

Abstract
The highly-conserved NOTCH signaling pathway plays many essential roles in the development of diverse cells, tissues and organs from Drosophila to humans, and dysregulated Notch signaling contributes to several disorders, including vascular and bone defects, as well as several cancers. Here we describe a novel mechanism of NOTCH regulation by reciprocal inhibition of two NOTCH downstream effectors: Deltex1 and HES1. This mechanism appears to regulate invasion of osteosarcoma cells, as Deltex1 blocks osteosarcoma invasiveness by downregulating NOTCH/HES1 signaling. The inhibitory effect of endogenous Deltex1 on NOTCH signaling is mediated through binding with the intracellular domain of NOTCH and ubiquitination and degradation of NOTCH receptors. Conversely, we show that the NOTCH target gene HES1 causes transcriptional inhibition of Deltex1 by directly binding to the promoter of Deltex1. A HES1 binding site is identified 400bp upstream of the transcription start site of Deltex1. HES1-mediated repression of Deltex1 requires the C-terminal H3/H4 and WRPW domains of HES1, which associate with the TLE/Groucho corepressors. Taken together, we define a molecular mechanism regulating NOTCH signaling by reciprocal inhibition of the NOTCH target genes HES1 and Deltex1 in mammalian cells. This mechanism may have important clinical implications for targeting NOTCH signaling in osteosarcoma and other cancers.
Keywords: NOTCH, HES1, Deltex1(DTX1), OSTEOSARCOMA, INVASION
Introduction
NOTCH signaling affects fundamental processes in cell-fate determination, including stem-cell maintenance, differentiation, proliferation and apoptosis (Matsuno et al., 2002). The roles of NOTCH signaling in cancer vary depending on cellular context. NOTCH may function as an oncogene, e.g. in T cell ALL or mammary adenocarcinoma, or as a tumor suppressor, e.g. in neuroendocrine and B-cell malignancies (Allenspach et al., 2002; Weng et al., 2004; Zweidler-McKay and Pear, 2004; Zweidler-McKay et al., 2005).
Ligand (Jagged1/2, Dll1/3/4) binding to membrane-localized NOTCH1-4 triggers a conformational change of NOTCH, exposing cleavage sites for ADAM10 and γ-secretase (Fortini, 2002). After cleavage, the intracellular domain of NOTCH (ICN) is released into the cytoplasm and translocates into the nucleus, binding to transcription factors CSL (CBF1, Su(H) and LAG-1), also known as RBPjk (Struhl and Adachi, 1998), initiating transcription. The primary targets of NOTCH are two basic helix-loop-helix (bHLH) transcriptional repressor families: the Hairy/enhancer-of-split (HES) and the Hairy/enhancer of split with YRWP motif (HERP) families (Iso et al., 2003). HES and HERP proteins normally form homodimers or heterodimers, inhibiting expression of specific downstream target genes in a tissue-specific manner. ICN also directly activates other target genes in a CSL-independent manner, including MAPK phosphatase LIP1, pre-T alpha and cyclin D1 (Allenspach et al., 2002).
NOTCH induces expression of Deltex1 (DTX1), a human ortholog of Drosophila Deltex, (Matsuno et al., 2002), but DTX1 expression is not strictly NOTCH-dependent (Matsuno et al., 1998). DTX1 contains a RING finger and two WWE domains, indicating that DTX1 may be an E3 ubiquitin ligase (Jackson et al., 2000). DTX1 can regulate T-cell activation through ubiquitination and degradation of MEKK1 in T-cells (Liu and Lai, 2005). Depending on the cellular context, Deltex can either promote or inhibit NOTCH signaling. In Drosophila, Deltex promotes NOTCH-dependent transcriptional activity, and human DTX1 can have a similar effect (Matsuno et al., 1998). However, Deltex also forms trimeric NOTCH-Deltex-Kurz (β-arrestin) protein complexes, mediating degradation of NOTCH receptors through a ubiquitination-dependent pathway (Mukherjee et al., 2005). DTX1 also can block NOTCH-dependent transcription in mammalian lymphoid cells and neuron cells (Itoh et al., 2003; Sestan et al., 1999). Izon et.al. suggested that DTX1 suppresses NOTCH1 induced gene expression in lymphoid progenitor cells by blocking coactivator recruitment (Izon et al., 2002). No clear role has been established for DTX1 in malignancy.
We previously reported that NOTCH/HES1 signaling promotes osteosarcoma invasion and metastasis (Zhang et al., 2008). In this report, we present a novel regulatory mechanism of NOTCH signaling in human cells. First, we demonstrate that endogenous DTX1 negatively regulates NOTCH-dependent transcriptional activity in osteosarcoma cells and that DTX1 physically binds to ICN1, inducing ubiquitination and degradation of ICN1. Next we show that the CSL-dependent NOTCH target gene HES1 transcriptionally inhibits DTX1 through direct binding to the promoter of DTX1. We identify a HES1 binding site (N-box) in the promoter region of DTX1, and show physical interaction of HES1 with this N-box. Finally, we show that this reciprocal inhibition of HES1 and DTX1 within the NOTCH pathway controls osteosarcoma invasiveness. Taken together, our results provide a novel mechanism of NOTCH signaling regulation by its downstream target genes, HES1 and DTX1. This mechanism may have important clinical implications to determine whether modulation of NOTCH signaling might benefit patients with osteosarcoma and other types of cancer.
Results
DTX1 negatively regulates NOTCH1/HES1 signaling and osteosarcoma invasiveness
To investigate whether DTX1 antagonizes NOTCH signaling in osteosarcoma, we overexpressed DTX1 in OS187 cells. DTX1 overexpression reduced ICN1 and HES1 protein (Figure 1A). DTX1 can be a transcription regulator or a ubiquitin ligase (Matsuno et al., 2002; Wu et al., 2001), so we wished to know which mechanism was downregulating NOTCH protein levels. RT-PCR and Q-PCR measured expression NOTCH1, HES1, DTX1 and actin mRNA in OS187 cells expressing DTX1 or empty vector (Figure 1B and 1C). NOTCH1 mRNA level was not significantly decreased by DTX1 overexpression, while HES1 mRNA was downregulated (p<0.05), suggesting that DTX1 downregulated NOTCH1 by affecting protein stability rather than gene transcription. Q-PCR showed that HES1 mRNA level was significantly decreased (30%, p<0.05) in cells overexpressing DTX1, presumably resulting from decreased ICN1 protein. Since Hes1 expression is affected by several promoters and not just ICN1, the reduction in expression by only 30% was not surprising. To measure directly the impact of DTX1 on ICN1-mediated gene activation, the CSL-responsive promoter (HES-AB) was co-transfected with Renilla luciferase control vector into OS187 cells stably overexpressing human DTX1 gene or empty vector. The HES-AB promoter is the 500bp promoter region upstream of the HES1 gene transcription start site, containing two ICN1-responsive CSL binding sites (Jarriault et al., 1995). Overexpression of DTX1 decreased the activity of the CSL-responsive promoter by 30% in OS187 cells (p<0.05), indicating that DTX1 inhibits NOTCH/CSL signaling (Figure 1D). Likewise, matrigel invasion was reduced by overexpression of DTX1 in OS187 cells, in parallel with downregulation of NOTCH signaling and HES1 (Figure 1E). However, constitutive HES1 expression in OS187 cells overexpressing DTX1 reversed the DTX1 inhibition on cell invasion (Fig 1E). This result demonstrates that DTX1-mediated inhibition of the Notch pathway acts upstream of HES1.
FIG 1.
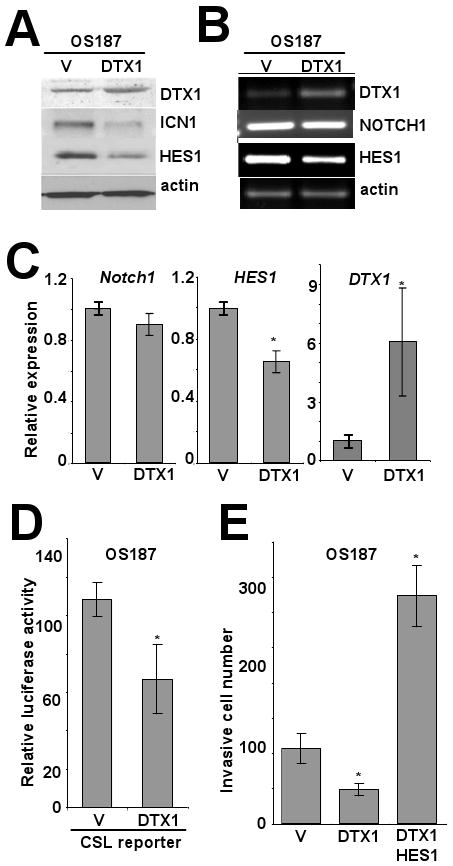
DTX1 negatively regulates NOTCH1/HES1 signaling and suppresses invasion of osteosarcoma cells. Immunoblot (A) and PCR (B) analysis of DTX1, NOTCH1/ICN1, HES1 and actin in OS187 cells stably expressing vector control or DTX1 gene. To confirm that the band identified by the western blot for ICN1 in fact represents the cleaved intracellular fragment of Notch1, we cultured OS 187 cells in 10 or 50 μM compound E, a gamma secretase inhibitor (GSI), and showed that the cleaved protein is reduced with GSI treatment (Supplemental figure 1). (C) Histograms represent quantitative PCR analysis of NOTCH1, HES1 and DTX1 from the same cells. Ct values were normalized to actin; vector control is defined as a relative expression of 1 (*, p<0.05). (D) Histograms represent luciferase emission from a CSL reporter (i.e., NOTCH-responsive) in OS 187 cells transduced with DTX1 or empty vector (*, p<0.05). (E) Histograms represent matrigel invasion of OS187 cells stably expressing empty vector, DTX1 or DTX1 and HES1. 1×104 stably transduced OS187 cells were seeded in each transwell and cell invasiveness was quantified by the invasive cell number after 48 hours incubation. Average of three wells, with error bars showing standard deviation, is shown (*, p<0.05).
DTX1 downregulates NOTCH signaling by ubiquitination of ICN
DTX1 contains a RING-H2 domain and two WWE domain structures, suggesting a E3 ubiquitin ligase function (Matsuno et al., 2002). To assess DTX1 regulatory function, we measured DTX1 interaction with ICN1 and induction of proteasome-dependent ICN1 downregulation. We treated COL cells, which have high endogenous DTX1, with 5mM EDTA for 15 minutes to induce NOTCH1 cleavage and ICN internalization, then immunoprecipated DTX1 from the treated cells (Figure 2A). Immunoprecipitation of DTX1 also brought down ICN1 protein, confirming physical interaction of endogenous DTX1 and ICN1.
FIG 2.
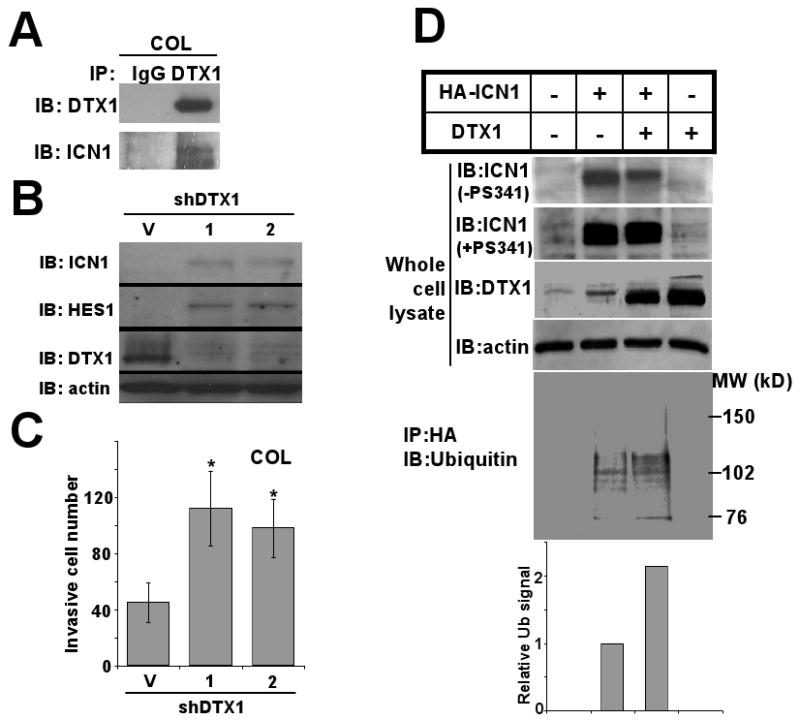
DTX1 physically interacts with ICN1 and induces ubiquitination and proteasome-mediated degradation of ICN1. (A) COL cells transduced with DTX1 or empty vector were treated with 5mM EDTA for 15 minutes to increase NOTCH1 cleavage and total ICN1 level. Immunoprecipitation (IP) of DTX1 from DTX1-transduced cells yielded both DTX1 and ICN1, as shown by immunoblot (IB). (B) Endogenous DTX1 was knocked down with either of 2 specific shRNA constructs (1) or (2); Immunoblot (IB) shows ICN1, HES1 and DTX1 protein from shRNA transduced cells compared to vector (V). (C) Histograms represent matrigel invasion of the COL cells shown in figure 1B. 1×104 COL cells with DTX1 shRNAs were seeded in each transwell and invasion was quantified after 48 hours; histograms shown the mean of three wells, and error bars show standard deviation. (*, p<0.05). (D) HA-ICN1 or DTX1, alone or in combination, were transiently overexpressed in 293T cells for 48 hours. After treatment with proteasome inhibitor PS341 (0.1μM) for 6 hours or no treatment, total cell lysates were prepared. Immunoblots (IB) of ICN1, DTX1 and actin in the whole cell extracts are shown (upper panels). After immunoprecipitation of ICN1 with anti-HA beads, ubiquitination of ICN1 was measured using anti-ubiquitin antibody (lower panel). Ubiquitination level of ICN1 was quantified by densitometry (bottom panel); histograms represent relative ubiquitination level.
To assess DTX1's impact on ICN1 protein stability, shRNAs targeting DTX1 were used to knock down endogenous DTX1 in COL cells. DTX1 shRNA suppressed DTX1 in COL cells and upregulated ICN1 and HES1 levels (Figure 2B), indicating that endogenous DTX1 decreased ICN1 protein level. COL invasiveness was increased by DTX1 shRNA (Figure 2C), suggesting that endogenous DTX1 reduces osteosarcoma cell invasion.
To study whether DTX1 induces ICN1 ubiquitination, HA-tagged ICN1 and DTX1 were overexpressed, either alone or together, in 293T cells (Figure 2D). Cells were pretreated by proteasome inhibitor PS341 or vehicle for 6 hours, and ICN1, DTX1 and actin examined by western blot (Figure 2D, upper panels). Without PS341, ICN1 was decreased by coexpressing DTX1. However, with proteasome inhibitor, ICN1 level was unchanged by coexpression of DTX1 in ICN1-expressing cells. These results suggested that DTX1 downregulated ICN1 through a proteasome dependent pathway. Immunoprecipitation of HA-ICN1 brought down DTX1 and vice versa (data not shown), suggesting the interaction of ICN1 and DTX1. The C-terminal PEST domain of ICN1 was not required for this interaction (Supplemental Figure 2). We next determined if overexpression of DTX1 promoted ubiquitination of ICN1. HA-ICN1 was precipitated by anti-HA antibody and probed with anti-ubiquitin antibody. Co-expression of DTX1 and HA-ICN1 increased ubiquitinated ICN1 compared to expression of HA-ICN1 alone, characterized by multiple ubiquitinized ICN1 protein bands (Figure 2D, lower panel). Compared to the total ICN1 protein precipitated in ICN1 only, DTX1 increased the ubiquitinated ICN1 2-fold, suggesting that DTX1 causes ICN ubiquitinization, leading to proteasome-mediated degradation.
Transcriptional regulation of NOTCH downstream target gene DTX1 in osteosarcoma cells
We wished to understand better the role of Notch pathway genes in regulating DTX1 transcription. In OS187 cells with stable overexpression of intracellular NOTCH1 (ICN1), dominant negative Mastermind (dnMAM, an inhibitor of Notch receptor-CSL interaction) or HES1, HES1 mRNA and protein were upregulated by ICN1 and downregulated by dnMAM (Figure 3A and 3B). ICN1 overexpression did not upregulate DTX1 mRNA or protein expression, while inhibiting Notch/CSL interaction via dnMAM upregulated DTX1 gene and protein expression in OS187 cells. Furthermore, overexpression of HES1 reduced DTX1 protein in OS187 cells, suggesting that HES1 suppresses DTX1 expression. To confirm the inhibition of DTX1 by HES1, we also transduced ICN1 and HES1 into COL cells, which have high endogenous DTX1 expression. Overexpression of ICN1 or HES1 in COL suppressed DTX1 mRNA and protein expression (Figure 3C). Q-PCR confirmed that DTX1 mRNA was reduced by ICN1 or HES1 (data not shown). This observation suggested that ICN1 induced HES1, therefore inhibiting DTX1 mRNA expression in osteosarcoma cells. We also assessed the impact of ICN1 or HES1 transduction on COL invasion (Figure 3D). Compared to vector control, transduction with ICN1 or HES1 significantly increased invasion of COL cells in matrigel, confirming our finding that HES1 is critical for osteosarcoma invasion.
FIG 3.
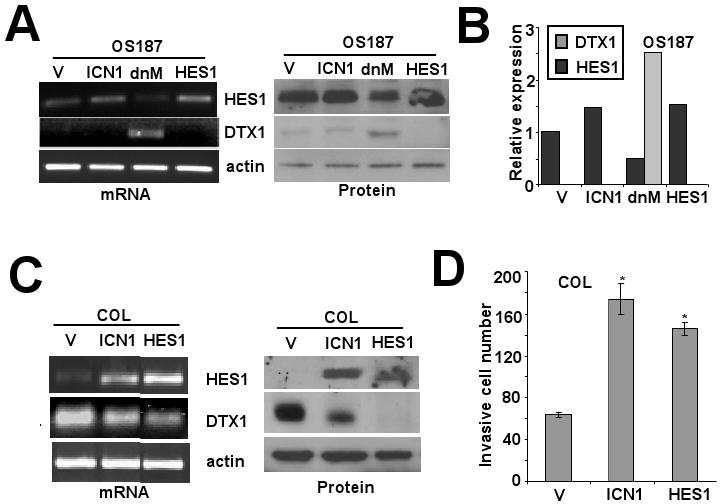
Impact of manipulation of NOTCH/HES1 signaling on DTX1 expression. (A) PCR (left panel) and western blot (right panel) analysis of HES1, DTX1 and actin expression in OS187 cells overexpressing ICN1, dnMAM or HES1 compared to vector control. (B) Quantitative PCR analysis of HES1 and DTX1 in the same cells at 3A. Analysis was performed as in figure 1C. (C) COL cells, which have high endogenous DTX1, were manipulated and analyzed as in 3A. Left panel: PCR analysis. Right panel: western blot analysis. (D) Histograms represent matrigel invasion of the COL cells from 3C (*, p<0.05). Assay and analysis were performed as in figure 1E.
Dominant negative HES1 increases DTX1 expression and blocks invasion of osteosarcoma cells
To further prove that HES1 negatively regulates DTX1 expression, we made a dominant negative human HES1 (dnHES1) by mutating three amino acids critical for DNA binding while leaving dimerization motifs intact. HES1 binds to and inhibits the activity of its own promoter, which can be shown with reporter construct HES1-AB (Castella et al., 2000; Takebayashi et al., 1994). In 293T cells, expression of HES1 suppressed HES1-AB promoter activity, but adding dnHES1 reversed HES1-mediated inhibition of Hes1-AB (Figure 4A). Similar results were found in OS187 cells (data not shown), confirming that dnHES1 blocks HES1 function. Then, we tested whether dnHES1 could block HES1-mediated inhibition of DTX1 transcription in osteosarcoma. Transduction with dnHES1 upregulated DTX1 transcription in three osteosarcoma cell lines (Figure 4B), indicating that HES1 suppressed DTX1 expression in osteosarcoma. Expression of HASH1, a known HES1 target (Axelson, 2004), also was upregulated by dnHES1 (Figure 4B and 4C) in parallel with DTX1.
FIG 4.
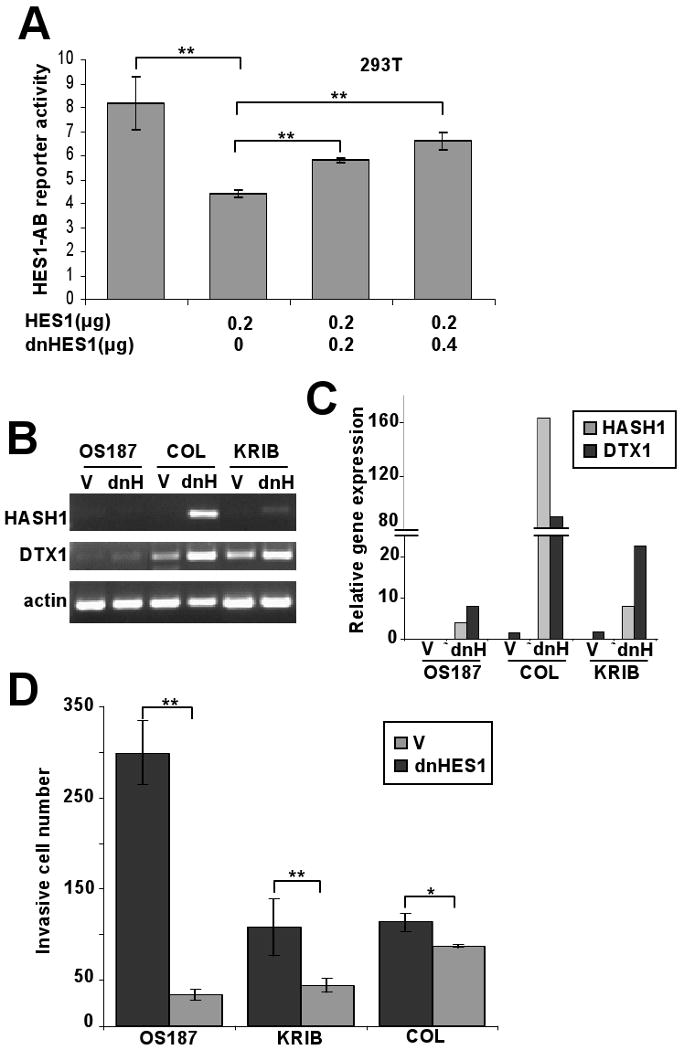
Dominant negative HES1 upregulates DTX1 expression. (A) The HES1-AB reporter is activated by CSL/NOTCH and inhibited by HES1. 293T cells were cotransfected with HES1-AB reporter (firefly luciferase) and a Renilla luciferase control together with various dosages of HES1 and/or dnHES1 as shown in the figure. Relative luciferase activity was measured 48 hours after transfection. (**, p<0.005) (B) PCR and (C) quantitative PCR analysis of changes in HASH1 and DTX1 expression induced by dnHES1. Osteosarcoma cells OS 187, COL and KRIB were transduced with empty vector (V) or dnHES1 (dnH). Gel image (B) shows the PCR amplification of HASH1, DTX1 and actin; histograms (C) represent relative Q-PCR results, normalized to a value of 1 for the vector-only cells. (D) Histograms represent matrigel invasion of the same cells described in 4B and 4C. Analysis was performed as in figure 1E. (**, p<0.005. *, p<0.05). V: empty vector control. dnH: dnHES1.
We previously showed that HES1 promotes osteosarcoma invasiveness (Zhang et al., 2008). We therefore examined matrigel invasion of osteosarcoma cells transduced with dnHES1. dnHES1 significantly suppressed osteosarcoma cell invasion (* p<0.05; ** p<0.005,) (Figure 4D), redemonstrating the essential role of HES1 for osteosarcoma invasiveness.
HES1 represses the DTX1 proximal promoter through direct binding to an N-box motif
We wished to identify the HES1 binding site(s) in the DTX1 promoter. Searching the 3kb human genomic sequence upstream of DTX1 transcription start site, we identified six potential HES1-binding motifs (N-box motif): CACGAG at -400/394 bp, CACCAG at -1833/1828, CACAAG at -2445/2440, CACGAG at -2652/2646, and CACCAG at -2861/2855 and -3154/3149. Because of evolutionary conservation and proximity to the start site, we focused on the most proximal of these: the N-box motif at -400bp (Figure 5A upper panel). This N-box is present in mouse, dog and human, suggesting that HES1 regulation of DTX1 is evolutionarily conserved and biologically important (Figure 5A lower panel). To prove HES1 repression of the DTX1 promoter, a luciferase reporter construct containing 1 Kb of the 5′UT of DTX1 was used. In 293T cells, which have low endogenous HES1, expression of HES1 decreased DTX1 promoter activity by 60% (p<0.005) (Figure 5B), confirming HES1 repression of DTX1 transcription.
FIG 5.
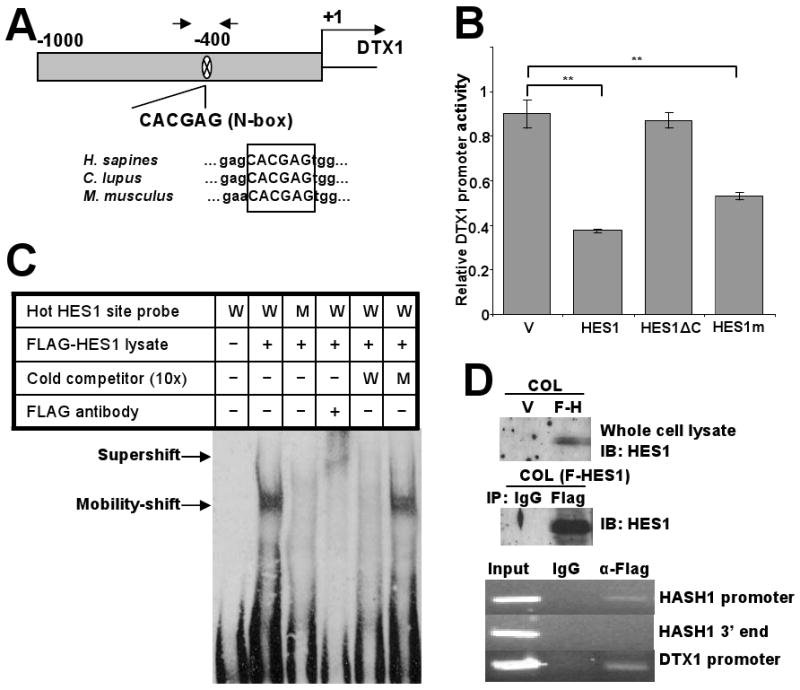
HES1 actively represses the DTX1 through binding to an N-box motif in the DTX1 promoter. (A) Schematic depiction of the 1kb DTX1 proximal promoter in the 5′ untranslated region. One potential HES1 binding site (N-box) was located at -400bp. Arrows represent the position of the PCR primers utilized for the ChIP analysis of the following panels (upper part). Sequence alignment of the DTX1 genes of human, dog and mouse showed the identical N-box in the similar position (lower part). (B) DTX1 promoter reporter (Renilla luciferase) was cotransfected with a firefly luciferase control into 293T cells transiently expressing vector control, full length HES1, a mutant HES1 with the C-terminus deleted (HES1ΔC) or HES1-WRPW mutant (HES1m), which is predicted to have less Groucho/TLE binding. Relative DTX1 promoter activity was determined after 48 hours. (**, p<0.005). (C) Gel mobility-shift analysis showing HES1 binding to the N-box motif in the DTX1 promoter. Nuclear protein extract (3μg) from 293T cells expressing FLAG-HES1 were incubated with biotin labeled probes containing WT N-box (W), mutant N-box (M), and anti-FLAG antibody, either alone or in combination, at room temperature for 15 min. As cold competitors, excess doses (10-fold) of non-biotinylated WT or mutant oligomers were included. Arrows indicate the specific N-box DNA-HES1 complex and the HES1 supershifted complex. (D) Chromatin immunoprecipitation (ChIP) assay: COL cells were transduced with FLAG-tagged HES1. Immunoblot (IB) confirmed expression of HES1 and effective immunoprecipitation (IP) of HES1 with anti-FLAG antibody (upper panels). Sheared chromatin from transfected cells was immunoprecipitated with anti-FLAG antibody or nonspecific IgG as control. Immunoprecipitated DNA was used as template in PCR for HASH1 promoter region (positive control), HASH1 3′ end region (negative control) or DTX1 promoter region (lower panels).
HES1 repressor activity is dependent on recruitment of co-repressors from the Groucho/TLE co-repressor family, which bind to a highly-conserved WRPW motif (Cinnamon and Paroush, 2008). The HES1 C-terminal orange domain, also called helix3-helix4, is involved in the transcriptional repression of target genes (Castella et al., 2000). To determine whether interaction of HES1 with Groucho/TLE co-repressors is critical for inhibition of DTX1, mutant HES1 constructs were used: a truncated HES1 with C-terminal deletion including the orange domain and WRPW motif (HES1-ΔC) and a WRPW domain mutant of HES1 (WRPW mutated to GRPG). Deletion of the entire C-terminus completely reversed HES1 inhibition of the DTX1 promoter, suggesting that active transcriptional repression is likely the mechanism of regulation (Figure 5B). However, the WRPW domain mutant still partially repressed DTX1 promoter activity, suggesting that the Groucho/TLE family of co-repressors may not be the only repressor interacting with HES1. Thus HES1 represses the DTX1 promoter through active transcriptional repression which may require recruitment of the Groucho-TLE co-repressor family through the WRPW motif and other corepressors.
To assess HES1 binding to the DTX1 -400 N-box, we performed an electrophoretic mobility shift assay (EMSA). A specific protein bound to the DNA oligomer containing the DTX1 promoter N-box motif in the nuclear extract of 293T cells transfected with FLAG-HES1 but not in untransfected cells (Figure 5C). A mutant N-box oligomer did not show binding. The oligo-bound protein band was supershifted by anti-FLAG antibody (Figure 5C, lane 4), confirming the identity of HES1 as the oligo-bound protein. Wild type cold oligomers competitively inhibited binding of HES1 to wild type biotin-labeled oligomers, while cold mutant oligomers did not inhibit binding (Figure 5C, lanes 5-6). These results demonstrate that HES1 directly binds to the -400 N-box of DTX1 promoter.
To confirm HES1 interaction with the N-box motif in genomic DNA of whole cells, a chromatin immunoprecipitation (ChIP) assay was performed using FLAG-tagged HES1. Sheared chromatin from osteosarcoma COL cells overexpressing FLAG-HES1 was immunoprecipitated with anti-FLAG or control antibody, followed by PCR amplification of the DTX1 promoter region containing the N-box motif. The human HASH1 promoter region and 3′ terminal region served as positive and negative controls respectively, since HES1 is known to bind the HASH1 promoter (Chen et al., 1997). Western analysis of HES1 in COL cells transduced with FLAG-HES1 confirmed expression of HES1 in whole cell lysate and immunoprecipitation of FLAG-HES1 (Figure 5D upper and middle panel). PCR showed that immunoprecipitation with anti-FLAG pulled down the N-box-containing region of DTX1 promoter, while control antibody did not (Figure 5D lower panels), confirming HES1 binding to this site.
Endogenous HES1 inhibits DTX1 promoter activity
To assess whether endogenous HES1 in osteosarcoma inhibits DTX1 expression by acting on the DTX1 promoter, we tested whether downregulation of HES1 by shRNA or dnHES1 would increase promoter activity, and whether mutation of the -400 N-box would reverse this suppression. OS187 cells were stably transduced with empty vector control or four distinct shRNA targeting HES1 (clone 5, 6, 7 and 8). The shRNA constructs decreased HES1 protein level, compared to vector control (Figure 6A left panels). OS187 cells expressing dnHES1 also were used (Figure 6A right panels). Western blot analysis of DTX1 in these cells showed that DTX1 expression was upregulated when HES1 expression was decreased by shRNA or dnHES1 (Figure 6A), demonstrating that endogenous HES1 represses DTX1 expression. We also transfected wild type (WT) or N-box mutated (mutN) DTX1 promoter reporter constructs in OS187 cells stably transduced with vector control, shHES1 or dnHES1. shRNA clone 6 had the greatest HES1 knock down (70%), and was chosen for promoter activity assay. The WT DTX1 promoter activity, measured by luciferase activity, showed that knockdown of HES1 by shHES1 increased promoter activity 70% (p<0.005) compared to vector control. Similarly, dnHES1 also activates DTX1 promoter by 70% (p<0.005) (Figure 6B). However, mutation of this putative HES1-binding site upregulated DTX1 promoter activity in the vector control and greatly reduced the impact of HES1 shRNA and dnHES1 on DTX1 promoter activity (Figure 6B). These results demonstrate that endogenous HES1 downregulates DTX1 transcription through this N-box motif. Furthermore, mutant DTX1 promoter activity was significantly higher (p<0.05) than wild type promoter, suggesting that endogenous HES1 normally represses this promoter (Figure 6B).
FIG 6.
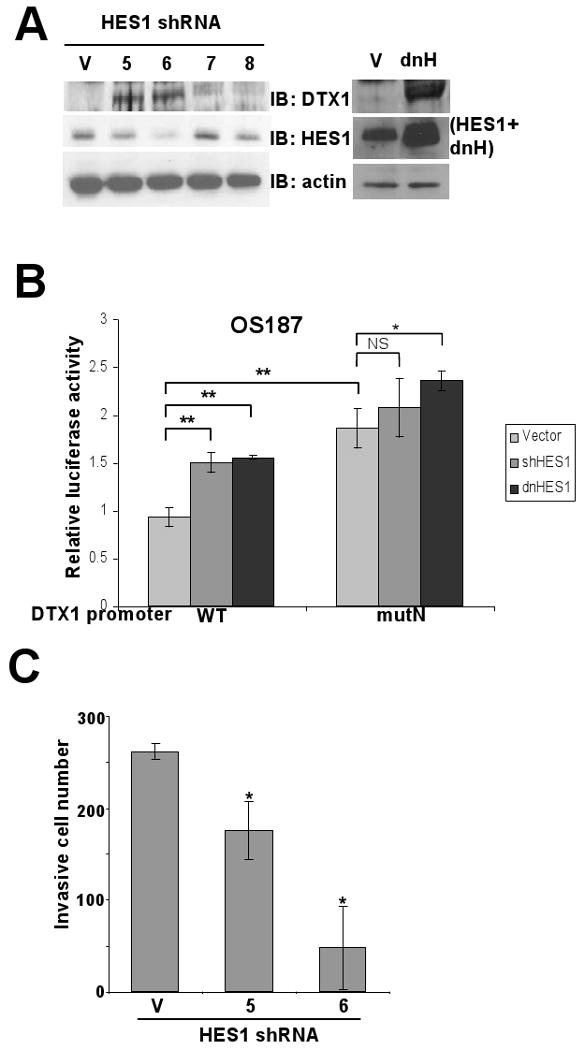
HES1 suppression increases DTX1 promoter activity through the N-box motif. (A) Immunoblot (IB) shows the impact of shRNA to HES1 or of dnHES1 on DTX1 and HES1 expression in OS187. Left panel: V indicates vector control; 5, 6, 7 and 8 indicate four distinct shRNAs specific for HES1. Actin serves as a loading control. Right panel: V indicates vector control; dhH indicates dominant negative HES1. Anti-HES1 antibody can detect both HES1 and dnHES1. (B) Wild type (WT) or N-box mutated (mutN) DTX1 promoter reporter construct (Renilla luciferase) was cotransfected with a firefly luciferase control into OS187 cells stably expressing empty vector, shHES1 or dnHES1. Relative luciferase activity was determined after 48-hr incubation. (*, p<0.05. **, p<0.005). (C) Histograms represent matrigel invasion of OS187 cells stably expressing empty vector or HES1-specific shRNA (clone 5 and 6). Analysis was performed as in figure 1E (*, p<0.05).
To investigate whether shRNAs targeting HES1 decreased osteosarcoma cell invasion, we measured the invasiveness of OS187 cell with vector control, clone 5 and clone 6 in matrigel transwells (Figure 6C). Our results showed that not only did shRNAs knock down HES1 expression and OS187 invasiveness, but the decrease of invasiveness in clone 5 and 6 was proportional to the decrease of HES1. This result confirmed the critical role of HES1 in osteosarcoma cell invasion and metastasis.
Expression of ICN1, DTX1 and HES1 in a panel of osteosarcoma cells
To investigate how generalizable the mutual inhibition of DTX1 and HES1 is, we examined the expression of ICN1, DTX1 and HES1 in human normal osteoblast cells and eight osteosarcoma cell lines (Figure 7A). Western analysis showed that expression of ICN1 and HES1 is positively correlated, while ICN1 and HES1 are negatively correlated to expression of DTX1. Normal osteoblast cells have the highest level of DTX1 and very low level of ICN1 and HES1, suggesting that downregulation of DTX1 and upregulation of NOTCH/HES1 may be an important step in osteosarcoma pathogenesis.
FIG 7.
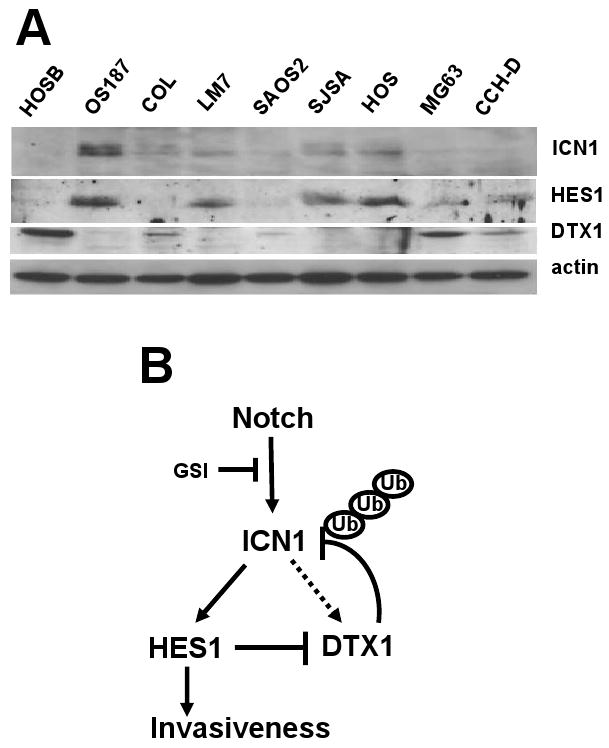
Expression of ICN1, HES1 and DTX1 in osteoblasts and osteosarcoma cells, and model for the regulation of NOTCH signaling by NOTCH target genes HES1 and DTX1. (A) Immunoblots demonstrate expression of ICN1, HES1 and DTX1 in normal human osteoblasts [HOSB] and a panel of human osteosarcoma cell lines. (B) Model: NOTCH1 is cleaved into ICN1 by γ–secretase, and then activates downstream target genes HES1 and DTX1. Inhibitors of γ-secretase can block this step. DTX1 causes negative feedback of ICN1 by targeting ICN1 for ubiquitination and proteasome-mediated degradation. Meanwhile, HES1 causes positive feedback of ICN1 by repressing DTX1 gene expression. HES1 promotes the invasion and metastasis of osteosarcoma. In osteosarcoma cells expressing high HES1 and low DTX1, NOTCH inhibition, such as that caused by GSI, may downregulate HES1, increasing DTX1, which further downregulates ICN1 through ubiquitination, reducing HES1-mediated invasiveness. However, in osteosarcoma cells with high DTX1 and low HES1, NOTCH inhibition may not reduce invasiveness. In fact, reducing DTX1 may stabilize low level ICN1 expression, increasing ICN1 signaling and HES1 expression, causing increased invasion, as happens with COL. Thus the impact of GSI on NOTCH signaling should take into account expression of both HES1 and DTX1.
Discussion
The importance of NOTCH signaling in cell growth, differentiation and survival is well established (Artavanis-Tsakonas et al., 1999). However, NOTCH signaling is highly cell-context dependent, with the potential to activate or inhibit a range of different downstream pathways (Radtke and Raj, 2003). It is likely that the effects of NOTCH signaling are specifically determined by which target genes, such as HES1-7, HERP1-3 and DTX1-4, are activated in a specific cell context. The regulation among these direct target genes and NOTCH receptors is critical for maintaining proper NOTCH signaling in specific cell environment. However, the mechanisms that regulate NOTCH target gene activation and NOTCH receptor stability are not fully understood. The present study reveals a molecular mechanism that regulates NOTCH signaling through reciprocal regulation of the NOTCH target genes HES1 and DTX1 in human cells. More importantly, our analysis has clinical implications for the use of NOTCH inhibitors for osteosarcoma and other cancers.
Our present studies suggest a novel mechanism of reciprocal regulation between the NOTCH target genes HES1 and DTX1: 1) DTX1 negatively regulates NOTCH/HES1 signaling, and 2) HES1 inhibits DTX1 mRNA expression in osteosarcoma cells. Based on our analysis, we propose a model for the regulation of NOTCH signaling by HES1 and DTX1 (Figure 7B). In our model, DTX1, a CSL-dependent NOTCH target gene that also has NOTCH-independent mechanisms of expression, provides negative feedback for NOTCH signaling by targeting ICN1 for ubiquitination and proteasome-mediated degradation. Since our system used a genetically-defined ICN fragment, we know that this mechanism can act on the GSI-cleaved fragment of Notch1. It is possible that DTX1 might also be able to act on full-length Notch1 as well. HES1 regulates this negative feedback through direct transcriptional repression of the DTX1 promoter. Consequently, the impact of NOTCH signaling is affected by a balance of HES1 and DTX1. In osteosarcoma cells, we previously showed that NOTCH signaling, and specifically HES1, promotes invasiveness and metastasis in vivo (Zhang et al., 2008). Inhibition of NOTCH signaling by γ–secretase inhibitor (GSI) blocked osteosarcoma cell invasion in OS187 cells. However, another osteosarcoma cell line, COL, is resistant to GSI treatment (Zhang et al., 2008). In this study, we suggest that invasion may be regulated by the reciprocal inhibition of HES1 and DTX1. If an osteosarcoma cell has high HES1 and low DTX1, such as OS187 cells (Zhang et al., 2008), GSI or other NOTCH inhibitors may repress HES1 expression, and tumor cell invasion. However, if an osteosarcoma has high DTX1 and lower HES1, such as COL, GSI may not suppress tumor cell invasion.
If the regulation of Notch signaling in humans is similar to drosophila, then the model may be even more complex. Matsuno et al. have shown that dominant negative Deltex can function upstream of ICN but downstream of full-length Notch, through binding of the proline-rich domain with the ankyrin repeats of Notch (Matsuno et al., 2002). Their further studies demonstrated that Deltex could mediate ligand-independent activation of Notch (Hori et al., 2004), and that the level of Deltex activity might dictate whether Deltex mediates cleavage and activation of Notch or ubiquitin-mediated lysosomal degradation of Notch and/or ICN (Wilkin et al., 2008). Our data in humans would provide direct mechanistic support for such a model in osteosarcoma.
In our study, the C-terminal region of HES1 was necessary for transcriptional repression of DTX1, but the WRPW motif was dispensible. Previous research has shown that WRPW domain is required for HES1 recruiting the corepressor TLE (Groucho in Drosophila) to the target gene promoter (Cinnamon and Paroush, 2008; Struhl and Adachi, 1998). Our data suggests that the Groucho/TLE binding to the WRPW motif is not essential for HES1 mediated repression of DTX1 in osteosarcoma. Further studies are needed to determine if other corepressors are also involved in the inhibition of DTX1 promoter, e.g. CtBP.
HES1 functions as transcription repressor by forming homodimers or heterodimers with HERP (Iso et al., 2003). dnHES1 may also form heterodimers with HERP protein and regulate HERP function. We found that dnHES1 can upregulate DTX1 gene expression even in COL cells which have low endogeous HES1. In this case, dnHES1 might regulate DTX1 by affecting other interacting partners for HES1 such as HERP2, which is present in COL (Zhang et al., 2008).
DTX1, while clearly a NOTCH target gene, has also been shown to have NOTCH regulatory functions. In Drosophila, the non-visual β–arrestin (Kurtz) is required for mediating the interaction of Dx (Drosophila homolog of DTX1) and NOTCH (Mukherjee et al., 2005). However, DTX1 prevented ICN1 from recruiting coactivators, limiting ICN1 CSL activation in mammalian cells (Jackson et al., 2000). Protein structure studies illustrate that the two WWE repeats in N-terminal domain of Dx form heterodimers with the ankyrin domain of NOTCH (Zweifel et al., 2005). Here we show that DTX1 physically binds to ICN1, inducing ubiquitination of ICN1. Similar to the Dx effects on Notch signaling (Mukherjee et al., 2005), human DTX1 inhibition on ICN1 is not very strong. However, we have not explored the role of human β–arrestin in the DTX1 and ICN1 regulation in human cells. Further studies are needed to examine whether β–arrestin will enhance the DTX1 inhibitory effects on ICN1 in human cells.
NOTCH function in cancers is complex, since NOTCH signaling can be oncogenic or act as a tumor suppressor in different cancer types. For those cancers in which NOTCH functions as an oncogene, NOTCH inhibitors may provide therapeutic benefit. Based on our results, we suggest that in these malignancies the potential benefit of a NOTCH inhibitor may depend on the relative expression of both HES1 and DTX1. GSI might only benefit osteosarcoma patients with tumors expressing high HES1 and low DTX1, but not those with high DTX1. Low DTX1 expression may be useful as a marker to select osteosarcoma patients who could benefit from NOTCH inhibitor treatment. This regulatory mechanism may exist in other types of cancer and should be evaluated, since NOTCH inhibitors such as GSI are being evaluated in clinical trials.
In summary, our study identifies a novel regulatory mechanism of NOTCH signaling by reciprocal inhibition of its own target genes, HES1 and DTX1 in human cells. Our data suggest that DTX1 also plays a role in regulation of osteosarcoma invasiveness, likely through NOTCH/HES1. We believe that this regulation is important not only for our understanding of NOTCH signaling itself but also for its implication on the selection of patients for clinical trials related to NOTCH for cancer.
Materials and Methods
Cell culture and invasion
Human osteosarcoma cell lines OS187, COL, SAOS2, LM7 and KRIB were cultured as described (Hughes et al., 2004). SJSA, MG63 and HOS were cultured in Modified Eagle's Medium (MEM) with 10% fetal bovine serum (FBS), penicillin-streptomycin and L-glutamine. HEK-293T and SH-EP cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) with 10% FBS, penicillin-streptomycin and L-glutamine. Culture of OS 187 cells in GSI Compound E (Calbiochem) and matrigel invasion assay were performed as described (Zhang et al., 2008).
Plasmid constructs and retroviral transduction
MigR1, MigR1-ICN1, MigR1-dnMAM and MigR1-HES1 were described previously (Zhang et al., 2008). HA-tagged human ICN1 and ICN1-Δ162 in pCS2 vectors were provided by Dr. Iannis Aifantis (Thompson et al., 2008). MigR1-dominant negative HES1 (dnHES1) was generated from MigR1-HES1 by based site-mutagenesis (Stratagene). Specific details for construction of the human dhHES1 are included in the supplemental materials.
Immunoprecipitation, western blot, and RT-PCR
Immunoprecipitation and western blot analysis were performed according to manufacturer's instructions. Cells were either untreated or treated with 5 mM EDTA for 15 minutes to cause pan-NOTCH activation, and transduced with retroviral vectors as noted. Antibodies used were: mouse anti-FLAG (Sigma), rabbit anti-HES1 (gift from Dr. Tetsuo Sudo) (13), rabbit anti-ICN (Cell Signaling), rabbit anti-DTX1 (Santa Cruz), goat anti-DTX1 (Santa Cruz), mouse anti-ubiquitin (Cell Signaling), rabbit anti-actin (Cell Signaling), protein A/G beads (Santa Cruz) and goat anti-mouse/rabbit IgG (GE Healthcare). RT-PCR analyses of NOTCH1, HES1, DTX1 and actin were performed as described (Zhang et al., 2008).
Dual-luciferase expression reporter plasmid with human DTX1 promoter
The 1kb fragment upstream from the transcription start site of DTX1 (chromosome 12, NT 009775, NCBI database) was amplified by PCR from human genomic DNA (BglII and NheI sites underlined): Forward primer: 5′-gcagatctagagacgatccagctcctggctt-3′ and reverse primer: 5′-gcgcgctagcgctggctcaggtagggagtga-3′. The promoter activity reporter system was modified from the pRL vector (Promega) by replacing the SV40 early enhancer and T7 promoter (1-684bp) with 1 Kb of the human DTX1 promoter. The HES1 binding site at -400bp was mutated using the QuickChange site-directed mutagenesis kit (Stratagene) and the following primers (mutated sequences underlined): forward primer 5′- cctggcaggtcgcgagagtcaatggggtggggcgtgcc-3′ and reverse primer 5′- ggcacgccccaccccattgactctcgcgacctgccagg-3′.
Transient transfection and luciferase assay
Cells in 6 or 24-well dishes, were transfected in triplicate using Lipofectamine 2000 (Invitrogen) with 3 or 0.6 μg, respectively, of specific plasmids. 48 hours after transfection, cells were washed with prechilled PBS and lysed in passive lysis buffer (Dual Luciferase, Promega). Firefly luciferase and Renilla luciferase activities were measured according to manufacturer's instruction using Monolight 3010 luminometer (Pharmingen). Renilla luciferase activity was normalized to firefly luciferase activity.
Chromatin immunoprecipitation assay (ChIP)
ChIP assay was performed using ChIP assay kit (Upstate) following manufacturer's instructions. COL cells were stably transduced with the FLAG-tagged HES1 construct and cross linked. Chromatin was prepared, sonicated to 0.5-1kb fragments and immunoprecipitated with anti-FLAG-M2 agarose (Sigma) or with control mouse IgG agarose. Immunoprecipitated DNA fragments were eluted and PCR amplified using gene specific primers. For amplification of the DTX1 N-box, the following primers were used: DTX1 promoter forward 5′-gcgcgagccggagccggagc-3′ and DTX1 promoter reverse 5′-aagccaggagctggatcgtctct-3′.
Electrophoresis mobility shift assay (EMSA)
Oligonucleotides containing wild type or mutant HES1 binding site and positive and negative control oligonucleotides for HES1 binding were 5′ labeled with biotin. Forward primer sequences are WT-DTX1: 5′-caggtcgcgagcacgagtggggtggggc-3′, mutant-DTX1: 5′-caggtcgcgagagtcaatggggtggggc-3′. Wild type and mutant HES1 binding sites are underlined. EMSA used the LightShift EMSA Optimization kit (Pierce) and Chemiluminescent Nucleic Acid Detection Module (Pierce) following manufacturer's instructions. Nuclear extract of 293T cells transfected with FLAG-tagged HES1 was prepared as described (Zhang et al., 2008). Binding reactions containing 10 μg of nuclear protein, 10 mM HEPES, 2 μg poly(dI•dC), 50 mM NaCl, 0.1 mM EDTA, 25% glycerol and 2 pmol of biotinylated oligonucleotide probe were incubated for 30 minutes at room temperature. For supershift experiments, anti-FLAG antibody was added and incubated for 30 minutes more. Protein DNA complexes were separated by electrophoresis on a 4% non-denaturing acrylamide gel in 0.5XTBE, transferred to positively charged nylon membranes, and visualized by streptavidin-horseradish peroxidase followed by chemiluminescent detection.
Supplementary Material
Acknowledgments
We thank Drs. Tetsuo Sudo, Anders Ström and Iannis Aifantis for providing reagents, and to Dr. Shufang Jia and Wendy Fang for technical assistance. This work is supported by Physician Scientist Awards from U.T. M.D. Anderson Cancer Center to Dr. D. Hughes and Dr. P. Zweidler-McKay and by NIH grant K08CA118730-04 to Dr. Dennis Hughes. Dr. P. Zhang was supported by the Jori Zemel Children's Bone Cancer Foundation (www.JoriZemel.com) and Hope Street Kids Foundation (www.hopestreetkids.org).
Footnotes
Conflict of Interest Statement: The authors declare no conflicts of interest.
References
- Allenspach E, Maillard I, Aster J, Pear W. Notch signaling in cancer. Cancer Biol Ther. 2002;1:466–76. doi: 10.4161/cbt.1.5.159. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Axelson H. The Notch signaling cascade in neuroblastoma: role of the basic helix-loop-helix proteins HASH-1 and HES-1. Cancer Lett. 2004;204:171–8. doi: 10.1016/S0304-3835(03)00453-1. [DOI] [PubMed] [Google Scholar]
- Castella P, Sawai S, Nakao K, Wagner JA, Caudy M. HES-1 Repression of Differentiation and Proliferation in PC12 Cells: Role for the Helix 3-Helix 4 Domain in Transcription Repression. Mol Cell Biol. 2000;20:6170–6183. doi: 10.1128/mcb.20.16.6170-6183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fischer WH, Gill GN. Regulation of the ERBB-2 Promoter by RBPJkappa and NOTCH. J Biol Chem. 1997;272:14110–14114. doi: 10.1074/jbc.272.22.14110. [DOI] [PubMed] [Google Scholar]
- Cinnamon E, Paroush Z. Context-dependent regulation of Groucho/TLE-mediated repression. Curr Opin Genet Dev. 2008;18:435–40. doi: 10.1016/j.gde.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Fortini M. Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol. 2002;3:673–84. doi: 10.1038/nrm910. [DOI] [PubMed] [Google Scholar]
- Hori K, Fostier M, Ito M, Fuwa TJ, Go MJ, Okano H, et al. Drosophila Deltex mediates Suppressor of Hairless-independent and late-endosomal activation of Notch signaling. Development. 2004;131:5527–5537. doi: 10.1242/dev.01448. [DOI] [PubMed] [Google Scholar]
- Hughes DPM, Thomas DG, Giordano TJ, Baker LH, McDonagh KT. Cell Surface Expression of Epidermal Growth Factor Receptor and Her-2 with Nuclear Expression of Her-4 in Primary Osteosarcoma. Cancer Res. 2004;64:2047–2053. doi: 10.1158/0008-5472.can-03-3096. [DOI] [PubMed] [Google Scholar]
- Iso T, Kedes L, Hamamori Y. HES and HERP families: Multiple effectors of the notch signaling pathway. Journal of Cellular Physiology. 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- Itoh M, Kim C, Palardy G, Oda T, Jiang Y, Maust D, et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev Cell. 2003;4:67–82. doi: 10.1016/s1534-5807(02)00409-4. [DOI] [PubMed] [Google Scholar]
- Izon DJ, Aster JC, He Y, Weng A, Karnell FG, Patriub V, et al. Deltex1 Redirects Lymphoid Progenitors to the B Cell Lineage by Antagonizing Notch1. Immunity. 2002;16:231–243. doi: 10.1016/s1074-7613(02)00271-6. [DOI] [PubMed] [Google Scholar]
- Jackson P, Eldridge A, Freed E, Furstenthal L, Hsu J, Kaiser B, et al. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 2000;10:429–39. doi: 10.1016/s0962-8924(00)01834-1. [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter E, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–8. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Liu WH, Lai MZ. Deltex Regulates T-Cell Activation by Targeted Degradation of Active MEKK1. Mol Cell Biol. 2005;25:1367–1378. doi: 10.1128/MCB.25.4.1367-1378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K, Eastman D, Mitsiades T, Quinn AM, Carcanciu ML, Ordentlich P, et al. Human deltex is a conserved regulator of Notch signalling. 1998;19:74–78. doi: 10.1038/ng0598-74. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Ito M, Hori K, Miyashita F, Suzuki S, Kishi N, et al. Involvement of a proline-rich motif and RING-H2 finger of Deltex in the regulation of Notch signaling. Development. 2002;129:1049–1059. doi: 10.1242/dev.129.4.1049. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Veraksa A, Bauer A, Rosse C, Camonis J, Artavanis-Tsakona S. Regulation of Notch signalling by non-visual beta-arrestin. Nat Cell Biol. 2005;7:1191–201. doi: 10.1038/ncb1327. [DOI] [PubMed] [Google Scholar]
- Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3:756–67. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- Sestan N, Artavanis-Tsakonas S, Rakic P. Contact-Dependent Inhibition of Cortical Neurite Growth Mediated by Notch Signaling. Science. 1999;286:741–746. doi: 10.1126/science.286.5440.741. [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Nuclear Access and Action of Notch In Vivo. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- Takebayashi K, Sasai Y, Sakai Y, Watanabe T, Nakanishi S, Kageyama R. Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. J Biol Chem. 1994;269:5150–5156. [PubMed] [Google Scholar]
- Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008;205:1395–1408. doi: 10.1084/jem.20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP, IV, Silverman LB, Sanchez-Irizarry C, et al. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Wilkin M, Tongngok P, Gensch N, Clemence S, Motoki M, Yamada K, et al. Drosophila HOPS and AP-3 complex genes are required for a Deltex-regulated activation of notch in the endosomal trafficking pathway. Dev Cell. 2008;15:762–72. doi: 10.1016/j.devcel.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, et al. SEL-10 Is an Inhibitor of Notch Signaling That Targets Notch for Ubiquitin-Mediated Protein Degradation. Mol Cell Biol. 2001;21:7403–7415. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Yang Y, Zweidler-McKay PA, Hughes DPM. Critical Role of Notch Signaling in Osteosarcoma Invasion and Metastasis. Clin Cancer Res. 2008;14:2962–2969. doi: 10.1158/1078-0432.CCR-07-1992. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zweidler-McKay P, Pear W. Notch and T cell malignancy. Semin Cancer Biol. 2004;14:329–40. doi: 10.1016/j.semcancer.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, Carpenter AC, et al. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood. 2005;106:3898–3906. doi: 10.1182/blood-2005-01-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel M, Leahy D, Barrick D. Structure and Notch receptor binding of the tandem WWE domain of Deltex. Structure. 2005;13:1599–611. doi: 10.1016/j.str.2005.07.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.