann Arbor, MI). CellTiter 96 AQ Non-Radioactive Cell Proliferation Assay kits were from Promega (Madison, WI) and oligofectamine was obtained from Invitrogen (Carlsbad, CA). SuperSignal West Pico Chemiluminescent Substrate and NE-PER Nuclear and Cytoplasmic Extract Reagents were from Pierce (Rockford, IL). Dulbecco’s Modified Eagle’s Medium (DMEM) was from Hyclone (Logan, UT) and Optimem was from Gibco/Invitrogen. AlexaFluor 555-conjugated goat anti-rabbit secondary antibody was from Molecular Probes (Eugene, OR). Mouse monoclonal anti-ubiquitin antibody, and rabbit polyclonal antibodies to CREB were from Cell Signaling Technology, Inc. (Beverly, MA). Goat polyclonal anti-phospho-Akt-1/2/3 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Rat monoclonal anti-BrdU antibody was purchased from Abcam (Cambridge, MA) and a rabbit polyclonal antibody to CK2 α and α′ catalytic subunits for immunohistochemistry was purchased from Bethyl Laboratories, Inc. (Montgomery, TX). A CK2 α/α′ rabbit polyclonal antibody for western blotting was from BD Bioscience (San Jose, CA). The CK2 Protein Kinase Assay Kit, 5-bromo-2′deoxyuridine (BrdU) and troglitazone were from Sigma (St. Louis, MO). γ-32P-ATP was obtained from GE Healthcare (Piscataway, NJ). Three double stranded siRNA oligo-nucleotides to CK2 α′ subunit (261-AACCTTCGTGGTGGAACAAAT-281, 320-AAAGACACCAGCTTTGGTATT-340, and 671-AAGCATGATATTCCGAAAGGA-691) and a control siRNA oligonucleotide to firefly luciferase (CGTACGCGGAATACTTCGA) were from Dharmacon (Lafayette, CO).
Immunostaining and microscopy
Immunostaining was performed on the same rat lung tissue used in previous studies in which ROSI was shown to block and reverse hypoxia-induced PA thickening (19). All animal procedures were performed with approval and in accordance with the guidelines of the University of Colorado Denver Center for Laboratory Animal Care. Basically, tissue was retrieved from male Wistar-Kyoto rats (6 wk old) randomized to normoxia (1600 m, 630 mmHg) or hypobaric hypoxia (5500m, 410 mmHg) and treated with and without the PPARγ agonist, ROSI at 5 mg/kg/day, for 3 weeks (n = 6 animals/treatment group). At the end of 3 weeks, the rats were anesthetized and then treated with the RhoA/Rho kinase inhibitor, Fasudil (10 mg/kg, IV) to dilate the vasculature prior to fixation. Lungs were fixed with 4% paraformaldehyde in PBS containing 5 mM EDTA by airway inflation at 30 cm H2O pressure. Pulmonary arteries were perfused with 4% paraformaldehyde/PBS/5 mM EDTA (to maintain vasodilatation) at a pressure similar to that measured in vivo. The heart and lungs were then removed en bloc. Lung tissue was fixed overnight in 4% paraformaldehyde and embedded in paraffin.
Five um sections of paraformaldehyde-fixed, paraffin-embedded lung tissue were deparaffinized with Hemo-D, and rehydrated in a graded ethanol/water series. Sections were subjected to antigen retrieval in citrate buffer in a microwaveable pressure cooker for 20 minutes. Sections were blocked with PBS containing 5% horse serum for 30 minutes at room temperature. The sections were incubated overnight in PBS/5% FBS at 4°C with the primary antibodies indicated in the figure legends. The sections were then washed and incubated with the indicated Alexa Fluor-conjugated secondary antibodies for 1 hour at room temperature. Sections were washed extensively in PBS and coverslips were affixed with Vectashield (Vector Laboratories, Burlingame, CA).
Staining for BrdU incorporation was performed on SMCs treated as indicated in the figure legends and fixed in PBS/4%paraformaldehyde. Slides were incubated in 0.5% hydrogen peroxide for 10 minutes to quench endogenous catalase and briefly rinsed in tap water. The slides were then incubated in 2 N HCl at 37 C for 30 minutes and then transferred to 0.1 N disodium tetraborate for 10 minutes. The slides were rinsed and immunostaining was conducted as described above using a rat monoclonal anti-BrdU primary antibody and an anti-rat secondary antibody conjugated to horseradish peroxidase. Slides were then developed with Vector NovaRed reagents. Proliferation was calculated as the number of red (BrdU-positive) nuclei divided by the total number of nuclei in each micrograph (BrdU labeling index).
Microscopy was performed on a Nikon TE2000-U inverted epifluorescent microscope. Brightfield, phase contrast, and fluorescent digital deconvolution images were captured to a personal computer with either a Spot RT/KE monochrome camera or Spot Insight color camera (Diagnostic Imaging, Sterling Heights, MI). Images were analyzed and processed with MetaMorph 6.1 Software (Molecular Devices, Sunnyvale, CA). The nuclear fluorescence intensity of CREB, phospho-Akt and CK2 staining was determined by inclusive thresholding for bright objects in each micrograph followed by computer-assisted fluorescence signal integration for nuclei located between internal and external elastic lammellae. The cellularity of the vascular media in each treatment group was evaluated by counting the number of nuclei (DAPI-stained) between the internal and external elastic lammellae. This number was divided by the lumen radius of the vessel to account for artery size.
PA SMC Isolation and Cell Culture
Five hundred um PAs were recovered from adult rat lungs. Segments of the PAs were cut open and mechanically stripped of adventitia and endothelium. The segments were then placed lumen-side-down into individual wells of a six well plate. Tissue explants were maintained in complete DMEM supplemented with 200 U/ml penicillin, 0.2 mg/ml streptomycin, and 10% fetal bovine serum (FCS).
Since our goal was to obtain pure subpopulations of SMCs we selectively isolated individual cell colonies with a distinct, although uniform, morphological appearance from primary culture using cloning cylinders. Expression of SMC-specific markers (α-SM actin and SM-myosin heavy chains) in each isolated cell subpopulation was examined. Only cell subpopulations with uniform morphological appearance and uniform patterns of expression of SMC markers were selected for further experimentation. Individual cell colonies growing from tissue explants in primary culture were isolated by placing cloning cylinders (5 to 10 mm in diameter, greased on the bottom) over each cell colony of interest. Cells within the ring were trypsinized and transferred to a 24-multiwell plate for expansion. Cells were passaged in DMEM containing 10% FCS. During experiments, the cells were placed in sealed Plexiglas chambers purged with filled with either normoxic (80% nitrogen, 20% oxygen) or hypoxic (97% nitrogen, 3% oxygen) gas mixtures. The gas mixture in each chamber was replaced every 24 hours. All studies were carried out using cells at passages 1 to 8. Cell cultures were tested for mycoplasma contamination using a Gen-Probe Mycoplasma T. C. Rapid Detection System (GenProbe Inc, San Diego, CA).
SiRNA treatments
SMCs were plated at 30–50% confluency on 6-well plates in complete medium. Twenty-four hours later the cells were transferred to serum-free DMEM for transfection. Double stranded CK2 siRNA oligonucleotides (equal amounts of the 3 CK2 siRNAs were mixed together) or the control luciferase-specific siRNA were complexed with Oligofectamine reagent and applied to the cells according to the manufacturer’s recommendations. After 3 hours, and equal volume of DMEM containing 30% FCS was added to the wells. Cells were allowed to recover for at least 48 hours before subsequent manipulations.
Proliferation and CK2 assays
SMC proliferation was measured with CellTiter 96 AQ Assay Kit according to the manufacturer’s direction. BrdU incorporation was measured by adding BrdU to cell culture medium to a final concentration of 10 uM 6 hours prior to fixation and immunostaining. CK2 activity was measured with a CK2 Protein Kinase Assay Kit using whole SMC lysates. To account for non-specific substrate phosphorylation, reactions were performed in the presence and absence of CK2 inhibitor (heparin). The activity measured in the reactions containing inhibitor was subtracted from corresponding reactions without inhibitor.
Subcellular fractionation and western blots
Cytosolic and nuclear fractions were prepared from SMCs using NE-PER reagents following the supplier’s instructions. After correcting for protein concentrations, cell lysates were mixed with an equal volume of Laemmli SDS loading buffer, resolved on 10% polyacrylamide-SDS gels and transferred to PVDF membranes. The blots were blocked with phosphate buffered saline (PBS) containing 5% dry milk and 0.1% Tween 20, and then treated with antibodies that recognize the target proteins indicated in each figure overnight at 4°C. The blots were washed and subsequently treated with appropriate secondary antibodies conjugated to horseradish peroxidase. After the blots are washed, specific immune complexes will visualized with SuperSignal West Pico Chemiluminescent Substrate.
Statistics
Data averaged from the number of samples or conditions noted in each figure legend we compared using the 2-tailed Student’s t test. Differences were considered statistically significant at p ≤ 0.05.
Results
ROSI attenuates hypoxia-induced PA remodeling and loss of SMC CREB
We have previously shown that CREB levels are diminished is medial SMCs of hypertensive, remodeled PAs from animals exposed to chronic hypoxia (18). Other studies demonstrated that the TZD, ROSI, prevented thickening of the PA wall in rats exposed to chronic hypoxia (27). Here we tested whether ROSI could not only attenuate PA remodeling, but also prevent hypoxia-induced CREB depletion in SMCs. We used the same preserved lung tissue from adult, male rats maintained under ambient normoxia, hypobaric hypoxia or hypoxia plus ROSI as used in our previous studies (19). Microscopic observation of lung tissue sections from the three groups of animals revealed substantial remodeling or thickening of the PA wall in lungs from rats exposed to hypoxia (average wall thickness/lumen radius = 0.56 ± 0.14) (Fig. 1A). No remodeling was detected in normoxic animals (average wall thickness/lumen radius = 0.18 ± 0.09), and thickening was reduced in hypoxic animals receiving ROSI (average wall thickness/lumen radius = 0.23 ± 0.11) compared to ROSI-naïve subjects. We also measured the overall cellularity of the arterial media in each treatment to determine whether medial SMC numbers were different. We found that the number of DAPI-stained nuclei present between the elastic lamellae was significantly increased in hypoxic animals (Fig. 1B), while minimal change was noted between normoxic and hypoxic plus ROSI groups.
Figure 1. ROSI prevents both PA remodeling and SMC CREB loss in rats exposed to chronic hypoxia.

Sections of lung tissue were prepared from adult male Wistar Kyoto rats were subjected to isobaric normoxia or hypobaric hypoxia for 3 weeks. Some rats exposed to hypoxia were fed chow impregnated with ROSI. At 3 weeks the animals were euthanized, and their lungs were inflated and perfused with PBS containing 4% paraformaldehyde. The sections were deparaffinized, rehydrated and subjected to immunohistochemistry for CREB. A) The top row shows representative phase contrast micrographs of intralobar pulmonary arteries (PA) adjacent to airways (AW). The middle row shows the corresponding fluorescence deconvolution images in which the signal for CREB (red) has been combined with the autofluorescence of the internal and external elastic lamellae (green). The bottom row shows enlargements of the regions within the blue squares in the middle row. Bar = 75 um. B) Cellularity of the medial compartment was quantitated by counting the number of DAPI-stained nuclei located between the inner and outer lammelae. This number was divided by the lumen radius to normalize for vessel size. Data were averaged for at least five vessels per animal from 6 animals. * indicates p ≤ 0.05. C) Nuclear CREB levels were quantitated by measuring fluorescence intensities of nuclei located between the inner and outer lammelae. Data were averaged for at least five vessels per animal from 6 animals. The data indicate that ROSI attenuates hypoxia-induced PA thickening and loss of SMC CREB. * indicates p ≤ 0.05.
For the analysis of CREB levels in medial SMCs, vessels were selected based on bright autofluorescence of the internal and external elastic lammellae to clearly delineate the medial compartment. This was necessary as heavily remodeled arteries (particularly in hypoxic lung) exhibited degraded or incomplete lammellae or multiple autofluorescent filaments throughout the vessel wall making it difficult to determine the extent of the arterial media. Relative CREB levels were evaluated by immunofluorescent staining and quantitated by integration of nuclear fluorescence intensity by computer-assisted morphometry (Fig. 1C). CREB was easily detected in medial SMCs and throughout the lung in sections from normoxic rats, but CREB levels were significantly lower in the media and adventitia in hypoxic animals. However, CREB levels in hypoxic rats treated with ROSI were comparable to those in normoxic animals. The data indicate that ROSI prevents both hypoxia-induced PA remodeling and loss of CREB in SMCs.
TRO represses PDGF-BB-induced SMC proliferation and CREB depletion in vitro
We previously reported that PDGF-BB, a potent mitogen produced in the PA wall in response to hypoxia (11, 12), induces CREB loss in purified PA SMCs (19). Given the protective impact of ROSI on PA remodeling and SMC CREB content in vivo, we tested the ability of a closely related TZD, troglitazone (TRO) to suppress PDGF-BB-induced SMC growth and CREB loss in vitro. Staining for BrdU incorporation in figure 2A shows that PDGF-BB stimulates proliferation of primary PA SMCs (red staining nuclei), but that this effect is inhibited by simultaneous addition of TRO. This was confirmed by quantitation of the BrdU-positive cells versus total cell number in each micrograph (Fig. 2B). PDGF increased proliferation by approximately 10-fold over untreated cells, while TRO almost completely blocked PDGF-induced growth. An MTT-based proliferation assay provided similar results (Fig. 2C). Cytoplasmic and nuclear CREB levels were also assessed in SMCs treated with PDGF-BB and/or TRO. PDGF-BB alone stimulated loss of nuclear CREB (Fig. 2D). However, in cells receiving both PDGF-BB and TRO, no loss of CREB was observed, and in some cases, CREB levels were slightly higher in PDGF/TRO treated cells than in untreated control cells. These data show that TRO inhibits PDGF-BB-induced SMC proliferation and depletion of CREB in PA SMCs.
Figure 2. TRO prevents both proliferation and CREB loss in PA SMCs treated with PDGF.

PA SMCs were propagated in DMEM containing 0.2% fetal calf serum. Some cells were left untreated as controls while others received PDGF (25 ng/ml) alone or in combination with 1 uM TRO for 48 hours. A) BrdU was added to the cells at a final concentration of 10 uM, 6 hours prior to fixation. The fixed cells were subjected to immunocytochemical staining for BrdU. The figure shows representative bright field micrographs of cells exposed to each treatment. BrdU-positive nuclei are red. B) The BrdU-positive (red) nuclei and total nuclei were counted in 10 random fields for each treatment. The number of BrdU-positive nuclei were divided by the number of total nuclei to obtain the BrdU Labeling Index as a measure of proliferation. The graph shows data averaged from three experiments. * indicates p ≤ 0.05. C) Cell number was determined using CellTiter 96 Aq reagents. The graph shows the average fold change in cell number from 4 experiments with 3 replicates relative to untreated control cells (Cntrl). * indicates p≤0.05. D) Cells were lysed and separated into cytosolic and nuclear fractions. Equal amounts of extract protein were resolved on SDS-polyacrylamide gels and transferred to PVDF membranes. Equal loading and transfer were verified by staining of the blot with Ponceau S. The membranes were then probed with a CREB-specific antibody. The results demonstrate that PDGF promotes SMC proliferation and loss of CREB from the nuclear fraction, but that TRO prevents CREB loss and inhibits proliferation.
TZDs do not suppress PI3-kinase/Akt signaling in PA SMCs
We have reported that PDGF-BB-induced depletion of CREB (which involved CREB ubiquitination and proteasomal degradation) in PA SMCs is blocked by inhibitors of PI3-kinase or Akt (19). Therefore, we tested whether TZDs could inhibit this signaling pathway in vitro and in vivo. For in vitro studies, primary PA SMCs were treated with PDGF-BB alone or in combination with TRO for various times. As expected, CREB levels diminished in SMCs exposed to PDGF-BB alone, commensurate with an increase in ubiquitinated CREB (Fig. 3A and B). However, in cells treated with PDGF-BB and TRO, no loss of CREB or increase in ubiquitinated CREB was observed. PDGF-BB also stimulated activation of PI3-kinase/Akt signaling as evidenced by increased activated P-Akt levels. However, TRO failed to block PDGF-induced Akt activation. Thus, although TRO inhibits CREB depletion, it does not appear to suppress signaling via PI3-kinase and Akt in vitro.
Figure 3. TRO inhibits CREB ubiquitination but not activation of Akt by PDGF.

PA SMCs were grown in DMEM containing 0.2% fetal calf serum and treated with 25 ng/ml PDGF with or without 1 uM TRO for the times shown above each lane. At each time point, cells were harvested, lysed and equal amounts of protein separated on polyacrylamide-SDS gels and transferred to PVDF membranes. A) Blots were probed with antibodies against total CREB or P-Akt as indicated. For the panel labeled IP:CREB/Blot:Ubiquitin, equal amounts of lysate protein were first subjected to immunoprecipitation with anti-CREB antibody and the precipitated material was separated on polyacrylamide-SDS gels and transferred to membranes. These blots were probed with an ubiquitin-specific antibody. B) Blots from three separate experiments were scanned into an Apple MacBook Pro computer and the band density and area at the 48 hour time point was measured with ImageJ software. The averaged data are shown relative to band densities for the 48 hour normoxic samples. * indicates p ≤ 0.05. The results show that PDGF stimulates CREB ubiquitination and depletion in SMCs concomitant with an increase in Akt activity. While TRO blocks CREB ubiquitination and loss, it has no effect on PDGF-stimulation of Akt activity.
We also examined activation of Akt in lung sections from rats exposed to normoxia, chronic hypoxia alone, or hypoxia and ROSI by immunofluorescent staining to P-Akt. Low levels of P-Akt staining were observed in the PA wall in normoxic animals. (Fig. 4A). Higher levels of P-Akt staining were present in the sections of hypoxic rat lung, most notably in the medial PA between the inner and outer elastic lammelae. Similarly, P-Akt staining was present in medial PA in sections from rats exposed to hypoxia and treated with ROSI. Quantitation of P-Akt nuclear fluorescence intensities confirmed these visual impressions (Fig. 4B). P-Akt levels were approximately 3-fold higher in both hypoxic and hypoxic/ROSI medial SMC nuclei than in normoxic controls. These data suggest that TZDs fail to block activation of Akt in vivo, analogous to their lack of effect in vitro.
Figure 4. ROSI attenuates hypoxia-induced PA remodeling, but does not inhibit hypoxia-induced Akt activation in PA SMCs in vivo.

Sections of lung tissue were prepared from adult male Wistar Kyoto rats were subjected to isobaric normoxia or hypobaric hypoxia for 3 weeks. Some rats exposed to hypoxia were fed chow impregnated with ROSI. The sections were deparaffinized, rehydrated and subjected to immunohistochemistry for P-Akt. The top row shows representative phase contrast micrographs of intralobar pulmonary arteries (PA) adjacent to airways (AW). The middle row shows the corresponding fluorescence deconvolution images in which the signal for P-Akt (red) has been combined with the autofluorescence of the internal and external elastic lamellae (green). The bottom row shows enlargements of the regions within the blue squares in the middle row. Bar = 75 um. B) Nuclear P-Akt levels were quantitated by measuring fluorescence intensities of nuclei located between the inner and outer lammelae. Data were averaged for at least five vessels per animal from 6 animals. * indicates p ≤ 0.05. The data indicate that ROSI attenuates hypoxia-induced PA thickening but does not block activation of Akt by hypoxia in arterial SMC.
Casein kinase 2 (CK2) promotes CREB loss in response to PDGF-BB
We have reported that PDGF-BB-induced CREB loss in PA SMCs requires the presence of 2 closely spaced serine residues (serines 103 and 107) in the CREB molecule that are putative targets for CK2 (19). Simultaneous mutation of both serines to alanines results in a CREB molecule refractory to PDGF-induced degradation. To determine whether CK2 mediated CREB depletion in response to PDGF-BB we first assessed CK2 catalytic subunit expression in SMCs. We found that CK2 α′ catalytic subunit expression was consistently increased in PDGF-BB treated cells (Fig. 5A and 6A). In some experiments the α catalytic subunit was also increased by PDGF (Fig. 6A), but this was not observed in all experiments. This is likely due to differences in α subunit expression in SMCs isolated from different animals, the heterogeneity of the medial SMC population, and possible expansion of different SMC subgroups in each isolate. Both basal and PDGF-stimulated CK2 α′ subunit expression was inhibited by the PI3-kinase inhibitor, LY294002. We next developed siRNA oligonucleotides to CK2 α′ subunit. Figure 5B shows that complete depletion of the CK2 subunit in SMCs transfected with the specific siRNA, but not in cells transfected with a control siRNA. We then measured CREB levels in cells transfected with each siRNA, and left untreated or exposed to PDGF-BB fro 48 hours. In PDGF-BB-naïve cells, CREB was present in both control and CK2 siRNA transfected SMCs (Fig. 5C). CREB was not detected in cells transfected with control siRNA and treated with PDGF-BB. However, in CK2 α′ siRNA transfected cells, CREB was detected in spite of PDGF-BB exposure. The results indicate that PDGF-BB stimulates CK2 expression α′ in a PI3-kinase-dependent manner, and that CK2 α′ is required for PDGF-BB-induced CREB depletion in PA SMCs.
Figure 5. PDGF induces CK2 catalytic subunit expression, which is required for PDGF-induced CREB depletion in PA SMCs.
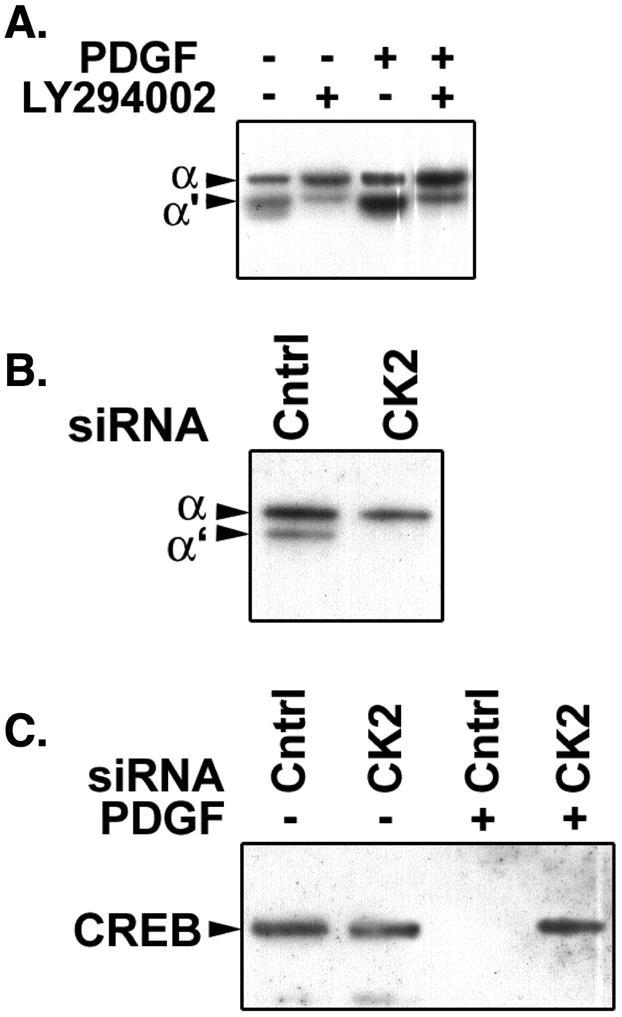
A) PA SMCs grown in DMEM containing 10% fetal calf serum were transferred to medium containing 0.2% fetal calf serum. The cells were treated with 25 ng/ml PDGF with or without the PI3Kinase inhibitor LY294002 for 48 hours. Control cells were left untreated. Equal amounts of whole cell lysate protein were resolved on polyacry-lamide-SDS gels, transferred to PVDF membranes, which were probed with CK2 catalytic subunit-specific antibody. The panel shows a representative western blot demonstrating that PDGF stimulates CK2 catalytic subunit expression, and that this is mediated by PI3Kinase signaling. B) PA SMCs were transfected with control (Cntrl) or CK2 catalytic subunit-specific siRNA. Forty-eight hours later the cells were lysed and equal amounts of lysate protein were resolved on polyacrylamide-SDS gels and transferred to PVDF membranes. Equal protein loading and transfer were verified by Ponceau S staining of the membranes prior to blotting. The membranes were probed with CK2-specific antibody. The panel shows a representative western blot demonstrating that the CK2-targeted siRNA effectively suppresses CK2 catalytic subunit expression. C) PA SMCs were transfected with control or CK2-catalytic subunit-specific siRNA. Twenty-four hours later the cells were transferred to DMEM containing 0.2% fetal calf serum. After an overnight incubation, PDGF was added to some of the cells at a final concentration of 25 ng/ml. Forty-eight hours later the cells were lysed and equal amounts of lysate protein were resolved on polyacrylamide-SDS gels and transferred to PDVF membranes. The membranes were probed with anti-CREB antibody. The representative blot shows that PDGF induces CREB loss in the cells transfected with control siRNA, but not in cells in which CK2 has been depleted with specific siRNA.
Figure 6. PDGF-stimulated CK2 catalytic subunit expression and activity are inhibited by TRO in PA SMCs.

PA SMCs were grown in DMEM containing 10% fetal calf serum, and transferred to medium containing 0.2% serum 24 hours prior to the of 25 ng/ml PDGF with and without 1 uM TRO. A) At the times indicated above each lane, equal amounts of lysate protein were resolved on polyacrylamide-SDS gels and transferred to PVDF membranes. The membranes were probed with antibody against CK2 catalytic subunit. The representative blot shows that PDGF stimulates CK2 catalytic subunit expression in time-dependent manner, but that this process is blocked by TRO B) Forty-eight hours post PDGF/TRO addition, cells were lysed and approximately 20 ug of lysate protein were assayed for CK2 activity as described in Experimental Procedures. Triplicate reactions were run in the presence or absence of CK2 inhibitor. The non-specific activity measured in the reactions containing inhibitor was subtracted from activity measured in reactions without inhibitor. The graph shows average kinase activity relative to reactions performed with lysate protein from cells not exposed to PDGF or TRO. * indicates p≤0.05.
TZDs suppress upregulation of CK2 α′ by PDGF-BB in vitro and by hypoxia in vivo
We next tested whether TZDs could inhibit CK2 catalytic subunit expression or activity in primary PA SMCs treated with PDGF-BB. We found that PDGF alone stimulated expression of CK2 catalytic subunits in a manner temporally consistent with the kinetics of CREB depletion (Fig. 6A). This increase in CK2 catalytic subunit expression was largely abrogated in cells treated with PDGF-BB and TRO. Changes in CK2 catalytic subunit expression were reflected in changes in CK2 kinase activity. PDGF-BB alone stimulated CK2 activity by slightly more than 2-fold (Fig. 6B). TRO inhibited both basal and PDGF-BB-stimulated CK2 activity.
In vivo, CK2 was difficult to detect in lung sections from rats held under normoxic conditions (Fig. 7A). CK2 catalytic subunit levels were elevated in nuclei throughout the lung (by more than 3-fold, Fig. 7B), and especially the medial PA SMC layer of animals exposed to chronic hypoxia. However, in animals simultaneously receiving ROSI and hypoxic exposure, CK2 levels were normalized.
Figure 7. ROSI inhibits hypoxia-induced PA remodeling and expression of CK2 catalytic subunit in vivo.

Sections of lung tissue were prepared from adult male Wistar Kyoto rats were subjected to isobaric normoxia or hypobaric hypoxia for 3 weeks. Some rats exposed to hypoxia were fed chow impregnated with ROSI. At 3 weeks the animals were euthanized, and their lungs were inflated and perfused with PBS containing 4% paraformaldehyde. The sections were deparaffinized, rehydrated and subjected to immunohistochemistry for CK2 catalytic subunit. The top row shows representative phase contrast micrographs of intralobar pulmonary arteries (PA) adjacent to airways (AW). The middle row shows the corresponding fluorescence deconvolution images in which the signal for CK2 (red) has been combined with the autofluorescence of the internal and external elastic lamellae (green). The bottom row shows enlargements of the regions within the blue squares in the middle row. Bar = 75 um. B) Nuclear CK2 levels were quantitated by measuring fluorescence intensities of nuclei located between the inner and outer lammelae. Data were averaged for at least five vessels per animal from 6 animals. * indicates p ≤ 0.05. The data indicate that ROSI attenuates hypoxia-induced PA thickening and increased CK2 catalytic subunit expression.
Blockade of CK2 α′ expression in vitro inhibits PDGF-BB-induced SMC proliferation
Since PDGF promotes CREB loss via CK2, and loss of CREB stimulates PA SMC proliferation, we tested whether forced depletion of CK2 α′ with specific siRNA would suppress PA SMC proliferation stimulated with PDGF-BB. Substantial proliferation (BrdU incorporation) was evident in PDGF-BB treated SMCs previously transfected with a control siRNA (Fig. 8A and B). However, PDGF-BB-stimulated SMC proliferation was much less in cells transfected with CK2 α′ subunit-specific siRNA.
Figure 8. Forced depletion of CK2 catalytic subunit suppresses PDGF-stimulated PA SMC proliferation.

PA SMCs were transfected with control (Cntrl) and CK2 catalytic subunit siRNA. The cells were transferred to medium containing 0.2% serum for 24 hours, and then exposed to 25 ng/ml PDGF for 48 hours. BrdU was added to the wells for the last 6 hours of the experiment. A) The SMCs were then fixed and stained for BrdU incorporation. The figure shows representative brightfield micrographs in which BrdU-positive nuclei are red. B) The BrdU-positive (red) nuclei and total nuclei were counted in 10 random fields for each treatment. The number of BrdU-positive nuclei were divided by the number of total nuclei to obtain the BrdU Labeling Index as a measure of proliferation. The graph shows data averaged from three experiments. * indicates p ≤ 0.05. The results indicate that forced depletion of CK2 catalytic subunit blocks PDGF-induced SMC proliferation. Bar = 50 um.
Discussion
We previously demonstrated that hypoxia-induced remodeling of PAs in rats and calves was associated with decreased levels of CREB in medial SMCs of the PA wall (18). Subsequent studies indicated that forced loss or inhibition of CREB in isolated PA SMCs stimulated basal and PDGF-induced proliferation, PDGF-induced migration, expression of extracellular matrix components, and loss of SM markers (19). Alternately, ectopic expression of wild type or constitutively active forms of CREB suppressed basal and mitogen-induced proliferation and migration (18, 28). These results were observed in both pulmonary and systemic SMC populations. The data suggest that CREB plays a key role in regulating SMC phenotype, and that loss of CREB contributes to the proliferative and synthetic SMC phenotype observed in vascular pathologies.
In other studies we demonstrated that the TZD ROSI could not only prevent hypoxia-induced PA remodeling, but reverse thickening due to previous exposure to hypoxia (29). Similar results were reported by Matsuda et al (30) who showed that the TZDs pioglitazone and TRO inhibited remodeling and decreased pulmonary vascular resistance in rats treated with monocrotaline. Recent studies by Rabinovitch and colleagues demonstrate a similar beneficial impact of TZDs on PH in apoE-null mice, underscoring the central role of PPARγ and its agonists in PA remodeling (31).
Here we show that ROSI not only suppresses PA remodeling, but also prevents depletion of PA SMC CREB in rats exposed to hypoxia. Since loss of CREB in isolated SMCs elicits changes consistent with those observed in arterial remodeling, our results suggest that the protective impact of ROSI is mediated at least in part by preventing CREB proteasomal degradation. We are currently developing SMC-targeted CREB loss-of-function mouse models to fully evaluate the impact of CREB loss on SMC phenotype and PA remodeling with and without hypoxic exposure. Given that CREB acts downstream of PPARγ activation, we predict that TZDs will fail to block PA remodeling in the SMC CREB deficient animals.
Our data also indicate that expression of CK2 catalytic subunit expression is regulated by hypoxia in vivo, and by PDGF through the PI3K/Akt signaling pathway in vitro in PA SMCs. Both the catalytic and regulatory subunits of CK2 are expressed in most tissues and levels of each are tightly controlled (32, 33). While levels of CK2 catalytic and regulatory subunits are refractory to most stimuli, increases and decreases in CK2 expression and activity have been reported in relation to cell proliferation and survival. For example, CK2 expression and activity increase upon entry to the cell cycle in many cell types (32), and elevated activity has been found in many leukemias and solid tumors (34, 35). Alternately, CK2 gene transcription is suppressed in senescent human lung fibroblasts and in tissues from aged rats (36).
There is now good evidence that CK2 plays important roles in cell proliferation and survival. Inhibition of CK2 with antisense oligonucleotides or pharmacologic agents attenuates cell cycle progress in mammalian cells (37, 38). Likewise, ectopic expression of a kinase-dead form of CK2 catalytic subunit α decreased proliferation of NIH-3T3 cells (39). On the other hand, CK2 catalytic subunit transgenic mice have a higher incidence of lymphoma, which is further aggravated by loss or mutation of the tumor suppressor p53 (40). Our results are consistent with these reports in that they demonstrate an increase in CK2 catalytic subunit in the hypertensive, remodeled PA wall in response to hypoxia, and increases in CK2 α′ subunit levels and enzyme activity in SMCs treated with PDGF. Importantly, forced depletion of CK2 α′ with siRNA blocked PDGF-induced SMC proliferation. It is interesting to speculate that anti-cancer therapies based on CK2 inhibition might also be effective agents to arrest PA remodeling in PH.
How do thiazolidinediones inhibit PDGF-stimulated CK2 expression? The regulation of CK2 gene transcription remains mysterious. The promoters for both α and α′ catalytic subunits contain binding sites for SP1, Ets and NFκB transcription factors (41) and binding of these factors to the promoter of the α gene has been confirmed by super-shift analysis and affinity chromatography. Interestingly, the promoters for the regulatory subunit genes contain a similar combination of regulatory sites, and work by Pyerin and Ackerman (33) indicate that transcription of the catalytic and regulatory subunits occurs in a coordinate fashion. It was recently reported that downregulation of CK2 α and α′ during cellular senescence is mediated by promoter hypermethylation (42). In addition, DNA methylation also resulted in downregulation of Sp1, Ets and NFκB levels. We are currently investigating changes in methylation of the CK2 α and α′ gene promoters to determine whether this mechanism might account for the impact of ROSI and TRO on CK2 catalytic subunit levels in PA SMCs.
Conclusion
TZDs inhibit CREB depletion in PA SMCs. and inhibit SMC proliferation in vitro and PA remodeling in vivo. TZDs do not appear to regulate PI3Kinase/Akt signaling, but rather prevent CREB loss by attenuating hypoxia- or PDGF-induced CK2 α′ subunit expression. Given the ability of CREB to modulate SMC phenotype in culture, we conclude that TZDs prevent PA remodeling in part by suppressing upregulation of CK2 and loss of CREB in PA SMCs.
Acknowledgments
This research was supported by grant NIH P01-HL014985 (D.J.K.)
Footnotes
None of the authors have financial interests or affiliations with institutions or companies mentioned in this manuscript.
References
- 1.Stenmark KR, McMurtry IF. Vascular Remodeling Versus Vasoconstriction in Chronic Hypoxic Pulmonary Hypertension: A Time for Reappraisal. Circulation Research. 2005;97:95–98. doi: 10.1161/01.RES.00000175934.68087.29. [DOI] [PubMed] [Google Scholar]
- 2.Jones R, Jacobson M, Steudel W. a-smooth-muscle actin and microvascular precursor cells in pulmonary hypertension. American Journal of Respiratory Cell and Molecular Biology. 1999;20:582–594. doi: 10.1165/ajrcmb.20.4.3357. [DOI] [PubMed] [Google Scholar]
- 3.Belknap JK, Orton EC, Ensley B, Tucker A, Stenmark KR. Hypoxia increase bromo-deoxyuridine labeling indices in bovine neonatal pulmonary arteries. American Journal of Respiratory Cell and Molecular Biology. 1997;16:366–371. doi: 10.1165/ajrcmb.16.4.9115746. [DOI] [PubMed] [Google Scholar]
- 4.Cornfield DN, Stevens T, McMurtry IF, Abman SH, Rodman DM. Acute hypoxia increases cytosolic calcium in fetal pulmonary artery smooth muscle cells. American Journal of Physiology. 1993;265:L53–L56. doi: 10.1152/ajplung.1993.265.1.L53. [DOI] [PubMed] [Google Scholar]
- 5.Salvaterra CG, Goldman WF. Acute hypoxia increases cytosolic calcium in cultured pulmonary arterial myocytes. American Journal of Physiology. 1993;264:L323–L328. doi: 10.1152/ajplung.1993.264.3.L323. [DOI] [PubMed] [Google Scholar]
- 6.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated by mitochondrial complex III stabilize hypoxia-inducible factor-1a during hypoxia. Journal of Biological Chemistry. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 7.Herget J, wilhelm J, Novotna J, Eckhardt A, Vytasek R, Mrazkova L, Ostadal M. A possible role of the oxidant tissue injury in the development of hypoxic pulmonary hypertension. Physiology Research. 2000;49:493–501. [PubMed] [Google Scholar]
- 8.Jones RD, Morice AH. Hydrogen peroxide - an intracellular signal in the pulmonary circulation: involvement in hypoxic pulmonary vasoconstriction. Pharmacology and Therapeutics. 2000;88:153–161. doi: 10.1016/s0163-7258(00)00089-9. [DOI] [PubMed] [Google Scholar]
- 9.Killilea DW, Hester R, Balczon R, Babal P, Gillespie MN. Free radical production in hypoxic pulmonary artery smooth muscle cells. American Journal of Physiology Lung Cell Molecular Physiology. 2000;279:L408–L412. doi: 10.1152/ajplung.2000.279.2.L408. [DOI] [PubMed] [Google Scholar]
- 10.Weissman N, Tadic A, Hanze J, Rose F, Winterhalder S, Nollen M, Schermuly RT, Ghofrani HA, Seeger W, Grimminger F. Hypoxic vasoconstriction in intact lungs: a role for NADPH oxidase-derived H2O2. American Journal of Physiology Lung Cell Molecular Physiology. 2000;279:L683–L690. doi: 10.1152/ajplung.2000.279.4.L683. [DOI] [PubMed] [Google Scholar]
- 11.Dawes KE, Peacock AJ, Gray AJ, Bishop JE, Laurent GJ. Characterization of fibroblast mitogens and chemoattractants produced by endothelial cells exposed to hypoxia. American Journal of Respiratory Cell and Molecular Biology. 1994;10:552–559. doi: 10.1165/ajrcmb.10.5.8179919. [DOI] [PubMed] [Google Scholar]
- 12.Faller DV. Endothelial cell responses to hypoxic stress. Clinical and Experimental Pharmacology and Physiology. 1999;26:74–84. doi: 10.1046/j.1440-1681.1999.02992.x. [DOI] [PubMed] [Google Scholar]
- 13.Shaul PW, Kinanae B, Farrar MA, Buja M, Magness RR. Prostacyclin production and mediation of adenylate cyclase activity in the pulmonary artery: alteration after prolonged hypoxia in the rat. Journal of Clinical Investigation. 1991;88:447–455. doi: 10.1172/JCI115324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bornfeldt KE, Campbell JS, Koyama H, Argast GM, Leslie CC, Raines EW, Krebs EG, Ross R. The mitogen-activated protein kinase pathway can mediate growth inhibition and proliferation in smooth muscle cells. Dependence on the availability of downstrean targets. Journal of Clinical Investigation. 1997;100:875–885. doi: 10.1172/JCI119603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graves LM, Bornfeldt KE, Raines EW, Potts BC, Macdonald SG, Ross R, Krebs EG. Protein kinase A antagonizes platlet-derived growth factor-induced signaling by mitogen-activated protein kinase in human arterial smooth muscle cells. Proceedings of the National Academy of Science USA. 1993;90:10300–10304. doi: 10.1073/pnas.90.21.10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vadiveloo PK, Filonzi EL, Stanton HR, Hamilton JA. G1 phase arrest of human smooth muscle cells by heparin, IL-4 and cAMP is linked to repression of cyclin D1 and cdk2. Atherosclerosis. 1997;133:61–69. doi: 10.1016/s0021-9150(97)00116-0. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi S, Morishita R, Matsushita H, Nakagami H, Taniyama Y, Nakamura T, Aoki M, Yamamoto K, Higaki J, Ogihara T. Cyclic AMP inhibited proliferation of human aortic vascular smooth muscle cells, accompanied by induction of p53 and p21. Hypertension. 2000;35:237–243. doi: 10.1161/01.hyp.35.1.237. [DOI] [PubMed] [Google Scholar]
- 18.Klemm DJ, Watson P, Frid MG, Dempsey EC, Schaack J, Colton LA, Nesterova A, Stenmark KR, Reusch JEB. cAMP response element-binding protein content is a molecular determinant of smooth muscle cell proliferation and migration. Journal of Biological Chemistry. 2001;276:46132–46141. doi: 10.1074/jbc.M104769200. [DOI] [PubMed] [Google Scholar]
- 19.Garat C, Fankell D, Erickson PF, Reusch JEB, Bauer NN, McMurtry IF, Klemm DJ. PDGF-BB Induces Nuclear Export and Proteosomal Degradation of CREB via PI3-Kinase/Akt Signaling in Pulmonary Artery Smooth Muscle Cells. Mol Cell Biol. 2006 doi: 10.1128/MCB.02477-05. Accepted for publication pending revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchanan TA, Meehan WP, Jeng YY, Yang DC, Chan TM, Nadler JL, Scott S, Rude RK, Hsueh WA. Blood pressure lowering by pioglitazone: evidence for a direct vascular effect. Journal of Clinical Investigation. 1995;96:354–360. doi: 10.1172/JCI118041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z, Ishibashi S, Perrey S, Osuga J, Gotoda T, Kitamine T, Tamura Y, Okazaki H, Yahagi N, Iizuka Y, Shionoiri F, Ohashi K, Harada K, Shimano H, Nagai R, Yamada N. Troglitazone inhibits atherosclerosis in apolipoprotein E knockout mice:pleiotropic effects in CD36 expression and HDL. Arterioscler Thromb Vasc Biol. 2001;21:372–377. doi: 10.1161/01.atv.21.3.372. [DOI] [PubMed] [Google Scholar]
- 22.Collins AR, Meehan WP, Kintscher U, Jackson S, Wakino S, Noh G, Palinski W, Hsueh WA, Law RE. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:365–371. doi: 10.1161/01.atv.21.3.365. [DOI] [PubMed] [Google Scholar]
- 23.Jawien A, Bowen-Pope DF, Linder V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. Journal of Clinical Investigation. 1992;89:507–511. doi: 10.1172/JCI115613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koshiyama H, Shimono D, Kuwamura N, Minamikawa J, Nakamura Y. Rapid communication: inhibitory effect of pioglitazone on carotid arterial wall thickness in type 2 diabetes. J Clin Endocrinol Metab. 2001;86:3452–3456. doi: 10.1210/jcem.86.7.7810. [DOI] [PubMed] [Google Scholar]
- 25.Langenfeld MR, Forst T, Hohberg C, Kann P, Lubben G, Konrad T, Fullert SD, Sachara C, Pfutzner A. Pioglitazone decreases carotid intimal-media thickness independently of glycemic control in patients with type 2 diabetes mellitus: results from a controlled randomized study. Circulation. 2005;111:2525–2531. doi: 10.1161/01.CIR.0000165072.01672.21. [DOI] [PubMed] [Google Scholar]
- 26.Law RE, Meehan WP, Xi XP, Graf K, Wuthrich DA, Coats W, Faxon D, Hsueh WA. Troglitazone inhibits vascular smooth muscle cell growth and intimal hyperplasia. Journal of Clinical Investigation. 1996;98:1897–1905. doi: 10.1172/JCI118991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crossno JJT, Garat CV, Reusch JEB, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol. 2007;292:L885–L897. doi: 10.1152/ajplung.00258.2006. [DOI] [PubMed] [Google Scholar]
- 28.Watson P, Nesterova A, Burant CF, Klemm DJ, Reusch JEB. Diabetes-related changes in cAMP response element-binding protein content enhance smooth muscle cell proliferation and migration. Journal of Biological Chemistry. 2001;276:46142–46150. doi: 10.1074/jbc.M104770200. [DOI] [PubMed] [Google Scholar]
- 29.Crossno JJT, Garat C, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone Attenuates Hypoxia-Induced Pulmonary Artery Remodeling. Am J Physiol Lung Cell Mol Physiol. 2006 doi: 10.1152/ajplung.00258.2006. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 30.Matsuda Y, Hoshikawa Y, Ameshima S, Suzuki S, Okada Y, Tabata T, Sugawara T, Matsumura Y, Kondo T. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats. Nihon Kokyuki Gakkai Zasshi. 2005;43:283–288. [PubMed] [Google Scholar]
- 31.Hansmann G, Wagner RA, Schellong S, de Jesus Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertensionis linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2006;115:1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 32.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pyerin W, Ackermann K. Transcriptional coordination of the genes encoding catalytic (CK2alpha) and regulatory (CK2beta) subunits of human protein kinase CK2. Mol Cell Biochem. 2001;227:45–57. [PubMed] [Google Scholar]
- 34.Gapany M, Faust RA, Tawfic S, Davis A, Adams GL, Ahmed K. Association of elevated protein kinase Ck2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med. 1995;1:659–666. [PMC free article] [PubMed] [Google Scholar]
- 35.Munsterman U, Fritz G, Seitz G, Lu YP, Schneider HR, Issinger OG. Casein kinase II is elevated in solid human tumors and rapidl proliferating neoplastic tissue. Eur J Biochem. 1990;180:251–257. doi: 10.1111/j.1432-1033.1990.tb15484.x. [DOI] [PubMed] [Google Scholar]
- 36.Ryu S-W, Woo JH, Kim Y-H, Lee Y-S, Park JW, Bae Y-S. Down-regulation of protein kinase CKII is associated with cellular senescence. FEBS Lett. 2006;580:988–994. doi: 10.1016/j.febslet.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 37.Pepperkok R, Lorenz P, Ansorge W, Pyerin W. Casein kinase II is required for transition of G0/G1, early G1, and G1/S phases of the cell cycle. J Biol Chem. 1994;269:6986–6991. [PubMed] [Google Scholar]
- 38.Pepperkok R, Lorenz P, Jakobi R, Ansorge W, Pyerin W. Cell growth stimulation by EGF: inhibition through antisense oligodeoxynucleotides demonstrates important role of casein kinase II. Exp Cell Res. 1991;197:245–253. doi: 10.1016/0014-4827(91)90429-x. [DOI] [PubMed] [Google Scholar]
- 39.Lebrin F, Chambaz EM, Bianchini L. A role for protein kinase CK2 in cell proliferation: evidence using a kinase-inactive mutant of CK2 catalytic subunit alpha. Oncogene. 2001;20:2010–2022. doi: 10.1038/sj.onc.1204307. [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Landesman-Bollag E, Channavajhala PL, Seldin DC. Murine protein kinase CK2: Gene and oncogene. Mol Cell Biochem. 1999;191:65–74. [PubMed] [Google Scholar]
- 41.Krehan A, Ansuini H, Bocher O, Grein S, Wirkner U, Pyerin W. Transcription factors Ets1, NFkappaB and Sp1 are major determinants of the promoter activity of the human protein kinase CK2 gene. J Biol Chem. 2000;275:18327–18336. doi: 10.1074/jbc.M909736199. [DOI] [PubMed] [Google Scholar]
- 42.Kim E-Y, Kang J-Y, Rho Y-H, Kim YS, Kim D-S, Bae Y-S. Silencing of the CKIIalpha and CKIIalpha’ genes during cellular senescence is mediated by DNA methylation. Gene. 2009;431:55–60. doi: 10.1016/j.gene.2008.10.020. [DOI] [PubMed] [Google Scholar]