

Abstract
Cancer cells have distinct metabolic needs that are different from normal cells and can be exploited for development of anti-cancer therapeutics. Activation of the tumor specific M2 form of pyruvate kinase (PKM2) is a potential strategy for returning cancer cells to a metabolic state characteristic of normal cells. Here, we describe activators of PKM2 based upon a substituted thieno[3,2-b]pyrrole[3,2-d]pyridazinone scaffold. The synthesis of these agents, structure activity relationships, analysis of activity at related targets (PKM1, PKR and PKL) and examination of aqueous solubility are investigated. These agents represent the second reported chemotype for activation of PKM2.
Keywords: Warburg effect, pyruvate kinase, cellular metabolism, anti-cancer strategies, small molecule activators
One of the most prominent distinctions between healthy and cancerous tissues is the differing energetic and nutritional needs associated with the rapid proliferative nature of cancer cells.1,2 The original observation that cancer cells maintain a different metabolic state relative to non-proliferating cells was made by Otto Warburg in the 1920’s and today the Warburg effect is a highly studied area of research.3,4 In normal cells, glucose is primarily metabolized by glycolysis and oxidative phosphorylation when oxygen is available. Glycolysis is a multistep process that ultimately converts glucose into two equivalents of pyruvate. Further metabolism by oxidative phosphorylation involves the conversion of pyruvate to acetyl-CoA and entry into the Krebs cycle with the potential of generating additional ATP. In cancer cells this process is altered and much of the pyruvate derived from glucose is instead converted to lactic acid even in aerobic conditions. The mechanism(s) that drive this altered metabolism in cancer cells are not fully understood. One contributor, however, is the differential expression of two isozymes of pyruvate kinase (PK).5 Tanaka and coworkers were the first to show that alternative RNA splicing yields the M1 and M2 forms of pyruvate kinase (PKM1 and PKM2).6,7,8 PKM2 is widely expressed in undifferentiated embryonic tissues and during development many differentiated tissues switch to express PKM1.8–10 A second gene produces two additional PK isozymes based on alternative splicing events to produce PKL and PKR which are expressed in specific adult tissues.11 An important realization in cancer biology was the recognition that all cancer cells express the PKM2 isozyme.5,10,12–15
The expression of PKM2 in cancer cells has been described as a clinical marker of malignancy for some time.16 Pyruvate kinase catalyzes the transformation of phosphoenolpyruvate (PEP) and ADP to pyruvate and ATP when the enzyme exists as a homotetramer (dimer of dimers). Three PK isozymes (PKM2, PKL and PKR) require the binding of fructose-1,6-bis-phosphate (FBP) at an allosteric site in the tetrameric complex for activity.17 In contrast, PKM1 does not require allosteric activation by FBP binding and retains a high affinity for PEP and high catalytic rate in its native state. Importantly, in cancer cells PKM2 exists as a less active dimer with little affinity for PEP and a low catalytic rate.5 It is theorized that down-regulation of the final step of glycolysis is a functional reason for the expression of PKM2 in cancer cells. Many glycolytic intermediates are starting points for amino acid, nucleic acid and lipid biosynthesis. Decreased pyruvate kinase activity is hypothesized to facilitate the shunting of glycolytic intermediates into these anabolic pathways required for cell growth. Hence, the expression of the less active PKM2 isozyme in cancer cells may enable anabolic processes necessary for cancer cell proliferation.
PKM1 expressing cells have a diminished capacity for tumor development in vivo relative to PKM2 expressing cells.18 One unique property that appears to give PKM2 expressing cells this proliferative advantage is that PKM2 is a phosphotyrosine binding protein.19 In cancer cells there is a general up-regulation of signalling events through the actions of protein kinases resulting in an elevated state of phosphorylated proteins relative to normal differentiated cells. It has been demonstrated that the binding of phosphorylated peptides to PKM2 is accompanied by the release of FBP and the further down-regulation of this enzyme. This down-regulation exacerbates the shift away from the catalytic levels associated with PKM1 and toward an anabolic state providing the needed resources for rapid proliferation.
The expression of PKM2 in cancer cells provides an attractive target for cancer therapy. Further, the observed down-regulation of PKM2 activity in cancer cells relative to the high PKM1 activity present in many normal cells suggests a therapeutic strategy whereby activation of PKM2 may restore normal cellular metabolism and, consequently, decreased cellular proliferation. To allow us to examine this possibility we have developed small molecule activators of PKM2 based upon a bis-sulfonamide scaffold previously reported20 and the substituted thieno[3,2-b]pyrrole[3,2-d]pyridazinones reported herein.
The development of the luminescent assay detection system for protein kinases and screening details were previously presented.20–22 Akin to our earlier report, to assess the activity of the compounds described here we utilized orthogonal assays that respond to ATP or pyruvate generation by PKM2 through measurement of either firefly luciferase activity or lactate dehydrogenase (LDH) activity, respectively. The data reported in Tables 1–3 was generated in the firefly luciferase assay system.23 A quantitative high-throughput screen (qHTS)24 of nearly 300,000 small molecules of the NIH Molecular Libraries Small Molecule Repository was performed utilizing the firefly luciferase assay system. Numerous small molecule activators of PKM2 were identified. We ultimately selected two chemotypes for advanced interrogation. These agents are represented by the substituted thieno[3,2-b]pyrrole[3,2-d]pyridazinone 1 and substituted N,N′-diarylsulfonamide 2 shown in Figure 1.
Table 1.
SAR of selected thieno[3,2-b]pyrrole[3,2-d]pyridazinones and thieno[3,2-b]pyrrole[3,2-d]pyrimidinones
| # | R1 | R2 | R3 | X | Y | hPK, M2 AC50(μM)a | hPK, M2 Max. Resb | |
|---|---|---|---|---|---|---|---|---|
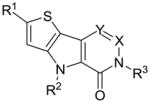 1,3–25 |
1 | Me | Me | 2-fluorobenzyl | N | CH | 0.063 | 122 |
| 3 | Et | Me | 2-fluorobenzyl | N | CH | 0.100 | 105 | |
| 4 | iPr | Me | 2-fluorobenzyl | N | CH | 0.142 | 106 | |
| 5 | H | Me | 2-fluorobenzyl | N | CH | 0.605 | 93 | |
| 6 | OMe | Me | 2-fluorobenzyl | N | CH | 0.086 | 107 | |
| 7 | SMe | Me | 2-fluorobenzyl | N | CH | 0.024 | 96 | |
| 8 | S(O)Me | Me | 2-fluorobenzyl | N | CH | 0.025 | 98 | |
| 9 | S(O)2Me | Me | 2-fluorobenzyl | N | CH | inactive | NA | |
| 10 | NO2 | Me | 2-fluorobenzyl | N | CH | 0.018 | 113 | |
| 11 | NHAc | Me | 2-fluorobenzyl | N | CH | >25 | 59 | |
| 12 | CN | Me | 2-fluorobenzyl | N | CH | 0.047 | 84 | |
| 13 | COOMe | Me | 2-fluorobenzyl | N | CH | 0.084 | 70 | |
| 14 | CHO | Me | 2-fluorobenzyl | N | CH | 0.016 | 100 | |
| 15 | CH2OH | Me | 2-fluorobenzyl | N | CH | 0.048 | 103 | |
| 16 | B(OH)2 | Me | 2-fluorobenzyl | N | CH | >10 | 101 | |
| 17 | COMe | Me | 2-fluorobenzyl | N | CH | 0.011 | 108 | |
| 18 | CHOH(Me) | Me | 2-fluorobenzyl | N | CH | 0.136 | 120 | |
| 19 | Me | H | 2-fluorobenzyl | N | CH | NA | 33 | |
| 20 | Me | Et | 2-fluorobenzyl | N | CH | 5.9 | 96 | |
| 21 | Me | iPr | 2-fluorobenzyl | N | CH | inactive | NA | |
| 22 | Me | Me | 2-fluorobenzyl | N | C(Me) | >30 | 56 | |
| 23 | H | Me | 2-fluorobenzyl | CH | N | >35 | 61 | |
| 24 | Me | Me | phenyl | N | CH | inactive | NA | |
| 25 | Me | Me | n-pentyl | N | CH | >35 | 47 |
AC50 values were determined utilizing the luminescent pyruvate kinase-luciferase coupled assay (ref. 21).
Max. Res. value represents the % activation at 57 μM of compound. Each value is the mean from three replicate experiments.
Table 3.
SAR and selected thieno[3,2-b]pyrrole[3,2-d]pyridazinones
| # | R1 | R3 | hPK, M2 AC50 (μM) | hPK, M2 Max. Res. | Solubilityc(μg/ml) | |
|---|---|---|---|---|---|---|
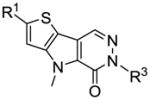 1,8,45–47 |
1 | Me | 2-fluorobenzyl | 0.063 | 122 | <0.2 |
| 8 | S(O)Me | 2-fluorobenzyl | 0.025 | 98 | 4.4 | |
| 45 | S(O)Me | 3-methoxybenzyl | 0.073 | 99 | 37.4 | |
| 46 | S(O)Me | 3 -(methyl)aniline | 0.092 | 93 | 29.6 | |
| 47 | S(O)Me | 3 -(methyl)phenol | 0.130 | 102 | 16.9 |
AC50 values were determined utilizing the luminescent pyruvate kinase-luciferase coupled assay (ref. 21) and the data represents the results from three separate experiments.
Max. Res. value represents the % activation at 57 μM of compound.
Kinetic solubility analysis was performed by Analiza Inc. and are based upon quantitative nitrogen detection as described (www.analiza.com). The data represents results from three separate experiments with an average intraassay %CV of 4.5%
Figure 1.

Chemical structures of the two lead activators of PKM2 substituted thieno[3,2-b]pyrrole[3,2-d]pyridazinone NCGC00031955 (1) and substituted N,N′-diarylsulfonamide NCGC00030335 (2).
In a similar manner as the recent report detailing NCGC00030335 (2)20, it was essential to establish the cooperative nature of these agents with the native substrates of PKM2. Given the allosteric activation of PKM2 by FBP, it was desirable to examine how our lead chemotypes affected the steady-state kinetics of PEP and ADP. In the absence of activator, hPK shows low affinity for PEP (KM ~ 1.5 mM). In the presence of NCGC00031955 (1) or FBP, the KM for PEP decreased 10-fold to 0.13±0.04 mM or 0.1±0.02 mM, respectively with lesser effects on Vmax (values of 245 pmols/min with or without FBP and 255 pmols/min with or without NCGC00031955). In contrast, variation of the concentration of ADP in the presence and absence of activators shows that the steady-state kinetics are not significantly affected (KM for ADP = 0.1 mM in either condition). Thus, NCGC00031955 (1) activates PKM2 by increasing the enzyme’s affinity for PEP and has little effect on ADP kinetics (Figure 2A). This is similar to what we observed for FBP (Figure 2B) which agrees with previous reports demonstrating increased affinity for PEP as the reason for activation of PKM2 by FBP.17
Figure 2.

A. Initial velocity of PKM2 as a function of PEP and ADP concentration in the presence (open squares) or absence (filled circles) of NCGC00031955 (1) (10 μM). B. Initial velocity as a function of PEP and ADP concentration in the presence (open circles) or absence (filled circles) of FBP (10 μM). Vo, initial rate in pmol/min as determined in the PK-LDH coupled assay (kinetic assays were carried out at [KCl] = 200 mM, [MgCl2] = 15 mM, and with either [ADP] or [PEP] = 4.0 mM; see supporting information).
The sequence required for the chemical synthesis of NCGC00031955 (1) was an assembly of literature procedures (Scheme 1). Several commercially available thiophene-2-carbaldehydes were reacted with ethyl 2-azidoacetate in sodium ethoxide at 0 °C to provide the corresponding 2-azido-3-(thiophen-2-yl)acrylates. Refluxing this intermediate in o-xylene provided the core thienopyrroles in good yields.25 Several techniques proved capable of installing the needed aldehyde functionality, but ultimately we relied upon a Vilsmeier-Haack reaction to form the substituted ethyl 6-formyl-4H-thieno[3,2-b]pyrrole-5-carboxylates.26 There was no indication of alternate regiochemical formyl installation. Through a series of experiments, we found that it was necessary to alkylate the pyrrole nitrogen before proceeding with the synthesis. This was accomplished via treatment with alkyl iodides in basic DMF. The remainder of the synthesis involved the formation of the pyridazinone via treatment with hydrazine in refluxing 2-ethoxyethanol and alkylation of the amide nitrogen with various alkyl and benzyl bromides in basic DMF.27 Arylation of the amide nitrogen was also explored through a copper catalyzed process developed by Buchwald and coworkers.28
Scheme 1.

Conditions and reagents: (i) Na, EtOH, 0 °C; (ii) o-Xylene, reflux; (iii) POCI3, DMF, 60 °C; (iv) R2I, K2C03, DMF, r.t.; (v) 2-Ethoxyethanol, hydrazine, reflux; (vi) Benzyl bromide or alkyl bromide, KOtert-Bu, DMF, r.t.; (vii) iodobenzene, Cul, trans-cyclohexane-1,2-diamine, 1,4-dioxane, reflux.
The utility of 5-bromothiophene-2-carbaldehyde as a starting reagent in this sequence (see supporting information) was a key to the synthetic elaboration of numerous analogues (Scheme 2). From the 2-bromo product 53 (shown in Scheme 2) we conducted numerous transformations. Treatment with sodium methoxide in refluxing 1,4-dioxane in the presence of copper iodide provided the 2-methoxy derivative 6 in good yield.29 Copper catalysis was again used for the installation of acetamide to provide direct access to the NHAc derivative 11.28 The nitrile analogue 12 was achieved through treatment of the bromide with CuCN in DMF at elevated temperatures.29 Palladium (0) catalysis, carbon monoxide and triethylamine in a MeOH/DMSO solution proved to be a successful strategy to install the methyl ester moiety of 13. Given the lack of commercially available of 5-alkyl substituted thiophene-2-carbaldehydes we were forced to explore alternate means to examine the SAR of this position. Either vinyl or isopropenylboronic acids/pinacol esters were entered into traditional Suzuki-Miyaura couplings to provide derivatives that, upon reduction, yielded the ethyl or isopropyl derivatives 3 and 4. Using standard reductive conditions on the starting bromide provided analogue 5 for study.
Scheme 2.

Conditions and reagents: (i) Na, MeOH, Cul, 1,4-dioxane, reflux; (ii) Acetamide, Cul, trans-cyclohexane-1,2-diamine, dioxane, reflux; (iii) CuCN, DMF, 140 °C; (iv) CO (1 atm), Pd(OAc)2,1,3-bis(diphenylphosphino)propane, Et3N, MeOH, DMSO, 65 °C. (v) vinyl or isopropenyboronic acid pinacol ester, Pd(PPh3)2CI2,1M Na2CO3/CH3CN, 120 °C, microwave; (vi) Pd/C, H2 (1 atm), MeOH, r.t.; (vii) iPrMgBr, tetramethylethylenediamine, THF, 15 °C, 20 min, then starting material, r.t., 25 min; (viii) B(OMe)3, 0 °C, then 0.1 N HCl; (ix) CH3CHO, 0 °C; (x) procedure ix followed by IBX, DMSO, r.t.; (xi) NaSMe, CuBr, DMF, 140 °C; (xii) mCPBA (1.5 eq.), CH2CI2, r.t.;
Preparation of the Grignard reagent was accomplished through metal-halogen exchange and exposure of this intermediate to trimethyl borate at 0 °C followed by work-up in 0.1 N aqueous HCl provided the boronic acid analogue 16.30 Alternatively, quenching of the Grignard reagent with acetaldehyde provided the secondary alcohol 18 which was further oxidized to the ketone 17 with IBX in DMSO. Treatment of 53 with sodium methanethiolate and copper(I)bromide in DMF at 140 °C provided the thiol ether 7 and mCPBA oxidation yielded sulfoxide 8 and sulfone 9 which were separable through chromatographic methods.31
Our attempts to synthesize an analogue bearing a nitro group at the 2-position of the thiophene ring of the final product were met with several challenges. First, the 5-nitrothiophene-2-carbaldehyde starting material could not be converted to the azido-acrylate intermediate. Nitration of the ethyl 4H-thieno[3,2-b]pyrrole-5-carboxylate intermediate lead to nitration at both the 2 and 6 positions. While the mixture could be separated, the 2-position nitrated product could not further undergo Vilsmeier-Haack reaction. Ultimately, we relied upon nitration following installation of the aldehyde moiety at the 6-position, thus driving the nitration to the appropriate 2-position of the heterocycle (Scheme 3). The remainder of the synthesis followed a similar course. However, we did find that the formation of the pyridazinone ring was more facile proceeding in ethanol at room temperature.
Scheme 3.

Conditions and reagents: (i) POCI3, DMF, 60 °C; (ii) Cu(NO3)2, Ac2O, 0 °C to r.t.; (iii) Mel, K2CO3, DMF; (iv) hydrazine, EtOH, r.t.; (v) 2-fluororobenzyl bromide, K2CO3, DMF, r.t..
We noted early that the solubility of these analogues would require improvement to generate appropriate compounds. Therefore, several mechanisms were explored to insert hydrogen bond donors into the core structure. One method for accomplishing this was to enter the un-substituted derivative 5 into a second Vilsmeier-Haack reaction at the 2-position of the thiophene ring to produce the aldehyde 14 (Scheme 4).32 Reduction of this agent with sodium borohydride in methanol provided the alcohol 15 for examination.
Scheme 4.

Conditions and reagents: (i) POCI3, DMF, CICH2CH2CI, reflux; (ii) NaBH4, MeOH.
One of our final SAR considerations involved changes directly on or to the pyridazinone ring. The 6-position of this ring system was the only position open for modification. To examine if substituents could be added at the lone un-substituted carbon, the aldehyde was converted to the methyl ketone through addition of a methyl Grignard reagent and IBX oxidation of the resulting secondary alcohol (Scheme 5). From this intermediate, steps iv through vi of Scheme 1 were used to produce the 6-methyl-pyridazinone version of our lead compound. A second consideration was changing from a pyridazinone to a pyrimidinone ring system (Scheme 6). To accomplish this, we took advantage of our previous observation that nitration of the ethyl 4H-thieno[3,2-b]pyrrole-5-carboxylate intermediate occurred on the 6-position of the pyrrole ring. Reduction of the nitro group was achieved via treatment with tin (II) chloride in acidic EtOH/H2O and the pyrimidinone ring was formed upon condensation with ammonia formate and formamide at elevated temperatures.33,34 The benzylation of the amide nitrogen occurred under similar conditions as previously described.
Scheme 5.

Conditions and reagents: (i) MeMgCI, THF, −78 °C; (ii) IBX, DMSO, r.t.. (iii) steps iv through vi (scheme 1).
Scheme 6.

Conditions and reagents: (i) Cu(NO3)2, Ac2O, 0 °C to r.t.; (ii) Mel, K2CO3, DMF; (iii) SnCI2, HCl, EtOH/H2O, 35 °C; (iv) NH2CHO, ammonium formate, 120°C; (v) 2-fluorobenzyl bromide, K2CO3, EtOH, reflux.
As a standard practice, NCGC00031955 (1) was re-synthesized and found to possess an AC50 value of 63 nM and maximum response of 122% in the ATP generation assay system and also showed good potency and efficacy in the LDH coupled reaction (AC50 value of 326 nM, maximum response of 224%). Our first SAR evaluations involved changes directly to the heterocyclic core structure while retaining the standard 2-fluorobenzyl substitution from the pyridazinone ring amide (Table 1). Steric expansions of the methyl group at the 2-position of the thiophene ring were typically well tolerated [for instance the ethyl and isopropyl analogues 3 (AC50 = 100 nM, maximum response = 105%) and 4 (AC50 = 142 nM, maximum response = 106%)]. In general, comparable potencies for these compounds were observed in the LDH assay, yet the efficacies were typically 2–3 fold higher (this was a general trend for all analogues). Removal of the methyl group resulted in a loss of potency and efficacy [see 5 (AC50 = 605 nM, maximum response = 93%)]. Insertions of heteroatoms (including oxygen and sulfur) typically resulted in improved potency including SMe [see 7 (AC50 = 24 nM, maximum response = 96%)] and S(O)Me [see 8 (AC50 = 25 nM, maximum response = 98%)]. Interestingly, oxidation past the sulfoxide to the sulfone resulted in a completely inactive analogue. Carbonyls and alcohols were examined and found to retain good potencies and maximum responses [for instance 14 (AC50 = 16 nM, maximum response = 100%), 15 (AC50 = 48 nM, maximum response = 103%) and 17 (AC50 = 11 nM, maximum response = 108%)]. In stark contrast to substitutions on the 2-position of the thiophene ring, the methyl group on the pyrrole ring nitrogen was found to be an absolute necessity. Alterations from the methyl to the ethyl and isopropyl groups were ineffective and lack of substitution resulted in an inactive analogue as well. Further, amides and sulfonamides were examined and were not tolerated (data not shown). Addition of a methyl group to the 6 position of the pyridazinone ring was also not allowed [see 22 (AC50 > 30 μM, maximum response <80%)]. Alteration from the pyridazinone to a pyrimidinone ring system was additionally problematic [see 23 (AC50 > 35 μM, maximum response <80%)]. The necessity of the benzyl substituent was proven through examination of the corresponding phenyl analogue 24 and the n-pentyl analogue 25, both of which had marked loss of potency.
Following the examination of the core heterocycle and selected appendages, a phenyl ring scan on the benzyl substituent was performed. The results suggest a less focused SAR for this moiety; however, selected trends did exist. For instance, bulky substituents were typically not successful at the para position of the ring [for instance 31 (AC50 = 326 nM, maximum response = 91%), 33 (AC50 = 553 nM, maximum response < 80%) and 43 (AC50 > 15 μM, maximum response <80%)]. Electron withdrawing substitutions were typically favored [for instance 35 (AC50 = 44 nM, maximum response = 96%), 36 (AC50 = 49 nM, maximum response = 94%)], however examples such as the 4-methoxybenzyl analogue 34 were exceptions (AC50 = 37 nM, maximum response = 96%).
With a better idea of the SAR for this chemotype, we next considered the aqueous solubility of these agents. Several of the most potent analogues were profiled by a commercial provider35 of solubility and it was determined that nearly all compounds examined had aqueous solubility levels (measured in both μg/mL and μM) below detectable limits. One of the few exceptions was the sulfoxide analogue 8 which had an aqueous solubility of 4.4 μg/mL. To expand upon this result, we synthesized numerous phenyl ring analogues that maintained the key sulfoxide moiety. We choose to expand our phenyl ring scan to include aniline and phenol derivatives in hopes of gaining additional solubility. Similarly to our previous studies with the bis-sulfonamide chemotype, there was a preference for meta-substituted analogues (Table 3). Additionally, several of these agents possessed improved aqueous solubility including 45 (AC50 = 73 nM, maximum response = 99%, aqueous solubility = 37.4 μg/mL), 46 (AC50 = 92 nM, maximum response = 93%, aqueous solubility = 29.6 μg/mL) and 47 (AC50 = 130 nM, maximum response = 102%, aqueous solubility = 16.9 μg/mL).
With the SAR and solubility assessments established, it was essential to consider the selectivity of these compounds versus PKM1, PKR and PKL. The N,N′-diarylsulfonamide chemotype presented in our previous manuscript possessed a high degree of selectivity for activation of PKM2. Gratifyingly, the substituted thieno[3,2-b]pyrrole[3,2-d]pyridazinones presented here were equally selective for PKM2 activation versus PKM1. Further, all analogues examined were inactive versus PKL and PKR (see PubChem AIDs 1541, 1542, 1543, 1780, 1781, and 1782). Figure 3 details the selectivity of NCGC00031955 (1) versus PKM2, PKM1, PKR and PKL.
Figure 3.

Selectivity assessment for NCGC00031955 (1) versus PKM2 (open circles), PKM1 (filled squares), PKL (open squares), and PKR (filled circles).
Warburg’s finding that cancer cells show altered cellular respiration and metabolism ranks as one of the earliest observations in cancer biology. A key realization associated with the Warburg effect is the re-expression of PKM2 in all cancer cells leading to increased availability of glycolytic intermediates for biosynthesis of the amino acid, nucleic acid and lipid building blocks of cellular construction. The native, down-regulated kinetics of PKM2 in combination with the allosteric control of PKM2 activity by FBP and binding to phosphotyrosine proteins is an important aspect of the altered metabolic state of cancer cells. Activation of PKM2 to levels comparable to PKM1 represents an intriguing potential strategy to halt the proliferative state of cancer cells by shuttling the required glycolytic intermediates/cellular building blocks away from an anabolic state of metabolism. Here we describe a novel chemotype based upon substituted thieno[3,2-b]pyrrole[3,2-d]pyridazinone scaffold for the activation of PKM2. This agent was expanded upon via chemical synthesis and numerous SAR aspects were explored. These agents were found to increase the affinity of PKM2 for phosphoenolpyruvate (PEP) and were capable of activation responses beyond that achieved by the native allosteric activator FBP. Several agents were found with potencies below 100 nM and maximum responses ≥ FBP-mediated activation and appropriate aqueous solubility including 45, 46 and 47. Explorations of these agents selectivity revealed that the thieno[3,2-b]pyrrole[3,2-d]pyridazinone chemotype was selective for PKM2 with little or no activity versus PKM1, PKL and PKR. These novel activators of PKM2 provide the necessary tool compounds to explore the hypothesis that PKM2 activation will ameliorate the Warburg effect, and thereby decrease cancer cell proliferation.
Supplementary Material
Table 2.
SAR scan of benzylated thieno[3,2-b]pyrrole[3,2-d]pyridazinones
| # | R3 | hPK, M2 AC50(μM)a | hPK, M2 Max. Resb | |
|---|---|---|---|---|
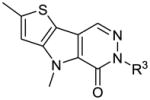 1, 26–44 |
1 | 2-fluorobenzyl | 0.063 | 122 |
| 26 | benzyl | 0.062 | 101 | |
| 27 | 3-fluorobenzyl | 0.225 | 92 | |
| 28 | 4-fluorobenzyl | 0.057 | 102 | |
| 29 | 2-chlorobenzyl | 0.298 | 96 | |
| 30 | 3-chlorobenzyl | 0.126 | 99 | |
| 31 | 4-chlorobenzyl | 0.326 | 91 | |
| 32 | 4-methylbenzyl | 0.356 | 84 | |
| 33 | 4-trifluoromethylbenzyl | 0.553 | 56 | |
| 34 | 4-methoxybenzyl | 0.037 | 96 | |
| 35 | 2,4-difluorobenzyl | 0.044 | 96 | |
| 36 | 2,6-difluorobenzyl | 0.049 | 94 | |
| 37 | 2,3-difluorobenzyl | 0.215 | 73 | |
| 38 | 2-chloro-6-fluorobenzyl | 0.060 | 93 | |
| 39 | 2,3,4-trifluorobenzyl | 0.174 | 69 | |
| 40 | 2,3,5,6-tetrafluorobenzyl | 0.345 | 59 | |
| 41 | 2-fluoro-3-methylbenzyl | 0.035 | 97 | |
| 42 | 2-fluoro-4-methylbenzyl | 0.108 | 81 | |
| 43 | 2-fluoro-4-trifluoromethylbenzyl | >15 | 59 | |
| 44 | 3 -fluoro-4-methoxybenzyl | 0.225 | 68 |
AC50 values were determined utilizing the luminescent pyruvate kinase-luciferase coupled assay (ref. 21).
Max. Res. value represents the % activation at 57 μM of compound. Each value is the mean from three replicate experiments.
Acknowledgments
The authors thank Jeremy Smith, Paul Shinn, and Danielle van Leer for assistance with compound management. We thank Ms. Allison Mandich for critical reading of this manuscript. This research was supported by the Molecular Libraries Program of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health and R03MH085679. The Structural Genomics Consortium is a registered charity (no. 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust.
Footnotes
Supporting Information Available: Assay protocols and experimental procedures and spectroscopic data (1HNMR, LC/MS and HRMS) are listed for all compounds. This material is available free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Devita VT Jr, Hellman S, Rosenberg SA, editors. Cancer Principles & Practice of Oncology. 7. Lippincott Williams & Wilkins; Philadelphia (PA): 2005. [Google Scholar]
- 2.Vander Heiden MG, Cantley LC, Thompson CB. Science. 2009;324:1029. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O. Science. 1956;123:309. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Warburg O. Science. 1956;124:269. [PubMed] [Google Scholar]
- 5.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Semin Cancer Biol. 2005;15:300. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Takenaka M, Noguchi T, Sadahiro S, Hirai H, Yamada K, Matsuda T, Imai E, Tanaka T. Eur J Biochem. 1991;198:101. doi: 10.1111/j.1432-1033.1991.tb15991.x. [DOI] [PubMed] [Google Scholar]
- 7.Takenaka M, Yamada K, Lu T, Kang R, Tanaka T, Noguchi T. Eur J Biochem. 1996;235:366. doi: 10.1111/j.1432-1033.1996.00366.x. [DOI] [PubMed] [Google Scholar]
- 8.Noguchi T, Inoue H, Tanaka T. J Biol Chem. 1986;261:13807. [PubMed] [Google Scholar]
- 9.Yamada K, Noguchi T. Biochem Biophys Res Commun. 1999;256:257. doi: 10.1006/bbrc.1999.0228. [DOI] [PubMed] [Google Scholar]
- 10.Reinacher M, Eigenbrodt E. Virchows Arch B Cell Pathol Incl Mol Pathol. 1981;37:79. doi: 10.1007/BF02892557. [DOI] [PubMed] [Google Scholar]
- 11.Noguchi T, Yamada K, Inoue H, Matsuda T, Tanaka T. J Biol Chem. 1987;262:14366. [PubMed] [Google Scholar]
- 12.Staal GEJ, Rijksen G. In: Biochemical and molecular aspects of selected cancers. Pretlow TG, Pretlow TP, editors. Academic Press; San Diego: 1991. p. 313. [Google Scholar]
- 13.Steinberg P, Klingelhöffer A, Schäfer A, Wüst G, Weisse G, Oesch F, Eigenbrotd E. Virchows Arch. 1999;434:313. doi: 10.1007/s004280050330. [DOI] [PubMed] [Google Scholar]
- 14.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. Cell Metab. 2008;7:11. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 15.DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB. Curr Opin Genet Dev. 2008;18:54. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eigenbrodt E, Basenau D, Holthusen S, Mazurek S, Fischer G. Anticancer Res. 1997;17:3153. [PubMed] [Google Scholar]
- 17.Dombrauckas JD, Santarsiero BD, Mesecar AD. Biochemistry. 2005;44:9417. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 18.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. Nature. 2008;452:230. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 19.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Nature. 2008;452:181. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 20.Boxer MB, Jiang JK, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, Inglese J, Cantley LC, Auld DS, Thomas CJ. J Med Chem. 2010;53:1048. doi: 10.1021/jm901577g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan F, Wood KV. Assay Drug Dev Technol. 2007;5:127. doi: 10.1089/adt.2006.053. [DOI] [PubMed] [Google Scholar]
- 22.Singh P, Harden BJ, Lillywhite BJ, Broad PM. Assay Drug Dev Technol. 2004;2:161. doi: 10.1089/154065804323056503. [DOI] [PubMed] [Google Scholar]
- 23.Luminescent pyruvate kinase-luciferase coupled assay. Production of a luminescent signal based on the generation of ATP by pyruvate kinase was determined by using the ATP-dependent enzyme firefly luciferase.22 Details for the development of this assay format are provided in reference 20. The primary qHTS data and confirmatory data are available in PubChem (AIDs: 1631, 1634, and 1751). Follow-up of synthesized analogs was determined using the same protocol except that for the isoforms PK M1, L and R the enzyme concentration was 1 nM, 0.1 nM, and 0.1 nM respectively (PubChem AIDs for M1, L and R bioluminescent assays are 1780, 1781, and 1782). Fluorescent pyruvate kinase-lactate dehydrogenase coupled secondary assay. All compounds were also tested in a kinetic mode by coupling the generation of pyruvate by pyruvate kinase to the depletion of NADH through lactate dehydrogenase.36 Details for the development of this assay format are provided in reference 20. The data has been deposited in PubChem (AID: 1540). Follow-up of synthesized analogs was determined using the same protocol (PubChem AIDs for L, M1 and R bioluminescent assays are 1541, 1542, and 1543). This assay was also used to determine the KM’s for PEP and ADP in the presence and absence of activator. Conversion of fluorescent units to pmols of NADH was performed using a standard curve of known NADH concentrations. Data was collected on the Perkin Elmer Viewlux. Data was fit in GraphPad.
- 24.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Proc Natl Acad Sci USA. 2006;103:11473. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eras J, Galvez C, Garcia F. J Heterocyclic Chem. 1984;21:215. [Google Scholar]
- 26.Bennasar M, Roca T, Ferrando F. J Org Chem. 2005;70:9077. doi: 10.1021/jo0514974. [DOI] [PubMed] [Google Scholar]
- 27.Gale WE, Scott AN, Snyder HR. J Org Chem. 1964;29:2160. [Google Scholar]
- 28.Klapars A, Antilla JC, Huang X, Buchwald SL. J Am Chem Soc. 2001;123:7727. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Cen J, Guo H. Faming Zhuanli Shenqing Gongkai Shuomingshu. 10035041 CN. 2008
- 30.Wang X, Sun X, Zhang L, Xu Y, Krishnamurthy D, Senanayake CH. Org Lett. 2006;8:305. doi: 10.1021/ol052704p. [DOI] [PubMed] [Google Scholar]
- 31.dit Chabert JF, Marquez B, Neville L, Joucla L, Broussous S, Bouhours P, David E, Pellet-Rostaing S, Marquet B, Moreau N, Lemaire M. Bioorg Med Chem. 2007;15:4482. doi: 10.1016/j.bmc.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 32.Frère P, Raimundo JM, Blanchard P, Delaunay J, Richomme P, Sauvajol JL, Orduna J, Garin J, Roncali J. J Org Chem. 2003;68:7254. doi: 10.1021/jo034319o. [DOI] [PubMed] [Google Scholar]
- 33.Marques MA, Doss RM, Urbach AR, Dervan PB. Helvetica Chimica Acta. 2002;85:4485. [Google Scholar]
- 34.Pandey A, Volkots DL, Seroogy JM, Rose JW, Yu J-C, Lambing JL, Hutchaleelaha A, Hollenbach SJ, Abe K, Giese NA, Scarborough RM. J Med Chem. 2002;45:3772. doi: 10.1021/jm020143r. [DOI] [PubMed] [Google Scholar]
- 35.Aqueous solubility was assessed at a pH of 7.4 at room temperature from stock DMSO solutions. http://www.analiza.com/
- 36.Hannaert V, Yernaux C, Rigden DJ, Fothergill-Gilmore LA, Opperdoes FR, Michels PA. FEBS Lett. 2002;514:255. doi: 10.1016/s0014-5793(02)02374-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.