

Abstract
Innate immunity is a crucial defense system against viral and bacterial pathogens, providing a rapid response to mitigate the effects of microbial attack. While more readily associated with respiration and metabolism, recent research has surprisingly identified a number of mitochondrial factors in the mammalian innate immune system. This review summarizes the novel mitochondrial proteins, such as MAVS and NLRX1, involved in this process and attempts to reconcile this new mitochondrial function with our previous knowledge of the organelle.
Keywords: MAVS, Innate immunity, NLRX1, STING, Mitochondria, Apoptosis
1. Introduction
The mammalian innate immune system acts as the first line of defense against microbial attack. On encountering viral or bacterial intruders, this system activates numerous signaling pathways that result in the production of anti-microbial compounds designed to mitigate damage, in effect by preventing both the replication and spread of the pathogen. Initiation of an immune response relies on numerous pattern-recognition proteins (PRPs) – a group of proteins shared by animals and plants (Janeway and Medzhitov, 2002) – which recognize the molecular markers of invaders (e.g. dsRNA, lipopolysaccharide, unmethylated CpG DNA) and signal to downstream members of the immune system. PRPs can be placed into two general groups, based on where they function. Extracellular pathogens are recognized by the toll-like receptor (TLR) proteins, which are either expressed on the cell membrane facing the extracellular matrix, or in endosomes (reviewed in Kawai and Akira, 2006). Once a pathogen is bound the TLR proteins signal through cytoplasmic adapter proteins, such as TRIF and MyD88, to initiate the production of nuclear encoded pro-immune cytokines. Intracellular pathogens are recognized by several groups of PRPs, including protein kinase R (PKR), the NOD-like protein family, and the more recently-identified RIG-I-like helicase (RLH) pathway (Janeway and Medzhitov, 2002, Meylan and Tschopp, 2008, Yoneyama and Fujita, 2009). The RLH pathway is composed of three RNA helicase proteins which bind virally-produced nucleic acids, and a number of downstream signaling molecules (Fig. 1 ). The prototypic member of the family is RIG-I, a protein which consists of twin N-terminal caspase activation and recruitment (CARD) domains, and a C-terminal RNA helicase domain (Yoneyama et al., 2004). RIG-I binds viral dsRNA at its helicase domain, which stimulates a conformational change that exposes the twin CARD domains, to allow downstream signaling through direct protein:protein interactions. The other two helicases are MDA-5, which is similar in structure and function to RIG-I, but which recognizes other viral substrates; and LGP2, which has a homologous helicase domain, but lacks the CARD domains and can act as a negative regulator of RLH signaling during infection by certain viruses, particularly those sensed by RIG-I (Yoneyama et al., 2004, Venkataraman et al., 2007, Yoneyama and Fujita, 2009).
Fig. 1.
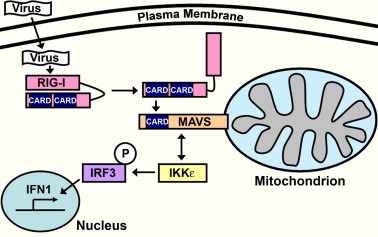
Overview of the RIG-I/MAVS signaling pathway. Intracellular microbial components, such as viral dsRNA, are recognized by cytosolic pattern-recognition proteins (PRPs) like the RNA helicase RIG-I. Binding of dsRNA to the helicase region of RIG-I induces a conformational change in the protein, which exposes twin caspase activation and recruitment (CARD) domains. The CARD domains on RIG-I can then interact with a similar CARD domain on MAVS, which is localized on the outer mitochondrial membrane, to initiate an immune signaling cascade. The RIG-I:MAVS interaction activates kinases (represented here by IKKε) that phosphorylate pro-immune transcription factors (e.g. IRF3), which then translocate to the nucleus to initiate the production of cytokines, such as type 1 interferon.
2. MAVS – the first mitochondrial protein identified in the innate immune system
2.1. Identification of the mitochondrial antiviral signaling protein, MAVS
In the year following the identification of RIG-I, four groups simultaneously reported a novel member of the RLH signaling pathway, with each group choosing a different name for the new protein. Kawai et al. (2005) found Interferon-β Promoter Stimulator 1 (IPS-1) by screening multiple cDNA pools for novel inducers of the IFN-β promoter. They reported that IPS-1 could activate the pro-immune transcription factors IRF3, IRF7 and NF-κB to induce type 1 interferon production, and that it interacted with RIG-I and MDA-5 through its CARD domain. Xu et al. (2005) made expression constructs of 12 unknown cDNAs from a previous study that had identified a large number of novel NK-κB activators (Matsuda et al., 2003). In their system, Virus-triggered IFN-β Signaling (VISA) was the only one of these cDNAs to significantly induce NK-κB signaling (Xu et al., 2005). Using a combination of mammalian coimmunoprecipitation and yeast two-hybrid studies, they found that VISA interacted with both TRAF2 and TRAF6, potentially implicating it in TLR-mediated pathways (Xu et al., 2005). Meylan et al. (2005) found CARD Adaptor Inducing IFN-β (CARDIF) using bioinformatic searches for novel CARD domain proteins. They showed that it could recruit the signaling kinases IKKα, IKKβ and IKKε, which led to the activation of the IRF3 and NK-κB transcription factors. The authors also employed an in vitro system to demonstrate that CARDIF is cleaved by a protease encoded by the hepatitis C virus, showing for the first time that viruses could specifically disrupt CARDIF signaling (Meylan et al., 2005). Finally, Seth et al. (2005) discovered the Mitochondrial Antiviral Signaling protein (MAVS) by performing sequence similarity searches, using the CARD domain of MDA-5 as bait. They reported that overexpression of MAVS activated IRF3 and NK-κB, and that its presence was required for a response to Sendai virus infection. Importantly, only Seth et al. (2005) in these primary papers localized this novel protein to mitochondria, therefore for this reason (and general simplicity) IPS-1/VISA/CARDIF/MAVS will be referred to as ‘‘MAVS’’ below.
2.2. Structure and function of MAVS
MAVS is a predicted 57 kDa protein consisting of three functional domains – a single N-terminal CARD domain (10–77 aa), a medial proline-rich region (PRR; 107–173 aa) and a C-terminal transmembrane domain (TM; 514–535 aa) that anchors it to the outer mitochondrial membrane (Seth et al., 2005). The N-terminal CARD domain shares high similarity to the CARD domains found in RIG-I and MDA-5 (Potter et al., 2008), and these proteins are predicted to bind with MAVS homotypically at this domain, following activation by viruses. As such, removal of the MAVS CARD domain appears to prevent this interaction and ablates its signaling function (Seth et al., 2005, Meylan et al., 2005, Kawai et al., 2005). The PRR section of MAVS contains consensus binding sites for many proline-associated immune proteins, such as members of the tumor necrosis factor receptor associated factor (TRAF) family like TRAF2, TRAF3 and TRAF6 (Xu et al., 2005, Saha et al., 2006). In the context of this review, perhaps the most interesting domain however is the C-terminal TM domain that directs MAVS to mitochondria. Removal of the TM prevents both its mitochondrial localization and signaling function, restricting the truncated protein to the cytosol (Seth et al., 2005, Xu et al., 2005). The TM domain has similarities to those found in the outer mitochondrial membrane proteins TOM20, Bcl-2 and Bcl-xL, and replacing the TM of MAVS with those from Bcl-2 or Bcl-xL restores both its function and mitochondrial localization (Seth et al., 2005). This suggests that the TM domain of MAVS has no other role in the antiviral function of the protein, other than to ensure correct placement at the outer mitochondrial membrane. When overexpressed, a construct containing just the CARD and TM domains of MAVS had a similar ability to activate the IFN-β promoter as the whole protein (Seth et al., 2005), indicating that these two domains are the most important for MAVS function.
2.3. Regulation of MAVS downstream signaling
There have been several (sometimes conflicting) reports of proteins that interact with MAVS, either to aid downstream signaling, or to regulate its function. Downstream signaling, resulting in the production of cytokines, has been linked to a number of pathways that converge on the pro-immune transcription factors IRF3, IRF7 and NF-κB. MAVS has been shown to associate with FADD and RIP1 (Matsuda et al., 2003, Lad et al., 2008), adapter proteins involved in the IKK complex that activates NF-κB. However, the requirement for RIP1 in MAVS-associated cytokine production is questioned by the finding that there is normal IFN-β activation in RIP1−/− mouse embryonic fibroblasts (MEFs; Seth et al., 2005). Binding of TRAF-family members (e.g. TRAF2, TRAF3 and TRAF6) to the proline-rich region of MAVS has been shown to be necessary for the downstream phosphorylation of NF-κB and IRF3/7 (Seth et al., 2005, Xu et al., 2005, Saha et al., 2006), although a deletion-mutant of MAVS without the binding sites still has the capacity to activate IFN-β (Seth et al., 2005). Kinases such as IKKε, have been shown to physically interact with MAVS at the outer mitochondrial membrane (Lin et al., 2006), and this leads to the phosphorylation and activation of IRF3. In contrast, the mitotic polo-like kinase 1 (PLK1) has been shown to inhibit MAVS signaling and IFN-β activation, by blocking the TRAF3 binding site on MAVS in a phospho-independent manner (Vitour et al., 2009).
Similar to other biological “arms race” scenarios, pathogenic persistence relies on the ability of microbes to overcome the host-cell defenses. Hepatitis C virus (HCV) encodes NS3/4A, a serine protease that blocks RLH signaling by cleaving MAVS from the outer mitochondrial membrane at Cys-508 (Meylan et al., 2005, Lin et al., 2006). Like other truncation mutants that prevent its mitochondrial localization (Seth et al., 2005, Lad et al., 2008), this cleavage prevents MAVS from signaling to downstream immune effectors. The evolutionarily-unrelated hepatitis A virus (HAV) cleaves MAVS from mitochondria at Gln-428, using its 3ABC cysteine protease (Yang et al., 2007), indicating that numerous viruses may have evolved strategies to block the immune system at this point.
Interestingly, two mammalian proteins have recently been shown to be involved in MAVS degradation in response to viral infection, potentially acting as a negative-feedback system to fine-tune interferon production. A subunit of the proteasome, PSMA7, interacts with MAVS in vivo and in vitro, and its overexpression leads to a reduction in endogenous MAVS levels (Jia et al., 2009). Importantly, there is also a transient increase in PSMA7 levels during viral infection, indicating that the MAVS degradation is not just experimental artifact. Potentially upstream of this protein is PCBP2, which interacts with MAVS and acts as a negative regulator of its function (You et al., 2009). PCBP2 is upregulated during viral infection, and appears to target MAVS for degradation via the ubiquitin–proteasome system. Following viral infection, PCBP2 recruits the HECT E3 ubiquitin ligase AIP4 to MAVS, resulting in its polyubiquitination and degradation (You et al., 2009).
2.4. MAVS, immunity and apoptosis
A familiar aspect of mitochondrial biology is the role the organelle plays in apoptosis. Mitochondria are an integral part of the intrinsic apoptotic pathway, being the location of many proteins involved in the early stages of cell death. For example, the Bcl-2 family of proteins (including the pro-apoptotic Bax and Bak, and the anti-apoptotic Bcl-2 and Bcl-xL) act at the outer mitochondrial membrane in response to stress stimuli, and help to regulate whether the cell enters the later stages of programmed cell death (for review, see Youle and Strasser, 2008). If the cell receives sufficient pro-apoptotic signals, Bax and Bak aid the release of cytochrome c from the mitochondrial inter membrane space, which leads to the induction of caspases and the orderly destruction of the cell. Apoptosis is also an important aspect of the immune system, as it can help to prevent the movement of pathogens between tissues, and many viruses (such as human immunodeficiency virus and cytomegalovirus) produce proteins that interfere with this process (Boya et al., 2003). For example, the 2′5′-oligoadenylate synthetase/RNaseL pathway induces apoptosis in response to viral infection, by using the endoribonuclease RNaseL to cleave all cellular RNA (host and viral), which shuts down protein synthesis and kills the cell (Castelli et al., 1998, Malathi et al., 2007).
In their initial paper, Seth et al. (2005) speculated that MAVS may have a role in apoptosis induction, as siRNA knockdown of protein levels led to the cleavage of PARP, a known marker of cell death; although a follow-up report by the same group found no difference in MAVS−/− MEFs following apoptosis (Sun et al., 2006). However, a number of recent studies have indicated that MAVS may have pro-apoptotic roles in immunity outside of cytokine production. Induction of apoptosis following reovirus infection is reduced in MAVS knockdown cells (Holm et al., 2007), while MAVS−/− MEFs show reduced apoptosis in response to infection by Sendai virus (Lei et al., 2009). These data are complimented by studies showing that overexpression of MAVS induces apoptosis by activation of caspase-3, -8 and -9 (Li et al., 2009, Lei et al., 2009, Yu et al., in press).
Given the role that MAVS may play in immune-related apoptosis, it is perhaps unsurprising that the protein is heavily regulated during apoptosis. Upstream members of the RLH pathway such as RIG-I and MDA-5 are degraded by caspases during apoptosis which, in the case of MDA-5, can be induced by poliovirus infection (Barral et al., 2007, Scott, 2009). MAVS is cleaved at Asp-429 by caspases during apoptosis, a process which can be blocked by the addition of the caspase inhibitor zVAD-fmk, or expression of the anti-apoptotic protein Bcl-xL (Scott and Norris, 2008, Rebsamen et al., 2008). Cleavage of these proteins during the early stages of cell death has the potential to aid viral replication, as it may prevent RLH proteins from signaling to downstream immune proteins, or delay completion of the apoptotic program. Initial studies of MDA-5 cleavage during poliovirus infection did not detect a benefit to the invading virus from inducing apoptosis, as there were no increases in viral replication or reduction in cytokine production (Barral et al., 2007). However, a recent study by Lei et al. (2009) has provided direct evidence of a viral protein targeting MAVS-induced apoptosis. The anti-apoptotic SARS-CoV protein NSP15 prevents apoptosis as a result of MAVS expression, but does not mediate apoptosis induced by staurosporine treatment, indicating that it has evolved as a MAVS-specific strategy (Lei et al., 2009). Further studies will help to elucidate the extent to which viruses target this interferon-independent aspect of MAVS biology.
3. NLRX1 – MAVS regulator or innocent bystander?
3.1. Discovery and conflicting functions of NLRX1
As mentioned above, one of the other groups of intracellular PRPs is the NOD-like receptor family of proteins. These leucine-rich repeat proteins are receptors involved in sensing numerous pathogen markers, such as bacterial lipopolysaccharide, leading to the activation of NF-κB; and also as part of the inflammasome complexes which regulate caspase-1 activation and production of inflammatory cytokines (Janeway and Medzhitov, 2002, O’Neill, 2008). Recently two research groups characterized a new member of the family, NLRX1, which was found to have a mitochondrial localization, in contrast to the cytosolic nature of the other family members. Moore et al. (2008) reported that NLRX1 was targeted to the mitochondrial outer membrane by a putative N-terminal localization sequence (Fig. 2 ), and had the ability to down-regulate RLH signaling by interacting with MAVS. This regulation was hypothesized to act as a brake on MAVS function, with the NACHT domain of NLRX1 reversibly binding to the CARD domain of MAVS – in effect blocking its ability to interact with RIG-I or MDA-5 (Moore et al., 2008). The second group also implicated NLRX1 in innate immunity, however with a greatly contrasting function. Tattoli et al. (2008) demonstrated that the mitochondrial localization of NLRX1 was dependent on a N-terminal addressing sequence, and that its expression did not lead to direct NF-κB activation. However, it was shown that NLRX1 could amplify reactive oxygen species production in response to Shigella infection and tumor necrosis factor α, which led to an increase in pro-immune NF-κB and JUN kinase signaling (Tattoli et al., 2008).
Fig. 2.
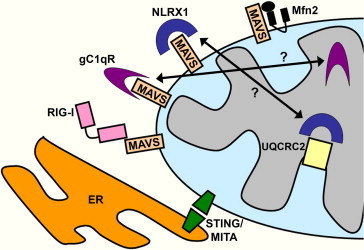
Mitochondrial proteins implicated in the innate immune response. A number of mitochondrial proteins have recently been implicated in the innate immune system, either through interaction with MAVS, or as part of an autonomous pathway. STING (also known as MITA) has been localized to both mitochondria and the endoplasmic reticulum, and may act downstream of RIG-I and MAVS in their initiation of pro-immune signaling kinases. NLRX1 and gC1qR have been implicated as regulators as MAVS function, possibly acting as physical barriers to prevent its interaction with RIG-I. However, there is strong evidence to suggest that the mature forms or both NLRX1 and gC1qR reside in the mitochondrial matrix, making a functional interaction with MAVS less likely. NLRX1 has recently been shown to physically interact with the respiratory chain complex III subunit UQCRC2, suggesting that it may be involved in reactive oxygen species production. Mfn2, an outer mitochondrial GTPase involved in mitochondrial fusion, has been shown to bind MAVS and negatively regulate immune signaling, raising questions about the role of mitochondrial dynamics in the immune response.
3.2. The matrix, not the membrane
Clearly the functions ascribed to NLRX1, as both a repressor and activator of innate immunity, pose some problems. While these may be accounted for by differences in experimental model or cell type, further research was required to elucidate NLRX1 function. Both groups reported that NLRX1 had a N-terminal mitochondrial targeting sequence, a feature which is normally associated with proteins that are imported into mitochondria via membrane translocases, as opposed to outer membrane proteins, which tend to have internal localization markers (Wiedemann et al., 2004). A thorough biochemical analysis of NLRX1 in HeLa and HEK293 cells indicated that NLRX1 is indeed imported into the mitochondrial matrix, a process which requires the removal of the N-terminal pre-sequence and an intact transmembrane potential (Arnoult et al., 2009). In addition, the authors demonstrated that NLRX1 coimmunoprecipitates with UQCRC2, a subunit of the respiratory chain complex III, which is localized on the matrix-facing side of the inner mitochondrial membrane. As complex III is involved in reactive oxygen species production, this finding substantiates both the matrix localization of NLRX1, and its involvement in reactive oxygen signaling (Arnoult et al., 2009).
Given the differences in localization between MAVS and NLRX1, at least in this system, it would appear unlikely that there could be a strong interaction between the two proteins in vivo. As noted by Arnoult et al. (2009), there is likely to be considerable physical distances between the NACHT domain of NLRX1 and the CARD domain of MAVS under normal physiological conditions, even at points where the inner and outer mitochondrial membranes are closely apposed. Further work will be required to understand if there is a change in NLRX1 localization under stressful conditions that would allow this molecular interaction to occur.
4. Other mitochondrial proteins and pathways in the innate immune system
Two other mitochondrial proteins have recently been linked to innate immunity (Fig. 2). STING/MITA was discovered simultaneously by two independent groups, and has been shown to interact with both RIG-I and MAVS (Ishikawa and Barber, 2008, Zhong et al., 2008). While both groups showed that it was required for downstream RLH-pathway signaling (by connecting upstream activator proteins like MAVS to downstream signaling kinases), and that it could activate reporter constructs for immune proteins like ISRE, they differed on the localization of the protein. Using similar techniques, the same protein was localized to mitochondria (MITA) and the endoplasmic reticulum (STING) by biochemical and microscopical analysis (Ishikawa and Barber, 2008, Zhong et al., 2008). Recent studies from two other groups have strengthened the case for the endoplasmic reticulum localization of STING/MITA (Sun et al., 2009, Castanier et al., in press); and it has been shown further that STING/MITA is involved in the response to intracellular viral DNA produced by herpes simplex-1 virus, where it translocates from the endoplasmic reticulum (with the signaling kinase TBK-1) to perinuclear vesicles during infection (Ishikawa et al., 2009). While these data suggest that the endoplasmic reticulum localization of STING/MITA is correct, there are many regions of close apposition between this organelle and mitochondria, known as mitochondria-associated membranes (MAMs; Vance, 1990), which can make unequivocal findings difficult. It is likely that these MAMs are important regions for the interaction between MAVS and STING/MITA (Castanier et al., in press).
The second protein implicated in regulating innate immunity is gC1qR, also known as p32, a receptor for the globular head component factor gC1. Overexpression of gC1qR has been shown to inhibit RLH signaling and IFN-β activation during Sendai virus infection, while knockdown of the endogenous protein by siRNA has the opposite effect (Xu et al., 2009). The authors show that the inhibition of RLH signaling occurs via the binding of gC1qR to MAVS at the outer mitochondrial membrane (Xu et al., 2009), presumably in a similar manner to that postulated for MAVS regulation by NLRX1 (Moore et al., 2008). However, several previous papers have shown that gC1qR is imported into the mitochondrial matrix using a N-terminal localization sequence (Muta et al., 1997, Dedio et al., 1998), which would keep it away from MAVS under physiological conditions. As with NLRX1, more work is required to further elucidate the connection between this protein and the RLH pathway.
5. Mitochondrial dynamics and innate immunity – where form meets function
A new avenue of research in this growing field is the interplay between mitochondrial morphology and the innate immune system. Mitochondria are dynamic organelles, undergoing constant fission and fusion in many cell types, and a balance between these two processes controls the overall mitochondrial morphology in any individual cell (for review, see Suen et al., 2008). A recent study has shown that there is an interaction between MAVS and Mfn2, a dynamin-like GTPase involved in mitochondrial fusion (Yasukawa et al., 2009), demonstrating the first potential connection between mitochondrial morphology and immunity. Overexpression of Mfn2 blocked MAVS signaling, while knockdown of the endogenous protein increased interferon production during viral infection, indicating that Mfn2 may act as a negative regulator of the RLH pathway. However, the authors suggested that the Mfn2-MAVS link may be separate from the role of Mfn2 in mitochondrial fusion (Yasukawa et al., 2009).
A more direct link between the two processes comes from an elegant study by Castanier et al. (2009), which found that activation of the RLH signaling pathway led to mitochondrial elongation. Altering mitochondrial morphology, by down-regulating mitochondrial fission and fusion genes using RNAi, regulated the antiviral response to Sendai virus infection. In cells with depleted mitochondrial fission (which led to elongated mitochondria), there was enhanced activation of the IFN-β and NF-κB promoters during infection; conversely, in cells with fragmented mitochondria (caused by the down-regulation of mitochondrial fusion genes), the antiviral response to infection was attenuated (Castanier et al., in press). The authors further showed that MAVS interacts with, and may regulate, the function of a mitochondrial fusion protein, Mfn1 (a homologue of Mfn2), and that this may be the cause of the observed mitochondrial elongation during infection. Finally, it was demonstrated that the change in mitochondrial morphology aided the interaction between MAVS and STING/MITA on the endoplasmic reticulum, suggesting that the elongation promotes the physical interaction of the two organelles during the antiviral response (Castanier et al., in press). In summary, it is clear that mitochondrial dynamics may play an important part in MAVS function, and it is likely that we have much more to learn about the role of mitochondrial morphology in this regard.
6. Conclusions
Prior to the discovery of MAVS, the closest links between mitochondria and the innate immune system came from mitochondrial involvement in apoptosis and reactive oxygen species production. However, it is now clear that mitochondria are more intimately linked to antiviral defenses than these important, yet slightly indirect processes. With MAVS and STING/MITA, we have direct and integral members of a crucial innate immune signaling pathway localized at the mitochondria; while the data on NLRX1 shows that a NOD protein (from a family that is involved in the immune systems of both plants and animals; Janeway and Medzhitov, 2002) has evolved to use mitochondria as a specific reactive oxygen species-producing factory to aid anti-microbial defenses. It will be interesting to see if there are other aspects of mitochondrial biology involved in the innate immune system, either involving the discovery of new mitochondrial proteins, or by finding a role for known mitochondrial processes in novel immune pathways.
Acknowledgements
The author would like to thank Michael Sack (NHLBI, NIH) and Richard J. Youle (NINDS, NIH) for their help and support. The author would also like to thank the two anonymous reviewers who suggested improvements to this manuscript. This research was supported by the intramural research program of the National Heart, Lung and Blood Institute, National Institutes of Health.
References
- Arnoult D., Soares F., Tattoli I. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 2009;122:3161–3168. doi: 10.1242/jcs.051193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral P.M., Morrison J.M., Drahos J. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007;81:3677–3684. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P., Roumier T., Andreau K. Mitochondrion-targeted apoptosis regulators of viral origin. Biochem. Biophys. Res. Commun. 2003;304:575–581. doi: 10.1016/s0006-291x(03)00630-2. [DOI] [PubMed] [Google Scholar]
- Castanier, C., Garcin, D., Vazquez, A. et al., in press. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. doi: 10.1038/embor.2009.258. [DOI] [PMC free article] [PubMed]
- Castelli J.C., Hassel B.A., Maran A. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998;5:313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- Dedio J., Jahnen-Dechent W., Bachmann M. The multiligand-binding protein gC1qR, putative c1q receptor, is a mitochondrial protein. J. Immunol. 1998;160:3534–3542. [PubMed] [Google Scholar]
- Holm G.H., Zurney J., Tumilasci V. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 2007;282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Ma Z., Barber G.N. STING regulates intracellular DNA-mediated, type 1 interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway C.A., Jr., Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Jia Y., Song T., Wei C. Negative regulation of MAVS-mediated innate immune response by PSMA7. J. Immunol. 2009;183:4241–4248. doi: 10.4049/jimmunol.0901646. [DOI] [PubMed] [Google Scholar]
- Kawai T., Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- Kawai T., Takahashi K., Sato S. IPS-1, an adaptor triggering RIG-I- and MDA5-mediated type 1 interferon induction. Nat. Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Lad S.P., Yang G., Scott D.A. Identification of MAVS splicing variants that interfere with RIG-I/MAVS pathway signaling. Mol. Immunol. 2008;45:2277–2287. doi: 10.1016/j.molimm.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Lei Y., Moore C.B., Liesman R.M. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One. 2009;4:e5466. doi: 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.M., Fujikura D., Harada T. IPS-1 is crucial for DAP3-mediated anoikis induction by caspase-8 activation. Cell Death Differ. 2009;16:1615–1621. doi: 10.1038/cdd.2009.97. [DOI] [PubMed] [Google Scholar]
- Lin R., Lacoste J., Nakhaei P. Dissociation of a MAVS/IPS-1/VISA/CARDIF-IKKe molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 2006;80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K., Dong B., Gale M., Jr. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda A., Suzuki Y., Honda G. Large-scale identification and characterization of human genes that activate NF-kB and MAPK signaling pathways. Oncogene. 2003;22:3307–3318. doi: 10.1038/sj.onc.1206406. [DOI] [PubMed] [Google Scholar]
- Meylan E., Tschopp J. NLRX1: friend or foe? EMBO Rep. 2008;9:243–245. doi: 10.1038/embor.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E., Curran J., Hofmann K. CARDIF is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Moore C.B., Bergstralh D.T., Duncan J.A. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- Muta T., Kang D., Kitajima S. P32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 1997;272:24363–24370. doi: 10.1074/jbc.272.39.24363. [DOI] [PubMed] [Google Scholar]
- O’Neill L.A. Innate immunity: squelching anti-viral signalling with NLRX1. Curr. Biol. 2008;18:R302–304. doi: 10.1016/j.cub.2008.02.021. [DOI] [PubMed] [Google Scholar]
- Potter J.A., Randall R.E., Taylor G.L. Crystal structure of human IPS-1/MAVS/VISA/CARDIF caspase activation recruitment domain. BMC Struct. Biol. 2008;8:11. doi: 10.1186/1472-6807-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M., Meylan E., Curran J. The antiviral adaptor proteins CARDIF and TRIF are processed and inactivated by caspases. Cell Death Differ. 2008;15:1804–1811. doi: 10.1038/cdd.2008.119. [DOI] [PubMed] [Google Scholar]
- Saha S.K., Pietras E.M., He J.Q. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and CARDIF. EMBO J. 2006;25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott I. Degradation of RIG-I following cytomegalovirus infection is independent of apoptosis. Microbes Infect. 2009;11:973–979. doi: 10.1016/j.micinf.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott I., Norris K.L. The mitochondrial antiviral signaling protein, MAVS, is cleaved during apoptosis. Biochem. Biophys. Res. Commun. 2008;375:101–106. doi: 10.1016/j.bbrc.2008.07.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R.B., Sun L., Ea C.K. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kB and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Suen D.F., Norris K.L., Youle R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Sun L., Liu H.H. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Sun W., Li Y., Chen L. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli I., Carneiro L.A., Jehanno M. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008;9:293–300. doi: 10.1038/sj.embor.7401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Venkataraman T., Valdes M., Elsby R. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 2007;178:6444–6455. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- Vitour D., Dabo S., Ahmadi Pour M. Polo-like kinase 1 (PLK1) regulates interferon (IFN) induction by MAVS. J. Biol. Chem. 2009;284:21797–21809. doi: 10.1074/jbc.M109.018275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N., Frazier A.E., Pfanner N. The protein import machinery of mitochondria. J. Biol. Chem. 2004;279:14473–14476. doi: 10.1074/jbc.R400003200. [DOI] [PubMed] [Google Scholar]
- Xu L.G., Wang Y.Y., Han K.J. VISA is an adapter protein required for virus-triggered IFN-b signaling. Mol. Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Xu L., Xiao N., Liu F. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc. Natl. Acad. Sci. USA. 2009;106:1530–1535. doi: 10.1073/pnas.0811029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Liang Y., Qu L. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa K., Oshiumi H., Takeda M. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009;2:ra47. doi: 10.1126/scisignal.2000287. [DOI] [PubMed] [Google Scholar]
- Yoneyama M., Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- Yoneyama M., Kikuchi M., Natsukawa T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- You F., Sun H., Zhou X. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009;10:1300–1308. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- Youle R.J., Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yu, C.Y., Chiang, R.L., Chang, T.H. et al., in press. The interferon stimulator MAVS facilitates cell death by disrupting the mitochondrial membrane potential and by activating caspases. J. Virol. doi:10.1128/JVI.02174-09. [DOI] [PMC free article] [PubMed]
- Zhong B., Yang Y., Li S. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]