

Abstract
Background
PL10 homologs exist in a wide range of eukaryotes from yeast, plants to animals. They share a DEAD motif and belong to the DEAD-box polypeptide 3 (DDX3) subfamily with a major role in RNA metabolism. The lineage-specific expression patterns and various genomic structures and locations of PL10 homologs indicate these homologs have an interesting evolutionary history.
Results
Phylogenetic analyses revealed that, in addition to the sex chromosome-linked PL10 homologs, DDX3X and DDX3Y, a single autosomal PL10 putative homologous sequence is present in each genome of the studied non-rodent eutheria. These autosomal homologous sequences originated from the retroposition of DDX3X but were pseudogenized during the evolution. In rodents, besides Ddx3x and Ddx3y, we found not only Pl10 but another autosomal homologous region, both of which also originated from the Ddx3x retroposition. These retropositions occurred after the divergence of eutheria and opossum. In contrast, an additional X putative homologous sequence was detected in primates and originated from the transposition of DDX3Y. The evolution of PL10 homologs was under positive selection and the elevated Ka/Ks ratios were observed in the eutherian lineages for DDX3Y but not PL10 and DDX3X, suggesting relaxed selective constraints on DDX3Y. Contrary to the highly conserved domains, several sites with relaxed selective constraints flanking the domains in the mammalian PL10 homologs may play roles in enhancing the gene function in a lineage-specific manner.
Conclusion
The eutherian DDX3X/DDX3Y in the X/Y-added region originated from the translocation of the ancient PL10 ortholog on the ancestral autosome, whereas the eutherian PL10 was retroposed from DDX3X. In addition to the functional PL10/DDX3X/DDX3Y, conserved homologous regions on the autosomes and X chromosome are present. The autosomal homologs were also derived from DDX3X, whereas the additional X-homologs were derived from DDX3Y. These homologs were apparently pseudogenized but may still be active transcriptionally. The evolution of PL10 homologs was positively selected.
Background
PL10 was first identified in mouse by using a human Y chromosome (Chr) derived probe [1] and is present in a wide range of eukaryotes from yeast, plants, and animals, including humans [2]. In the mouse, Pl10 has been shown to encode a functional protein with an important DEAD motif (Asp-Glu-Ala-Asp), which plays essential roles in spermatogenesis [3]. In eutherian mammals, PL10 has two closely-related paralogs, DDX3X (DEAD box polypeptide 3, X-linked) and DDX3Y (DEAD box polypeptide 3, Y-linked), located on the sex Chrs. PL10, DDX3X and DDX3Y share the DEAD motif and constitute the DDX3/DED1 (ATP-dependent DEAD-box RNA helicase) subfamily under the DEAD-box helicase family [4] with a major function related to RNA metabolism [5]. The DDX3/DED1 subfamily is involved in diverse cellular process including tissue differentiation at distinct developmental stages, embryogenesis, asexual reproduction, cell regeneration, tumorigenesis and immune response [2,6-8], which have been reviewed comprehensively by Rosner et al. [2].
Interestingly, the biological roles of the eutherian members in DDX3 subfamily appear to be varied and lineage-dependent although they share domain structures and highly similar sequences. In eutheria, the DDX3X has been shown to elicit immunoresponse because the DDX3X can interact with TANK-binding kinase 1 (TBK1) to induce the type I interferon (IFN) promoter and the downstream immune pathway [6]. In addition, DDX3X also plays a role in HIV infection and becomes an important target in antiviral therapy [9]. On the other hand, the human DDX3Y lies within the azoospermia factor a (AZFa) region on the proximal Yq11.21 and the deletion of human DDX3Y resulted in the oligozoospermia, azoospermia and the male sertoli-cell only syndrome [10,11]. In spite of their high amino acid (aa) similarity (91%), DDX3X cannot rescue the loss-of-function of DDX3Y in human [2], signifying the functional diversification between DDX3X and DDX3Y. The human DDX3Y is believed to be one of the essential genes involved in human spermatogenesis and male fertility [12]. In contrast to the human ortholog, the pivotal role of Ddx3y in spermatogenesis has been replaced by the autosomal Pl10 in mice [3]. The mouse Pl10 is believed to evolve from Ddx3x through the retroposition mechanism [13]. More interestingly, the bovine PL10 has also been proved to be active at the transcription level even though it may lose protein-coding potential [14]. In addition to the lineage-dependent functionality of PL10, the tissue specificity of DDX3X/DDX3Y homologs has also been shown to vary in mouse and human [15,3]. The lineage-specific expression patterns and the diverse genomic structures and locations of PL10 homologs suggest that the PL10 homologs regulate biological process via divergent mechanisms and evolved differently. However, previous studies focused mainly on elucidating the function rather than the evolution of PL10, which elicited our interest to investigate the evolutionary history behind PL10, DDX3X, and DDX3Y. Here, we report the results from a phylogenetic analysis of the PL10 homologs in 19 different species.
Results
The identification of PL10 homologous sequences
To obtain deep insight into PL10 evolution, we collected the PL10 related genes deposited in NCBI [16] and detected its potential homologs by comparing the mouse Pl10 mRNA sequence against the UCSC genome database [17]. In addition to the 22 annotated sequences for PL10, DDX3X and DDX3Y, we identified 15 PL10 putative homologous regions (coverage > 50%) in the genomes of mammals (Table 1). These putative homologs occupied the genomes with two major patterns in terms of their spanning size (2~4 and 10~14 Kb). The large-size homologs are located in the sex Chrs containing intron-exon structures, while the small-size ones are mostly autosomal and intronless (Table 1).
Table 1.
PL10 homologs in 19 species.
| Species (build version) | Chr | Gene | Homologous region | Span | Accession Number | |
|---|---|---|---|---|---|---|
| Human (37.1) | Y | DDX3Y | 15016838 | 15030444 | 14229 | NM_004660.3 |
| X | DDX3X | 41193484 | 41207386 | 14657 | NM_001356.3 | |
| X | 73340837 | 73351755 | 18744 | |||
| 4 | 104493233 | 104495627 | 3122 | |||
| Chimp (2.1) | Y | DDX3Y | 18024925 | 18030276 | 5352 | NM_001008986.1 |
| X | DDX3X | 41567709 | 41578379 | 10637 | ENSPTRT00000048707 | |
| X | 73472677 | 73479677 | 7000 | |||
| 4 | 106890486 | 106891981 | 1496 | |||
| Orangutan (2.0.2) | X | DDX3X | 41920594 | 41934087 | 13494 | ENSPPYT00000023631 |
| X | 71584239 | 71585574 | 1336 | |||
| 4 | 108027398 | 108029797 | 2400 | |||
| Mouse (37) | Y | Ddx3y | 599654 | 615438 | 15785 | NM_012008.1 |
| X | Ddx3x | 12858220 | 12869030 | 11577 | NM_010028.3 | |
| 1 | Pl10 | 188791295 | 188794506 | 3212 | NM_033077.2 | |
| 1 | 28046742 | 28049045 | 2304 | |||
| Rat (RGSC 3.4) | X | Ddx3x | 21497214 | 21508627 | 11414 | XM_228701.4 |
| 13 | Pl10 | 103083154 | 103086327 | 3174 | NM_001108858.1* | |
| 19 | 5498280 | 5501578 | 3299 | |||
| Dog (2.0) | X | DDX3X | 35708607 | 35722852 | 14282 | XM_856175.1 |
| 22 | 15373292 | 15375479 | 2188 | |||
| Horse (EquCab2.0) | X | DDX3X | 33503944 | 33514664 | 10721 | XM_001491432.2 |
| 17 | 31837579 | 31839780 | 2202 | |||
| Cow (Btau_4.0) | Y | DDX3Y | 86Δ | 5279Δ | 5194 | [14] |
| X | DDX3X | 68833 | 82540 | 13708 | [14] | |
| 15 | PL10 | 186070 | 189757 | 3688 | [14] | |
| Opossum (MonDom5) | 4 | DDX3 | 22331869 | 22343917 | 12049 | ENSMODT00000026845 |
| Chicken (2.1) | 1 | DDX3 | 115610539 | 115617788 | 7250 | NM_001030800.1 |
| X. tropicalis (4.1) | DDX3 | 940555 | 947979 | 7425 | BC063374 | |
| Zebrafish (Zv7) | 6 | PL10 | 25945 | 42249 | 16304 | NM_130941 |
| Clamworm | PL10a | AM048813.1 | ||||
| Flatworm | DjVLGA | AB017002.1 | ||||
| Hydra | CnPL10 | AB047381.1 | ||||
| Rice | DEAD-box RNA Helicase | NM_001074753.1 | ||||
| Arabidopsis | DEAD-box RNA Helicase | NM_129813.4 | ||||
| Fission Yeast | DED1 | AJ237697.1 | ||||
| Yeast | DBP1 | X55993.1 | ||||
| DED1 | X57278.1 | |||||
* The corresponding protein entry is NP_001102328.1.
Δ The position was annotated based on NW_001496707.1.
We extracted the sequences from these putative homologous regions and conducted a gene prediction using GENSCAN [18] to identify whether these homologous sequences maintain protein-coding potential. Based on gene similarity, structures and chromosomal locations, we obtained the predicted DDX3X in chimp and orangutan, and Pl10 on Chr 13 in rat (RNO13). The predicted chimp and orangutan DDX3X matched the predicted coding proteins in ENSEMBL [19] (Table 1). Compared to the human DDX3X protein of 662 aa, the predicted peptide is much shorter in the chimp with only 438 aa because of the incomplete sequence. The predicted rat Pl10 matched to the entry, NP_001102328.1, in the NCBI database, and we concluded that it is the rat Pl10 based on its intronless structure and high sequence similarity (96%) with the mouse Pl10. In opossum, the predicted DDX3 peptide matched to ENSMODT00000026845 in ENSEMBL [19] (Table 1). The remaining 11 homologs either do not have an open reading frame (ORF) or have a premature stop codon (Table 2). Thus, they are pseudogenes.
Table 2.
Pairwise comparison between mouse Pl10 (mPl10) and the non-annotated homologous regions in eutheria.
| Genomic Position of Non-annotated PL10 Homologs | Identity with m Pl10 (%) | Alignment Coverage with m Pl10 (%)* | Aligned Segment NumberΔ | Putative Peptide Length (aa)# | |
|---|---|---|---|---|---|
| Human Chr4 | (HSA4) | 80.08 | 100.00 | 1 | 84 |
| Human ChrX | (HSAX) | 78.52 | 98.38 | 2 (Ins: 8958 bp) | 241(DDX3Y) |
| Chimp Chr4 | (PTR4) | 74.83 | 81.93 | 1 | 84 |
| Chimp ChrX | (PTRX) | 78.37 | 98.34 | 2 (Ins: 1134 bp; Ns: 3010 bp) | 472(DDX3X) |
| Orangutan Chr4 | (PPY4) | 79.80 | 74.18 | 2 (Ns: 463 bp) | 132 |
| Orangutan ChrX | (PPYX) | 80.94 | 67.42 | 1 | 124(DDX3Y) |
| Mouse Chr1 | (MMU1) | 79.45 | 95.41 | 1 | 349(DDX3X) |
| Rat Chr19 | (RNO19) | 80.27 | 51.08 | 3 (Ns: 1509 bp; Ins: 316 bp; Gap: 962 bp) | 192(DDX3X) |
| Dog Chr22 | (CFA22) | 73.00 | 58.00 | 1 | N/A |
| Horse Chr17 | (ECA17) | 75.98 | 96.82 | 1 | N/A |
| Cow Chr15 | (BTA15) | 81.00 | 72.52 | 1 | N/A |
* The alignment coverage was calculated based on the pairwise alignment between the mouse Pl10 and identified homologous regions.
Δ Ns: the homologous region contains incomplete sequences. Ins: the homologous region is interrupted by non-homologous sequences. Gap: part of mPl10 was not alignable with the detected homologous sequences.
# The peptides were predicted via GENSCAN [18]. The protein name in parenthesis indicated the matched entries with lowest e-value in blastp analysis. N/A: not applied.
The analyses of PL10 phylogeny
Using the 22 PL10 related entries from NCBI together with 15 previously-described putatively homologous sequences, we constructed a phylogenetic tree to investigate the evolutionary relationship among these homologs. The tree clearly indicated several evolutionary clusters (Fig. 1). The first cluster is the PL10/DDX3X cluster, within which the putative homologous sequences on primate Chr4 were in the same clade and clustered with the primate DDX3X. The autosomal homologous regions in ruminants and carnivores, including the bovine PL10 pseudogene [14], were also in the same cluster and grouped with the DDX3X counterparts as in primates (Fig. 1). No apparent insertions were detected in these homologous regions. The mouse and rat Pl10 were in the same branch. However, an additional putative homolog of DDX3X was detected in mouse (MMU1) and rat (RNO19), which was grouped with its corresponding DDX3X gene, respectively, before clustering them together into a single group. It is noteworthy that all homologs of DDX3X identified in mammals are intronless (Table 1). Since the mammalian DDX3X contains an intron-exon structure, we reasoned that these intronless homologs are most likely the evolutionary trace after the DDX3X retroposition.
Figure 1.

The bootstrap consensus tree of PL10 homologous sequences. The evolutionary tree was built based on the Neighbor-Joining method implemented in MEGA4 [55,62]. The bootstrap consensus tree is inferred from 1000 replicates and the branches corresponding to partitions reproduced in less than 65% bootstrap replicates are collapsed. The bootstrap values are shown as percentages next to the branches. The evolutionary distances were computed using the Maximum Composite Likelihood method [63] and in the units of the number of base substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 0.91). All positions containing alignment gaps and missing data were eliminated by pairwise deletion. A total of 3944 positions were in the final dataset [Additional File 6]. The branches leading to the non-annotated autosomal homologous clusters of PL10 are highlighted in blue; the branches leading to the rodent Pl10 are highlighted in green; the branches leading to the non-annotated X-homologs are highlighted in red. The PL10/DDX3X cluster and the DDX3Y cluster are marked by vertical lines on the right.
In addition to the functional eutherian DDX3X, we detected another putative homologous region on primate ChrX which was present on the same branch with the DDX3Y instead of the DDX3X (Fig. 1). In contrast to the autosomal homologs, these ChrX putative homologs contain one or more insertions that appear to fit with the typical GT/AG splicing rule (Table 2), raising the possibility that these additional X homologs may have derived from a transposition event before the primate divergence (Fig. 1). Furthermore, in opossum and chicken, only a single homologous region with the intron-exon structure was detected on opossum Chr4 (MDO4) and chicken Chr1 (GGA1), respectively (Table 1).
Positive selection test for the PL10 related genes
We compared the one-ratio model with the free-ratio model to test the lineage-specific positive selection for the PL10 related functional homologs in our dataset using PAML4 package [20]. The one-ratio model assumes the same Ka/KS (w) ratio for all the lineages. The log-likelihood value under this model was l0 = -22217.097 with 58 parameters where the transition/transversion rate ratio was k = 1.685 and w = 0.041. The w was computed as the average of all codon sites and lineages. The free-ratio model assumes an independent w ratio for each branch and the number of parameters was increased to 104 for our dataset in this model. The likelihood value under this model was l1 = -22105.980. The comparison of the likelihood value, 2Δl = 2(l1-l0), was 222.234 as determined by the X2 distribution with degree of freedom (df) of 46 (p < 0.001), allowing us to reject the one-ratio model and conclude that the w ratios are varied among lineages (Fig. 2). In mammals, the estimates of w ratios were all lower than 0.1 on the branches leading to the lineages for PL10 and DDX3X, whereas the w ratios were higher on average (0.5) among the lineages for DDX3Y (Fig. 2). Furthermore, the primate lineages for DDX3Y in human and chimp were detected to be subject to positive selection (Fig. 2). The branches leading to the mammalian PL10 homologs clade (Fig. 2) also showed w ratios larger than 1, suggesting that the evolution of PL10/DDX3X/DDX3Y was under positive selection.
Figure 2.

The tree of the DDX3 family established for the positive selection test based on Maximum Likelihood approach. The branch length was estimated in the unit of the number of nucleotide substitutions per nucleotide. Values larger than 0.1 are denoted in bold. The numbers in the parenthesis represent the estimated numbers of nonsynonymous substitutions against synonymous substitutions of the specific branch. Scale bar = 0.05 unit.
Since some lineages were positively selected, especially in the case of DDX3Y, we further used a small dataset containing only the mammalian homologous coding sequences to examine the positively selected sites. The test statistic of likelihood ratio test (LRT) between the one-ratio model (M0) and the discrete model (M3) was 128.182 that is greater than the critical value 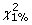 = 13.28 when df = 4 [Additional File 1 and File 2]. This suggested that the selective pressure is diverse among the codons. Three site classes calculated under model M3 have prior probability of p0 = 0.887, p1 = 0.108, and p3 = 0.005 with the Ka/Ks ratios of w0 = 0.025, w1 = 0.316 and w2 = 2.549 [Additional File 1]. The posterior probabilities of site classes calculated in model M3 are shown in Fig. 3. However, the LRT of the other two pairs of models, M1a (Nearly Neutral)/M2a (Selection) and M7 (beta)/M8 (beta & w), generated an incongruent result. The test statistic of the M1a/M2a is insignificant (p > 1), whereas the M7/M8 generated a significant result with a LRT value of 16.97 greater than the critical value at df = 2, = 9.1 (p < 0.01) [Additional File 1 and File 2], which together gave rise to the marginal prediction of the codon sites with relaxed selective constraints. Four (9A, 10L, 24S, 425S) and six (9A, 10L, 24S, 425S, 608A, 609S) sites were inferred to contain increased w ratios under models M2a and M8, respectively. Four of the six inferred sites in model M8 coincided with the result of model M2a, including Ala9, Leu10, Ser24, Ser425, in which the Ser24 and Ser425 have posterior probability higher than 0.9 under model M8 [Additional File 1]. All of the inferred sites are located in the non-domain regions.
= 13.28 when df = 4 [Additional File 1 and File 2]. This suggested that the selective pressure is diverse among the codons. Three site classes calculated under model M3 have prior probability of p0 = 0.887, p1 = 0.108, and p3 = 0.005 with the Ka/Ks ratios of w0 = 0.025, w1 = 0.316 and w2 = 2.549 [Additional File 1]. The posterior probabilities of site classes calculated in model M3 are shown in Fig. 3. However, the LRT of the other two pairs of models, M1a (Nearly Neutral)/M2a (Selection) and M7 (beta)/M8 (beta & w), generated an incongruent result. The test statistic of the M1a/M2a is insignificant (p > 1), whereas the M7/M8 generated a significant result with a LRT value of 16.97 greater than the critical value at df = 2, = 9.1 (p < 0.01) [Additional File 1 and File 2], which together gave rise to the marginal prediction of the codon sites with relaxed selective constraints. Four (9A, 10L, 24S, 425S) and six (9A, 10L, 24S, 425S, 608A, 609S) sites were inferred to contain increased w ratios under models M2a and M8, respectively. Four of the six inferred sites in model M8 coincided with the result of model M2a, including Ala9, Leu10, Ser24, Ser425, in which the Ser24 and Ser425 have posterior probability higher than 0.9 under model M8 [Additional File 1]. All of the inferred sites are located in the non-domain regions.
Figure 3.

Posterior probabilities of three site classes with different selective pressures (measured by the w ratio) for codon sites along the mammalian PL10 homologs under the site model M3. The X-axis represents the codon positions which were labeled based on the human DDX3X amino acids. The probabilities of the site classes are indicated in the Y-axis. The DEAD/DEAH-box helicase domain and helicase conserved C-terminal domain are underlined.
Conservation of PL10 homologs
We conducted a multiple alignment for all the analyzed sequences to investigate the domain conservation in PL10, DDX3X and DDX3Y, and found that the DEAD/DEAH box helicase domain (Pfam: PF00270) and helicase conserved C-terminal domain (Pfam: PF00271) of the DDX3 genes are highly conserved [Additional File 3]. We evaluated the degree of conservation by ConSurf (Fig. 4A) [21], which assigned the conservation score to each site of the provided DDX3X structures (PDB: 2I4I) [22] based on the empirical Bayesian method [21]. After mapping the conservation score to the structure, we found that the highly conserved codons concentrated in the cleft where the adenosine monophosphate (AMP) and RNA substrates interact with the DDX3 proteins (Fig. 4).
Figure 4.

The conserved domain and ATP binding site of the human DDX3X. A. The conservation score distribution on the human DDX3X (PDB: 2I4I) was assigned based on the empirical Bayesian method by ConSurf [21]. The domain regions are highlighted in dot-yellow halos. B. The ATP binding cleft depicted in PDBsum [64] corresponds to the conserved region in A.
Discussion
In the non-eutherian lineages, PL10 is the sole member of the DDX3 subfamily, whereas in eutheria, the ancient PL10 gene is located on the ancestral sex Chrs, resulting in the sex Chr-linked orthologs, DDX3X and DDX3Y (Fig. 1). Molecular evolutionary studies in recent years have established that the mammalian sex Chrs originated from a pair of ordinary autosomes, and most ancestral genes on that pair were still maintained on the X Chr but degenerated on the Y Chr due to the lack of recombination [23-25]. However, the Y Chr intends to maintain the functional genes that are beneficial to the male, such as those genes involved in spermatogenesis including DDX3Y [26]. Like the non-eutherian PL10, DDX3X and DDX3Y comprise the intron-exon structures, supporting the concept that DDX3Y and DDX3X are the evolutionary relics of the ancestral autosomal PL10. In opossum, the PL10 homologous sequence was detected only on MDO4 but not on the sex Chrs. The opossum Chr4 homolog also contains the intron-exon structure with predicted peptide close to DDX3X. Similarly, the single homologous sequence detected in chicken was located on the autosomes and it contains introns. A recent study for the gene cluster in the X/Y-added region of mammalian sex Chrs, XAR and YAR, revealed that the gene cluster and the gene order of this region are the same on chicken GGA1 but separated on opossum MDO4 and MDO7 [15], suggesting a single translocation event gave rise to the different chromosomal locations of the gene cluster among chicken and opossum. The DDX3X/DDX3Y also reside within the XAR/YAR, which allowed us to reach the parallel conclusion that the translocation generated the PL10 homologs on the chicken Chr1 and opossum Chr4, and DDX3X/DDX3Y on the eutherian sex Chrs (Table 1, Fig. 1).
Of particular interest is the occurrence of the mouse Pl10, an intronless gene and the only demonstrated functional autosomal ortholog in mammals to date. Consistent with a previous deduction [13], our result supported that the rodent Pl10 was derived from the retroposition of the DDX3X genes [27]. Retroposition is a crucial mechanism of gene duplication [28] and generates many new genes in new genomic positions through the reverse transcription of a parental gene [27,29,30]. The parental gene usually contains introns, whereas the processed retrocopy is intronless [27]. Thus, the other detected putative autosomal PL10 homologous sequences without apparent intron-exon structure in cattle, horse, dog and primates may have also evolved through the retroposition mechanism. These intronless homologous sequences on autosomes were consistently detected in eutherians but not in opossum, suggesting that the retroposition occurred after the divergence of eutherian and other mammals around 150 to 170 million years ago [31]. This raised an interesting question, why does the autosomal retroposition of the PL10 occur specifically in eutheria? It may be partially explained by the important functional role of mouse Pl10 and the recently discovered bovine PL10. The mouse Pl10 has been evidenced to be a central gene regulating the spermatogenesis and replace the role of DDX3Y [3]. The bovine PL10, albeit pseudogenized during the evolution, has also been proven to be active transcriptionally and may be involved in the regulatory coordination of bovine spermatogenesis [14]. Although the coding potential of the autosomal PL10 homologous sequences in eutheria, except in mouse, is diminished, we cannot exclude the possibility that these homologous sequences may be involved in regulating some biological process at the transcriptional level. Indeed, previous studies suggested that the pseudogenes may regulate the expression of the functional paralogous genes by producing antisense RNA [32,33]. Therefore, it is valuable to investigate whether these homologous sequences are transcriptable and their potential function in the future.
The maximum likelihood ratio test (LRT) for different lineages indicated that the Ka/Ks ratios in the PL10 homologs are varied among the evolutionary lineages. The Ka/Ks ratio along the branches among the mammalian lineages showed that the evolution of the mammalian PL10 homologs were not subject to positive selection, except for the human and chimp DDX3Y that are positively selected (w > 1). In addition to the chimp and human DDX3Y, we found that the ratios for the other eutherian lineages for DDX3Y appear to be higher when compared to those for mammalian PL10 and DDX3X, which is in line with a finding by Wilson and Makova [15]. These elevated w ratios can be explained by either the effect of relaxed selective constraints for the lineages containing DDX3Y due to the absent recombination of the ChrY or a weak positive selection operating on the Y-homologs [20]. The latter may still continue to refine the male-specific function for the Y-homologs [34]. In contrast, the mammalian lineages for DDX3X and PL10 with extremely low w ratios suggested that purifying selection may act strongly on the mammalian PL10 and X-homologs. Furthermore, the w ratios of the branches leading to the avian and mammalian lineages were larger than 1, indicating that the emergence of eutherian PL10 homologs was selected positively to acquire species-specific gene function and purifying selection acted on the DDX3X and PL10 homologs to preserve their crucial biological function and avoid their divergence.
A DDX3X/DDX3Y-specific multi-residue insertion (EALRAMKENG) has been observed to form an important positively charged cavity with the neighboring positive residues to increase the RNA binding surface in humans [35]. After incorporating PL10 and the homologous regions, we found that the insertion was highly conserved in the PL10 homologs of fish, frog, chicken, and other mammals, suggesting that the functional constraints occurred along the cavity region in the related homologs. Conversely, this insertion was not well-conserved in plants and invertebrates. Furthermore, the DDX3X displayed no activity with the RNA substrate when different flanking regions surrounding the domains were removed [35]. A similar effect was proven in other DEAD-box helicase related genes, such as the UAP56 [36] and DP103 [37], where the deletion of either the N-terminal or C-terminal flanking sequences outside the domain core regions impacts their helicase and ATPase activity, signifying the regulatory roles of the flanking regions. As shown from the analysis of positively selected amino acids, all the marginally inferred selected sites were located in the non-domain regions (Fig. 3) and may have served as the targets for improving the gene function during the evolution. One of the inferred sites, Ser425, occurred in the hinge region between two domains, suggesting its potential role in the adaption of the PL10 protein conformations and ligand-binding specificity. The slightly elevated w ratios were observed mostly in the non-domain regions as depicted in Fig. 3, indicating the purifying selection may attenuate in these regions to allow the functional accommodations of the PL10 homologs. Moreover, several sites in the flanking regions of the human DDX3X have been shown to undergo epigenetic modifications, including Ser2[38], Tyr69[39,40], Ser74[41], Ser76[41], Ser78[41], Tyr104[39], Ser125[41], Ser590[42], Ser594[43,42] and Ser612[43]. Meanwhile, two sites in the flanking sequences of the human DDX3Y, Tyr69 [39,44] and Ser592 [45], have been shown to be phosphorylated, and they are conserved with the modified sites at Tyr69 and Ser594 in the human DDX3X. The mouse Pl10 also has two phosphorylated sites at Tyr282 and Tyr465, but both of them were located in the domains [46,47]. The sequence comparison showed that the selection force has limited the divergence in the regions flanking the domains of PL10 related genes in fish, frog, bird and mammals, and most of the epigenetically modified sites were highly conserved among these species. Interestingly, despite the high degree of conservation, Ser76 exists specifically in the PL10 and DDX3X homologs but not in the DDX3Y orthologs. This distinction and different epigenetic modification pattern may partly contribute to the functional divergence between the eutherian DDX3Y, DDX3X and PL10. The functional specificity of PL10 homologs appear to be determined multifactorially, including the sequence elements located in the non-conserved regions, the factors controlling the diverse temporal and spatial expression patterns [14], and the distinct epigenetic modification patterns [35].
This study was limited to the availability of complete genomes and the accuracy of the genome assembly. Even though the number of finished genome projects in diverse species is growing, incomplete sequences of PL10 homologs still exist in the published genomes, especially for the challenges in sequencing and assembly of the Y Chr due to its highly repetitive nature. Further understanding of the evolution of sex Chr linked genes largely relies on the clarification of the diverse genomes in species other than primates.
Conclusion
Our analyses revealed that several conserved putative PL10 homologous regions, in addition to the functional PL10/DDX3X/DDX3Y, are present on the autosome and mammalian X Chr. These homologs share high similarity (> 70%) and coverage (> 50%) with mouse Pl10 but contain premature stop codons or indels, resulting in shorter putative peptides and/or frameshifts, suggesting their pseudogenization during the course of evolution [27,48]. The eutherian DDX3X/DDX3Y located in XAR/YAR were derived from the translocation of the orthologs on the ancestral autosome [15]. The identified putative autosomal homologs in mammals in the present study were retroposed from the DDX3X while the additional X-homologs in primates were transposed from the DDX3Y. These translocation events are lineage-specific. Like the bovine PL10, these homologs may still be active transcriptionally. Positive selection appears to operate on the PL10 homologs during the evolution. In addition to the highly conserved domain regions, several sites in the non-domain regions of functional PL10 homologs may play roles in enhancing the gene function in a lineage-specific manner.
The results reported in this study not only increase our knowledge regarding the molecular evolution of PL10 homologs, which will facilitate the future functional characterization of PL10 homologs, but also provide a valuable model to investigate the origin and evolution of the mammalian sex Chrs and the mechanisms behind lineage-specific gene duplication and functionality.
Methods
Sequence retrieval
By blating [49] the mouse Pl10 mRNA sequence against the genomes in UCSC genome database [17], we detected several putative homologous regions in mammals, birds and amphibians. We retrieved the homologous sequences from UCSC and conducted the pairwise alignment by the Bl2seq (NCBI Blast package) program using the mouse Pl10 mRNA sequence as the subject to filter out the sequences which cover < 50% of the mouse Pl10. We excluded the homologous sequences for the species without clear chromosomal annotations from our study. In addition, we downloaded the PL10 homologous sequences for other species from the NCBI nucleotide database based on the literature and database mining [2]. Afterwards, we used blastx [50] to confirm the identity of each sequence and retrieved the corresponding entries from the NCBI when the query sequences matched these entries perfectly. Alternatively, for sequences without a perfect hit in the database, we used the collected genomic sequences for gene prediction using the GENSCAN [18]. The predicted peptides were used for blastp [50] analysis to clarify their identities. Whenever the predicted proteins can match to the entries in the database, we used the deposited sequences in our analyses. The results were summarized in Tables 1 and 2. Several purpose-designed scripts were coded in C++ to facilitate the analysis.
Phylogenetic analysis
We performed multiple sequence alignments to investigate the conservation of the domain regions by ClustalW [51]. In order to ensure alignment quality, we first pre-aligned the annotated homologous sequences using their translated amino acid sequences in the coding regions. The parameters of the ClustalW for multiple alignment stage were modified to 3.0 for the gap opening penalty and 1.8 for the gap extension penalty to improve the alignment. Following that, we aligned the non-annotated homologous sequences based on the nucleotide sequences, which was further refined by manual adjustment. The positions of DEAD and the helicase domain were defined based on the annotation of Pfam [52] for the human DDX3X. The alignment was visualized through Jalview [53]. The degree of conservation was calculated by the empirical Bayesian method implemented in ConSurf [21] to investigate the highly conserved sites in the published human DDX3X structures (PDB: 2I4I) covering the important interacting domains involved in the RNA metabolism of the PL10 related homologs [35]. The conserved degree was represented through different colors using Jmol [54] as shown in Fig. 4A. In addition, we used the alignment to establish the Neighbor-Joining (NJ) phylogenic tree to study the relationship between PL10, DDX3X, and DDX3Y by MEGA4 [55] with the Maximum Composition Likelihood approach and 1000 bootstrap replicates (Fig. 1). The rate variation among sites was modeled with a gamma distribution (shape parameter = 0.91, estimated by the Model Selection of TOPALi (version 2.5 [56]). In Fig. 1, the branches corresponding to partitions reproduced in less than 65% bootstrap replicates are collapsed. The reason for using the NJ method is that the average pairwise Jukes-Cantor (JC) distance of the dataset is 0.362 smaller than 1.0, which is suitable for making the NJ trees [57]. We applied the pairwise deletion to remove gaps as our sequence lengths are varied, and a complete removal of the gaps is not a good choice as it eliminates a large portion of phylogenetically meaningful sites from consideration. Further, we used the Maximum Composite Likelihood model, recommended by the author of MEGA4 as a better evolutionary model. We also used the PHYML [58] and MrBayes [59] implemented in TOPALi to generate the phylogenetic trees [Additional File 4 and File 5]. The models used in PHYML and MrBayes are general-time-reversible (GTR+Gamma) selected via the TOPALi.
Potential pseudogenes were excluded from the positive selection test since the selective constraint may not act on them anymore. We prepared an amino acid alignment and the corresponding cDNA alignment for the complete set of 25 sequences for the lineage test and a small dataset (14) containing only the functional mammalian PL10 homologs for the site-specific test. The Codeml [20] in PAML4 was applied for the following analyses. The sites and lineages subject to positive selection were detected based on the maximum likelihood approach [60]. We compared the log likelihood values (l) derived from pairs of models to testify if there was a significant difference between model pairs by LRT. Each pair of the models contains a simple model, where the Ka/Ks ratios of the sites are limited, and a complex model, where the Ka/Ks ratios can be varied. We can infer the occurrence of lineage-specific and site-specific positive selection when the estimated Ka/Ks ratio in the complex model is larger than 1.0 and the calculated test statistic (2Δl) is significantly larger than the critical values of the X2 distribution at the corresponding degrees of freedom. We used M0 (one-ratio) and M1 (free-ratio) to test whether the lineage-specific positive selection occurred. The pairs of models used for the site-specific test were M0 (One Ratio)/M3 (Discrete), M1a (Nearly Neutral)/M2a (Positive Selection), M7 (beta)/M8 (beta & w) [20]. Each test was repeated to ensure the reproducible statistical results. The detailed assumptions and descriptions of each model were illustrated by Yang et al. [61,20].
Authors' contributions
TCC assisted in designing the study, collected the data, carried out the analyses and drafted the manuscript. WSL conceived of the study, participated in the experimental design and edited the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Selection test result for functional mammalian PL10 homologs. Log-likelihood and parametric estimates of the site-specific positive selection for functional mammalian PL10 homologs.
Test statistics of site-specific positive selection test. Likelihood ratio statistics (2Δl) of the site-specific positive selection test.
The multiple alignment and conserved regions of the PL10 related sequences. Only the domain regions are shown in this figure. The newly identified homologs are shaded in grey while the DEADc and Helicase C-terminal conserved domain are boxed in red. The blue color residues indicate the sites with conserved identity over 85%. The DDX3X/Y and PL10 specific insertion and the extended DDX3 unique positive residues are highlighted in green.
The Maximum-likelihood tree built for the PL10 related homologous sequences. The evolutionary tree was built based on the Maximum-likelihood method implemented in TOPALi [56]. The bootstrap values (1000 replicates) are shown next to the branches. The evolutionary model used was GTR+G. The tree has a similar topology to Fig. 1. Compared to Fig. 1, a swap occurred between the branches leading to the hydra, clamworm and flatworm homologs, and another swap observed between the branches leading to the putative rat Chr19 homologous region and other rodent homologous regions. The branches leading to the non-annotated autosomal homologous clusters of PL10 in primate are highlighted in blue; the branches leading to the rodent Pl10 are highlighted in green; the branches leading to the non-annotated X-homologs are highlighted in red. The PL10/DDX3X cluster and the DDX3Y cluster are marked by vertical lines on the right.
The Bayesian inference tree built for the PL10 related homologous sequences. The evolutionary tree was built based on the Bayesian inference method implemented in TOPALi [56]. The bootstrap values (1000 replicates) are shown next to the branches. The evolutionary model used was GTR+G. This tree is very much similar to the Supplementary Fig. 2.
The multiple sequence alignment of the putative PL10 homologs. A total of 37 sequences from 19 species were included in the alignment. The alignment length is 3944 bp. The number of phylogenetic informative sites is 2954 (79.4%).
Contributor Information
Ti-Cheng Chang, Email: txc272@psu.edu.
Wan-Sheng Liu, Email: wul12@psu.edu.
Acknowledgements
We are grateful to Dr. Daniel Hagen, Department of Dairy and Animal Science at the Pennsylvania State University, for his valuable comments. This work was supported by grants from USDA-CSREES (No. 2005-35205-18653 and No. 2010-65205-20362) and start-up funds from the Pennsylvania State University to Liu, W-S.
References
- Leroy P, Seboun E, Mattei MG, Fellous M, Bishop CE. Testis-specific transcripts detected by a human Y-DNA-derived probe. Development. 1987;101(Suppl):177–183. doi: 10.1242/dev.101.Supplement.177. [DOI] [PubMed] [Google Scholar]
- Rosner A, Rinkevich B. The DDX3 subfamily of the DEAD box helicases: divergent roles as unveiled by studying different organisms and in vitro assays. Curr Med Chem. 2007;14:2517–25. doi: 10.2174/092986707782023677. [DOI] [PubMed] [Google Scholar]
- Vong QP, Li Y, Lau YC. Structural characterization and expression studies of Dby and its homologs in the mouse. J Androl. 2006;27:653–61. doi: 10.2164/jandrol.106.000471. [DOI] [PubMed] [Google Scholar]
- UniProt. http://www.uniprot.org/
- Linder P. Dead-box proteins: a family affair--active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34:4168–80. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulat D, Bürckstümmer T, Westermayer S. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008;27:2135–2146. doi: 10.1038/emboj.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botlagunta M, Vesuna F, Mironchik Y. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene. 2008;27:3912–3922. doi: 10.1038/onc.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner A, Paz G, Rinkevich B. Divergent roles of the DEAD-box protein BS-PL10, the urochordate homologue of human DDX3 and DDX3Y proteins, in colony astogeny and ontogeny. Dev Dyn. 2006;235:1508–1521. doi: 10.1002/dvdy.20728. [DOI] [PubMed] [Google Scholar]
- Kwong AD, Rao BG, Jeang K. Viral and cellular RNA helicases as antiviral targets. Nat Rev Drug Discov. 2005;4:845–853. doi: 10.1038/nrd1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo P, Lin Y, Teng Y. Transcriptional levels of four Y chromosome-linked AZF genes in azoospermic men and their association with successful sperm retrieval. Urology. 2004;63:131–6. doi: 10.1016/j.urology.2003.08.048. discussion 136. [DOI] [PubMed] [Google Scholar]
- Foresta C, Moro E, Ferlin A. Prognostic value of Y deletion analysis. The role of current methods. Hum Reprod. 2001;16:1543–7. doi: 10.1093/humrep/16.8.1543. [DOI] [PubMed] [Google Scholar]
- Lardone MC, Parodi DA, Valdevenito R. Quantification of DDX3Y, RBMY1, DAZ and TSPY mRNAs in testes of patients with severe impairment of spermatogenesis. Mol Hum Reprod. 2007;13:705–712. doi: 10.1093/molehr/gam057. [DOI] [PubMed] [Google Scholar]
- Mazeyrat S, Saut N, Sargent CA. The mouse Y chromosome interval necessary for spermatogonial proliferation is gene dense with syntenic homology to the human AZFa region. Hum Mol Genet. 1998;7:1713–24. doi: 10.1093/hmg/7.11.1713. [DOI] [PubMed] [Google Scholar]
- Liu W, Wang A, Yang Y. Molecular characterization of the DDX3Y gene and its homologs in cattle. Cytogenet and Genome Res. 2009. [DOI] [PubMed]
- Wilson MA, Makova KD. Evolution and survival on eutherian sex chromosomes. PLoS Genet. 2009;5:e1000568. doi: 10.1371/journal.pgen.1000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI. http://www.ncbi.nlm.nih.gov/
- UCSC Genome Browser Home. http://genome.ucsc.edu/
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Ensembl Genome Browser. http://www.ensembl.org/index.html
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RCSB Protein Data Bank. http://www.rcsb.org/pdb/home/home.do
- Bachtrog D. A dynamic view of sex chromosome evolution. Curr Opin Genet Dev. 2006;16:578–85. doi: 10.1016/j.gde.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Graves JAM. Sex chromosome specialization and degeneration in mammals. Cell. 2006;124:901–14. doi: 10.1016/j.cell.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Wallis MC, Waters PD, Graves JAM. Sex determination in mammals--before and after the evolution of SRY. Cell Mol Life Sci. 2008;65:3182–3195. doi: 10.1007/s00018-008-8109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall Graves JA. Human Y chromosome, sex determination, and spermatogenesis- a feminist view. Biol Reprod. 2000;63:667–676. doi: 10.1095/biolreprod63.3.667. [DOI] [PubMed] [Google Scholar]
- Emerson JJ, Kaessmann H, Betrán E, Long M. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- Betrán E, Thornton K, Long M. Retroposed New Genes Out of the X in Drosophila. Genome Research. 2002;12:1854–1859. doi: 10.1101/gr.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betran E, Wang W, Jin L, Long M. Evolution of the Phosphoglycerate mutase Processed Gene in Human and Chimpanzee Revealing the Origin of a New Primate Gene. Mol Biol Evol. 2002;19:654–663. doi: 10.1093/oxfordjournals.molbev.a004124. [DOI] [PubMed] [Google Scholar]
- Brosius J. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene. 1999;238:115–134. doi: 10.1016/S0378-1119(99)00227-9. [DOI] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- Korneev SA, Park JH, O'Shea M. Neuronal expression of neural nitric oxide synthase (nNOS) protein is suppressed by an antisense RNA transcribed from an NOS pseudogene. J Neurosci. 1999;19:7711–7720. doi: 10.1523/JNEUROSCI.19-18-07711.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. Pseudogenes: are they "junk" or functional DNA? Annu Rev Genet. 2003;37:123–51. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- Gerrard DT, Filatov DA. Positive and negative selection on mammalian Y chromosomes. Mol Biol Evol. 2005;22:1423–1432. doi: 10.1093/molbev/msi128. [DOI] [PubMed] [Google Scholar]
- Högbom M, Collins R, Berg S van den. Crystal structure of conserved domains 1 and 2 of the human DEAD-box helicase DDX3X in complex with the mononucleotide AMP. J Mol Biol. 2007;372:150–159. doi: 10.1016/j.jmb.2007.06.050. [DOI] [PubMed] [Google Scholar]
- Shi H, Cordin O, Minder CM, Linder P, Xu R. Crystal structure of the human ATP-dependent splicing and export factor UAP56. Proc Natl Acad Sci USA. 2004;101:17628–17633. doi: 10.1073/pnas.0408172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Mouillet J, Ou Q, Sadovsky Y. A novel domain within the DEAD-box protein DP103 is essential for transcriptional repression and helicase activity. Mol Cell Biol. 2003;23:414–423. doi: 10.1128/MCB.23.1.414-423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagüe J, Alvarez I, Rognan D. An N-acetylated natural ligand of human histocompatibility leukocyte antigen (HLA)-B39. Classical major histocompatibility complex class I proteins bind peptides with a blocked NH(2) terminus in vivo. J Exp Med. 2000;191:2083–2092. doi: 10.1084/jem.191.12.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J, Moritz A, Lee KA. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- Tao WA, Wollscheid B, O'Brien R. Quantitative phosphoproteome analysis using a dendrimer conjugation chemistry and tandem mass spectrometry. Nat Methods. 2005;2:591–598. doi: 10.1038/nmeth776. [DOI] [PubMed] [Google Scholar]
- Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci USA. 2007;104:2199–2204. doi: 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Zhu Z, Chan KC. Improved titanium dioxide enrichment of phosphopeptides from HeLa cells and high confident phosphopeptide identification by cross-validation of MS/MS and MS/MS/MS spectra. J Proteome Res. 2007;6:4150–4162. doi: 10.1021/pr070152u. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill LM, Salomon AR, Ficarro SB. Robust phosphoproteomic profiling of tyrosine phosphorylation sites from human T cells using immobilized metal affinity chromatography and tandem mass spectrometry. Anal Chem. 2004;76:2763–2772. doi: 10.1021/ac035352d. [DOI] [PubMed] [Google Scholar]
- Daub H, Olsen JV, Bairlein M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol Cell. 2008;31:438–448. doi: 10.1016/j.molcel.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Cao L, Yu K, Banh C. Quantitative time-resolved phosphoproteomic analysis of mast cell signaling. J Immunol. 2007;179:5864–5876. doi: 10.4049/jimmunol.179.9.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BA, Carey GR, Sunyaev SR, Gygi SP. Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J Proteome Res. 2008;7:311–318. doi: 10.1021/pr0701254. [DOI] [PubMed] [Google Scholar]
- Majumdar M, Bharadwaj A, Ghosh I, Ramachandran S, Datta K. Evidence for the Presence of HABP1 Pseudogene in Multiple Locations of Mammalian Genome. DNA Cell Bio. 2002;21:727–735. doi: 10.1089/104454902760599708. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLAST: Basic Local Alignment Search Tool. http://blast.ncbi.nlm.nih.gov/Blast.cgi
- Larkin MA, Blackshields G, Brown NP. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jmol: an open-source Java viewer for chemical structures in 3D. http://jmol.sourceforge.net/
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Milne I, Lindner D, Bayer M. TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics. 2009;25:126–127. doi: 10.1093/bioinformatics/btn575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. 1. Oxford University Press, USA; 2000. [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Yang Bielawski. Statistical methods for detecting molecular adaptation. Trends Ecol Evol (Amst) 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Swanson WJ, Vacquier VD. Maximum-likelihood analysis of molecular adaptation in abalone sperm lysin reveals variable selective pressures among lineages and sites. Mol Biol Evol. 2000;17:1446–1455. doi: 10.1093/oxfordjournals.molbev.a026245. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution. 1985;39:791–783. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009;37:D355–359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Selection test result for functional mammalian PL10 homologs. Log-likelihood and parametric estimates of the site-specific positive selection for functional mammalian PL10 homologs.
Test statistics of site-specific positive selection test. Likelihood ratio statistics (2Δl) of the site-specific positive selection test.
The multiple alignment and conserved regions of the PL10 related sequences. Only the domain regions are shown in this figure. The newly identified homologs are shaded in grey while the DEADc and Helicase C-terminal conserved domain are boxed in red. The blue color residues indicate the sites with conserved identity over 85%. The DDX3X/Y and PL10 specific insertion and the extended DDX3 unique positive residues are highlighted in green.
The Maximum-likelihood tree built for the PL10 related homologous sequences. The evolutionary tree was built based on the Maximum-likelihood method implemented in TOPALi [56]. The bootstrap values (1000 replicates) are shown next to the branches. The evolutionary model used was GTR+G. The tree has a similar topology to Fig. 1. Compared to Fig. 1, a swap occurred between the branches leading to the hydra, clamworm and flatworm homologs, and another swap observed between the branches leading to the putative rat Chr19 homologous region and other rodent homologous regions. The branches leading to the non-annotated autosomal homologous clusters of PL10 in primate are highlighted in blue; the branches leading to the rodent Pl10 are highlighted in green; the branches leading to the non-annotated X-homologs are highlighted in red. The PL10/DDX3X cluster and the DDX3Y cluster are marked by vertical lines on the right.
The Bayesian inference tree built for the PL10 related homologous sequences. The evolutionary tree was built based on the Bayesian inference method implemented in TOPALi [56]. The bootstrap values (1000 replicates) are shown next to the branches. The evolutionary model used was GTR+G. This tree is very much similar to the Supplementary Fig. 2.
The multiple sequence alignment of the putative PL10 homologs. A total of 37 sequences from 19 species were included in the alignment. The alignment length is 3944 bp. The number of phylogenetic informative sites is 2954 (79.4%).