

Abstract
Myocardial ischemia-reperfusion injury is a major cause of morbidity and mortality in developed countries. To date, the only treatment of complete ischemia is to restore blood flow; thus the search for new cardioprotective approaches is absolutely necessary to reduce the mortality associated with myocardial ischemia.
Ischemia has long been considered to result in necrotic tissue damage but the reduction in oxygen supply can also lead to apoptosis. Therefore, in the last few years, mitochondria have become the subject of growing interest in myocardial ischemia-reperfusion since they are strongly involved in the regulation of the apoptotic process. Indeed, during ischemia-reperfusion, pathological signals converge in the mitochondria to induce permeabilization of the mitochondrial membrane. Two classes of mechanisms, which are not mutually exclusive, emerged to explain mitochondrial membrane permeabilization. The first occurs via a non-specific channel known as the mitochondrial permeability transition pore (mPTP) in the inner and the outer membranes causing disruption of the impermeability of the inner membrane, and ultimately complete inhibition of mitochondrial function. The second mechanism, involving only the outer membrane, induces the release of cell death effectors. Thus, drugs able to block or to limit mitochondrial membrane permeabilization may be cytoprotective during ischemia-reperfusion. The objective of this review is to examine the pharmacological strategies capable of inhibiting mitochondrial membrane permeabilization induced by myocardial ischemia-reperfusion.
Keywords: Adenosine Triphosphatases, metabolism, Apoptosis, Mitochondrial Membrane Transport Proteins, antagonists & inhibitors, metabolism, Mitochondrial Membranes, drug effects, metabolism, Myocardial Reperfusion Injury, drug therapy, prevention & control, Permeability, bcl-2 Homologous Antagonist-Killer Protein, metabolism
Keywords: mitochondria, heart, ischemia-reperfusion, necrosis, apoptosis, mitochondrial membrane permeability.
1. Introduction
Myocardial ischemia is a leading cause of death in developed countries. After an acute myocardial infarction, the only available treatment to reduce infarct size and to improve the clinical outcome is to restore blood flow (reperfusion) to the ischemic myocardium by thrombolysis, primary percutaneous coronary intervention or cardiac surgery. It is now well-known that reperfusion can induce injury and is at least as deleterious as the oxygen deficit (for a complete review on the mechanisms of the disease see [1]).
Cell death during myocardial ischemia-reperfusion has been assumed to occur primarily by necrosis. However, several studies indicate now that cell death following myocardial ischemia has features of apoptosis [2,3]; autophagy has also been suggested to play a role in myocardial ischemia-reperfusion injury [4,5]. Given that recent data indicate that the different forms of cell death are regulated and are probably interrelated [6,7], the better strategy to develop cardioprotective agents is not to define the precise mode of cell death and its proportion occuring during ischemia-reperfusion, but to identify mediators active in all forms of cell death. In this context, the mitochondrion has emerged as a relevant candidate. More importantly, the change in mitochondrial membrane permeability appears to be a major regulator of both necrotic and apoptotic cell death; the mode of death likely depends on the severity of the insult and on the ability of the cell to maintain ATP synthesis.
In this review, we will examine the role of mitochondria during myocardial ischemia-reperfusion and we will show that pharmacological inhibition of mitochondrial membrane permeability represents a relevant strategy to protect the myocardium from ischemia-reperfusion. We will address this new pharmacological concept of cardioprotection using to two approaches:
The pharmacological strategies acting directly on the mitochondrial membrane by targeting mitochondrial channels (section 3).
The use of pharmacological agents acting indirectly on mitochondrial membrane permeability, by preventing conditions promoting membrane permeabilization (e.g., oxidative stress or by stimulation of the survival pathways referred to as the “reperfusion injury salvage kinases (RISK)” pathways [8]) (section 4).
2. Mitochondrial dysfunctions are at the crossroad of the deleterious events caused by myocardial ischemia-reperfusion
Ischemia results in insufficient oxygen availability for mitochondrial respiration and oxidative phosphorylation and, rapidly leads to interruption of aerobic ATP synthesis (Fig. (1) and for extensive information concerning the cellular and mitochondrial events occurring during cardiac ischemia see reviews [9–12]). The rapid depletion of intracellular creatine kinase and the concomitant increase in inorganic phosphates (Pi) stimulate anaerobic ATP generation with an increase in glycolysis and lactate production. This temporarily supplies the deficiency of the aerobic pathway in the first period of the ischemic process. When hypoxic conditions continue, the intracellular acidification induced by lactate production and the hydrolysis ATP give rise to activation of the Na+/H+ exchanger (as the cell tries to restore intracellular pH), Na+/K+ ATPase inhibition (because of the drop in ATP levels) and activation of the Na+/Ca2+ exchanger (due to the increase in intracellular Na+ concentration). This results in a profound ionic imbalance, cell membrane depolarization and accumulation of cytosolic Ca2+ (with depletion of the sarcoplasmic reticulum Ca2+). Elevated cytosolic Ca2+ concentrations may contribute to cellular damage by activation of degrading enzymes such as phospholipases, proteases and nucleases, cumulating in the destruction of the membrane integrity and leading to cell death if the ischemic period is of sufficient duration.
Fig. (1). Scheme illustrating the mitochondrial sequence of events triggered by myocardial ischemia-reperfusion.
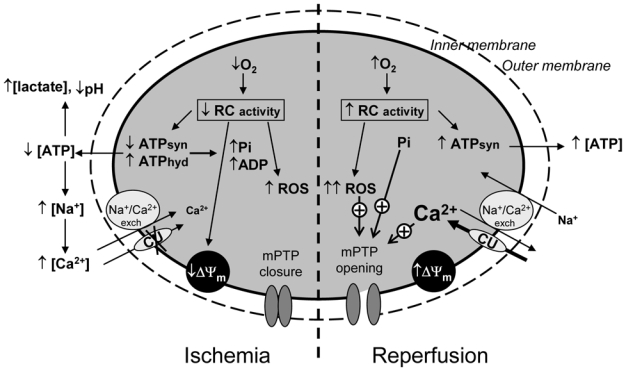
The scheme summarizes the processes described in the text, showing the relationships between oxygen consumption, ATP production, Ca2+ accumulation, reactive oxygen species (ROS) generation and the initiation of cell death through activation of the mitochondrial permeability transition pore (mPTP).
During ischemia, cellular acidosis along with a high concentration of ADP prevents mPTP opening although favourable conditions prevail (ΔΨm decrease, high Pi, slight increase in ROS). During reperfusion, high matrix concentrations of Ca2+ and Pi associated with a burst of ROS favour mPTP opening despite the antagonizing effect of ΔΨm recovery.
Mitochondria are initially able to buffer cytosolic Ca2+ by storing the excess, but this process is limited since mitochondria become at least partly de-energized, inhibiting the Ca2+ uniporter, the main route of entry of Ca2+ into mitochondria. However, during prolonged ischemia, Ca2+ entry may occur via reversal of the Na+/Ca2+ exchanger in the heart [12]. Temporally, the maintenance of a low mitochondrial Ca2+ concentration associated with the cytosolic acidosis and a high concentration of ADP prevents the opening of the mitochondrial transition pore, and thus protects mitochondrial membrane impermeability and potential. If the ischemic event continues, it may contribute to myocardial damage during reperfusion, since the electron transfer chain is altered, resulting in the increased production of superoxide anions from complexes I and III. This sets the stage for an increase in reactive oxygen species (ROS) production during reperfusion [13]. In addition, the F1F0-ATPsynthase now hydrolyses ATP. Jennings et al. [14] established that 35% of ATP utilization observed during the first 90 minutes of complete ischemia in the canine heart is due to F1F0-ATPase activity. Mitochondria are believed to hydrolyse ATP under conditions of oxygen deficit to maintain the mitochondrial membrane potential. However, this effect destroys any available ATP and favours Ca2+ accumulation, which may contribute to the reduced performance of the heart (stunning) during reperfusion.
Some reports have also observed decreases in NADH dehydrogenase and adenine translocase activities, a reduction of the protective mechanisms against oxygen toxicity, and decreases in superoxide dismutase (SOD) activity and glutathione content. The final extent of the damages depends on the duration and the severity of ischemic event, which emphasises the crucial time for the onset of the reperfusion (which can be combined with a pharmacological intervention). This is a critical juncture which may limit or even reverse at least some of the changes in mitochondrial functions.
Ischemic damage can paradoxically be amplified by reperfusion that corresponds to the restoration of oxygenated blood flow to the ischemic tissue. This is mainly due to the generation of ROS, which can result from the reactivation of respiratory complexes blocked in a reduced state without functional coordination (inefficient transfer of electrons generating superoxide anions). ROS have direct effects on the respiratory chain components, resulting in decreased efficiency of oxidative phosphorylation; complex I appears to be especially sensitive to ischemic injury [13,15]. Changes in complexes III and IV were also observed, but this would occur later during the deleterious process [11]. They also cause inhibition of the enzymes of the Krebs cycle (i.e. aconitase) [16,17]. ROS cause non-specific damages to lipids, proteins and mitochondrial DNA, and induce peroxidation of cardiolipin, a major constituent of the inner membrane, which increases the inhibition of oxidative phosphorylation [18,19].
ROS production is exacerbated by the high concentrations of Ca2+, which is driven from the cytosol into the mitochondria by restoration of the mitochondrial membrane potential. These conditions favour an increase in mitochondrial membrane permeability and the induction of the mitochondrial permeability transition pore (mPTP), which cause swelling, collapse of membrane potential, and ultimately total inhibition of mitochondrial functions [10]. This phenomenon is now considered to play a central role in different kinds of cell death; Crompton et al. [20] were the first to report that mPTP opening plays a crucial role in myocardial ischemia-reperfusion injury. It is generally accepted that mPTP opening is associated with postischemic reperfusion [21] but some studies have suggested that mPTP opening might also occur during ischemia [22]. It should be noted that mPTP is not the only mechanism by which mitochondrial membranes can be permeabilized, and the search for cardioprotective agents must consider all the mitochondrial targets that can prevent mitochondrial membrane permeabilization.
3. Pharmacological strategies acting by direct inhibition of mitochondrial membrane permeability by targeting mitochondrial channels
During ischemia-reperfusion, cell death occurs by necrosis and apoptosis, both being induced by the change in mitochondrial membrane permeability. Mitochondrial membrane permeability is regulated by stress signals originating in mitochondria but also those of cytosolic origin such as proteins of the Bcl-2 family. Two classes of mechanisms (Fig. (2)), which are not mutually exclusive, have been proposed to explain mitochondrial membrane permeabilization (for review see [23]).
Fig. (2). Hypothetical models of the increase in mitochondrial membrane permeability during ischemia-reperfusion.
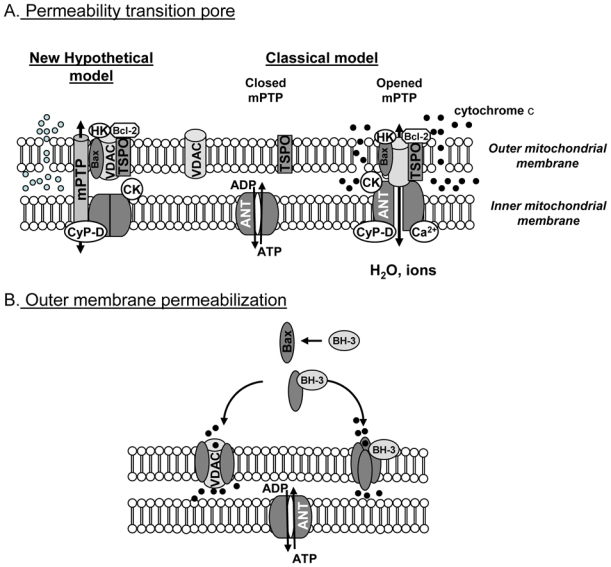
A: mitochondrial permeability transition pore (mPTP). Classical model: mPTP is formed by the interaction of VDAC, ANT and CyP-D at the contact sites between the inner and the outer membrane and can be modulated by other proteins such as creatine kinase (CK), hexokinase (HK), translocator protein (TSPO) and proteins of the Bcl-2 family. New hypothetical model: recent genetic manipulations suggest that mPTP might be an unidentified channel regulated by the proteins constitutive of the classical model.
B: outer membrane permeability can be due to the formation of pores induced by the activation of Bax by BH-3 only proteins and its translocation to the mitochondrial membrane. It should also be caused by the interaction of Bax with VDAC.
It should be noted that VDAC alone could regulate outer membrane permeability and that other mechanisms than Bax and BH-3 only proteins have been involved in outer membrane permeability. However, they were not described during myocardial ischemia-reperfusion.
The first one involves the participation of both the inner and the outer membranes and corresponds to the mPTP. The second mechanism involves only outer mitochondrial membrane and the formation of channels across the membrane.
Although there is a controversy concerning the structure, the regulation and the definite role of these different channels, strong evidences indicate that proteins of the Bcl-2 family contribute to both mechanisms [24,25].
Triggering mitochondrial membrane permeabilization induces the release of cell death effectors and the loss of mitochondrial functions which are fundamental for cell survival. Thus, drugs able to block or to limit mitochondrial membrane permeabilization might be cytoprotective during ischemia-reperfusion and this pharmacological approach is reviewed in this section (Fig. (3)).
Fig. (3). The inhibition of mitochondrial permeabilization as a pharmacological approach to limit myocardial ischemia-reperfusion injury.
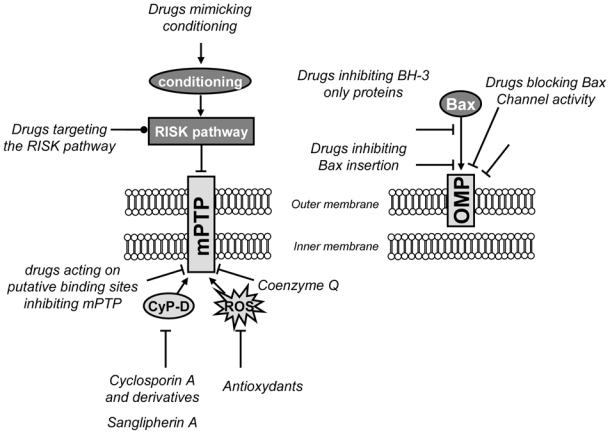
Schematic representation of the pharmacological strategies which have been or might be used to limit mitochondrial membrane permeabilization. They can target directly or indirectly the outer mitochondrial membrane permeability (OMP) or both the outer and the inner membrane (mPTP).
CyP-D: cyclophilin D; mPTP: permeability transition pore; VDAC: voltage-dependent anion channel.
➔ : activation
⊥: inhibition
 : activation or inhibition
: activation or inhibition
3.1. The mitochondrial transition pore as a pharmacological target
The mitochondrial respiratory chain catalyzes the oxidation of substrates by oxygen and couples electron flux to H+ pumping to establish an H+ electrochemical gradient across the inner membrane. ATP is synthesized by the enzyme F1F0-ATPsynthase which utilizes the free energy released by the H+ backflow from intermembrane to mitochondrial matrix. Therefore, the maintenance of the H+ gradient and the impermeability of the inner membrane to ions, especially to H+, is vital to the cell. However, under stress conditions, the permeability of the membrane may increase with the formation of a voltage-dependent nonspecific pore, known as the mPTP, which allows the influx of water and molecules up to 1.5 kD and therefore induces swelling of the organelle matrix. An extensive swelling results in the rupture of the outer membrane and in the release of proapoptotic proteins into the cytosol. The exact composition of this pore remains uncertain although it was formerly thought to include adenine nucleotide translocase (ANT) modulated by cyclophilin D (CyP-D) in the inner membane and a voltage-dependent anion channel (VDAC) in the outer membrane [10,26]. CyP-D is a member of the cyclophilin family which displays peptidyl-prolyl cis-trans isomerase activity and resides in the mitochondrial matrix. During mPTP formation, CyP-D interacts with the inner membrane and is thought to induce a conformational change of ANT, leading to an increase in inner membrane permeability (Fig. (2)).
In vivo experiments confirm the role of CyP-D-dependent mitochondrial mPTP for mediating Ca2+- and oxidative damage-induced cell death: CyP-D-deficient mice are protected from ischaemia-reperfusion-induced cell death in vivo, whereas CyP-D-overexpressing mice show mitochondrial swelling and spontaneous cell death [27–29]. These genetic experiments also provided significant information about the mechanism of cell death triggered by mPTP. CyP-D-deficient cells responded to various apoptotic stimuli, but showed resistance to necrotic cell death induced by ROS and Ca2+ overload. This led to the conclusion that CyP-D and thus mPTP do not play a major role in the apoptotic process, but rather are major components of the necrotic death pathway.
Studies on mitochondria lacking these putative components of the mPTP did not permit definit conclusions about the identity of its membrane components. Indeed, a functional mPTP can form in the absence of ANT in mice [30], and it was suggested that ANT may not be the protein which binds CyP-D [31]. Juhaszova et al. [32] suggest that ANT may be a non-essential structural component of the mPTP but may have a regulatory role in mPTP induction (see Fig. (2A)). VDAC also may not be an essential component of the mPTP as the pore can be induced in mitochondria devoid of all VDAC isoforms [33]. It should be added that other proteins such as hexokinase, creatine kinase or the mitochondrial translocator protein (TSPO), have been proposed to be involved in pore formation, but to date it is not known whether these proteins are structural or regulatory components of the pore.
Taken together, these data show that the molecular composition of mPTP remains a source of debate, but nevertheless, strong evidence suggests that mPTP contributes to cellular injury during myocardial ischemia-reperfusion: (1) mPTP opening is favored by conditions prevailing during ischemia-reperfusion (2) mPTP might control cell death by releasing apoptogenic factors [10,34,35], (3) mPTP is regulated by the pro- and the anti-apoptotic members of the Bcl-2 family [36,37], (4) increasing lines of evidence suggest that mPTP is involved in the cardioprotective effect of ischemic pre- and post-conditioning (see section 4.1.).
Therefore, targeting mPTP appears as a promising pharmacological approach in ischemia-reperfusion and numerous studies show that almost any procedure that reduces mPTP opening affords protection against ischemia-reperfusion injury.
The first demonstration of the relevance of this approach was provided by the immunosuppressant agent cyclosporin A (CsA) which binds to CyP-D with high affinity and precludes the interaction of this protein with a component of the mPTP, thus inhibiting its opening. CsA protected against hypoxia-reoxygenation-mediated cell death in isolated cardiomyocytes [38,39] and reduced infarct size in ex-vivo models of cardiac ischemia-reperfusion [40,41]. Shanmuganathan et al. [42] have also demonstrated that inhibition of mPTP opening by CsA at the onset of reoxygenation protected the human myocardium against lethal hypoxia-reoxygenation injury. The clinical use of CsA is hampered by pharmacokinetic and pharmacodynamic parameters [43] and by the fact that CsA fails to inhibit pore opening when mitochondria are exposed to strong stimuli [44]. However, a recent clinical study showed that an intravenous bolus of 2.5 mg/kg CsA just before reperfusion reduced infarct size in patients suffering from acute myocardial infarction [45].
Similar cardioprotective effects were obtained with non-immunosuppressant derivatives of CsA, such as [N-methyl-ala6]CsA, [N-methyl-Val4]CsA, Debio 025 or NIM811, [21,40,46,47]) confirming the relevance of this approach. Sanglifehrin A also prevents myocardial ischemia-reperfusion injury [48,49]. This drug is unrelated to CsA but potently inhibits mPTP by inhibiting the peptidyl-prolyl cis-trans isomerase activity of CyP-D (Fig. (3) and (4)).
Fig. (4).
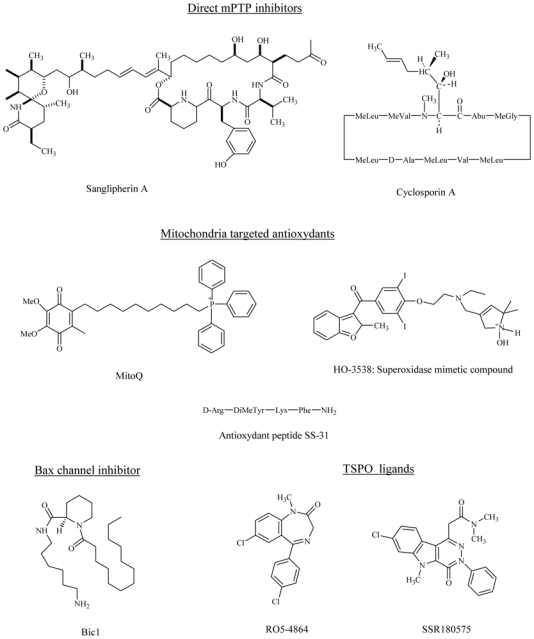
Chemical structures of some pharmacological agents capable of limiting mitochondrial membrane permeabilization.
It should be added that the inhibition of mPTP opening by CsA was also effective in a particular case of ischemia-reperfusion, the hypothermic preservation of the heart before transplantation. Indeed, CsA improved functional recovery after long-time hypothermic heart preservation [50].
Although the molecular structure of mPTP remains unknown, other approaches have been developed to find new mPTP inhibitors. Binding sites involved in the regulation of mPTP were described [51]. Clinically approved drugs, including tricyclic antidepressants, antipsychotics and antiarythmic agents were found to bind to these sites and to inhibit mPTP in vitro [51,52]. Some of these drugs reduced neurological impairment in mice subjected to middle cerebral artery occlusion/reperfusion [52], but, to our knowledge, have not been tested in myocardial models of ischemia-reperfusion. Most of these drugs are amphiphilic cations which are known to interact with biological membranes which could change mitochondrial membrane potential and thus possibly induces closure of the pore [53].
Another approach to design new pore inhibitors was provided by the results of Bernardi and colleagues who demonstrated that the coenzyme Q family in addition to its antioxidant effects and its role in the regulation of electron transfer modulates mPTP opening by acting on a specific site [54]. Exogenous coenzyme Q0 (ubiquinone 0) and coenzyme Q10 (decylubiquinone) are very potent inhibitors of mPTP opening induced by Ca2+ overload, whereas coenzyme Q1 (ubiquinone 5), which did not inhibit pore opening per se, counteracted the effects of coenzyme Q0 and decylubiquinone [54]. As a putative component of mPTP, VDAC was suggested to be one of the target of ubiquinone analogues [55], but since liver mitochondria from mice lacking VDAC1 exhibit normal mPTP opening and inhibition by ubiquinone analogues [56], this hypothesis seems unlikely. It should be noticed that the design of new mPTP inhibitors from coenzyme Q may be difficult since minor structural changes profoundly modify the effects of quinones on the mPTP [57].
To our knowledge, it has not been established whether this is what is responsible for the antiischemic effect of coenzymes Q [58] and data indicate that coenzyme Q0 even impairs myocardial performance following ischemia-reperfusion [44]. However, the antioxidant properties of coenzyme Q may contribute to mPTP inhibition as decylubiquinone was shown to inhibit redox-activated permeability transition [59].
Taken together, these data suggest that the direct inhibition of mPTP opening constitutes a relevant objective in cardioprotection. This requires the search for new direct mPTP inhibitors [55,60] with good specificity and selectivity and the identification of the molecular site of action of these compounds to further elucidate mPTP structure.
3.2. The MAC/Bak channel
It is now well-established that mPTP is not the only mechanism by which the mitochondrial membrane can be permeabilized [61]. Indeed, cytochrome c release was observed in the absence of mitochondrial depolarization [62] and without loss of mitochondrial outer membrane integrity (i.e., independently of mPTP opening), indicating that permeabilization of the outer membrane is a regulated phenomenon occurring via the formation of a pore [24]. This type of release has been observed during cardiac and liver ischemia [63–65].
Mitochondrial membrane permeabilization is under the control of the Bcl-2 family of proteins (for a recent review see [66]). Proapoptotic members (e.g. Bax, Bak or the BH3-only subfamily proteins Bid, Bad, Bnip3) facilitate membrane permeabilization and promote the release of cytochrome c and other intermembrane space components. Cytochrome c triggers formation of the apoptosome complex and activation of caspase-9 [61]. This is not the only pathway which can be activated by outer membrane permeabilization, since two other proteins, apoptosis inducing factor (AIF) and endonuclease G, which are also released after outer membrane permeabilization, can induce apoptosis in a caspase-independent manner [67,68]. Two main possibilities have been proposed to be responsible for governing this increase in permeability. Bax resides in the cytosol in normal, healthy cells but, once activated, translocates to mitochondria, where 1) it incorporates into the outer membranes and oligomerizes to form permeable pores [3,61] and/or 2) interacts with existing channels such as VDAC [3,69]. The activation of Bax is regulated by the BH3-only proteins, which facilitate Bax channel formation. It should be pointed out that the Bax channel appears to be identical to the mitochondrial apoptosis-induced channel (MAC channel; [24]), which was first detected on mitochondria in patch-clamp experiments. The anti-apoptotic members Bcl-2 and Bcl-xL are mainly localized to the mitochondrial outer membrane where they antagonize the pro-apoptotic effect of Bax and Bak [70].
The importance of the Bcl-2 family proteins in myocardial ischemia-reperfusion injury was demonstrated in mouse models. Indeed, Bax deletion in mice, interference with Bax activation, or over-expression of Bcl-2 levels have been shown to attenuate apoptosis and reduce infarct size, while reduction of Bcl-2 levels (via antisense oligonucleotides) suppressed protection against injury [72–76]. Therefore, anti-apoptotic interventions targeting Bcl-2 family proteins provide opportunities for possible antiischemic therapies [77], either through a gain of antiapoptotic function or loss of proapoptotic function. Polster et al. [78] demonstrated that the well-known drugs propranolol and dibucaine inhibited Bax-induced permeability changes and cytochrome c release from mitochondria through a direct interaction with the lipid membrane. A similar approach allowed others to identify 3,6-dibromocarbazole piperazine derivatives of 2-propanol as inhibitors of cytochrome c release triggered by induction of the Bax channel [79]. In the same way, a pentapeptide derived from the Bax-modulating Ku70 protein inhibited Bax-dependent apoptosis [80]. More recently, Hetz et al. [81] identified two blockers of Bax channel activity, bci1 and bci2 (Fig. (4)), which prevented cytchrome c release in vitro and in vivo, and protected neurons in a gerbil model of global brain ischemia at reperfusion. It is a promising pharmacological strategy, but to our knowledge, these compounds have not been evaluated during myocardial ischemia.
3.3. BH3-only Bcl-2 proteins
The activation of Bax and Bak is regulated by the class of Bcl-2 proteins that contain single BH3 domains. These proteins control apoptotic signals upstream of mitochondria and transmit them directly or indirectly to Bax and Bak [66], thus they represent specific targets for preventing myocardial ischemia-reperfusion injury. This appears to be an excellent point of intervention because only activation of Bax and Bak would be affected, minimizing cell apoptosis. This approach would also facilitate maintenance of mitochondrial integrity.
Among these proteins Bid (BH3-Interacting Domain death agonist) is one of the most abundant in mammalian tissues, including the heart [82]. Bid is subjected to a protein cleavage by caspase-8, granzyme, or calpain, and truncated Bid (tBid) translocates to the mitochondria where it is involved in Bax/Bak activation and/or Bax/Bak channel formation [66]. Thus, Bid is at the crossroad of the intrinsic and the extrinsic death pathway. Bid-mediated apoptosis has been shown to contribute to myocardial ischemic injury [83] and Bid-deficient mice have reduced infarct size and improved cardiac functions after ischemia-reperfusion [3]. In addition, calpain inhibitors reduce infarct size in animal models of isolated perfused heart [84] and slow the progression of heart failure in rats [85,86]. These data suggest that Bid is an attractive target during myocardial ischemia-reperfusion, and pharmacological inhibitors which are effective in vitro assays have already been designed [87].
Other BH3-only Bcl-2 proteins have been implicated in cardiac ischemia-reperfusion injury [88]. Bnip3 (Bcl-2/adenovirus E1B 19kDa interacting protein 3) has been shown to mediate mitochondrial dysfunction and cell death of ventricular myocytes subjected to hypoxia [89–90]. It was also reported to contribute to myocardial ischemia-reperfusion injury through a mitochondrial death pathway [91–92]. Interestingly, a TAT-fusion protein encoding a carboxyl terminal transmembrane deletion mutation in of Bnip3 conferred protection against myocardial ischemia-reperfusion injury, improved cardiac function, and protected mitochondrial integrity [92], confirming that Bnip3 could constitute a relevant therapeutic target.
In the same way, Puma (p53-Upregulated Modulator of Apoptosis) was upregulated in response to hypoxia/reoxygenation in isolated cardiomyocytes and Puma-deficient mice have reduced infarct size and improved cardiac function after myocardial ischemia-reperfusion [93]. Until now, pharmacological strategies targeting selectively Puma have not been described.
The pharmacological strategy inhibiting mitochondrial outer membrane permeability, and thus apoptosis, by manipulation of the Bcl-2 family proteins to protect the myocardium against ischemia-reperfusion is very recent. It has already provided interesting results, which support the idea that a clinical benefit might be obtained in the near future.
3.4. The voltage-dependent-anion channel (VDAC) as a pharmacological target
Another potential mechanism for outer mitochondrial membrane permeabilization might involve VDAC which is the major permeability pathway for metabolites through the mitochondrial outer membrane [94]. Different isoforms of VDAC have been identified [95,96] and the role of VDAC in cell death induced by mitochondria seems to be isoform specific. Indeed, VDAC1 has been implicated in the mitochondrial release of proapototic protein whereas VDAC2 displays antiapoptotic properties [97,98].
It is important to note that much of the evidence involving VDAC opening and closing in proapototic protein release has arisen from in vitro experiments, and it is not clearly established whether this can be observed in vivo [99]. However, VDAC is a hypothetical component of mPTP, interacts with pro- and anti-apoptotic proteins of the Bcl-2 family members, and VDAC has been also proposed to control the release of cytochrome c without mPTP opening [97,100,101].
Taken together, these data make VDAC a putative target in ischemia-reperfusion. Interestingly, a strategy developed to inhibit VDAC was shown to reduce myocardial ischemia-reperfusion injury. Perfusion of rat hearts with a cell permeable peptide corresponding to the BH4 domain of Bcl-XL, which had been reported to close VDAC and to prevent the VDAC-mediated release of cytochrome c [102], was shown to inhibit creatine kinase release and to reduce myocardial cell death [84]. In the same way, the Ca2+-dependent actin-regulatory protein gelsolin was suggested to inhibit apoptosis by blocking mitochondrial VDAC activity [103]. This is in disagreement with recent results that indicate that closure of VDAC, not opening, leads to mitochondrial outer membrane permeabilization and apoptosis [104]. It is clear that a better knowledge of the role of VDAC, if any, in ischemic injury, will take time, given the lack of proven pharmacological compounds specifically targeting VDAC.
3.5. Mitochondrial potassium (mitoKATP) channels
MitoKATP channels were first identified in liver mitoplasts (mitochondria stripped of the outer membrane) by Inoue et al. [105] in patch-clamp experiments, and then by light scattering experiments in liver and heart isolated mitochondria [106–107]. Evidence for the involvement of mitoKATP channels in cardioprotection was provided by the observation that mitoKATP openers, like diazoxide or nicorandil, can protect the myocardium against ischemia-reperfusion injury [108–110] and mimic ischemic pre-conditioning, while blockers inhibit [111–113]. In the same way, recent data show that the myocardial protection conferred by ischemic post-conditioning is blocked by the mitoKATP channel blocker 5-hydroxydecanoate and thus achieved at least in part by opening of mitoKATP channels [114]. The mechanism for mitoKATP channels opening during pre-and post-conditioning is currently the subject of intense investigation.
Several recent studies have linked the protein kinase C epsilon (PKCε), which translocation to mitochondria seems important for pre-conditioning [115], to mitoKATP channel opening. Other kinases have also been involved [116–117].
The mechanism by which activation of mitoKATP results in cardioprotection is not well-understood, but implies a limitation in mitochondrial membrane permeability. Several hypotheses have been proposed. The first one considers that the beneficial effect of mitoKATP activation could be the consequence of a partial dissipation of the membrane potential caused by the net influx of K+ [118]. This would reduce Ca2+ entry or release an excess of Ca2+, preventing Ca2+ overload [119] and thus mPTP opening during reperfusion. This is consistent with the results of Korge et al. [120] and Facundo et al. [121], who reported that diazoxide prevents opening of the mPTP induced by elevated Ca2+ concentrations in isolated mitochondria. However, this hypothesis has been disputed, because mitochondrial membrane depolarization generated by K+ flux does not appear to be sufficient to affect Ca2+ transport [122] and has not been observed in intact cardiac myocytes [123]. Moreover, a higher depolarization may lead to deleterious phenomena more frequently associated with cell death, than t protection (such as decreased ATP synthesis and even mPTP opening).
The second hypothesis proposes that perturbed ROS production is responsible for mitoKATP channel effect. Depending on the conditions, pre-conditioning phase, ischemia or reperfusion, mitoKATP channel openings could either increase or attenuate ROS generation [121,124], but the link between mitoKATP channels, ROS and the induction of a cytoprotective effect is not clear. In this context, Hausenloy et al. [125] suggested that the cardioprotection induced by diazoxide is ROS-dependent and due to transient mPTP opening-mediated ROS release during the preconditioning phase. In this scheme, mPTP opening may act as a protective mechanism in response to different stimuli during the pre-conditioning period to inhibit its deleterious opening during the reperfusion phase. This is an attractive mechanism but is still highly controversial [126]. Similarly, Costa et al. [127] have reported that mPTP inhibition by mitoKATP opening requires the generation of ROS.
The third hypothesis, which is not mutually exclusive, considers the only well-defined physiological function of mitochondrial K+ transporters, i.e. the regulation of the matrix volume [128]. Dos Santos et al. [129] hypothesized that opening of the mitoKATP during ischemia resulted in a slight mitochondrial swelling which preserved the structure of the intermembrane space and thus maintained VDAC in a low permeability state for adenine nucleotides. This would reduce glycolytic ATP entry into mitochondria and, therefore, ATP hydrolysis by F1F0-ATPase, and would preserve ADP for phosphorylation upon reperfusion (for review see [130]). Thus, the mechanism by which mitoKATP channels exert their cardioprotective effect is still a source of debate, but most of the studies suggest that opening of mitoKATP channels prevents cellular death by decreasing mitochondrial ROS release and Ca2+ overload during the reperfusion, thus inhibiting mPTP.
Based on these experimental data, clinical studies have been performed. Some of them reported cardioprotection with intravenous administration of nicorandil (a mixed mitoKATP channels opener and nitric oxide donor) in patients with acute myocardial infarction [131,132], but another study failed to demonstrate this effect in the same clinical setting [133].
However, the full demonstration of the involvement of mitoKATP channels in cardioprotection is hampered by the absence of selective agents for the mitoKATP channel; the mitoKATP channel openers and inhibitors which have been used to demonstrate the cardioprotective effects of these channels (most notably diazoxide and 5-hydroxydecanoate), are claimed to be specific for this channel, but numerous studies argue against this specificity (see the review of Halestrap et al. [134]).
A better understanding of the cellular mechanisms and of the role of mitoKATP channels will require the development of more specific and more selective pharmacological entities; however, they still appear to be a promising target for the development of new cardioprotective agents.
3.6. The mitochondrial translocator protein (TSPO)
TSPO (formerly known as Peripheral Benzodiazepine Receptor [135]) is a ubiquitous high affinity cholesterol binding 18 kDa protein primarily located on the outer mitochondrial membrane, where it is associated with VDAC [136]. It has been implicated in numerous biological functions [135], including regulation of mPTP opening [136,137] but the exact physiological role of TSPO remains unclear (except in steroid-producing tissues where TSPO mediates the transport of cholesterol from the outer to the inner mitochondrial membrane and promotes pregnenolone synthesis [138]). TSPO is also present in non steroidogenic tissues, especially in the heart, where its function is unknown. Several reports suggest that TSPO ligands might modulate apoptotic responses [139] and play an anti-apoptotic role in oxidative stress conditions such as ischemia-reperfusion [140]. In this context, TSPO expression correlated with the quality of kidney preservation, indicating that it might serve as an index of kidney and mitochondrial viability during storage [141]. Thus, one means to improve resistance of the cells to oxidative stress generated during ischemia would be to increase TSPO expression levels using pharmacological tools to increase TSPO transcription. Leducq et al. [142] found that the irreversible TSPO ligand SSR180575 prevented the cellular damages resulting from oxidative stress and cardiac injuries induced by ischemia-reperfusion in rodents, and Obame et al. [143] showed that the specific TSPO ligand 4′-chlorodiazepam (Fig. (4)) reduced infarct size in both global and regional models of myocardial ischemia-reperfusion in rats. The protective mechanism involved limiting mitochondrial membrane permeabilization, notably a resistance of mitochondria to mPTP opening, associated with a reorganization of the balance between pro- and anti-apoptotic proteins of the Bcl-2 family proteins at the level of mitochondrial membranes [143]. Taken together, these data demonstrate a role for TSPO as a modulator of necrotic and apoptotic cell death induced by ischemia-reperfusion, suggesting that TSPO could constitute a possible target for cardioprotection.
4. Pharmacological strategies acting indirectly on mitochondrial membrane permeability
4.1. Ischemic pre- and post-conditioning
Ischemic pre-conditioning (IPC) is one of the more powerful processes to protect the heart from lethal ischemia-reperfusion injury. IPC was originally described by Murry et al. [144], who demonstrated that a sequence of successive short periods of ischemia reduced infarct size produced by a subsequent long ischemic period. Studies revealed that IPC caused the release of G protein-coupled receptors agonists such as adenosine, bradykinin or opioids that activated a cascade of cardioprotective kinases referred to as “Reperfusion Injury Salvage Kinase” (RISK) pathway [145]. This cascade includes PKC, the PI3kinase-AKT pathway, the ERK1/2 pathway, and downstream proteins such as glycogen synthase kinase-3β (GSK-3β), PKG, Bad or endothelial nitric oxide synthase (eNOS). PKCε is a primary cardioprotective PKC isoform, whereas PKCδ promotes injury. Activation of AKT and ERK1/2 and phosphorylation of GSK-3β appear essential for IPC in numerous studies, although this has been questioned by Nishino et al. [146]. More recently, Zhao et al. [147] demonstrated that brief repetitive ischemic periods of ischemia followed by reperfusion after a lethal ischemic period results in a robust reduction in infarct size, termed ischemic post-conditioning (IPOC). Subsequent studies have shown that IPC and IPOC use a similar signalling pathway, i.e. the RISK pathway, and that classical ligands able to trigger IPC, such as adenosine or opioids, were also able to induce IPOC [148]. For a detailed characterization of the signalling pathways involved in IPC and IPOC see [115]. From a clinical point of view the discovery of IPOC is fundamental as it can be applied in patients [149].
The cellular mechanisms governing IPC and IPOC protection are not fully understood, but increasing lines of evidence suggest that indirect inhibition of mitochondrial membrane permeability, through inhibition of mPTP opening during reperfusion, may be the target of the RISK pathway [150–153]. The mechanism by which the RISK pathway inhibits mPTP is unclear, but several candidates have emerged which may act in concert to mediate mPTP inhibition (Fig. (5)). These include:
Fig. (5).
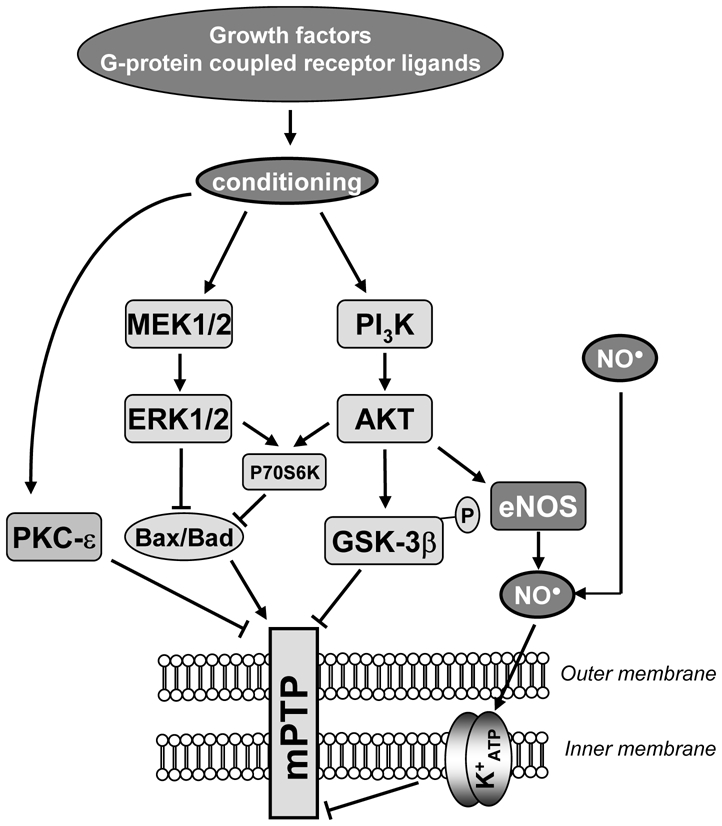
Hypothetical scheme showing the cardioprotective pathways activated at the time of reperfusion by both ischemic pre- and post-conditioning. Ligands of receptors coupled to G-proteins or growth factors initiate cardioprotection by activating a cascade of kinase leading to mPTP inhibition and thus protect the cell against apoptotic and necrotic death.
mPTP indicates permeability transition pore; K+ ATP: mitochondrial potassium channel. ERK1/2, extracellular regulated kinase; MEK1/2, mitogen-activated protein kinase/ERK1/2 kinase; PI3K, Phosphoinositide 3-kinase; GSK-3β, glycogen synthase kinase 3β; eNOS, endothelial NOS; P70S6K, p70 ribosomal S6 protein kinase
➔ : activation
⊥: inhibition
eNOS generation of NO, which alone [154], or through the PKG-PKCε-mitoKATP channel can limit mPTP opening [115,127],
phosphorylation and/or complex formation with cytosolic proteins like PKCε can interact with mPTP [155,156], whereas the activity of PKCδ and its translocation to mitochondria at the onset of reperfusion mediates apoptosis [157],
phosphorylation and inhibition of GSK-3β, a downstream target of AKT, which have been demonstrated to inhibit mPTP [150,153]. Nishihara et al. [158] suggested that the binding of GSK-3β to ANT might be responsible for mPTP inhibition and the myocardial protection afforded by IPC,
inhibition of downstream signals of the RISK pathway, such as Bax translocation from cytoplasm to mitochondria [159] or hexokinase II activation [160].
However, whatever the mechanism(s) connecting the RISK pathway to mPTP inhibition, the activation of this cascade of kinases offers the opportunity to promote mPTP inhibition and thus cardioprotection at reperfusion. This property can be exploited therapeutically as described in the following studies.
4.1.1. Targeting of the RISK pathway by ischemic post-conditioning
IPOC was recently described, but small clinical studies have already been initiated [161–163]. In these studies, IPOC was induced in patients by a series of successive episodes of inflation-deflation of the angioplasty balloon during percutaneous coronary intervention. IPOC significantly protected the heart against ischemia-reperfusion-induced injury and, importantly, the protective effect is of significant duration [163].
4.1.2. Targeting the RISK pathway via pharmacological agents
The administration of high doses of adenosine at reperfusion in acute myocardial infarction resulted in myocardial protection in two small clinical studies [164,165]. A reduction in infarct size was also observed in larger randomized controlled studies in which adenosine was administrated as an adjunct to thrombolytic therapy after the beginning of reperfusion [166,167]. However, this pharmacological strategy did not improve clinical outcomes [167].
Morphine is another G protein-coupled receptor agonist which is known to activate the RISK pathway and to cause potent cardioprotection when administrated upon reperfusion [168]. Recently, Obame et al. [153] have shown that morphine provides protection against myocardial infarction through inhibition of mPTP opening. Thus, the use of morphine, a well-known drug which can be administrated acutely in safe conditions, as an adjunct of myocardial reperfusion, may be a useful clinical strategy in acute myocardial infarction.
It is also possible to act upstream in the RISK pathway, as several groups have shown that inhibitors of GSK-3β reduced infarct size in mice and rats, and that this effect was associated with the inhibition of mPTP opening [153,168,169].
Similarly, beside their lipid-lowering actions, statines (3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors) have been shown to exhibit cardioprotective properties [170,171]. The mechanism seems to involve stimulation of several kinases in the RISK pathway but also activation of the mitoKATP channel through NO generation and inhibition of mPTP opening [172].
Another possible strategy is to directly target PKC via PKCε activators or PKCδ inhibitors. A recent small clinical study of acute myocardial infarction using the PKCδ inhibitory peptide, KAI-9803, which was delivered via intracoronary injection during percutaneous coronary intervention, revealed signs of cardioprotection. Unfortunately, the observed improvements were not significant, probably because of the small size of the sample [173]
4.2. Mitochondria-targeted antioxidants in limiting mitochondrial membrane permeability
In physiological conditions, ROS are formed within mitochondria and are eliminated by powerful antioxidant systems [174]. Under pathological conditions, especially during post-ischemic reperfusion, the sudden influx of oxygen to the mitochondrial respiratory chain leads to a burst of ROS (as a superoxide anion; [175]) which can overwhelm endogenous antioxidant systems and deplete the reducing compounds protecting mitochondria against oxidative insults. It should be noted that the respiratory chain is not the only source of ROS in mitochondria [174]. For instance, MAO-A has been shown to represent an important source of hydrogen peroxide in rat heart [176]. ROS cause non-specific damage to lipids, proteins and mitochondrial DNA. When associated with mitochondrial Ca2+ overload and adenine nucleotide depletion, conditions that pertain during myocardial reperfusion, high levels of ROS can induce opening of the mPTP [44]. ROS generated during early reperfusion have been found to be primary activators of mPTP and cardiomyocyte death [177], and Clarke et al. [178] have reported that IPC-induced inhibition of mPTP is probably mediated by a reduction in oxidative stress. This property might also contribute to the cardioprotective effect of some therapeutic drugs such as pyruvate [179], the β-adrenoceptor blocking agent carvedilol [180] or the anaesthetic propofol, frequently used for cardiac surgery [181].
These data highlight the major role of ROS in the increase in mitochondrial membrane permeability. Thus, any antioxidant treatment is likely to protect mitochondria and ultimately the myocardium at reperfusion. Numerous pharmacological strategies have been developed to scavenge ROS or to increase mitochondrial ROS degradation [182]. Recently, Bognar et al. [183] have developed a novel superoxide dismutase mimetic drug which was able to eliminate ROS in the microenvironment of the mPTP and to inhibit its opening, leading to the protection of the myocardium against myocardial ischemia-reperfusion (Fig. (4)).
An interesting result was also obtained with the free radical scavenger edavarone which was shown to inhibit mPTP opening and to prevent cardiac reperfusion injury [184]. In a recent clinical study, edavarone delivered via intravenous injection during percutaneous coronary intervention reduced infarct size and reperfusion arrhythmias, and improved short term clinical outcomes in acute myocardial infarction [185]. However, most of the clinical studies with conventional antioxidants have yielded disappointing results [186].
Several reasons can be evoked to explain this failure; among them, the main reason may be the difficulty of delivering the drug to mitochondria in situ. To overcome these limitations, investigators have synthetized amphiphilic cations from well-established antioxidant compounds, which are sufficiently lipophilic to cross lipid bilayer membranes and which can be selectively taken up into the mitochondria by the large, negative inside, inner membrane potential [187,188]. These drugs include alpha tocopherol (MitoVit E) and ubiquinone (MitoQ) (Fig. (4)). A recent study demonstrated that feeding rats with MitoQ significantly limited cardiac reperfusion-injury [189]; this approach might provide a promising pharmacological strategy.
Using a similar idea, a novel class of small cell-permeable antioxidant peptides (Fig. (4)) that target mitochondria have been reported [190]. Their antioxidant effect is due to the presence of basic amino acids, tyrosine and dimethyltyrosine, which are effective in scavenging ROS. These peptides enter the mitochondria in a potential-independent manner (contrary to MitoQ and MitoVitE) because they include a sequence motif targeted to mitochondria. These peptides reduce mitochondrial ROS production, inhibit mPTP opening, prevent the release of cytochrome c from mitochondria [190] and protect against myocardial ischemia-reperfusion ex vivo and in vivo [191].
Another alternative antioxidant approach would be to prevent mitochondrial ROS production to protect mitochondrial membrane. A first possibility would be the development of specific MAO-A inhibitors, since MAO-A inhibition has been shown to reduce myocardial damage induced by ischemia-reperfusion [192]. This can also be achieved by mild uncoupling [193], decreasing ROS generation by dissipating the electrochemical potential of H+, allowing an increase in electron flux at the expense of ubiquinol. Mild uncoupling might represent an antioxygen defense mechanism during the reperfusion period, preventing the sudden increase in membrane potential, the overproduction of ROS, and the increase in mitochondrial membrane permeability. Experimental results have confirmed the relevance of this approach. Decreased ROS formation associated with myocardial protection has been demonstrated using uncouplers [194,195], or by overexpressing uncoupling proteins [196,197], and the data of McLeod et al. [198] support a role for UCP2 in delayed IPC induced cardioprotection.
5. Concluding remarks
This manuscript highlights that mitochondria represent a pertinent target to develop cardioprotective pharmacological therapies and the crucial role of the preservation of mitochondrial membrane integrity to protect cardiac cells during myocardial ischemia-reperfusion. Beside their essential roles in energy production, Ca2+ homeostasis and ROS production, mitochondria control necrotic and apoptotic myocardial cell death by regulating the permeability of its membranes. Indeed, mPTP may be considered as a central event in necrosis (although a controversy persists concerning the molecular composition of mPTP), whereas the permeabilization of mitochondrial outer membranes by proteins of the Bcl-2 family leads to apoptosis. On the whole, the studies presented above reveal that both anti-necrotic and anti-apoptotic strategies targeting mitochondrial permeability are able to protect cardiomyocytes from death during ischemia-reperfusion. However, the signaling mechanisms mediating mitochondrial membrane permeabilization in ischemia-reperfusion are diverse, so it seems likely that a combination of pharmacological therapies, i.e. compounds combining different pharmacological effects or co-administration of several drugs acting simultaneously on distinct targets, should afford a better protection against myocardial ischemia-reperfusion injury.
Acknowledgments
R. Assaly was supported by a doctoral grant from region Ile-de France and S. Paradis was supported by the Ministère de la Recherche et de la Technologie. We gratefully acknowledge Dr W. S. Neckameyer (Department of Pharmacological and Physiological Science, Saint-Louis University School of Medicine, St Louis MO, USA) for reading the manuscript.
References
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Eng J Med. 2007;107:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 2.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 3.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 4.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takagi H, Matsui Y, Sadoshima J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal. 2007;9:1373–1381. doi: 10.1089/ars.2007.1689. [DOI] [PubMed] [Google Scholar]
- 6.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 8.Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Stanley WC, Lopaschuk GD, Hall GL, Mc Cormack JG. Regulation of myocardialcarbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997;33:243–257. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 10.Crompton M. The mitochondrial transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 11.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 12.Suleiman MS, Halestrap AP, Griffiths EJ. Mitochondria: a target for myocardial protection. Pharmacol Ther. 2001;89:29–46. doi: 10.1016/s0163-7258(00)00102-9. [DOI] [PubMed] [Google Scholar]
- 13.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 14.Jennings RB, Reimer KA, Steenbergen C. Effect of inhibition of the mitochondrial ATPase on net myocardial ATP in total ischemia. J Mol Cell Cardiol. 1991;23:1383–1395. doi: 10.1016/0022-2828(91)90185-o. [DOI] [PubMed] [Google Scholar]
- 15.Lucas DT, Szweda LI. Declines in mitochondrial respiration during cardiac reperfusion: age-dependent inactivation of alpha-ketoglutarate dehydrogenase. Proc Natl Acad Sci U S A. 1999;96:6689–6693. doi: 10.1073/pnas.96.12.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadek HA, Humphries KM, Szweda PA, Szweda LI. Selective inactivation of redox-sensitive mitochondrial enzymes during cardiac reperfusion. Arch Biochem Biophys. 2002;406:222–228. doi: 10.1016/s0003-9861(02)00446-0. [DOI] [PubMed] [Google Scholar]
- 17.Zini R, Berdeaux A, Morin D. The differential effects of superoxide anion, hydrogen peroxide and hydroxyl radical on cardiac mitochondrial oxidative phosphorylation. Free Radic Res. 2007;41:1159–1166. doi: 10.1080/10715760701635074. [DOI] [PubMed] [Google Scholar]
- 18.Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 2003;17:714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 19.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 20.Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245:915–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borutaite V, Jekabsone A, Morkuniene R, Brown GC. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J Mol Cell Cardiol. 2003;35:357–366. doi: 10.1016/s0022-2828(03)00005-1. [DOI] [PubMed] [Google Scholar]
- 23.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 24.Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007;12:857–868. doi: 10.1007/s10495-007-0722-z. [DOI] [PubMed] [Google Scholar]
- 25.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 26.Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial permeability transition and cell death. Biochim Biophys Acta. 2006;1757:1297–1300. doi: 10.1016/j.bbabio.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 27.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 29.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung AWC, Halestrap AP. The cyclophilin-D binding protein of the mitochondrial permeability transition pore may not be the adenine nucleotide translocase. J Mol Cell Cardiol. 2007;42:S117. [Google Scholar]
- 32.Juhaszova M, Wang S, Zorov DB, Nuss HB, Gleichmann M, Mattson MP, Sollott SJ. The identity and regulation of the mitochondrial permeability transition pore: where the known meets the unknown. Ann N Y Acad Sci. 2008;1123:197–212. doi: 10.1196/annals.1420.023. [DOI] [PubMed] [Google Scholar]
- 33.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 36.Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366:127–137. doi: 10.1016/s0005-2728(98)00108-x. [DOI] [PubMed] [Google Scholar]
- 37.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 38.Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J Mol Cell Cardiol. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- 39.Xu M, Wang Y, Hirai K, Ayub A, Ashraf M. Calcium preconditioning inhibits mitochondrial permeability transition and apoptosis. Am J Physiol Heart Circ Physiol. 2001;280:H899–H908. doi: 10.1152/ajpheart.2001.280.2.H899. [DOI] [PubMed] [Google Scholar]
- 40.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 41.Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Béjui F, Robert D, Ovize M. Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc Res. 2004;61:115–122. doi: 10.1016/j.cardiores.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Shanmuganathan S, Hausenloy DJ, Duchen MR, Yellon DM. Mitochondrial permeability transition pore as a target for cardioprotection in the human heart. Am J Physiol Heart Circ Physiol. 2005;289:H237–H242. doi: 10.1152/ajpheart.01192.2004. [DOI] [PubMed] [Google Scholar]
- 43.Morin D, Hauet T, Spedding M, Tillement J. Mitochondria as target for antiischemic drugs. Adv Drug Deliv Rev. 2001;49:151–174. doi: 10.1016/s0169-409x(01)00132-6. [DOI] [PubMed] [Google Scholar]
- 44.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 45.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 46.Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Post-conditioning inhibits mitochondrial permeability transition. Circulation. 2005;111:194–197. doi: 10.1161/01.CIR.0000151290.04952.3B. [DOI] [PubMed] [Google Scholar]
- 47.Gomez L, Thibault H, Gharib A, Dumont JM, Vuagniaux G, Scalfaro P, Derumeaux G, Ovize M. nhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1654–H1661. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- 48.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277(38):34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 49.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 50.Rajesh KG, Sasaguri S, Ryoko S, Maeda H. Mitochondrial permeability transition-pore inhibition enhances functional recovery after long-time hypothermic heart preservation. Transplantation. 2003;76:1314–1320. doi: 10.1097/01.TP.0000085660.93090.79. [DOI] [PubMed] [Google Scholar]
- 51.Morin D, Elimadi A, Sapena R, Crevat A, Carrupt PA, Testa B, Tillement JP. Evidence for the existence of [3H]-trimetazidine binding sites involved in the regulation of the mitochondrial permeability transition pore. Br J Pharmacol. 1998;123:1385–1394. doi: 10.1038/sj.bjp.0701755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stavrovskaya IG, Narayanan MV, Zhang W, Krasnikov BF, Heemskerk J, Young SS, Blass JP, Brown AM, Beal MF, Friedlander RM, Kristal BS. Clinically approved heterocyclics act on a mitochondrial target and reduce stroke-induced pathology. J Exp Med. 2004;200:211–222. doi: 10.1084/jem.20032053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broekemeier KM, Pfeiffer DR. Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry. 1995;34:16440–16449. doi: 10.1021/bi00050a027. [DOI] [PubMed] [Google Scholar]
- 54.Fontaine E, Ichas F, Bernardi P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J Biol Chem. 1998;273:25734–25734. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- 55.Cesura AM, Pinard E, Schubenel R, Goetschy V, Friedlein A, Langen H, Polcic P, Forte MA, Bernardi P, Kemp JA. The voltage-dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. J Biol Chem. 2003;278:49812–49818. doi: 10.1074/jbc.M304748200. [DOI] [PubMed] [Google Scholar]
- 56.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1(−/−) mitochondria. Biochim Biophys Acta. 2006;1757:590–595. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Walter L, Miyoshi H, Leverve X, Bernard P, Fontaine E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic Res. 2002;36:405–412. doi: 10.1080/10715760290021252. [DOI] [PubMed] [Google Scholar]
- 58.Langsjoen PH, Langsjoen AM. Overview of the use of CoQ10 in cardiovascular disease. Biofactor. 1999;9:273–284. doi: 10.1002/biof.5520090224. [DOI] [PubMed] [Google Scholar]
- 59.Armstrong JS, Whiteman M, Rose P, Jones DP. The Coenzyme Q10 analog decylubiquinone inhibits the redox-activated mitochondrial permeability transition: role of mitochondrial complex III. J Biol Chem. 2003;278:49079–49084. doi: 10.1074/jbc.M307841200. [DOI] [PubMed] [Google Scholar]
- 60.Fuks B, Talaga P, Huart C, Henichart JP, Bertrand K, Grimee R, Lorent G. In vitro properties of 5-(benzylsulfonyl)-4-bromo-2-methyl-3(2H)-pyridazinone: a novel permeability transition pore inhibitor. Eur J Pharmacol. 2005;519:24–30. doi: 10.1016/j.ejphar.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 61.Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U, Green DR. Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science. 2005;310:66–67. doi: 10.1126/science.1117105. [DOI] [PubMed] [Google Scholar]
- 62.Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Czerski LW, Szweda PA, Szweda LI. Dissociation of cytochrome c from the inner mitochondrial membrane during cardiac ischemia. J Biol Chem. 2003;278:34499–34504. doi: 10.1074/jbc.M302021200. [DOI] [PubMed] [Google Scholar]
- 64.Morin D, Pires F, Plin C, Tillement JP. Role of the permeability transition pore in cytochrome C release from mitochondria during ischemia-reperfusion in rat liver. Biochem Pharmacol. 2004;68:2065–2073. doi: 10.1016/j.bcp.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 65.Lundberg KC, Szweda LI. Preconditioning prevents loss in mitochondrial function and release of cytochrome c during prolonged cardiac ischemia/reperfusion. Arch Biochem Biophys. 2006;453:130–134. doi: 10.1016/j.abb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 66.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2007;292:C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 67.Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prévost MC, Leber B, Andrews D, Penninger J, Kroemer G. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–739. [PubMed] [Google Scholar]
- 68.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 69.Granville DJ, Gottlieb RA. The mitochondrial voltage-dependent anion channel (VDAC) as a therapeutic target for initiating cell death. Curr Med Chem. 2003;10:1527–1533. doi: 10.2174/0929867033457214. [DOI] [PubMed] [Google Scholar]
- 70.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 71.Brocheriou V, Hagège AA, Oubenaïssa A, Lambert M, Mallet VO, Duriez M, Wassef M, Kahn A, Menasché P, Gilgenkrantz H. Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J Gene Med. 2000;2:326–333. doi: 10.1002/1521-2254(200009/10)2:5<326::AID-JGM133>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 72.Hattori R, Hernandez TE, Zhu L, Maulik N, Otani H, Kaneda Y, Das DK. An essential role of the antioxidant gene Bcl-2 in myocardial adaptation to ischemia: an insight with antisense Bcl-2 therapy. Antioxid Redox Signal. 2001;3:403–413. doi: 10.1089/15230860152409059. [DOI] [PubMed] [Google Scholar]
- 73.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–H2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 74.Hochhauser E, Kivity S, Offen D, Maulik N, Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub V, Tobar A, Vidne BA. Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2003;284:H2351–H2359. doi: 10.1152/ajpheart.00783.2002. [DOI] [PubMed] [Google Scholar]
- 75.Gustafsson AB, Tsai JG, Logue SE, Crow MT, Gottlieb RA. Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J Biol Chem. 2004;279:21233–21238. doi: 10.1074/jbc.M400695200. [DOI] [PubMed] [Google Scholar]
- 76.Huang J, Nakamura K, Ito Y, Uzuka T, Morikawa M, Hirai S, Tomihara K, Tanaka T, Masuta Y, Ishii K, Kato K, Hamada H. Bcl-xL gene transfer inhibits Bax translocation and prolongs cardiac cold preservation time in rats. Circulation. 2005;112:76–83. doi: 10.1161/CIRCULATIONAHA.105.535740. [DOI] [PubMed] [Google Scholar]
- 77.Reed JC. Apoptosis-regulating proteins as targets for drug discovery. Trends Mol Med. 2001;7:314–319. doi: 10.1016/s1471-4914(01)02026-3. [DOI] [PubMed] [Google Scholar]
- 78.Polster BM, Basanez G, Young M, Suzuki M, Fiskum G. Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J Neurosci. 2003;23:2735–2743. doi: 10.1523/JNEUROSCI.23-07-02735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B, Halazy S. 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J Med Chem. 2003;46:4365–4368. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- 80.Sawada M, Hayes P, Matsuyama S. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat Cell Biol. 2003;5:352–357. doi: 10.1038/ncb955. [DOI] [PubMed] [Google Scholar]
- 81.Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC, Antonsson B. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280:42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- 82.Esposti MD. The roles of Bid. Apoptosis. 2002;7:433–440. doi: 10.1023/a:1020035124855. [DOI] [PubMed] [Google Scholar]
- 83.Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- 84.Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–29186. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi M, Tanonaka K, Yoshida H, Koshimizu M, Daicho T, Oikawa R, Takeo S. Possible involvement of calpain activation in pathogenesis of chronic heart failure after acute myocardial infarction. J Cardiovasc Pharmacol. 2006;47:413–421. doi: 10.1097/01.fjc.0000210074.56614.3b. [DOI] [PubMed] [Google Scholar]
- 86.Saez ME, Ramirez-Lorca R, Moron FJ, Ruiz A. The therapeutic potential of the calpain family: new aspects. Drug Discov Today. 2006;11:917–923. doi: 10.1016/j.drudis.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 87.Becattini B, Culmsee C, Leone M, Zhai D, Zhang X, Crowell KJ, Rega MF, Landshamer S, Reed JC, Plesnila N, Pellecchia M. Structure-activity relationships by interligand NOE-based design and synthesis of antiapoptotic compounds targeting Bid. Proc Natl Acad Sci U S A. 2006;103:12602–12606. doi: 10.1073/pnas.0603460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Webster KA, Graham RM, Thompson JW, Spiga MG, Frazier DP, Wilson A, Bishopric NH. Redox stress and the contributions of BH3-only proteins to infarction. Antioxid Redox Signal. 2006;8:1667–1676. doi: 10.1089/ars.2006.8.1667. [DOI] [PubMed] [Google Scholar]
- 89.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 90.Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci U S A. 2002;99:12825–12830. doi: 10.1073/pnas.202474099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007;405:407–415. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 93.Toth A, Jeffers JR, Nickson P, Min JY, Morgan JP, Zambetti GP, Erhardt P. Targeted deletion of Puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;291:H52–H60. doi: 10.1152/ajpheart.01046.2005. [DOI] [PubMed] [Google Scholar]
- 94.Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem. 2004;256:107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 95.Anflous K, Blondel O, Bernard A, Khrestchatisky M, Ventura-Clapier R. Characterization of rat porin isoforms: cloning of a cardiac type-3 variant encoding an additional methionine at its putative N-terminal region. Biochim Biophys Acta. 1998;1399:47–50. doi: 10.1016/s0167-4781(98)00088-8. [DOI] [PubMed] [Google Scholar]
- 96.Cesar Mde C, Wilson JE. All three isoforms of the voltage-dependent anion channel (VDAC1, VDAC2, and VDAC3) are present in mitochondria from bovine, rabbit, and rat brain. Arch Biochem Biophys. 2004;422:191–196. doi: 10.1016/j.abb.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 97.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 98.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 99.Blachly-Dyson E, Forte M. VDAC channels. IUBMB Life. 2001;52:113–118. doi: 10.1080/15216540152845902. [DOI] [PubMed] [Google Scholar]
- 100.Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell. 1999;3:159–167. doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- 101.Madesh M, Hajnóczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol. 2001;155:1003–1015. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kusano H, Shimizu S, Koya RC, Fujita H, Kamada S, Kuzumaki N, Tsujimoto Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene. 2000;19:4807–4814. doi: 10.1038/sj.onc.1203868. [DOI] [PubMed] [Google Scholar]
- 104.Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–142. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- 105.Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- 106.Beavis AD, Lu Y, Garlid KD. On the regulation of K+ uniport in intact mitochondria by adenine nucleotides and nucleotide analogs. J Biol Chem. 1993;268:997–1004. [PubMed] [Google Scholar]
- 107.Grover GJ, Garlid KD. ATP-Sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- 108.Iwai T, Tanonaka K, Koshimizu M, Takeo S. Preservation of mitochondrial function by diazoxide during sustained ischaemia in the rat heart. Br J Pharmacol. 2000;129:1219–1227. doi: 10.1038/sj.bjp.0703148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Iwai T, Tanonaka K, Motegi K, Inoue R, Kasahara S, Takeo S. Nicorandil preserves mitochondrial function during ischemia in perfused rat heart. Eur J Pharmacol. 2002;446:119–127. doi: 10.1016/s0014-2999(02)01645-x. [DOI] [PubMed] [Google Scholar]
- 110.Carreira RS, Monteiro P, Kowaltowski AJ, Gonçalves LM, Providência LA. icorandil protects cardiac mitochondria against permeability transition induced by ischemia-reperfusion. J Bioenerg Biomembr. 2008;40:95–102. doi: 10.1007/s10863-008-9133-2. [DOI] [PubMed] [Google Scholar]
- 111.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 112.Peart JN, Gross GJ. Sarcolemmal and mitochondrial K(ATP) channels and myocardial ischemic preconditioning. J Cell Mol Med. 2002;6:453–464. doi: 10.1111/j.1582-4934.2002.tb00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hausenloy DJ, Wynne AM, Yellon DM. Ischemic preconditioning targets the reperfusion phase. Basic Res Cardiol. 2007;102:445–452. doi: 10.1007/s00395-007-0656-1. [DOI] [PubMed] [Google Scholar]
- 114.Mykytenko J, Reeves JG, Kin H, Wang NP, Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J, Zhao ZQ. Persistent beneficial effect of postconditioning against infarct size: role of mitochondrial K(ATP) channels during reperfusion. Basic Res Cardiol. 2008;103:472–484. doi: 10.1007/s00395-008-0731-2. [DOI] [PubMed] [Google Scholar]
- 115.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ahmad N, Wang Y, Haider KH, Wang B, Pasha Z, Uzun O, Ashraf M. Cardiac protection by mitoKATP channels is dependent on Akt translocation from cytosol to mitochondria during late preconditioning. Am J Physiol Heart Circ Physiol. 2006;290:H2402–H2408. doi: 10.1152/ajpheart.00737.2005. [DOI] [PubMed] [Google Scholar]
- 117.Gross ER, Hsu AK, Gross GJ. Delayed cardioprotection afforded by the glycogen synthase kinase 3 inhibitor SB-216763 occurs via a KATP- and MPTP-dependent mechanism at reperfusion. Am J Physiol Heart Circ Physiol. 2008;294:H1497–H1500. doi: 10.1152/ajpheart.01381.2007. [DOI] [PubMed] [Google Scholar]
- 118.Szewczyk A, Czyz A, Wojcik G, Wojtczak L, Nalecz MJ. ATP-regulated K+ channel in mitochondria: pharmacology and function. J Bioenerg Biomembr. 1996;28:147–152. doi: 10.1007/BF02110645. [DOI] [PubMed] [Google Scholar]
- 119.Holmuhamedov EL, Wang L, Terzic A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J Physiol (Lond) 1999;519:347–360. doi: 10.1111/j.1469-7793.1999.0347m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Korge P, Honda HM, Weiss JN. Protection of cardiac mitochondria by diazoxide and protein kinase C: implications for ischemic preconditioning. Proc Natl Acad Sci U S A. 2002;99:3312–3317. doi: 10.1073/pnas.052713199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Facundo HT, de Paula JG, Kowaltowski AJ. Mitochondrial ATP-sensitive K+ channels prevent oxidative stress, permeability transition and cell death. J Bioenerg Biomembr. 2005;37:75–82. doi: 10.1007/s10863-005-4130-1. [DOI] [PubMed] [Google Scholar]
- 122.Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H649–H657. doi: 10.1152/ajpheart.2001.280.2.H649. [DOI] [PubMed] [Google Scholar]
- 123.Lawrence CL, Billups B, Rodrigo GC, Standen NB. The KATP channel opener diazoxide protects cardiac myocytes during metabolic inhibition without causing mitochondrial depolarization or flavoprotein oxidation. Br J Pharmacol. 2001;134:535–542. doi: 10.1038/sj.bjp.0704289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.O’Rourke B. Myocardial K(ATP) channels in preconditioning. Circ Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- 125.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- 126.Halestrap AP. Does the mitochondrial permeability transition have a role in preconditioning? Circulation. 2004;110:e303. doi: 10.1161/01.CIR.0000141458.28925.D2. [DOI] [PubMed] [Google Scholar]
- 127.Costa AD, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD. The mechanism by which the mitochondrial ATP-sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J Biol Chem. 2006;281:20801–20808. doi: 10.1074/jbc.M600959200. [DOI] [PubMed] [Google Scholar]
- 128.Garlid KD. In: Integration of mitochondrial function. Lemaster JJ, Hackenbrock CR, Thurman RG, Westerhoff HV, editors. Plenum Publ. Corp; New York: 1998. pp. 257–276. [Google Scholar]
- 129.Dos Santos P, Kowaltowski AJ, Laclau MN, Seetharaman S, Paucek P, Boudina S, Thambo JB, Tariosse L, Garlid KD. Mechanisms by which opening the mitochondrial ATP- sensitive K(+) channel protects the ischemic heart. Am J Physiol Heart Circ Physiol. 2002;283:H284–H295. doi: 10.1152/ajpheart.00034.2002. [DOI] [PubMed] [Google Scholar]
- 130.Garlid KD, Dos Santos P, Xie ZJ, Costa AD, Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K(+) channel in cardiac function and cardioprotection. Biochim Biophys Acta. 2003;1606:1–21. doi: 10.1016/s0005-2728(03)00109-9. [DOI] [PubMed] [Google Scholar]
- 131.Ono H, Osanai T, Ishizaka H, Hanada H, Kamada T, Onodera H, Fujita N, Sasaki S, Matsunaga T, Okumura K. Nicorandil improves cardiac function and clinical outcome in patients with acute myocardial infarction undergoing primary percutaneous coronary intervention: role of inhibitory effect on reactive oxygen species formation. Am Heart J. 2004;148:E15. doi: 10.1016/j.ahj.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 132.Ishii H, Ichimiya S, Kanashiro M, Amano T, Imai K, Murohara T, Matsubara T. Impact of a single intravenous administration of nicorandil before reperfusion in patients with ST-segment-elevation myocardial infarction. Circulation. 2005;112:1284–1288. doi: 10.1161/CIRCULATIONAHA.104.530329. [DOI] [PubMed] [Google Scholar]
- 133.Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, Seguchi O, Myoishi M, Minamino T, Ohara T, Nagai Y, Nanto S, Watanabe K, Fukuzawa S, Hirayama A, Nakamura N, Kimura K, Fujii K, Ishihara M, Saito Y, Tomoike H, Kitamura S J-WIND investigators. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483–14936. doi: 10.1016/S0140-6736(07)61634-1. [DOI] [PubMed] [Google Scholar]
- 134.Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapère JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 136.McEnery MW. The mitochondrial benzodiazepine receptor: evidence for association with the voltage-dependent anion channel (VDAC) J Bioenerg Biomembr. 1992;24:63–69. doi: 10.1007/BF00769532. [DOI] [PubMed] [Google Scholar]
- 137.Hirsch T, Decaudin D, Susin SA, Marchetti P, Larochette N, Resche-Rigon M, Kroemer G. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp Cell Res. 1998;241:426–434. doi: 10.1006/excr.1998.4084. [DOI] [PubMed] [Google Scholar]
- 138.Lacapere JJ, Papadopoulos V. Peripheral-type benzodiazepine receptor: structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids. 2003;68:569–585. doi: 10.1016/s0039-128x(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 139.Bono F, Lamarche I, Prabonnaud V, Le Fur G, Herbert JM. Peripheral benzodiazepine receptor agonists exhibit potent antiapoptotic activities. Biochem Biophys Res Commun. 1999;265:457–461. doi: 10.1006/bbrc.1999.1683. [DOI] [PubMed] [Google Scholar]
- 140.Carayon P, Portier M, Dussossoy D, Bord A, Petitpretre G, Canat X, Le Fur G, Casellas P. nvolvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood. 1996;87:3170–3178. [PubMed] [Google Scholar]
- 141.Hauet T, Han Z, Wang Y, Hameury F, Jayle C, Gibelin H, Goujon JM, Eugene M, Papadopoulos V. odulation of peripheral-type benzodiazepine receptor levels in a reperfusion injury pig kidney-graft model. Transplantation. 2002;74:1507–1515. doi: 10.1097/00007890-200212150-00006. [DOI] [PubMed] [Google Scholar]
- 142.Leducq N, Bono F, Sulpice T, Vin V, Janiak P, Fur GL, O’Connor SE, Herbert JM. Role of peripheral benzodiazepine receptors in mitochondrial, cellular, and cardiac damage induced by oxidative stress and ischemia-reperfusion. J Pharmacol Exp Ther. 2003;306:828–837. doi: 10.1124/jpet.103.052068. [DOI] [PubMed] [Google Scholar]
- 143.Obame FN, Zini R, Souktani R, Berdeaux A, Morin D. Peripheral benzodiazepine receptor-induced myocardial protection is mediated by inhibition of mitochondrial membrane permeabilization. J Pharmacol Exp Ther. 2007;323:336–345. doi: 10.1124/jpet.107.124255. [DOI] [PubMed] [Google Scholar]
- 144.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 145.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 146.Nishino Y, Webb IG, Davidson SM, Ahmed AI, Clark JE, Jacquet S, Shah AM, Miura T, Yellon DM, Avkiran M, Marber MS. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–314. doi: 10.1161/CIRCRESAHA.107.169953. [DOI] [PubMed] [Google Scholar]
- 147.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. nhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 148.Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: united at reperfusion. Pharmacol Ther. 2007;116:173–191. doi: 10.1016/j.pharmthera.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 149.Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, Cung TT, Bonnefoy E, Angoulvant D, Aupetit JF, Finet G, André-Fouët X, Macia JC, Raczka F, Rossi R, Itti R, Kirkorian G, Derumeaux G, Ovize M. Long-term benefit of postconditioning. Circulation. 2008;117:1037–1044. doi: 10.1161/CIRCULATIONAHA.107.729780. [DOI] [PubMed] [Google Scholar]
- 150.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006;38:414–419. doi: 10.1016/j.biocel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 152.Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc Res. 2006;69:178–185. doi: 10.1016/j.cardiores.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 153.Obame FN, Plin-Mercier C, Assaly R, Zini R, Dubois-Randé JL, Berdeaux A, Morin D. ardioprotective effect of morphine and a blocker of glycogen synthase kinase 3 beta, SB216763 [3-(2,4-dichlorophenyl)-4(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione], via inhibition of the mitochondrial permeability transition pore. J Pharmacol Exp Ther. 2008;326:252–258. doi: 10.1124/jpet.108.138008. [DOI] [PubMed] [Google Scholar]
- 154.Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:579–599. doi: 10.1089/ars.2007.1845. [DOI] [PubMed] [Google Scholar]
- 155.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res. 2003;92:873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 157.Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem. 2004;279:47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 158.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol. 2007;43:564–570. doi: 10.1016/j.yjmcc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 159.Tsuruta F, Masuyama N, Gotoh Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem. 2002;277:14040–14047. doi: 10.1074/jbc.M108975200. [DOI] [PubMed] [Google Scholar]
- 160.Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289:H496–H499. doi: 10.1152/ajpheart.01182.2004. [DOI] [PubMed] [Google Scholar]
- 161.Laskey WK. Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: a pilot study. Catheter Cardiovasc Interv. 2005;65:361–367. doi: 10.1002/ccd.20397. [DOI] [PubMed] [Google Scholar]
- 162.Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, Aupetit JF, Bonnefoy E, Finet G, André-Fouët X, Ovize M. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- 163.Yang XC, Liu Y, Wang LF, Cui L, Wang T, Ge YG, Wang HS, Li WM, Xu L, Ni ZH, Liu SH, Zhang L, Jia HM, Vinten-Johansen J, Zhao ZQ. Reduction in myocardial infarct size by postconditioning in patients after percutaneous coronary intervention. J Invasive Cardiol. 2007;19:424–430. [PubMed] [Google Scholar]
- 164.Marzilli M, Orsini E, Marraccini P, Testa R. Beneficial effects of intracoronary adenosine as an adjunct to primary angioplasty in acute myocardial infarction. Circulation. 2000;101:2154–2159. doi: 10.1161/01.cir.101.18.2154. [DOI] [PubMed] [Google Scholar]
- 165.Quintana M, Hjemdahl P, Sollevi A, Kahan T, Edner M, Rehnqvist N, Swahn E, Kjerr AC, Näsman P ATTACC investigators. Left ventricular function and cardiovascular events following adjuvant therapy with adenosine in acute myocardial infarction treated with thrombolysis, results of the ATTenuation by Adenosine of Cardiac Complications (ATTACC) study. Eur J Clin Pharmacol. 2003;59:1–9. doi: 10.1007/s00228-003-0564-8. [DOI] [PubMed] [Google Scholar]
- 166.Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, Orlandi C, Blevins R, Gibbons RJ, Califf RM, Granger CB. denosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34:1711–1720. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- 167.Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW AMISTAD-II Investigators. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45:1775–1780. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 168.Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ Res. 2004;94:960–966. doi: 10.1161/01.RES.0000122392.33172.09. [DOI] [PubMed] [Google Scholar]
- 169.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–2768. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- 170.Pan W, Pintar T, Anton J, Lee VV, Vaughn WK, Collard CD. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation. 2004;110(11 Suppl 1):II45–II49. doi: 10.1161/01.CIR.0000138316.24048.08. [DOI] [PubMed] [Google Scholar]
- 171.Efthymiou CA, Mocanu MM, Yellon DM. Atorvastatin and myocardial reperfusion injury: new pleiotropic effect implicating multiple prosurvival signaling. J Cardiovasc Pharmacol. 2005;45:247–252. doi: 10.1097/01.fjc.0000154376.82445.06. [DOI] [PubMed] [Google Scholar]
- 172.Jones SP, Teshima Y, Akao M, Marbán E. Simvastatin attenuates oxidant-induced mitochondrial dysfunction in cardiac myocytes. Circ Res. 2003;93:697–699. doi: 10.1161/01.RES.0000097262.21507.DF. [DOI] [PubMed] [Google Scholar]
- 173.Direct Inhibition of delta-Protein Kinase C Enzyme to Limit Total Infarct Size in Acute Myocardial Infarction (DELTA MI) Investigators. Bates E, Bode C, Costa M, Gibson CM, Granger C, Green C, Grimes K, Harrington R, Huber K, Kleiman N, Mochly-Rosen D, Roe M, Sadowski Z, Solomon S, Widimsky P. Circulation. 2008;117:886. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 174.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 175.Zweier JL. Measurement of superoxide-derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J Biol Chem. 1988;263:1353–1357. [PubMed] [Google Scholar]
- 176.Maurel A, Hernandez C, Kunduzova O, Bompart G, Cambon C, Parini A, Francés B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am J Physiol Heart Circ Physiol. 2003;284:H1460–H1467. doi: 10.1152/ajpheart.00700.2002. [DOI] [PubMed] [Google Scholar]
- 177.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 178.Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ Res. 2008;102:1082–1090. doi: 10.1161/CIRCRESAHA.107.167072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dobsak P, Courderot-Masuyer C, Zeller M, Vergely C, Laubriet A, Assem M, Eicher JC, Teyssier JR, Wolf JE, Rochette L. Antioxidative properties of pyruvate and protection of the ischemic rat heart during cardioplegia. J Cardiovasc Pharmacol. 1999;34:651–659. doi: 10.1097/00005344-199911000-00005. [DOI] [PubMed] [Google Scholar]
- 180.Oliveira PJ, Gonçalves L, Monteiro P, Providencia LA, Moreno AJ. Are the antioxidant properties of carvedilol important for the protection of cardiac mitochondria? Curr Vasc Pharmacol. 2005;3:147–158. doi: 10.2174/1570161053586903. [DOI] [PubMed] [Google Scholar]
- 181.Javadov SA, Lim KH, Kerr PM, Suleiman MS, Angelini GD, Halestrap AP. Protection of hearts from reperfusion injury by propofol is associated with inhibition of the mitochondrial permeability transition. Cardiovasc Res. 2000;45:360–369. doi: 10.1016/s0008-6363(99)00365-x. [DOI] [PubMed] [Google Scholar]
- 182.Morin D, Papadopoulos V, Tillement JP. Prevention of cell damage in ischemia: novel molecular targets in mitochondria. Expert Opin Ther Targets. 2002;6:315–334. [Google Scholar]
- 183.Bognar Z, Kalai T, Palfi A, Hanto K, Bognar B, Mark L, Szabo Z, Tapodi A, Radnai B, Sarszegi Z, Szanto A, Gallyas F, Jr, Hideg K, Sumegi B, Varbiro G. A novel SOD-mimetic permeability transition inhibitor agent protects ischemic heart by inhibiting both apoptotic and necrotic cell death. Free Radic Biol Med. 2006;41:835–848. doi: 10.1016/j.freeradbiomed.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 184.Rajesh KG, Sasaguri S, Suzuki R, Maeda H. Antioxidant MCI-186 inhibits mitochondrial permeability transition pore and upregulates Bcl-2 expression. Am J Physiol Heart Circ Physiol. 2003;285:H2171–H2178. doi: 10.1152/ajpheart.00143.2003. [DOI] [PubMed] [Google Scholar]
- 185.Tsujita K, Shimomura H, Kaikita K, Kawano H, Hokamaki J, Nagayoshi Y, Yamashita T, Fukuda M, Nakamura Y, Sakamoto T, Yoshimura M, Ogawa H. Long-term efficacy of edaravone in patients with acute myocardial infarction. Circ J. 2006;70:832–837. doi: 10.1253/circj.70.832. [DOI] [PubMed] [Google Scholar]
- 186.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation. 2003;108:2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4. [DOI] [PubMed] [Google Scholar]
- 187.Smith RA, Kelso GF, Blaikie FH, Porteous CM, Ledgerwood EC, Hughes G, James AM, Ross MF, Asin-Cayuela J, Cocheme HM, Filipovska A, Murphy MP. Using mitochondria-targeted molecules to study mitochondrial radical production and its consequences. Biochem Soc Trans. 2003;31:1295–1299. doi: 10.1042/bst0311295. [DOI] [PubMed] [Google Scholar]
- 188.Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 200;1777:1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 189.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 190.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 191.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. Antioxid Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 192.Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, Seguelas MH, Nistri S, Colucci W, Leducq N, Parini A. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation. 2005;112:3297–3305. doi: 10.1161/CIRCULATIONAHA.104.528133. [DOI] [PubMed] [Google Scholar]
- 193.Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta. 1998;1363:100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- 194.Ganote CE, Armstrong SC. Effects of CCCP-induced mitochondrial uncoupling and cyclosporin A on cell volume, cell injury and preconditioning protection of isolated rabbit cardiomyocytes. J Mol Cell Cardiol. 2003;35:749–759. doi: 10.1016/s0022-2828(03)00114-7. [DOI] [PubMed] [Google Scholar]
- 195.Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res. 2006;72:313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 196.Bienengraeber M, Ozcan C, Terzic A. Stable transfection of UCP1 confers resistance to hypoxia/reoxygenation in a heart-derived cell line. J Mol Cell Cardiol. 2003;35:861–865. doi: 10.1016/s0022-2828(03)00147-0. [DOI] [PubMed] [Google Scholar]
- 197.Teshima Y, Akao M, Jones SP, Marbán E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res. 2003;93:192–200. doi: 10.1161/01.RES.0000085581.60197.4D. [DOI] [PubMed] [Google Scholar]
- 198.McLeod CJ, Aziz A, Hoyt RF, Jr, McCoy JP, Jr, Sack M. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J Biol Chem. 2005;280:33470–33476. doi: 10.1074/jbc.M505258200. [DOI] [PubMed] [Google Scholar]