

Abstract
Cerebral vascular endothelial cell (CEC) degeneration significantly contributes to blood–brain barrier (BBB) breakdown and neuronal loss after cerebral ischemia. Recently, emerging data suggest that peroxisome proliferator-activated receptor δ (PPARδ) activation has a potential neuroprotective role in ischemic stroke. Here we report for the first time that PPARδ is significantly reduced in oxygen-glucose deprivation (OGD)-induced mouse CEC death. Interestingly, PPARδ overexpression can suppress OGD-induced caspase-3 activity, Golgi fragmentation, and CEC death through an increase of bcl-2 protein levels without change of bcl-2 mRNA levels. To explore the molecular mechanisms, we have identified that upregulation of PPARδ can alleviate ODG-activated microRNA-15a (miR-15a) expression in CECs. Moreover, we have demonstrated that bcl-2 is a translationally repressed target of miR-15a. Intriguingly, gain- or loss-of-miR-15a function can significantly reduce or increase OGD-induced CEC death, respectively. Furthermore, we have identified that miR-15a is a transcriptional target of PPARδ. Consistent with the in vitro findings, we found that intracerebroventricular infusion of a specific PPARδ agonist, GW 501516 (2-[2-methyl-4-[[4-methyl-2-[4-(trifluoromethyl)phenyl]-1,3-thiazol-5-yl]methylsulfanyl]phenoxy]acetic acid), significantly reduced ischemia-induced miR-15a expression, increased bcl-2 protein levels, and attenuated caspase-3 activity and subsequent DNA fragmentation in isolated cerebral microvessels, leading to decreased BBB disruption and reduced cerebral infarction in mice after transient focal cerebral ischemia. Together, these results suggest that PPARδ plays a vascular-protective role in ischemia-like insults via transcriptional repression of miR-15a, resulting in subsequent release of its posttranscriptional inhibition of bcl-2. Thus, regulation of PPARδ-mediated miR-15a inhibition of bcl-2 could provide a novel therapeutic strategy for the treatment of stroke-related vascular dysfunction.
Introduction
Cerebral vascular endothelial cells (CECs) are the basic components of the blood–brain barrier (BBB) and play a critical role in maintaining cerebral homeostasis in physiological conditions. There is increasing evidence showing that ischemia-induced cerebral endothelial injury or death increases vascular permeability and disruption of the BBB, leading to primary brain damage and postischemic secondary injury (Sandoval and Witt, 2008). Therefore, protection of the cerebral endothelium becomes an important therapeutic target for stroke. However, the molecular mechanisms of cerebral endothelial injury after cerebral ischemia have not been well defined.
Peroxisome proliferator-activated receptors (PPARs) belong to the nuclear receptor family of ligand-activated transcription factors (Blanquart et al., 2003). Three isoforms of PPARs (α, γ, and δ) have been identified, displaying distinct physiological and pharmacological functions. PPARδ is highly expressed in diverse tissues including the vasculature and brain, but its function outside of metabolic effects is less understood compared with PPARα and PPARγ (Chen et al., 2003; Hamblin et al., 2009). It is noteworthy that PPARδ has been shown recently to contribute to vascular remodeling, angiogenesis, and both vascular and neuronal protection. In the vascular endothelium, PPARδ activation is shown to induce the proliferation of endothelial cells (ECs) (Stephen et al., 2004) and protect them from apoptosis (Liou et al., 2006; Han et al., 2008). PPARδ cytoprotective effects are further highlighted in the brain by recent findings that PPARδ null mice exhibited significantly greater infarct sizes than wild-type (WT) animals after focal cerebral ischemia (Arsenijevic et al., 2006). However, mechanisms of PPARδ-mediated protection after ischemic insults remain unclear.
MicroRNAs (miRs) are a novel family of non-protein-coding short RNA molecules that negatively regulate protein expression in various organisms (Bartel, 2004; Kim, 2005). The discovery of miRs has shed light on how noncoding RNAs can play important roles in cell differentiation, proliferation, migration, and apoptosis (Jovanovic and Hengartner, 2006; Schickel et al., 2008; Eacker et al., 2009). Several miRs have been found recently to regulate proapoptotic and antiapoptotic genes (Jovanovic and Hengartner, 2006; Schickel et al., 2008). For example, miR-15a is able to reduce the antiapoptotic protein bcl-2 by directly binding to the 3′ untranslated region (UTR) of bcl-2 mRNA and inhibiting its translation (Cimmino et al., 2005).
In the present study, we have used oxygen-glucose deprivation (OGD) and a transient focal cerebral ischemia model to explore the effects and molecular mechanisms of PPARδ on ischemia-induced cerebrovascular injury. We have identified for the first time that miR-15a is a novel target of PPARδ trans-repression, directly regulates bcl-2, an antiapoptotic protein in a posttranscriptional manner, and contributes to PPARδ-mediated vascular protection against ischemia-like insults.
Materials and Methods
All chemicals and reagents were purchased from Sigma-Aldrich, and cell culture supplies were purchased from Invitrogen unless specified.
Cell culture.
Mouse CECs were prepared as described previously. Briefly, mouse cerebral cortex from adult male C57BL/6J mice (body weight, 25–30 g; 3–4 months old) (The Jackson Laboratory) was homogenized, filtered, and sequentially digested with collagenase B, then collagenase/dispase (Roche Molecular Biochemicals), followed by centrifugation in a 40% Percoll solution. The second band containing microvessels was collected and plated onto collagen-coated dishes. Mouse CECs (4–15 passages, >95% purity based on expression of factor VIII and exhibiting bradykinin receptor function) were grown to 85–95% confluency before use (Yin et al., 2002b).
Oxygen-glucose deprivation.
To mimic ischemia-like conditions in vitro, cell cultures were exposed to OGD for various times (Yin et al., 2002b). Briefly, confluent mouse CECs were transferred into a temperature-controlled (37 ± 1°C) anaerobic chamber (Forma Scientific) containing a gas mixture composed of 5% CO2 and 95% N2. The culture medium was replaced with deoxygenated glucose-free HBSS, and cells were maintained in the hypoxic chamber for 0, 4, and 15 h. Control mouse CECs were not exposed to OGD.
Mouse model of transient focal cerebral ischemia.
Male C57BL/6 mice were purchased from The Jackson Laboratory. Focal cerebral ischemia was induced by intraluminal middle cerebral artery occlusion (MCAO) using a nylon monofilament suture as described previously (Yang et al., 1997; Yin et al., 2010). Briefly, mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). After a midline skin incision, the left common carotid artery was exposed, and then its branches were electrocoagulated. A 2 cm length of 6-0 rounded tip nylon suture was gently advanced from the external carotid artery up to the internal carotid artery until regional cerebral blood flow (CBF) was reduced to <16% of baseline. After 60 min of proximal MCA occlusion, blood flow was restored by removing the suture. Changes in CBF at the surface of the cortex were recorded using a laser-Doppler flowmetry monitor (BPM2 System; Vasamedic). Sham control animals were subjected to similar operations to expose the carotid arteries without occlusion of the middle cerebral artery. After 60 min of MCAO, the mice were allowed to recover for 24 h. Arterial blood gases, mean arterial pressure, and heart rate were also monitored in selected animals 30 min before, during, and 30 min after MCAO. The rectal temperature was controlled at 37.0 ± 0.5°C during surgery with a feedback-regulated heating pad (Harvard Apparatus). After ischemic insult, mice were kept in an air-ventilated incubator at 24.0 ± 0.5°C. The animals were killed at 24 h of reperfusion, and the brains were quickly removed for biochemical assays as well as infarct determination. All procedures using laboratory animals were approved by the University of Michigan Animal Care and Use Committee.
Treatment of mouse CECs with an miR-15a mimic or inhibitor.
In certain experiments, mouse CECs were plated in 24-well or 6-well multiwell plates and were transfected with an miR-15a mimic, mimic control, miR-15a inhibitor, or inhibitor control at a final concentration of 50 nm (miRIDIAN mimic; Dharmacon) using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. The cells were then subjected to OGD after 48 h and harvested for detection of cell death, caspase-3 activity, and Golgi fragmentation.
Assessment of mouse CEC death.
The extent of mouse CEC death was assessed by the trypan blue exclusion and lactate dehydrogenase (LDH) assays as described previously (Yin et al., 2002a,b).
Intracerebroventricular infusion of GW 501516.
The procedure was performed as described previously (Dharap et al., 2009). Briefly, C57BL/6 mice were anesthetized and fixed in a stereotaxic apparatus. The brain infusion cannula was stereotaxically implanted into the left lateral ventricle of the brain (bregma, −0.22 mm; dorsoventral, 3 mm; lateral, 1 mm) (Paxinos et al., 1985) and was affixed to the skull. The micro-osmotic pump (Alzet 1003D; Durect Corporation) connected with a brain infusion cannula was placed in a subcutaneous pocket on the neck of the mouse. Continuous intracerebroventricular infusion of a specific PPARδ agonist, GW 501516 (10 μg/μl, at a rate of 1 μl/h; Enzo Life Science), or vehicle was started 24 h before MCA occlusion. Transient focal cerebral ischemia was induced in the left MCA territory for 1 h, and all mice were killed 24 h after MCAO. Mouse brains were removed and then subjected to 2,3,5-triphenyltetrazolium chloride (TTC) staining for measurement of infarct volume (n = 6), Evans Blue (EB) assay for detection of BBB permeability (n = 6), and isolation of cerebral microvessels for quantitative PCR, Western blot, caspase-3 activity assay, and DNA laddering (n = 3). GW 501516 and vehicle reagent were filled into micro-osmotic minipumps and incubated at 37°C overnight before implantation.
Cerebral microvessel isolation.
Cerebral microvessel isolation used previously described methods with modifications (Pardridge et al., 1985; Zlokovic et al., 1993; Yin et al., 2006a). Briefly, mice were killed by decapitation under anesthesia. The brains were immediately removed from the skull and immersed in ice-cold buffer A (in mm: 103 NaCl, 4.7 KC1, 2.5 CaC12, 1.2 KH2PO4, 1.2 MgSO4, and 15 HEPES, pH 7.4). The brain was homogenized in a fivefold volume excess of buffer B (103 mm NaCl, 4.7 mm KC1, 2.5 mm CaC12, 1.2 mm KH2PO4, 1.2 mm MgSO4, 15 mm HEPES, 25 mm HCO3, 10 mm glucose, 1 mm sodium pyruvate, and 1 g/100 ml dextran, pH 7.4) with a Teflon homogenizer. The homogenate was suspended in an equal volume of 25% BSA and was centrifuged at 5800 × g at 4°C for 30 min. The pellet was resuspended in 10 ml of buffer B and passed over an 85 μm nylon mesh (Tetko). This filtrate was then passed over a 3 × 4 cm glass bead column (0.45 mm glass beads) with a 44 μm nylon mesh at the bottom and then washed with 400 ml of buffer B. Microvessels adhered to the glass beads whereas contaminants passed unimpeded. Microvessels were recovered by repeated gentle agitation of the glass beads in buffer B. The supernatant with microvessels was decanted and spun at 500 × g for 5 min, and the final pellet was stored at −80°C until various biochemical assays were performed.
Measurement of infarct volume.
Infarct volume was measured using 2% TTC staining as described previously. Mouse brains were removed at 24 h after MCAO and sliced into eight coronal sections (1 mm thick) by a mouse brain matrix. The slices were stained with 2% TTC for 15 min at 37°C and scanned, and the infarct area was estimated by MetaMorph software. The infarct volume was calculated using a derived formula (Swanson et al., 1990) in which infarct volume as a percentage of the contralateral hemisphere was calculated as 100 (contralateral hemisphere volume − non-infarct ipsilateral hemisphere volume)/contralateral hemisphere volume.
Real-time PCR.
A quantitative real-time reverse-transcriptase (RT)-PCR was performed with a Bio-Rad thermocycler and an SYBR green kit (Bio-Rad) according to the recommendations of the manufacturer. Specific primers used for the reaction are as follows: PPARδ forward, 5′-AGGCCCGGGAAGAGGAGAAAGAGG-3′; PPARδ reverse, 5′-CGCGTGGACCCCGTAGTGGA-3′; bcl-2 forward, 5′-GCGCAAGCCGGGAGAACAGGGTAT-3′; bcl-2 reverse, 5′-CCGGCGGAGGGTCAGATGGA-3′. The relative PPARδ or bcl-2 mRNA expression was normalized by 18S RNA levels.
TaqMan miRNA assay for identification of miR-15a.
Total RNA including miRNA was extracted from treated mouse CEC cultures or isolated cerebral microvessels by using an miRNeasy Mini kit (Qiagen). Reverse transcription was performed using the specific TaqMan MiRNA Reverse Transcription kit (Applied Biosystems). Equal amounts of total RNA (10 ng) were reverse transcribed with 100 mm dNTPs (with dTTP), 50 U of reverse transcriptase, 0.4 U of RNase inhibitor, and specific microRNA reverse transcriptase primers at a condition of 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min. PCR reactions were then conducted using the TaqMan MiRNA Assay kit (Applied Biosystems) at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Each PCR reaction contained 1.33 μl of the RT reaction product, 10 μl of TaqMan 2× Universal PCR Master Mix, and 1 μl of 20× TaqMan MicroRNA Assay reagent for miR-15a in a total volume of 20 μl. The relative microRNA levels were normalized to endogenous SnoRNA 202 expression for each sample. PCR experiments were repeated three times, each using separate sets of cultures (Yin et al., 2006b).
Caspase-3 activity assay.
Caspase-3 activity in treated CECs was detected by a commercial Caspase-3 Colorimetric Activity Assay kit (Millipore Corporation). Cultured CECs after various treatments or isolated cerebral microvessels were lysed in 200 μl of 1× cell lysis. After incubation for 10 min on ice, the lysate was spun at 10,000 × g for 10 min at 4°C, and the supernatant was used to detect caspase-3 activity. Cell lysate containing 50 μg of protein in 100 μl of assay buffer was mixed with the 10 μl substrate of caspase-3 and incubated for 2 h at 37°C. The samples were read at 405 nm in a microtiter plate reader. Fold increase in caspase-3 activity was determined by comparing optical density readings from the induced apoptotic samples with the levels of the controls. Data were presented as the percentage of controls.
Detection of DNA fragmentation.
A genomic DNA isolation kit from Promega was used for the extraction of DNA from isolated cerebral microvessels. The vessel samples were lysed by the addition of 1.5 ml of lysis solution and treated with RNase A solution. Proteins were precipitated by a solution provided with the DNA isolation kit, and DNA was hydrated. The DNA samples (10 μg/lane) were electrophoresed at 80 V for 1.5 h in 1% agarose gel containing 0.4 μg/ml ethidium bromide in a Tris–acetate buffer (0.4 mol/L Tris, 0.25 mol/L sodium acetate, and 0.22 mmol/L EDTA, pH 7.8). DNA was visualized through ultraviolet transillumination and photographed (Xu et al., 2000).
Western blot analysis.
Total protein was extracted from mouse CECs or isolated cerebral microvessels as described previously (Yin et al., 2006a,b). Samples (20–40 μg of protein) were electrophoresed onto a 10–15% SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were blocked in TBST buffer containing 20 mm Tris-HCl, 5% nonfat milk, 150 mm NaCl, and 0.05% Tween 20, pH 7.5, for 1 h at room temperature. Thereafter, the blot was incubated, respectively, with primary rabbit anti-PPARδ antibody (1:200; Santa Cruz Biotechnology), rabbit anti-GRASP65 antibody (1:200; Santa Cruz Biotechnology), rabbit anti-bcl-2 antibody (1:100; Santa Cruz Biotechnology), rabbit anti-bcl-xl antibody (1:100; Santa Cruz Biotechnology), or mouse anti-actin antiserum (1:500; Santa Cruz Biotechnology) for 1–2 h at room temperature. The membrane was washed with TBST three times at 10 min intervals, incubated with the secondary antibody [1:5000; anti-rabbit, anti-mouse, or anti-goat IgG conjugated with alkaline phosphatase (Promega)] at room temperature for 1 h, and then washed three times each at 10 min intervals with TBST and two times each for 10 min with TBS (TBST without Tween 20). The color reaction was developed by the Blot AP System (Promega).
Immunofluorescence staining.
Mouse CECs grown on coverslips, with or without infection with adenoviral PPARδ or corresponding controls for 48 h, were subjected to OGD for various times. The cells were then fixed with 4% paraformaldehyde for 30 min and washed three times with 0.1 m PBS, pH 7.4. The cells were incubated with a primary rabbit anti-PPARδ antibody or rabbit anti-GRASP65 antibody (1:50; Santa Cruz Biotechnology) overnight at 4°C. On the following day, the cells were incubated with fluorescein-conjugated anti-goat IgG (1:100; Vector Laboratories) for 1 h. CECs were counterstained with 1 μg/ml propidium iodide (Invitrogen) to visualize nuclear morphology. Slides were washed, wet mounted, and examined with an Olympus fluorescence microscope (Yin et al., 2002a, 2006b).
Quantitation of Evans Blue extravasation.
For analysis of cerebrovascular permeability after MCAO, mice were injected with 100 μl of 4% EB (Sigma-Aldrich) 23 h after MCAO. One hour later, animals were perfused with PBS, and the brains were removed and separated into hemispheres ipsilateral and contralateral to the MCAO. Each hemisphere was then homogenized in N,N-dimethylformamide (Sigma-Aldrich) and centrifuged for 45 min at 25,000 relative centrifugal force. The supernatants were collected, and quantitation of EB extravasation was performed as described previously (Su et al., 2008). Briefly, EB levels in each hemisphere were determined from the formula (A620 nm − ((A500 nm + A740 nm)/2))/mg wet weight. Background EB levels in the non-ischemic hemisphere were subtracted from the ischemic hemisphere ipsilateral to the MCAO.
Functional analysis of miR-15a interaction with 3′-UTR of bcl-2.
A 566 bp segment from the 3′-UTR of the bcl-2 gene containing miR-15a binding sites was amplified by PCR from mouse genomic DNA and then cloned into the SacI/HindIII site of a pMIR-REPORT Luciferase vector (Applied Biosystems). The following primer sets were used to generate specific fragments: bcl-2-UTR forward, 5′-AGAGCTCGCCTTCAGAGGGTTGCTT-3′; bcl-2-UTR reverse, 5′-CAAGCTTTACTGTCCAGTTCTAGGGTCC-3′. We also generated a mutant 3′-UTR of the bcl-2 gene with substitutions of 3 bp, respectively, from the site of perfect complementarity by using the QuikChange XL Site-Directed Mutagenesis kit (Stratagene). The sequence of the mutant bcl-2 3′-UTR segment contained 5′-GAATAAGAAACCCTGTGGCCCTATCCTGCCAAGATCATTG-3′ (the three italic bold nucleotides are mutated). Wild-type and mutant inserts were confirmed by sequencing analysis. HEK293 cells were plated at 0.5 × 105 cells per well in 24-well plates. The following day, cells were cotransfected with an 800 ng of pMIR-REPORT Luciferase vector, including the 3′-UTR of bcl-2 (with either wild-type or mutant miR-15a binding sites) and an miR-15a mimic or mimic control at a final concentration of 50 nm by using Lipofectamine 2000 (Invitrogen). Luciferase assays were performed by using the dual luciferase reporter assay system (Promega) 48 h after transfection.
Adenovirus-mediated gain-of-PPARδ function.
We used a previously described method that facilitates adenoviral vector construction (He et al., 1998). Briefly, a ∼1.4 kb segment of a mouse PPARδ gene coding sequence was amplified by RT-PCR from mouse total RNA and then cloned into pCMVTrack, an adenovirus shuttle vector. This plasmid was cotransformed with AdEasy, a backbone of the adenovirus, into Escherichia coli. The clone from in vivo recombination was isolated, digested, and used for transfection in the packaging cell line HEK293. Transfected cells were monitored for green fluorescent protein (GFP) expression and collected 7–10 d later by scraping and pelleting along with any floating cells in the culture. After amplification in HEK293, viruses were purified by a CsCl gradient; final yields were generally 1011 to 1012 plaque-forming units. The generated adenovirus was used to infect CECs or HEK293 cells, and the expression levels of the transgene determined by immunoblotting. Comparisons were made using control adenoviral vectors with empty inserts or GFP.
Chromatin immunoprecipitation assay.
Mouse CECs were infected with an adenovirus carrying mouse PPARδ and GFP. After 48 h, cells were fixed and a chromatin immunoprecipitation (ChIP) assay was performed by using the EZ-ChIP assay kit from Millipore Corporation. The size of the sonicated DNA fragments subjected to immunoprecipitation was 0.5–1 kb as determined by ethidium bromide gel electrophoresis. Purified chromatin was immunoprecipitated using an anti-PPARδ antibody (Santa Cruz Biotechnology). Eluted DNA fragments were purified to serve as templates for PCR amplification. The input fraction corresponded to 2% of the chromatin solution before immunoprecipitation. By using bioinformatics software to analyze the miR-15a promoter, a potential PPARδ binding site was identified, which is located −1593/−1573 bp [PPAR response element (PPRE)] upstream of the transcription start site. Primers used to amplify the area containing this PPARδ binding site are as follows: forward primer, 5′-TTCAAAAAGTATCCCATTATTCTG-3′ and reverse primer, 5′-TGTCCTATTTCCCCCTTCAA-3′, resulting in a 197 bp fragment. To monitor the specificity of each assay, no observable DNA binding was found in normal IgG immunoprecipitates as background control, whereas strong DNA binding was confirmed in anti-acetyl-histone H3 immunoprecipitates as a positive control.
Plasmid constructs and Luciferase assays.
A 1887 bp segment from the promoter region (−1824/+62) of the mouse miR-15a gene (miR-15a 1.9 kb WT) was amplified by PCR from mouse genomic DNA and then cloned into the KpnI/XhoI site of the pGL 4.10 Luciferase vector (Promega). The following primer sets were used to generate specific fragments: forward, 5′-CGGGGTACCACTAGAATAACAGCCATGGGAGACAC-3′; reverse, 5′-CCGCTCGAGAGTATGGCCTGCACCT-TTTCAACA-3′. According to bioinformatics analysis, a potential PPAR binding site at a location of −1593/−1573 bp (PPRE) was identified in this promoter fragment. Thus, we also generated mutant miR-15a promoter constructs (miR-15a 1.9 kb PPRE mutant) with substitutions of 3 bp, respectively, from the PPRE site of perfect complementarity by using the QuikChange XL Site-Directed Mutagenesis kit (Stratagene). The sequence of the mutant PPRE miR-15a promoter segment contained 5′-GAATTAATATTAGAAGTTTTATAAGG-CGCAAAGTAAATGTTTCTAAAG-3′ (the three italic underlined nucleotides are mutated). Wild-type and mutant inserts were confirmed by sequencing.
miR-15a promoter plasmids, including miR-15a 1.9 kb WT and PPRE mutant together with the Renilla luciferase control reporter vector (pRL-TK), were transfected into HEK293 cells with Lipofectamine 2000 (Invitrogen) for 4 h and then changed to growth medium for 48 h. HEK293 cells were also coinfected with an adenovirus carrying mouse PPARδ or GFP for 48 h. Promoter activity was examined using the Dual-Luciferase Assay kit (Promega) with a TD-20/20 luminometer (Turner Designs). miR-15a promoter–firefly–luciferase activity was normalized to the TK promoter–Renilla–luciferase activity.
Statistical analysis.
Quantitative data are expressed as mean ± SD or SEM based on at least three independent experiments of triplicate samples. Differences among groups were statistically analyzed by one-way ANOVA, followed by Bonferroni's post hoc test. Comparisons between two experimental groups were based on a two-tailed t test. A p value <0.05 was considered significant.
Results
The expression of PPARδ is reduced in OGD-induced CEC death
OGD causes a rapid reduction in PPARδ mRNA and protein expression in mouse CECs, decreasing at 4 h and maintaining a lower level at 16 h after OGD onset (Fig. 1A,B). The decrease in PPARδ expression is consistent with CEC death after OGD exposure, starting at 4 h and even worsening at 16 h after OGD stimuli (Fig. 1C). The cellular distribution of PPARδ after OGD was further examined by immunofluorescent staining. As shown in Figure 1D, normal nuclear localization of PPARδ expression is initially observed in CECs, and this nuclear pattern changed to a smaller, dense nuclear distribution after 4 h of OGD exposure. Finally, nuclear localization of PPARδ disappears at 16 h of OGD insult.
Figure 1.

The expression of PPARδ in OGD-induced CEC death. A, B, OGD causes a time-dependent decrease in PPARδ mRNA expression (A) and protein levels (B), starting at 4 h after OGD and persisting at least to 16 h after OGD. C, D, Exposure to OGD results in obvious mouse CEC death detected by the LDH assay (C) and trypan blue exclusion-based viable cell counts (D), which initiated at 4 h and persisted to at least 16 h. E, OGD-induced PPARδ reduction is further confirmed by immunofluorescent staining, showing gradual loss of nuclear localization. Scale bar,10 μm. *p < 0.05 versus non-OGD controls. Representative data from three separate experiments with similar results are shown.
OGD induces fragmentation of the Golgi apparatus in CECs
Golgi fragmentation and subsequent collapse are an essential process in the development of cell death (Gonatas et al., 2006). To explore whether Golgi fragmentation occurs in OGD-induced CEC death, we used immunofluorescent staining and Western blotting to evaluate its temporal profiles (Fig. 2A–G). As demonstrated in Figure 2C, the Golgi apparatus appears to be ribbon-like structures adjacent to the nuclei in normal conditions. After OGD, the morphology of Golgi apparatus changes to debris-like structures scattered in the cytoplasm (Fig. 2D). These alterations are further confirmed by Western blotting analysis showing that GRASP65, a Golgi structural protein that is cleaved in apoptosis (Lane et al., 2002), is significantly decreased after OGD. The reduction begins as early as 4 h after OGD exposure and further enhances after 16 h treatment (Fig. 2G).
Figure 2.
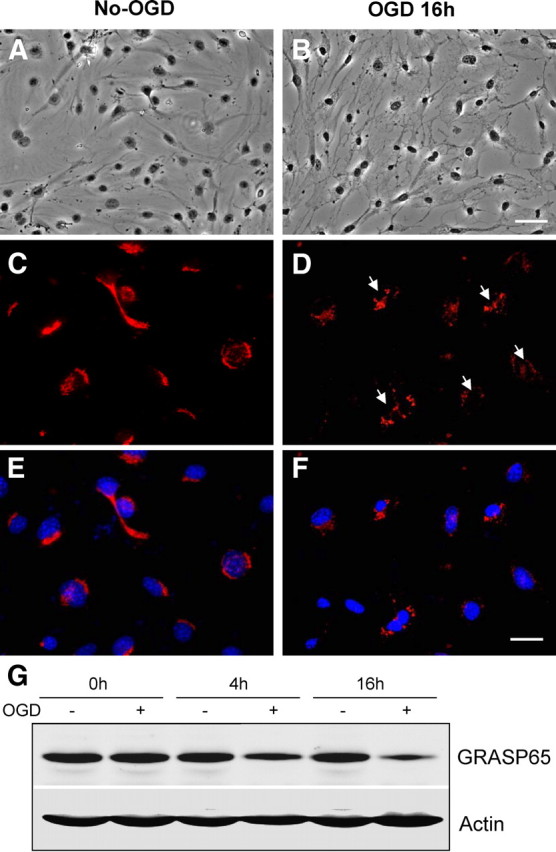
Fragmentation of the Golgi apparatus in CECs after OGD. A, B, Phase-contrast light microscopy shows normal mouse CECs (A) and CECs with OGD for 16 h (B). Note the obvious changes in cellular morphology and nuclear shrinkage after 16 h OGD. C, D, Immunofluorescent stain using an antibody against Golgi marker GRASP65 (red color) reveals normal Golgi apparatus (C) and Golgi fragmentation (D). Arrows indicate disassembled Golgi apparatus. E, F, The same areas of C and D (blue color) were counterstained with 4′,6′-diamidino-2-phenylindole and show nuclei with normal and smaller shapes. G, Western blot analysis shows the time course of GRASP65 expression in the indicated time. Note the gradual reduction of this Golgi structural protein during OGD exposure. Actin is used as a loading control. Scale bars: A, B, 20 μm; C–F, 10 μm.
Overexpression of PPARδ decreases OGD-induced cell death, caspase-3 activity, and Golgi fragmentation in CECs
Given the recent evidence suggesting that PPARδ activation may protect against neural cell loss after focal cerebral ischemia (Arsenijevic et al., 2006), we investigated this potential link by upregulating PPARδ gene expression using adenoviral gene transfer. The success of this gain-of-PPARδ function approach is demonstrated by the high percentage of infected CECs (supplemental Fig. A, available at www.jneurosci.org as supplemental material), significantly increased PPARδ protein levels and PPRE-driven transactivation (supplemental Figs. B, C, available at www.jneurosci.org as supplemental material). In addition, overexpression of PPARδ alleviates CEC death attributable to 16 h of OGD exposure, as evidenced by LDH quantitation (Fig. 3A) and live cell counts (Fig. 3B). Adenoviral GFP gene transfer into CECs has no effect, confirming the specific action of adenovirus-mediated PPARδ gene upregulation.
Figure 3.

PPARδ regulation of cell death, caspase-3 activity, and fragmentation of the Golgi apparatus in CECs after OGD. A, B, Infection of CECs with adenoviral PPARδ (Ad.PPARδ) but not adenovirus GFP (Ad.GFP) effectively attenuates OGD-induced CEC death detected by LDH assay (A) and trypan blue exclusion-based viable cell counts (B). C, Overexpression of PPARδ in mouse CECs reduces OGD-induced caspase-3 activity. D, Upregulation of PPARδ in mouse CECs reverses OGD-induced GRASP65 reduction analyzed by Western blotting. Infection of CECs with adenovirus GFP or no transfection control (NTC) has no cytoprotective effect after OGD. *p < 0.05 versus Ad.GFP or NTC. Results shown are representative of three separate experiments with similar results.
Previous work by our laboratory and others has reported that apoptosis is one of the key mechanisms for CEC death after ischemic or hypoxic insult (Xu et al., 2000; Yin et al., 2002b). Recently, PPARδ activation appears to prevent apoptosis by inhibiting caspases (Liou et al., 2006), and some specific proteins in the Golgi apparatus have been shown to be substrates of several executive caspases (Mancini et al., 2000; Lane et al., 2002). Therefore, we examined the effects of PPARδ upregulation on caspase-3 activity and Golgi fragmentation in CECs after OGD. Sixteen hours of OGD increases caspase-3 activity, and this increase is significantly reduced by adenovirus-mediated overexpression of PPARδ (Fig. 3C). Moreover, gain-of-PPARδ function by adenovirus effectively reverses OGD-induced reduction of full-length GRASP65 protein levels (Fig. 3D) and also fragmentation of the Golgi apparatus (data no shown). Adenoviral GFP gene transfer into CECs has no effect on caspase-3 activity and Golgi fragmentation. These results suggest that PPARδ may confer a cytoprotective action in CECs after OGD treatment.
Overexpression of PPARδ increases bcl-2 protein levels and inhibits miR-15a expression in CECs after OGD
To further identify the molecular mechanisms responsible for PPARδ-mediated protective roles in OGD-induced CEC death, we examined PPARδ regulatory effects on the expression of one critical antiapoptotic gene, bcl-2. As shown in Figure 4, bcl-2 protein expression is significantly decreased in CECs after OGD, starting at 4 h and persisting at least to 16 h after OGD stimuli (Fig. 4A). Moreover, gain-of-PPARδ function in mouse CECs by adenovirus-mediated gene transfer partially reverses OGD-induced reduction of bcl-2 protein levels (Fig. 4B). However, upregulation of PPARδ has no effect on bcl-2 mRNA levels (Fig. 4C), suggesting that the increased bcl-2 protein levels by PPARδ may not be at the transcriptional but rather at the translational level.
Figure 4.

PPARδ regulation of bcl-2 and miR-15a levels. A, Western blotting shows bcl-2 protein levels are significantly decreased after OGD, which began at 4 h and persisted up to 16 h after OGD. B, Western blotting data demonstrate that upregulation of adenovirus PPARδ (Ad.PPARδ or Ad.Pδ), but not adenovirus GFP (Ad.GFP), reverses OGD-induced reduction of bcl-2 protein levels in CECs. C, Quantitative PCR data demonstrate that upregulation of adenovirus PPARδ has no effect on bcl-2 mRNA levels in CECs. D, Quantitative PCR data show that the expression of miR-15a is gradually increased after OGD stimuli. E, Infection of CECs with adenovirus PPARδ, but not adenovirus GFP, effectively mitigates OGD-induced miR-15a detected by quantitative PCR. *p < 0.05 versus non-OGD or Ad.GFP and no transfection control (NTC). Data are expressed as mean ± SEM from three separate experiments in triplicate.
The discovery of miRs that mediate posttranscriptional silencing of specific target genes has shed light on how noncoding RNAs can play important roles in apoptosis. miRs have been found recently to regulate both proapoptotic and antiapoptotic genes in apoptotic pathways (Jovanovic and Hengartner, 2006; Schickel et al., 2008). For example, Cimmino et al. (2005) showed that miR-15a negatively regulates bcl-2 protein levels, activates the intrinsic apoptosis pathway, and causes cell death. In this study, we noticed that miR-15a expression is significantly increased in the same OGD temporal profile, elevating at 4 h and maintaining an even higher level at 16 h after OGD onset (Fig. 4D). Of significance, gain-of-PPARδ function in mouse CECs by adenovirus-mediated gene transfer effectively alleviates OGD-activated miR-15a expression (Fig. 4E). As expected, adenoviral GFP gene transfer into CECs has no effect on the expression of miR-15a.
These data imply that PPARδ may execute a cytoprotective action in CECs in response to ischemic insult by posttranscriptionally enhancing bcl-2, an antiapoptotic protein that is translationally inhibited by miR-15a.
miR-15a regulates OGD-induced caspase-3 activity, Golgi fragmentation, and cell death in CECs
To further explore the contributory role of miR-15a in the regulation of CEC death after OGD exposure, gain- or loss-of-miR-15a function was achieved by transfecting an miR-15a mimic or inhibitor into CECs. As demonstrated in Figure 5, treatment with the miR-15a mimic at a final concentration of 50 nm increases CEC death after 16 h of OGD (Fig. 5A,C), whereas the miR-15a inhibitor effectively reduces OGD-induced CEC death compared with nontransfected control groups (Fig. 5B,D). These results demonstrate that in vitro ischemia-like stimuli induce miR-15a expression, resulting in CEC death.
Figure 5.

miR-15a regulation of CEC death, caspase-3 activity, and Golgi fragmentation after OGD. A, C, Quantitative PCR data demonstrate that an miR-15a mimic, but not miR-15a mimic control, aggravates OGD-induced neuronal death detected by an LDH assay (A) and viable cell counts (C). E, G, The miR-15a mimic also causes an increase in caspase-3 activity (E) and aggravates OGD-induced loss of the Golgi structural protein GRASP65 (G) in CECs. B, D, F, H, In contrast, the miR-15a inhibitor, but not inhibitor control, alleviates OGD-induced CEC loss (B, D), activation of caspase-3 (F), and Golgi fragmentation (H). Treatment with the miR-15a mimic or inhibitor control in CECs has no effect. *p < 0.05 versus mimic or inhibitor control. Results shown are representative of three separate experiments with similar results. Ctl, Control.
Also, we further examined the regulatory role of miR-15a in the regulation of OGD-induced caspase-3 activity and Golgi fragmentation in CEC cultures. As shown in Figure 5, the miR-15a mimic significantly aggravates OGD-induced activation of caspase-3 activity and reduction in the content of the GRASP65 protein (Fig. 5E,G), whereas the miR-15a inhibitor effectively reverses these effects (Fig. 5F,H). Treatment of CECs with the miR-15a mimic or inhibitor negative control has no effect on OGD-induced caspase-3 activity, Golgi fragmentation, or cell death in mouse CECs.
bcl-2 is a target gene of miR-15a in CECs after OGD
Recent research has revealed that miR15a can directly bind to a specific sequence in the 3′-UTR region of bcl-2 and inhibit translation of bcl-2 in a posttranscriptional manner (Cimmino et al., 2005). To confirm the causative role of miR-15a in the regulation of bcl-2, both gain- and loss-of-miR-15a function were applied by transfecting CECs with an miR-15a mimic or inhibitor reagent. After 48 h of treatment, the transfected CEC cultures were subjected to 16 h OGD, and then the expression of bcl-2 was examined by Western blot. As shown in Figure 6, the miR-15a inhibitor effectively increases (Fig. 6A) whereas the mimic decreases (Fig. 6B) the expression of bcl-2 in mouse CECs after ODG. Of note, the miR-15a mimic or inhibitor has no effect on the expression of bcl-xl, another member of the antiapoptotic bcl-2 family of proteins, suggesting the specificity of miR-15a suppression of bcl-2.
Figure 6.

bcl-2 is a target gene of miR-15a in mouse CECs. A, Treatment of mouse CECs with an miR-15a inhibitor effectively increases bcl-2 protein levels after OGD. B, In contrast, an miR-15a mimic reduces protein expression of bcl-2 after OGD. The miR-15a mimic and inhibitor control have no effect on the expression of bcl-2. Of note, the expression of bcl-xl, a bcl-2 family protein, is not affected in mouse CECs after treatment with either an miR-15a mimic or inhibitor. C, miR-15a binding site within the 3′-UTR of bcl-2 mediates miR-15a translational repression. Design of an miR-15a reporter vector containing a CMV promoter driving the expression of luciferase cDNA fused to the bcl-2 3′-UTR (pMIR–bcl2–3′-UTR) or to a mutated bcl-2 3′-UTR (pMIR–bcl2–m3′-UTR). miR-15a and the miR-15a-binding site in the 3′-UTR of bcl-2 are shown. Mouse CECs were transfected with a pMIR–bcl2–3′-UTR and a pMIR–bcl2–m3′-UTR, respectively. The cells were also cotransfected with an miR-15a mimic or mimic control. Compared with the mimic control, the miR-15a mimic decreases expression of luciferase containing a WT miR-15a binding site (left 2 columns) but not a mutant binding site (right 2 columns). Data are expressed as mean ± SEM. *p < 0.05 versus mimic control. Results shown are representative of three separate experiments with similar results. Ctl, Control.
To further verify that miR-15a may repress bcl-2 by binding directly to the predicted binding site in its 3′-UTR region, we cloned a reporter vector in which cytomegalovirus (CMV)-driven luciferase cDNA was followed by a fragment of the 3′-UTR of bcl-2 mRNA containing the conserved miR-15a binding sequence (Fig. 6C). We also generated a luciferase reporter construct fused to the bcl-2 3′-UTR but with a mutant miR-15a binding sequence (Fig. 6C). We then transfected this luciferase reporter vector with a wild-type or mutant miR-15a binding sequence into HEK293 cells. We also cotransfected these cells with an miR-15a mimic or mimic control and measured luciferase activity. As shown in Figure 6C, the miR-15a mimic at a concentration of 50 nm decreases luciferase activity of the reporter vector containing the miR-15a binding sequence (Fig. 6C). In contrast, the miR-15a mimic has no significant effect on a reporter vector with a mutated miR-15a binding sequence. Together, these results suggest that miR-15a decreases bcl-2 translation by directly acting on an miR-15a-specific response element in the 3′-UTR region of bcl-2 mRNA.
PPARδ binds directly to the PPRE site in the miR-15a promoter region and trans-represses its expression
To further study the possibility that PPARδ may suppress miR-15a expression in a transcriptional manner, we performed a functional analysis of potential PPREs in the mouse miR-15a promoter. As demonstrated in Figure 7A, we identified a putative PPARδ binding site in the promoter of the miR-15a at the location of −1593 to −1573 bp. To determine the functionality of this PPRE in the miR-15a promoter, we cloned a 1887 bp (−1824/+62) promoter of the mouse miR-15a gene into a luciferase reporter vector, and the transcriptional response of the miR-15a promoter to PPARδ was performed using a luciferase transcriptional assay in vitro. HEK293 cells were transfected with a 1.9 kb promoter (miR-15a 1.9 kb WT) or the corresponding site-directed mutated promoter on the identified PPRE (miR-15a 1.9 kb PPRE mutant). Cells were coinfected with adenoviral PPARδ to overexpress PPARδ or two controls including no transfection control and adenoviral GFP. As shown in Figure 7B, PPARδ overexpression significantly reduces the transcriptional activity of the miR-15a 1.9 kb promoter. Moreover, luciferase activities from the miR-15a 1.9 kb promoter driven by the mutated PPRE did not respond to PPARδ overexpression, indicating that the predicted PPRE site in the promoter region of miR-15a is responsible for PPARδ trans-repression of miR-15a expression in vitro.
Figure 7.
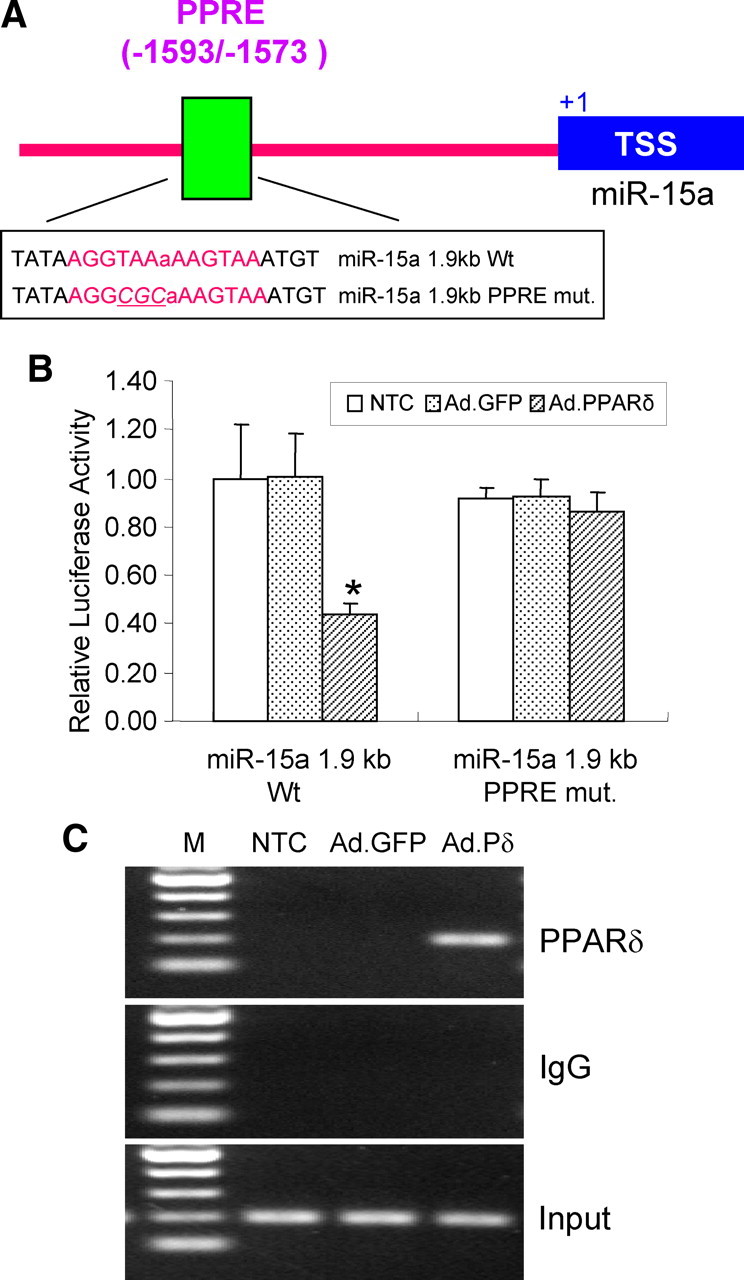
Functional analysis of a PPRE site located in the mouse miR-15a promoter. A, Schematic representation of the mouse miR-15a promoter region. A putative PPARδ binding site was demonstrated in the promoter of miR-15a at locations of −1593/−1573 bp (PPRE). The translation start site (TSS) is numbered as +1. B, Luciferase reporter assays were performed by transfecting HEK293 cells with an miR-15a 1.9 kb wild-type promoter (miR-15a 1.9 kb Wt) versus a PPRE mutated miR-15a promoter (miR-15a 1.9 kb PPRE mut.). Transcriptional activity is reduced during overexpression of PPARδ in the miR-15a wild-type but not in the PPRE mutant promoter. C, A ChIP assay showing PPARδ binds to the putative PPRE site in the mouse miR-15a promoter. Mouse CECs were infected with an adenovirus carrying GFP (Ad.GFP) or PPARδ (Ad.PPARδ or Ad.Pδ) for 48 h. DNA from PPARδ immunoprecipitated chromatin and input were subjected to PCR analysis using a pair of primer covering regions containing the PPRE site in the mouse miR-15a promoter. The experiments were repeated at least three times. Data were expressed as mean ± SD. *p < 0.05 versus Ad.GFP or no transfection control (NTC). Results shown are representative of three separate experiments with similar results.
Next, chromatin domains in the miR-15a promoter were scanned for PPARδ binding by ChIP analysis in mouse CECs. As shown in Figure 7C, consistent with PPRE-driven transcriptional activity, adenovirus-mediated overexpression of PPARδ detectably binds to the PPRE site located between −1593 and −1573 bp upstream of the transcription start site. An isotypic IgG antibody was used as a negative immunoprecipitation control. These data strongly suggest that, in the context of chromatin, the PPAR site (PPRE) between nucleotides −1593 and −1573 appears to be functional, showing binding of PPARδ to the miR-15a promoter.
Together, these data suggest that the miR-15a gene is a novel target of PPARδ transcriptional repression.
Activation of PPARδ inhibits microvascular miR-15a levels and downstream proapoptotic activities and attenuates ischemic cerebrovascular damage in vivo
To further determine the possible role of PPARδ regulation of miR-15a in ischemic cerebrovascular injury in vivo, we used a micro minipump technique to continuously deliver a specific PPARδ agonist, GW 501516 (10 μg/μl, at a rate of 1 μl/h), or vehicle reagent into the cerebral lateral ventricle. The treated mice were subjected to MCAO 24 h later and allowed to survive for an additional 24 h before taking measurements of cerebrovascular permeability, infarct volume, and biochemical assays. As shown in Figure 8A–C, GW 501516 effectively attenuates ischemic brain infarction (Fig. 8A,B) and improves cerebrovascular permeability, as analyzed by Evans Blue extravasation (Fig. 8C). As expected, treatment with GW 501516 also significantly attenuated ischemia-triggered upregulation of miR-15a levels (Fig. 8D), reversed ischemia-induced reduction of bcl-2 protein levels (Fig. 8E), and inhibited ischemia-activated caspase-3 activity (Fig. 8F) and DNA fragmentation (Fig. 8G) in isolated cerebral microvessels ipsilateral to MCAO. These results suggest that activation of cerebral PPARδ provides a vascular protective role during ischemic brain insult by directly targeting miR-15a and its downstream bcl-2-related antiapoptotic signaling cascade in the cerebral microvasculature.
Figure 8.
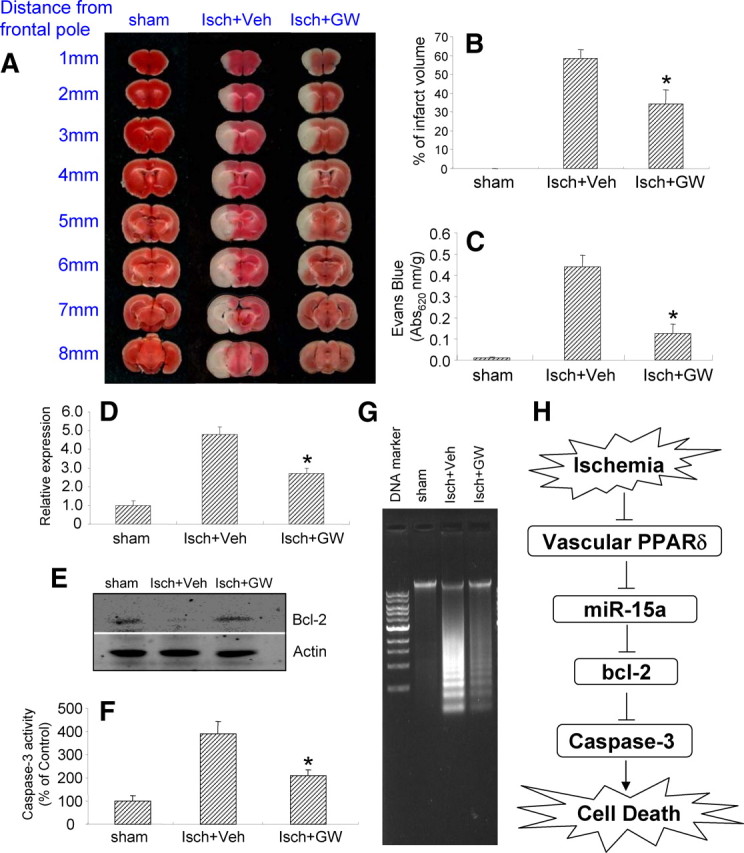
The effect of PPARδ activation on ischemia-induced cerebrovascular permeability and potential mechanisms in vivo. Mice were pretreated with a specific PPARδ agonist, GW 501516, or vehicle reagent for 24 h via minipump-assisted cerebroventricular infusion and then subjected to 1 h MCA occlusion and 24 h reperfusion. A, The 2% TTC-stained coronal sections were shown at different brain levels posterior to the frontal pole. B, C, Quantitative analysis was made on brain infarct volume (B), and cerebrovascular permeability was determined 1 h after injection from Evans Blue extravasation as described in Materials and Methods (C) (n = 6) in mice after stroke. Compared with the vehicle control, treatment of GW 501516 significantly attenuates ischemic brain infarction and improves cerebrovascular permeability. D–G, Intracerebroventricular infusion of GW 501516 also significantly reduced ischemia-induced miR-15a upregulation (D), increased bcl-2 protein levels (E), and attenuated caspase-3 activation (F) and DNA fragmentation (G) in the cerebral microvasculature in mice after MCAO. Data are expressed as mean ± SD. *p < 0.05 versus the ischemia plus vehicle (Isch+Veh) group. H, Schematic representation of PPARδ control of miR-15a in the regulation of OGD-induced CEC death signaling. In mouse brain vasculature, ischemic stimuli induce PPARδ dysfunction, featuring attenuated PPARδ trans-suppression of miR-15a and subsequent activation of miR-15a expression. miR-15a may bind to the 3′-UTR region of bcl-2 and further inhibit its antiapoptotic activity through post-transcriptional gene silencing, thereby resulting in a series of mitochondrial apoptotic cascades, such as activation of caspase-3, fragmentation of the Golgi apparatus, and final cell death.
Discussion
The major finding of our study is that PPARδ inhibits proapoptotic miR-15a expression in a transcriptional manner and subsequently increases the translation of the antiapoptotic protein bcl-2, resulting in a reduction of caspase-3 activity, fragmentation of the Golgi apparatus, and cell death in mouse cerebral endothelium after in vitro and in vivo ischemic insults (Fig. 8H). To our knowledge, our experiments have provided the first evidence that PPARδ, a nuclear receptor, controls apoptotic cell death and cerebrovascular dysfunction via a microRNA-mediated mechanism.
PPARs and their ligands are closely involved in apoptotic regulation of ECs after various stimuli. In contrast to the conflicting effects of PPARγ on apoptosis in ECs (Ameshima et al., 2003; Chen et al., 2004; Kim and Cheon, 2006), PPARδ activation has been reported recently to stimulate proliferation and attenuate apoptosis in endothelial progenitor cells through Akt-dependent signaling cascades (Han et al., 2008). Results from another study also showed that PPARδ may upregulate 14-3-3ε expression to protect ECs from H2O2-induced apoptosis (Liou et al., 2006). In this study, we report for the first time that PPARδ plays a cytoprotective role in CEC culture after OGD, an ischemia-like insult that has been reported to induce CEC apoptotic as well as necrotic cell death (Xu et al., 2000; Yin et al., 2002b). We further identify the cerebrovascular protective effects of PPARδ in vivo after transient focal cerebral ischemia in mice, showing evidence that pharmacological activation of PPARδ by intracerebroventricular infusion of GW 501516 effectively inhibits cerebral endothelial degeneration and improves BBB permeability. Until now, little is known about the role of PPARδ in the regulation of ischemia-induced BBB dysfunction and breakdown, which is a hallmark of stroke-induced brain damage. Our data here clearly define PPARδ as a novel mediator in cerebrovascular protection after ischemic stroke, implying that PPARδ-mediated vascular protection is one of the important mechanisms for protection from ischemia/reperfusion brain injury. On the other hand, our results document that GW 501516 significantly reduces ischemic brain infarction, suggesting that PPARδ activation may also have a neuroprotective role in ischemic brain insults. This finding is consistent with several previous animal studies that showed manipulations of PPARδ by genetic mutation increases infarction volume, whereas a PPARδ-specific agonist provides neuroprotection in the brain after ischemic injury (Arsenijevic et al., 2006; Iwashita et al., 2007). Together, it is possible that PPARδ and its ligands may contribute to brain protection in stroke by both vascular and neuroprotective mechanisms.
Generally, the structural integrity of the Golgi apparatus is maintained by complicated mechanisms, and disturbances often result in Golgi fragmentation (Marsh and Howell, 2002), which is involved in many neurodegenerative diseases, including Alzheimer's disease (Gonatas et al., 2006), stroke, or hypoxia-related brain injury (Dux et al., 1996; Hayashi and Abe, 2004). Until now, the potential interaction between PPARs and fragmentation of the Golgi apparatus is poorly understood (de Figueiredo and Brown, 1999). In this study, our data suggest that PPARδ protects CECs against OGD-induced apoptosis by inhibiting Golgi fragmentation, indicating a role for the Golgi apparatus in mediating PPARδ-related antiapoptotic effects in CECs after ischemia-like insults.
PPARδ-related vascular protection may involve multiple mechanisms, including regulation of the bcl-2 family (Love, 2003; Youle and Strasser, 2008). Indeed, bcl-2 has been shown to protect ischemic brain or vascular injury in rodent stroke models through genetic or pharmacological approaches (Martinou et al., 1994; Linnik et al., 1995; Hata et al., 1999). Consistent with these previous reports, we found that the bcl-2 protein is reduced in a time-dependent manner in CECs after ODG exposure as well as in the cerebral vasculature after cerebral ischemia, and this reduction is associated with CEC death or BBB disruption. Of significance, gain-of-PPARδ function by adenovirus or GW 501516 protects CECs or cerebral microvasculature against ischemic injury, and this PPARδ-mediated vascular protection results from upregulated bcl-2 protein levels. However, gain-of-PPARδ function does not affect bcl-2 mRNAs in CECs, implying that PPARδ may not directly transactivate bcl-2 expression. Thus, PPARδ posttranscriptional regulation of bcl-2 may be responsible for its cytoprotective role.
miRs are one novel class of noncoding short RNAs that mediate posttranscriptional silencing of specific target genes by binding to their 3′-UTRs (Bartel, 2004; Kim, 2005). The discovery of miRs has exhibited a new field to study how small RNAs control apoptosis. Various miRs appear to regulate both proapoptotic and antiapoptotic genes involved in apoptotic pathways (Jovanovic and Hengartner, 2006; Schickel et al., 2008; Eacker et al., 2009). miR-21 and the miR-17-92 cluster have been reported recently to be upregulated in various cancers and function as antiapoptotic miRs (Hayashita et al., 2005; Frankel et al., 2008). In contrast, miR-15a and miR-16 promote apoptosis by posttranscriptional gene silencing of bcl-2 (Cimmino et al., 2005). In this study, we have found that the expression of miR-15a is significantly increased in CEC cultures and the cerebral vasculature after in vitro and in vivo ischemic insults, and this increase is negatively related to OGD or cerebral ischemia-induced reduction of bcl-2 levels. Of significance, in vitro loss-of-miR-15a function results in a reduction in caspase-3 activity and CEC death, whereas gain-of-miR-15a function has the opposite effect. Moreover, we further found that fusion of the bcl-2 3′-UTR fragment to the luciferase reporter vector results in a functional expression in vitro, and this activity is significantly suppressed by treatment of exogenous miR-15a. Furthermore, upregulation of miR-15a leads to an even stronger reduction of bcl-2 expression after OGD exposure, whereas downregulation of miR-15a increases bcl-2 translation. Together, our experimental data extend the previous findings and further identify that miR-15a can repress bcl-2 translation by directly binding to the 3′-UTR of bcl-2 and subsequently suppress the downstream apoptotic cascades, including caspase-3 activation, Golgi fragmentation, and eventual cell loss in the cerebral microvasculature after ischemic insults. To our knowledge, we are the first to document that the miR-15 family has a causative role in the regulation of apoptosis by directly targeting bcl-2 in ischemic vascular injury.
PPARs and miRs are essential regulators in both cell physiological and pathological processes (Chen et al., 2003; Bartel and Chen, 2004; Hamblin et al., 2009). Theoretically, there is a reciprocal regulation between PPARs and miRs. It has been documented that miRs have the capacity to inhibit the expression and function of PPARs by directly targeting their 3′-UTR to trigger posttranscriptional gene silencing. One recent study (Iliopoulos et al., 2008) reported that miR-22 is able to negatively regulate the expression of PPARα protein levels in normal and osteoarthritic chondrocytes. Also, miR-27 blockade of PPARγ expression contributes to inhibition of adipocyte formation in the pathogenesis of obesity (Lin et al., 2009). Conversely, PPARs may act as transcription factors to transactivate or trans-repress various miRs by directly binding to their promoter regions. Shah et al. (2007) report that PPARα is a major regulator of hepatic miR expression, such as let-7c, which in turn activates a let-7C downstream signaling cascade responsible for PPARα agonist-induced liver proliferation and tumorigenesis. In the present study, we examine the interaction of PPARδ and miRs by studying the role of miR-15a in the regulation of PPARδ-mediated cell survival in CECs after ischemic stimuli. We found that gain-of-PPARδ function by adenoviral and pharmacological approaches significantly reduce the expression of miR-15a in cultured CECs and cerebral microvessels after in vitro and in vivo ischemic stimuli. These treatments also effectively reverse ischemia-induced reduction of bcl-2 levels in cerebral endothelium. Importantly, our data are the first to show that PPARδ can bind directly to a putative PPRE site in the miR-15a promoter region and in turn inhibit miR-15a expression through transcriptional repression. Thus, we are the first to reveal that miR-15a is a novel downstream target of PPARδ trans-repression and miR-15a mediates PPARδ-triggered vascular-protective effects. However, it should be emphasized that other mechanisms may also be involved in PPARδ-mediated vascular protection after ischemic brain insults, in addition to the miR-15a/bcl-2/caspase-3 signaling cascade. These mechanisms, including anti-inflammatory (Polak et al., 2005; Barish et al., 2008; Takata et al., 2008), antioxidant (Arsenijevic et al., 2006), pro-cell survival (Smith et al., 2004; Iwashita et al., 2007), proangiogenic actions (Fan and Yang, 2007; Müller-Brüsselbach et al., 2007), and anti-cellular adhesion (Lampen et al., 2005; Del Zoppo et al., 2006; Fan et al., 2008), have been reported recently to be responsible for the neuroprotective and vascular protective effects related to PPARδ. Thus, future studies will be needed to further investigate their contributions to PPARδ-mediated cerebrovascular protection during ischemic stroke.
In conclusion, the mechanisms of PPARδ-mediated protection in endothelial damage elucidated in this study are important for understanding the pathogenesis of cerebral ischemia. Our data demonstrate a cerebrovascular protective role of PPARδ in both in vitro and in vivo stroke models. We also identify a unique microRNA, miR-15a, as a contributor to the pathogenesis of ischemic vascular injury through direct inhibition of the antiapoptotic gene bcl-2. Furthermore, we demonstrate that PPARδ attenuates ischemia-induced increases in miR-15a. Therefore, either pharmacological activation of PPARδ or inhibition of miR-15a may be potentially suitable therapeutic options for stroke-induced vascular damage.
Footnotes
This work was partially funded by National Institutes of Health Grants HL68878, HL89544, HL92421, and GM087364. K.J.Y. and J.Z. are supported by the American Heart Association National Scientist Development Grants 0630209N and 0835237N, respectively. M.H. is supported by National Institutes of Health Postdoctoral Fellowship Grant T32 HL007853. Y.E.C. is an established investigator of the American Heart Association under Grant 0840025N.
References
- Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–1169. doi: 10.1161/01.RES.0000073585.50092.14. [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, de Bilbao F, Plamondon J, Paradis E, Vallet P, Richard D, Langhans W, Giannakopoulos P. Increased infarct size and lack of hyperphagic response after focal cerebral ischemia in peroxisome proliferator-activated receptor beta-deficient mice. J Cereb Blood Flow Metab. 2006;26:433–445. doi: 10.1038/sj.jcbfm.9600200. [DOI] [PubMed] [Google Scholar]
- Barish GD, Atkins AR, Downes M, Olson P, Chong LW, Nelson M, Zou Y, Hwang H, Kang H, Curtiss L, Evans RM, Lee CH. PPARdelta regulates multiple proinflammatory pathways to suppress atherosclerosis. Proc Natl Acad Sci U S A. 2008;105:4271–4276. doi: 10.1073/pnas.0711875105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C. Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J Steroid Biochem Mol Biol. 2003;85:267–273. doi: 10.1016/s0960-0760(03)00214-0. [DOI] [PubMed] [Google Scholar]
- Chen J, Li D, Zhang X, Mehta JL. Tumor necrosis factor-alpha-induced apoptosis of human coronary artery endothelial cells: modulation by the peroxisome proliferator-activated receptor-gamma ligand pioglitazone. J Cardiovasc Pharmacol Ther. 2004;9:35–41. doi: 10.1177/107424840400900i106. [DOI] [PubMed] [Google Scholar]
- Chen YE, Fu M, Zhang J, Zhu X, Lin Y, Akinbami MA, Song Q. Peroxisome proliferator-activated receptors and the cardiovascular system. Vitam Horm. 2003;66:157–188. doi: 10.1016/s0083-6729(03)01005-7. [DOI] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo P, Brown WJ. Clofibrate inhibits membrane trafficking to the Golgi complex and induces its retrograde movement to the endoplasmic reticulum. Cell Biol Toxicol. 1999;15:311–323. doi: 10.1023/a:1007667802497. [DOI] [PubMed] [Google Scholar]
- Del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Koziol JA. Vascular matrix adhesion and the blood-brain barrier. Biochem Soc Trans. 2006;34:1261–1266. doi: 10.1042/BST0341261. [DOI] [PubMed] [Google Scholar]
- Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab. 2009;29:675–687. doi: 10.1038/jcbfm.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dux E, Oschlies U, Uto A, Kusumoto M, Hossmann KA. Early ultrastructural changes after brief histotoxic hypoxia in cultured cortical and hippocampal CA1 neurons. Acta Neuropathol. 1996;92:541–544. doi: 10.1007/s004010050559. [DOI] [PubMed] [Google Scholar]
- Eacker SM, Dawson TM, Dawson VL. Understanding microRNAs in neurodegeneration. Nat Rev Neurosci. 2009;10:837–841. doi: 10.1038/nrn2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Yang GY. Therapeutic angiogenesis for brain ischemia: a brief review. J Neuroimmune Pharmacol. 2007;2:284–289. doi: 10.1007/s11481-007-9073-3. [DOI] [PubMed] [Google Scholar]
- Fan Y, Wang Y, Tang Z, Zhang H, Qin X, Zhu Y, Guan Y, Wang X, Staels B, Chien S, Wang N. Suppression of pro-inflammatory adhesion molecules by PPAR-delta in human vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:315–321. doi: 10.1161/ATVBAHA.107.149815. [DOI] [PubMed] [Google Scholar]
- Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283:1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- Gonatas NK, Stieber A, Gonatas JO. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci. 2006;246:21–30. doi: 10.1016/j.jns.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Hamblin M, Chang L, Fan Y, Zhang J, Chen YE. PPARs and the cardiovascular system. Antioxid Redox signal. 2009;11:1415–1452. doi: 10.1089/ars.2008.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JK, Lee HS, Yang HM, Hur J, Jun SI, Kim JY, Cho CH, Koh GY, Peters JM, Park KW, Cho HJ, Lee HY, Kang HJ, Oh BH, Park YB, Kim HS. Peroxisome proliferator-activated receptor-delta agonist enhances vasculogenesis by regulating endothelial progenitor cells through genomic and nongenomic activations of the phosphatidylinositol 3-kinase/Akt pathway. Circulation. 2008;118:1021–1033. doi: 10.1161/CIRCULATIONAHA.108.777169. [DOI] [PubMed] [Google Scholar]
- Hata R, Gillardon F, Michaelidis TM, Hossmann KA. Targeted disruption of the bcl-2 gene in mice exacerbates focal ischemic brain injury. Metab Brain Dis. 1999;14:117–124. doi: 10.1023/a:1020709814456. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Abe K. Ischemic neuronal cell death and organellae damage. Neurol Res. 2004;26:827–834. doi: 10.1179/016164104X3770. [DOI] [PubMed] [Google Scholar]
- Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T. A polycistronic microRNA cluster, miR-17–92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- He X, Goldsmith CM, Marmary Y, Wellner RB, Parlow AF, Nieman LK, Baum BJ. Systemic action of human growth hormone following adenovirus-mediated gene transfer to rat submandibular glands. Gene Ther. 1998;5:537–541. doi: 10.1038/sj.gt.3300622. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Malizos KN, Oikonomou P, Tsezou A. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE. 2008;3:e3740. doi: 10.1371/journal.pone.0003740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashita A, Muramatsu Y, Yamazaki T, Muramoto M, Kita Y, Yamazaki S, Mihara K, Moriguchi A, Matsuoka N. Neuroprotective efficacy of the peroxisome proliferator-activated receptor delta-selective agonists in vitro and in vivo. J Pharmacol Exp Ther. 2007;320:1087–1096. doi: 10.1124/jpet.106.115758. [DOI] [PubMed] [Google Scholar]
- Jovanovic M, Hengartner MO. miRNAs and apoptosis: RNAs to die for. Oncogene. 2006;25:6176–6187. doi: 10.1038/sj.onc.1209912. [DOI] [PubMed] [Google Scholar]
- Kim KY, Cheon HG. Antiangiogenic effect of rosiglitazone is mediated via peroxisome proliferator-activated receptor gamma-activated maxi-K channel opening in human umbilical vein endothelial cells. J Biol Chem. 2006;281:13503–13512. doi: 10.1074/jbc.M510357200. [DOI] [PubMed] [Google Scholar]
- Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- Lampen A, Grimaldi PA, Nau H. Modulation of peroxisome proliferator-activated receptor delta activity affects neural cell adhesion molecule and polysialyltransferase ST8SiaIV induction by teratogenic valproic acid analogs in F9 cell differentiation. Mol Pharmacol. 2005;68:193–203. doi: 10.1124/mol.104.009340. [DOI] [PubMed] [Google Scholar]
- Lane JD, Lucocq J, Pryde J, Barr FA, Woodman PG, Allan VJ, Lowe M. Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J Cell Biol. 2002;156:495–509. doi: 10.1083/jcb.200110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. A role of miR-27 in the regulation of adipogenesis. FEBS J. 2009;276:2348–2358. doi: 10.1111/j.1742-4658.2009.06967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnik MD, Zahos P, Geschwind MD, Federoff HJ. Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke. 1995;26:1670–1674. doi: 10.1161/01.str.26.9.1670. discussion 1675. [DOI] [PubMed] [Google Scholar]
- Liou JY, Lee S, Ghelani D, Matijevic-Aleksic N, Wu KK. Protection of endothelial survival by peroxisome proliferator-activated receptor-delta mediated 14-3-3 upregulation. Arterioscler Thromb Vasc Biol. 2006;26:1481–1487. doi: 10.1161/01.ATV.0000223875.14120.93. [DOI] [PubMed] [Google Scholar]
- Love S. Apoptosis and brain ischaemia. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:267–282. doi: 10.1016/S0278-5846(03)00022-8. [DOI] [PubMed] [Google Scholar]
- Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, Casciola-Rosen LA, Rosen A. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol. 2000;149:603–612. doi: 10.1083/jcb.149.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ, Howell KE. The mammalian Golgi–complex debates. Nat Rev Mol Cell Biol. 2002;3:789–795. doi: 10.1038/nrm933. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- Müller-Brüsselbach S, Kömhoff M, Rieck M, Meissner W, Kaddatz K, Adamkiewicz J, Keil B, Klose KJ, Moll R, Burdick AD, Peters JM, Müller R. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARbeta-deficient mice. EMBO J. 2007;26:3686–3698. doi: 10.1038/sj.emboj.7601803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM, Eisenberg J, Yamada T. Rapid sequestration and degradation of somatostatin analogues by isolated brain microvessels. J Neurochem. 1985;44:1178–1184. doi: 10.1111/j.1471-4159.1985.tb08741.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- Polak PE, Kalinin S, Dello Russo C, Gavrilyuk V, Sharp A, Peters JM, Richardson J, Willson TM, Weinberg G, Feinstein DL. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;168:65–75. doi: 10.1016/j.jneuroim.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–5974. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol Cell Biol. 2007;27:4238–4247. doi: 10.1128/MCB.00317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Monteith GR, Robinson JA, Venkata NG, May FJ, Roberts-Thomson SJ. Effect of the peroxisome proliferator-activated receptor beta activator GW0742 in rat cultured cerebellar granule neurons. J Neurosci Res. 2004;77:240–249. doi: 10.1002/jnr.20153. [DOI] [PubMed] [Google Scholar]
- Stephen RL, Gustafsson MC, Jarvis M, Tatoud R, Marshall BR, Knight D, Ehrenborg E, Harris AL, Wolf CR, Palmer CN. Activation of peroxisome proliferator-activated receptor delta stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004;64:3162–3170. doi: 10.1158/0008-5472.can-03-2760. [DOI] [PubMed] [Google Scholar]
- Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, Gao Y, Pietras K, Mann K, Yepes M, Strickland DK, Betsholtz C, Eriksson U, Lawrence DA. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–737. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Takata Y, Liu J, Yin F, Collins AR, Lyon CJ, Lee CH, Atkins AR, Downes M, Barish GD, Evans RM, Hsueh WA, Tangirala RK. PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci U S A. 2008;105:4277–4282. doi: 10.1073/pnas.0708647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, He L, Ahmed SH, Chen SW, Goldberg MP, Beckman JS, Hsu CY. Oxygen-glucose deprivation induces inducible nitric oxide synthase and nitrotyrosine expression in cerebral endothelial cells. Stroke. 2000;31:1744–1751. doi: 10.1161/01.str.31.7.1744. [DOI] [PubMed] [Google Scholar]
- Yang GY, Zhao YJ, Davidson BL, Betz AL. Overexpression of interleukin-1 receptor antagonist in the mouse brain reduces ischemic brain injury. Brain Res. 1997;751:181–188. doi: 10.1016/s0006-8993(96)01277-2. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Lee JM, Chen SD, Xu J, Hsu CY. Amyloid-beta induces Smac release via AP-1/Bim activation in cerebral endothelial cells. J Neurosci. 2002a;22:9764–9770. doi: 10.1523/JNEUROSCI.22-22-09764.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Chen SD, Lee JM, Xu J, Hsu CY. ATM gene regulates oxygen-glucose deprivation-induced nuclear factor-kappaB DNA-binding activity and downstream apoptotic cascade in mouse cerebrovascular endothelial cells. Stroke. 2002b;33:2471–2477. doi: 10.1161/01.str.0000030316.79601.03. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Hsu CY, Hu XY, Chen H, Chen SW, Xu J, Lee JM. Protein phosphatase 2A regulates bim expression via the Akt/FKHRL1 signaling pathway in amyloid-β peptide-induced cerebrovascular endothelial cell death. J Neurosci. 2006a;26:2290–2299. doi: 10.1523/JNEUROSCI.5103-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, Xu J, Hsu CY, Mills JC, Holtzman DM, Lee JM. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-β peptide catabolism. J Neurosci. 2006b;26:10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J, Chen YE. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis. 2010;38:17–26. doi: 10.1016/j.nbd.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Mackic JB, Wang L, McComb JG, McDonough A. Differential expression of Na,K-ATPase alpha and beta subunit isoforms at the blood-brain barrier and the choroid plexus. J Biol Chem. 1993;268:8019–8025. [PubMed] [Google Scholar]