

Abstract
The evolutionarily conserved amphiphysin-like genes Bin1 and Bin3 function in membrane and actin dynamics, cell polarity, and stress signaling. Recent genetic studies in mice discriminate non-essential roles in endocytic processes commonly ascribed to amphiphysins from essential roles in cancer suppression. Bin1 acts in default pathways of apoptosis and senescence that are triggered by the Myc and Raf oncogenes in primary cells, and Bin1 gene products display a ‘moonlighting function’ in the nucleus where several other ‘endocytic’ proteins are also found. Together, genetic investigations in yeast, flies, and mice suggest that amphiphysin-like adapter proteins may suppress cancer by helping integrate cell polarity signals generated by actin and vesicle dynamics with central regulators of cell cycle arrest, apoptosis, and immune surveillance.
Keywords: tumor suppressor; modifier; lung cancer; vesicle trafficking; indoleamine 2,3-dioxygenase; c-myc; ras; rho
Introduction
BAR (Bin/Amphiphysin/Rvs) adapter proteins have been implicated in many cellular processes, including endocytosis, vesicle fusion and trafficking, specialized membrane organization, actin organization, cell polarity, stress signaling, transcription, and tumor suppression [1]. The signature fold that characterizes this group of adapter proteins, termed the BAR domain [2], mediates oligomerization to a banana-shaped dimer that can bind curved membranes, small GTPases, and other proteins [1, 3, 4]. Biochemical and structural studies indicate that BAR domains deform and tubulate membranes and can facilitate interactions with the actin cytoskeleton [3, 5]. However, like many other proteins implicated in membrane dynamics and endocytosis [6], certain BAR proteins also localize to the nucleus [2, 7-9]. BAR adapter proteins are now recognized to be part of a larger superfamily of structurally related proteins that includes the so-called F-BAR and I-BAR proteins [10-13].
Bin1 and Bin3 are the archetypal members of the BAR adapter gene family that are conserved throughout evolution from yeast to man. Figure 1 presents the primary structure of several ubiquitous and tissue-specific members of the BAR adapter family. Like most other genes that are evolutionarily well conserved, Bin1 and Bin3 are expressed ubiquitiously in mammalian cells with certain splice variants expressed in neurons, muscle cells or tumor cells (Fig. 2 and Suppl. Fig. 1). Bin1, also known as Amphiphysin II (Amph II), Amphl, ALP, or SH3P9, was described by several groups who cloned various splice isoforms on the basis of their amphiphysin-like structure, SH3 domain, or binding to the c-Myc or c-Abl oncoproteins [2, 14-18]. Although Bin1 was identified after Amph I the expression of the latter gene is primary restricted to neurons such that Bin1 is the germane amphiphysin-like function in most cells (Fig. 2). Of the >10 isoforms of Bin1 described, the ubiquitous and muscle-specific isoforms localize to nucleus as well as cytosol, a property essential for anticancer functions [8, 19-31]. A recent study suggests that cell polarity is a key determinant in controlling Bin1 nuclear localization [32].
Figure 1. BAR family of adapter proteins.
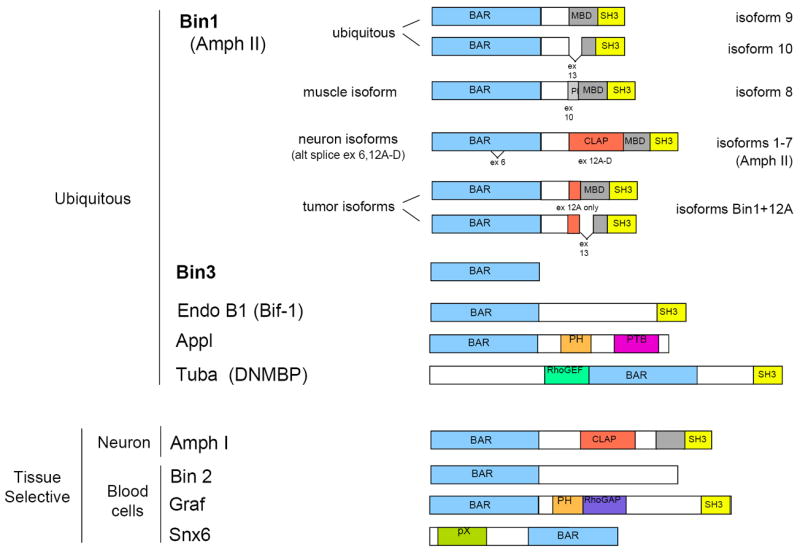
The primary structure of amphiphysin-like proteins and other selected ubiquitous and tissue-specific members of the BAR adapter family with relevance to cancer are presented. The organization and nomenclature of the various splice isoforms of Bin1 discussed in the text are noted. Exons 6, 10, 13, and 12A-D as defined by Wechsler-Reya et al. are alternately spliced [137]. While alternate splice isoforms for the other genes are known there has been generally little if any functional analysis reported, in contrast to Bin1 isoforms of Bin1. SH3, Src homology 3 domain; MBD, Myc binding domain; CLAP, clathrin-AP2 binding region; PH, pleckstrin homology region (PI binding); PTB, phosphotyrosine binding region; RhoGEF, Rho guanine nucleotide exchange function; RhoGAP, Rho guanine nucleotide activating protein function; pX, phox homology region (PI binding).
Figure 2. Patterns of Bin1, Amph I, and Bin3 expression in mouse tissues.
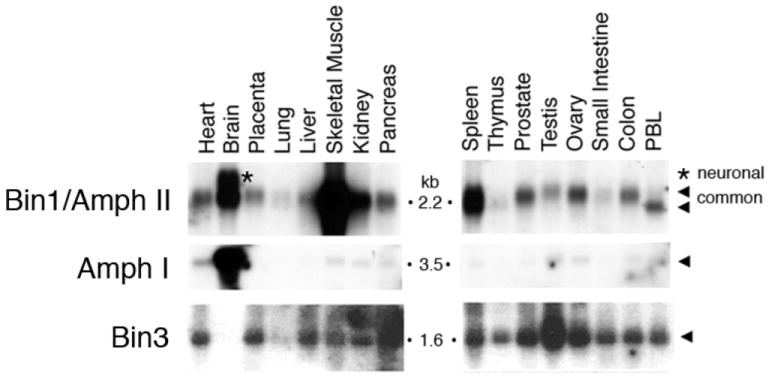
Northern analysis was performed of total RNAs isolated from the adult murine tissues indicated. Figure is adapted from Routhier et al. [92].
Bin3 was identified on the basis of structural similarity to its homolog RVS161 in budding yeast [33]. Bin3 is expressed oppositely to Amph I. Whereas Amph I is expressed mainly in the brain, Bin3 is expressed in all tissues but poorly in the brain (Fig. 2). Alternate splice isoforms which differentially include 5’ exons of Bin3 exist but nothing is known about their specific roles as yet. Bin3 proteins localize to vesicular membranes that are distinct from lysosomal membranes but partially overlap with mitochondrial membranes [34] (Fig. 3).
Figure 3. Bin3 localizes to vesicular membranes that overlap partially with mitochondria but not lysosomes.
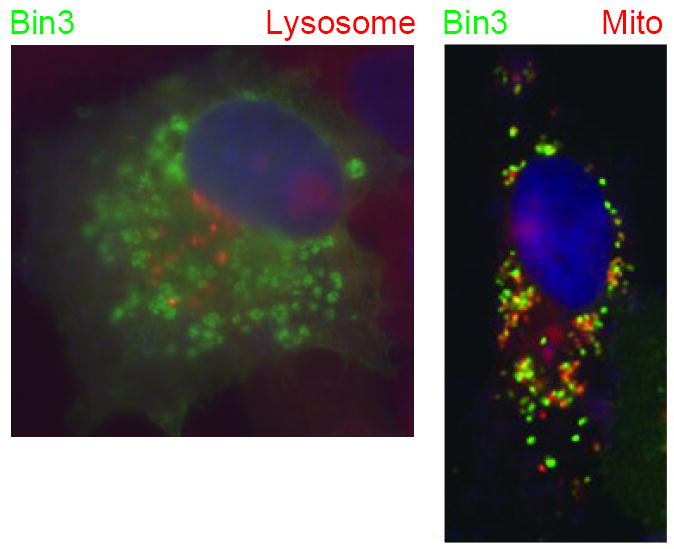
COS cells transfected with a human Bin3 expression vector were fixed and processed for indirect immunofluorescence with a Bin3 antibody [33] and simultaneously stained with DAPI to visualize nuclei plus LysoTracker or MitoTracker, to visualize lysosomes or mitochondria, respectively. Figure is adapted from Ramalingam et al. [34].
Although not similarly conserved in evolution, there are other BAR adapter proteins that reinforce interest in this gene family in cancer and their canonical roles in membrane dynamics, actin regulation, and signaling processes (Fig. 1 and Suppl. Fig. 1). Three ubiquitous expressed BAR family members of particular interest in cancer are Bif-1, APPL, and Tuba. Bif-1, also known as the vesicular protein endophilin B1 or the product of the SH3GLB1 gene, encodes Bax-interacting factor-1, a pro-apoptotic adapter protein that binds to Bax and promotes its conformational change at sites of mitochondrial scission, each of which are important to trigger apoptosis [35-37]. Of note to discussions below, Bif-1 interacts with Bin1 [38]. APPL encodes nuclear-shuttling adapter proteins that bind Rab5 and Akt2 on vesicles and that function in the EGF pathway to coordinate signaling and transcriptional repression via binding to the chromatin modification complex NuRD/Mi-2 in the nucleus [9, 39, 40]. Tuba (aka DNMBP) encodes an adapter protein that stimulates N-WASP-dependent actin assembly and is critical to drive the invasive motility of cancer cells [41, 42]. In contrast to Bif-1, APPL, Bin1 and Bin3, all of which exert tumor suppressor functions, Tuba may contribute positively to malignant progression by supporting invasion.
Several non-conserved BAR family genes of some interest to cancer are the tissue-specific genes Amphiphysin (Amph I), which is normally expressed only in neurons, and Graf and Snx6, which are expressed mainly in hematopoietic cells [43-45]. Amph I proteins are thought to contribute to synaptic vesicle recycling. In rare cases of breast and lung cancer, Amph I has been identified as paraneoplastic autoantigen underlying the neurological condition Stiff-man syndrome [46]. Although the meaning of this relationship is unclear, it is intriguing given evidence that the closely related amiphiphysin-like gene Bin1 has an essential role in supporting tumoral immune surveillance [27]. While amphiphysin-like orthologs in yeast and Drosophila are referred to as amphiphysin homologs in the literature, considerations of structural and functional homology make it clear that they likely derived from Bin1 rather than Amph I, the latter of which probably arose later in evolution. Graf (ARHGAP26) encodes a Rho GTPase activating protein (RhoGAP) that binds to the integrin effector kinase FAK and regulates Cdc42/Rho activity [47]. Prooncogenic chromosomal fusions involving Graf occur in juvenile myelomonocytic leukemia [44], perhaps helping deregulate FAK signals needed to drive cancer cell survival, motility, and crosstalk with the inflammatory microenvironment [48, 49]. Snx6 is a vesicle trafficking protein that interacts with TGF-ß receptors and is delivered to the nucleus by Pim oncoproteins that are required for Abl-mediated cell transformation [43, 50, 51]. Given the propensity of BAR adapter proteins to form heterodimers, the tissue-specific members of this family may associate with Bin1 or Bin3 to specify or integrate their functions in certain settings. For example, BAR domain-medated heterodimers of Bin2-Bin1 are found in hematopoietic cells and Amph I-Bin1 are found in neurons [22, 52], although the essential functions of each has yet to be understood.
Excellent recent reviews of the canonical function of BAR adapter proteins in membrane deformation, vesiculation processes, and actin organization have been published elsewhere [1, 11]. The purpose of the present review is to summarize the literature on amphiphysin-like genes in cancer, which has not been done before. These genes has been considered mainly in light of their roles in actin dynamics and endocytotic processes however, genetic studies in fission yeast, flies, and mice argue that amphiphysin-like genes are non-essential for endocytosis. We consider less studied but evolutionarily conserved roles of these genes in cell polarity and stress signaling that relate to cancer. Cell polarity signaling is closely integrated with vesicle trafficking and actin dynamics, such that amphiphysin-like proteins have an integrative role. In this review, we focus on the role of conserved amphiphysin-like genes in cell polarity signaling and stress signaling that are presently less studied or understood at present but proposed to be linked to their essential functions in cancer suppression.
Yeast amphiphysin-like orthologs function in cell polarity and stress signaling
Bin1 and Bin3 orthologs have been investigated extensively in budding yeast S. cerevisiae, where they are termed respectively RVS167 and RVS161, and also to a lesser extent in fission yeast S. pombe, where they are termed respectively hob1+ and hob3+. RVS genes were identified initially in budding yeast by a screen for mutants that exhibited reduced viability upon nutrient starvation [53, 54], revealing an essential function in stress signaling. RVS genes were also identified in screens for mutants that were defective in actin delocalization and internalization of endocytic vesicles [55], highlighting functions in actin and vesicular dynamics. More recently, a generalized function in vesicle trafficking has been defined based on evidence of a essential role in supporting ER-Golgi transport [56]. In addition to the extensive genetic and biochemical evidence linking RVS function to actin organization and vesiculation processes, suppressor screens reveal that rvs mutants can be rescued by sphingolipid alterations [1, 57]. The Rvs167p and Rvs161p proteins function in a stable heterodimeric complex mediated by BAR interaction [58-60]. However, a similar situation does not exist in mammalian cells, fission yeast, or Drosophila (which appears to lack a Bin3 homolog), where, as discussed further below, functional homologies diverge and analogous stable complexes are not seen. RVS167 is phosphorylated by the Pho85p kinase that helps integrate cell polarity and cell cycle control in budding yeast [61, 62]. In mammalian cells, the cell cycle-related kinase Cdk5 is an ortholog of Pho85p that phosphorylates Amph I protein in neurons [63, 64]. This is a specific connection insofar as Cdk5 does not phosphorylate Bin1 (Amph II) proteins which form heterodimers with Amph I in neurons (J. DuHadaway and G.C.P., unpublished observations). Like Amph I, Cdk5 is implicated in synaptic vesicle trafficking and its expression is confined mainly to neurons where it is controlled by the neuron-specific Cdk5 regulatory partner p35 [65-67]. Together these findings suggest that RVS167 may encompass functions in vesiculation processes akin to Amph I more than Bin1.
Rvs mutants exhibit a variety of cell polarity defects marked by aberrant budding patterns and actin organization [54, 68]. Rvs167 mutants accumulate late secretory vesicles at polar sites of membrane and cell wall construction [69]. During vegetative growth, Rvs167p is located at cortical actin patches but during mating it moves to the so-called shmoo tip of the yeast cell where cell-cell fusion occurs [70]. Rvs167p likely delivers Rvs161p to the cell fusion region, where Rvs161p binds to Fus2p to exert an actin-independent function needed for successful fusion and mating [71]. Reinforcing an important role in coordinating cell polarity-informed processes, protein-protein interaction studies argue that RVS167 is a nodal point for integrating cell polarity signaling [72].
One difficulty that arises in interpreting the information gained from budding yeast studies is that neither Bin1 nor Amph I are able to genetically complement the defects produced by deletion of RVS167 in budding yeast [73]. This speaks to an important difference in function not revealed by studies in budding yeast. The functional divergence along with regulatory differences prompted investigations of the BAR family genes in the fission yeast S. pombe. This yeast is diverged similarly from humans and budding yeast, but for certain processes such as cell division cycle control it is closer to mammalian cells. Similar to budding yeast, the fission yeast genome includes two BAR adapter family orthologs termed hob1+ and hob3+ that are closely related to Bin1 and Bin3 [33, 73].
hob1+ was defined as the homolog of Bin1 in S. pombe by sequence similarity but also by functional complementation [33, 73]. In support of the notion of some functional drift in BAR adapters during evolution, mutations in RVS167 and hob1+ produced somewhat different phenotypes in the two yeasts. Upon nutrient starvation or genotoxic stress produced by treatment with the DNA damaging agent phleomycin, hob1Δ mutant cells exhibited a cdc25-like phenotype marked by abnormal cell elongation and defective growth arrest followed by cell death [73]. In contrast, hob1Δ mutant lacked apparent defects in endocytosis or osmolar sensitivity in the manner of rvs167 mutant cells in budding yeast. Growth control could be rescued by Bin1 but not Amph I or RVS167 [73]. Thus, a specific homology existed between Bin1 and hob1+ that was not shared with Amph I despite its structural similarity [73]. The concept that hob1+ acts differently than RVS167 in how it influences actin organization and cell polarity has been extended by a further study [74]. Recently, analysis of the survival defect in hob1Δ mutant cells after phleomycin treatment suggests that hob1+ supports a Rad6/Set1-mediated pathway of chromatin modification that drives transcriptional repression [75]. In another study, evidence of genetic interaction between hob1+ and pku70+ encoding the DNA end-binding Ku70 was obtained, prompted by findings that Bin1 binds to Ku in mammalian cells [76]. Regarding the relationship between Bin1 and c-Myc in mammalian cells, these findings are interesting in light of evidence that c-Myc binds Ku and that c-Myc antagonizes the Rad/Set1 transcriptional repression pathway which is supported by hob1+ in fission yeast [77, 78]. Together, these findings are consistent with other evidence that in fission yeast the cell division cycle is more conserved to mammalian cells than in budding yeast.
hob3+ was defined as the homolog of Bin3 through sequence similarity but also through functional complementation [33]. hob3Δ mutant cells lacked any evident defects in endocytosis [33]. In contrast, under normal growth conditions many cells exhibited a profound disruption in polarized actin organization and an elongated, multinucleated phenotype associated with accumulation of cell wall material at sites of defective cell division [33]. This phenotype was strongly exacerbated by nutrient starvation, deepening the phenotype, but only limited effects on viability were seen. A comparison of hob3+, Rvs161, and Bin3 revealed that these genes could complement each other in budding and fission yeasts, arguing that their functional homology was conserved more closely than the Bin1/Amph homologs in evolution. hob3Δ mutant cells did not display sensitivity to phleomycin, however, loss of hob3+ relieved the senstitivity of hob1Δ cell to phleomycin, arguing that hob3+ may interact with hob1+ at some level in this response [73]. However, neither the Hob1p and Hob3p proteins nor the Bin1 and Bin3 proteins appear to interact in a stable complex like Rvs167 and Rvs161 (A. Ramalingam and G.C.P., unpublished observations). Notably, the defect in cell division in hob3Δ cells reflects a need for Hob3p to recruit and activate the Rho small GTPase Cdc42p to sites of cell division where it is required to mediate cytokinesis [79]. Included in the Hob3p-Cdc42p complex is a guanine nucleotide exchange factor termed Gef1p that Hob3p supports to stimulate Cdc42p activity in this role. There are two pathways leading to Cdc42 activation in cytokinesis in fission yeast but the Hob3p-Gef1p pathway is non-essential unless the second Cdc42 activating pathway is absent or defective, indicating a redundancy in this mechanism of cell division. The spatial regulation of Cdc42p in cytokinesis depends upon actin-dependent polarity cues [80]. Overall, genetic studies on the hob genes indicate that they coordinate cell polarity and cell division under under conditions of cell stress. In summary, fission yeast studies argue that the essential functions of amphiphysin-like genes that are evolutionarily conserved to mammals relate more to cell polarity signaling than endocytotic trafficking.
Drosophila ortholog of Bin1 is dispensable for endocytotic processes but essential to localize the cell polarity complex Scribble/Discs Large/Lethal Giant Larvae (Scr/Dlg/Lgl)
The fruit fly Drosophila encodes a gene termed Amphiphysin (dAmph) that is structurally related to the mammalian Bin1 and Amph I genes. In contrast, no Bin3 ortholog is found in flies. Mutants that lack dAmph are viable, lacking evident defects in endocytosis or neurotransmission, but they exhibit an altered synaptic physiology and are flightless due to a muscle defect [81, 82]. Consistent with a non-essential role in endocytosis the fly protein DAmph does not bind clathrin, however, it tubulates lipids and is localized to the specialized T tubule system in muscle cells [81, 82]. Additionally, it is broadly expressed at actin-rich membrane domains in many other cell types, such as at the apical membranes in polarized epithelial cells, the apical rhabdomere membranes of photoreceptor neurons and the postsynaptic density of neuromuscular junctions [82]. Mutant flies have a severely disorganized T-tubule/sarcoplasmic reticulum system, defining an essential function in organizing the membraneous compartments of the excitation-contraction coupling machinery in muscles [81]. Thus, the fly gene dAmph is implicated in muscle function like the mammalian Bin1 gene consistent with the notion that they are functional homologs [25, 30, 31].
In support of an important role in cell polarity, mutant fly larvae and adults exhibit defects in cellular locomotion and mislocalization of the cell polarity complex composed of the Scribble (Scr), Discs Large (Dlg), and Lethal Giant Larvae (Lgl) proteins [82]. In flies the Scr/Dlg/Lgl complex acts as a tumor suppressor [83]. Notably, inactivation of this complex cooperates with Ras to drive formation of invasive tumors in the same way that inactivation of Bin1 cooperates with Ras to drive invasive tumors in mice [84-86]. It is not known if inactivation of the dAmph gene similarly cooperates with Ras [28, 85]. However, even aside from considerations of homology, this question is interesting to consider in light of other information. In epithelial cells, vesicle trafficking maintains cell polarity that relies upon Scr/Dlg/Lgl function. Intriguingly, investigations of cell competition in Drosophila wing epithelial cells suggest a potential role for vesicle dynamics in coordinating cell division and survival. In particular, one study found that Myc can be phenocopied by Rab5 in cells that outcompete neighboring cells [87]. In mammalian cells, Bin1 binds to Rin2 and Rin3, two Rab GTP exchange factors (RabGEFs) that bind Ras and stimulate Rab5 activity [88]. Additionally, Bin1 binds to Myc and inhibits its oncogenic activity [2, 28, 85]. If Bin1 restricts the activities of Myc and Rab5, in the latter case by sequestering RabGEFs, then inactivation of Bin1 may cooperate with Ras by relieving restraints to Myc and Rab5 needed to license the division of polarized cells. In summary, studies of the Bin1-like gene dAmph in Drosophila reinforce the concept that the key evolutionarily conserved function of amphiphsyin-like genes relates primarily to cell polarity signaling, with hints to the mechanism of how its action may integrate polarity with cell growth and survival controls.
Bin1 exerts a tumor suppressor function that is widely attenuated in human cancer
Various isoforms of Bin1 that are produced by alternate RNA splicing were discovered in different laboratories on the basis of similarity to Amph I, the presence of an SH3 domain, or the ability to bind the Myc and Abl oncoproteins [89]. These diverse efforts, reflecting the complexity of Bin1 splicing in mammalian cells, has led to a complex nomenclature in the literature for the gene and its isoforms (which have also been termed Amphiphysin II or 2, Amph II or 2, Amphl, ALP, and SH3P9). In this review we have used HUGO-approved nomenclature and the NCBI Entrez Gene nomenclature to refer to genes and splice isoforms [90]. When not specifically defined in the text, Bin1 refers to the two ubiquitious isoforms 9 and 10 (aka Bin1-10 and Bin1-10-13), which localize in cells with preference to the nucleus [31, 91, 92] or cytosol [16, 91-93], respectively. CNS-specific Bin1 isoforms 1-7 are exclusively cytosolic and include the clathrin-binding domains found in Amph I proteins [14, 15, 92, 94]. The muscle-specific isoform 8 localizes to T tubules or nucleus [31, 92, 95]. Lastly, cancer-specific variants of the ubiquitous isoforms 9 and 10, which we refer to as Bin1+12A isoforms due to aberrant inclusion of the CNS-specific exon 12A [18], are exclusively cytosolic and lack tumor suppressor activity [22]. Notably, exon 12A missplicing in the cancer-specific Bin1+12A isoforms results form activation of the oncogenic RNA splicing factor SF2/ASF [96]. Bin1+12A isoforms are observed in many tumor cells and tumor cell lines, representing one of the most common missplicing events occuring in human cancer generally [97, 98].
Our group initially identified Bin1 through its ability to bind to c-Myc and inhibit its primary cell transforming activity with Ras [2]. The robust antitransforming activity of Bin1 displayed in classical primary cell assays of oncogene co-transformation is dependent on an intact Myc binding domain (MBD), present only in the muscle isoform 8 and ubiquitous isoform 9 which can localize to the nucleus [2, 7, 91, 92]. Further studies confirming the c-Myc-Bin1 interaction have provided evidence that the SH3 domain in Bin1 contributes to Myc binding and that the cytosolic isoform 10 lacking an intact MBD binds Myc poorly [26, 89]. Myc Box regions I and II, which are crucial for the oncogenicity and transcriptional activity of all Myc proteins, are also essential for Bin1 binding [2]. Bin1 suppresses the transcriptional transactivation activity of c-Myc and the BAR domain is sufficient for transcriptional repression when independently tethered to a promoter by the yeast Gal4 DNA binding domain [20]. T58 mutations in the Myc Box I region, which are found in all viral myc genes and many human tumors [99], permit Myc to escape the antitransforming effects of Bin1 in primary cells [2]. Moreover, inhibition of Myc-Bin1 interaction in cells via MBD overexpression promotes transformation and blunts apoptosis by Myc in primary cells [2, 19]. This effect is specific, because while the MBD is essential for Bin1 to suppress the co-transforming activity of Myc, it is non-essential to suppress the co-transforming activity of adenovirus E1A or mutant p53 plus Ras [2, 8], which appears to involve a distinct Rb-dependent mechanism [100]. The anti-transforming and pro-apoptotic effects of Bin1 are strongest in primary cells; during establishment of rodent cells in tissue culture, Bin1 tends to be attenuated at the level of expression, missplicing, or both (J. DuHadaway, A. Muller, and G.C.P., unpublished observations). This phenomenon is interesting as it parallels the disabling of polarity signaling which occurs in primary cells as they become established into cell lines in tissue culture. Supporting the in vivo relevance of Bin1 for helping restrict the oncogenicity of Myc, loss of Bin1 cooperates with Ras to drive malignant progression in mice [85]. Taken together, these findings offer genetic evidence of an critical role for Bin1 in helping safeguard normal cells from the powerful oncogenic effects of Myc overexpression.
In a separate line of work, Bin1 isoform 10 was identified through interaction with the tyrosine kinase Abl [16]. This interaction was dependent on the SH3 domains in each protein and it was later confirmed that all Bin1 isoforms can bind Abl in cells (D. Sakamuro and G.C.P., unpublished observations). In the initial study, overexpression of isoform 10 was found to cause an Abl-dependent morphological transformation of established NIH3T3 fibroblasts, suggesting that isoform 10 might mediate cytoskeletal functions of Abl kinase [16]. This finding was not extended, however, and later studies conducted in primary fibroblasts indicated that isoform 10 could suppress tumor formation by oncogene-transformed primary cells [28]. One study suggested that Bin1 isoform 10 (termed there Amph IIm) was critical for phagocytosis in macrophages [93], however, this finding was based on a weakly controlled dominant negative strategy and it was later refuted by studies employing genetically null cells [52]. Thus, not only Bin1 isoform 10 but all Bin1 isoforms are dispensable for phagocytosis.
Bin1 losses at the level of expression or missplicing occur frequently in human tumors of the breast, prostate, brain, colon, skin, brain, and lung, and efforts to restore expression in tumor cell lines causes growth inhibition and/or cell suicide [2, 21-24, 29, 101-103]. These effects are specific as Bin1 does not similarly compromise the growth or survival of normal primary cells or nontransformed cells [20, 22, 24, 31]. The human gene is located at human 2q14, within a mid-2q region deleted ~42% of metastatic prostate cancer that encompasses Bin1 [23, 104]. As noted above, Bin1 is often attenuated by missplicing of CNS-specific exon 12A [22] and Bin1+12A isoforms can no longer access the nucleus [20-22, 24, 102], bind to Myc [26], or activate cell suicide [20-22, 24, 102]. The high frequency of Bin1 missplicing in many human tumors and tumor cell lines that has been documented [97, 98] means that cancer microarray expression analyses defining Bin1 expression must be interpreted carefully, given the likelihood that tumors may accumulate mRNAs encoding Bin1+12A isoforms which lack tumor suppressor activity [22].
Several studies document a role for Bin1 in programmed cell death (PCD) in transformed or tumor cell lines [19-22, 24, 28, 85, 102, 105]. In panels of melanomas and breast carcinomas where Bin1 was misspliced or attenuated in expression, ectopic expression of Bin1 isoform 8 triggered PCD associated with cell detachment, rounding, and DNA degradation [22, 24]. These effects were not correlated with Myc overexpression. In contrast, in neuroblastomas Bin1 was found to be misspliced or attenuated only in neuroblastomas with amplified N-Myc, and ectopic expression facilitated apoptosis by serum deprivation or cytotoxic drug treatment [29, 102]. A detailed analysis of PCD induced in human hepatocarcinoma cells indicated that Bin1 engaged a caspase-independent process characterized by cell shrinkage, substratum detachment, vacuolated cytoplasm, and limited DNA degradation with nuclear margination [20]. PCD induction was relieved by mutation of the BAR domain or by exon 12A missplicing. p53 was dispensable and PCD was not blocked by either Bcl-2 or inhibition of the Fas pathway. In contrast, serine protease inhibitors delayed DNA degradation and SV40 large T antigen completely blocked PCD [20]. The latter finding was consistent with earlier evidence that co-transformation of primary cells by T antigen+Ras is refractory to suppression by Bin1 [2, 8]. Autophagy was ruled out based on the lack of autophagic vesicles in electron micrographs and the lack of sensitivity the autophagic inhibitor 3-methyl-adenine, but this should be be re-evaluated given recent advances in the field. Notably, electron micrography revealed a close resemblance between the PCD phenotype triggered by Bin1 and the non-apoptotic PCD triggered in cells by Myc when caspases are inhibited [106].
Bin1 is essential for default pathways to apoptosis or senescence that are triggered by oncogenes in primary cells where cell polarity signaling is intact
Precedents exist for tumor suppressor genes that support multiple apoptosis pathways while nevertheless triggering caspase-independent PCD when expressed in tumor cells, for example, the tumor suppressor gene Pml [107, 108]. In the case of Bin1, this dichotomy might be rectified by recognizing that in tumor cells polarity signaling and apoptotic signaling are both grossly altered. For example, in cells where polarity is intact, Myc-mediated apoptosis is influenced by the integrity of the polarity factor Lkb1 [109, 110] or the polarity complex Dgl/Lgl/Scr (S. Muthuswamy, pers. comm.). Indeed, genes such as Myc or adenovirus E1A that ‘epithelialize’ mesenchymal cells alter their sensitivity to apoptosis [111-113]. In frank tumor cells where apoptosis and polarity pathways are both altered, restoring a polarity function that normally licenses a default pathway of apoptosis triggered by oncogenic stress might instead manifest only as a non-apoptotic PCD. In this context, it is intriguing to note that studies of the proteomic response to Myc have revealed a cytoskeletal regulatory function involving integrins and actin regulators which play important roles in polarity signaling [114]. Overall, hints of the potential connections between Myc, Bin1 and polarity complexes such as Dgl/Lgl/Scr reinforce the notion that polarity signaling may license a default pathway of apoptosis that is triggered by oncogene stress in primary cells. Such pathways are vitally important in primary cells, in contrast to established cell lines which with rare exceptions have grossly impaired cell polarity signaling.
In support of this concept, other studies indicate that Bin1 provides a critical support to default pathways of apoptosis or senescence triggered by oncogenes in primary cells. In chick embryo cells, a classical model of one-step transformation by Myc, blocking Bin1 expression or Myc-Bin1 interaction did not affect cell proliferation or transformation but it specifically inhibited Myc-mediated apoptosis [19, 115]. Investigations in this system implicated Bin1 in the autocrine production of a secreted factor required for Myc to mediate apoptosis [115]. Similar effects were obtained in primary baby rat kidney (BRK) cells immortalized by c-Myc [19]. In contrast, in established or tumor cell lines, where polarity signaling is expected to be impaired, efforts to ablate Bin1 did not alter Myc-mediated apoptosis. While Myc can sensitize cells to apoptosis by a variety of mechanisms [116], one generally critical step seems to be its ability to trigger a configurational change in Bax needed to allow it to insert into the mitochondria outer membrane, where Bax must go to trigger cytochrome c release, caspase activation, and cell suicide [117, 118]. Precisely how Myc influences Bax configuration remains obscure at present. Notably, a recent report implicates the BAR adapter protein Bif-1 (endophilin B1) in activating the Bax configurational change that is needed for its mitochondrial recruitment [36]. In light of the evidence that Bif-1 and Bin1 can interact with each other [38], it is conceivable that the titration of Bin1 by Myc or other factors might relieve a restraint to Bif-1, thereby facilitating its liberation to promote Bax activation. In any case, the evidence that Bin1 is germane only in primary cell settings is consistent with the notion that its ability to facilitate apoptosis may rely upon intact cell polarity signaling at some level.
Additional studies performed in primary mouse cells where Bin1 has been deleted reinforce the idea that it supports a default pathway of apoptosis which is sensitized by oncogenic stress. Cell transformation by Myc or E1A is well-known to sensitize cells to apoptosis by tumor necrosis factor (TNF) or related factors such as TRAIL [118-121]. Similar effects occur in polarized cells, where TNF cooperates with Myc to trigger apoptosis [109]. In primary cells transformed by E1A+Ras, which are very sensitive to TNF, deletion of Bin1 was sufficient to abolish apoptosis [28]. This loss of sensitization was associated with precocious nuclear localization and DNA binding activity of NF-κB [28]. Together these results implied that Bin1 supported the apoptotic sensitivity of transformed cell by restricting NF-κB activity at some level. While the mechanism was not defined, effects on the canonical pathway of NF-κB were ruled out. The concept that Bin1 supports a ‘death sensitization pathway’ was extended in another study which illustrated a specific requirement in apoptosis induced by farnesyl transferase inhibitors [105], a class of targeted therapeutic drugs with a well-documented selectivity for transformed cells [122]. Together, these studies reinforced the conclusion that Bin1 supports a default pathway apoptosis that is triggered by oncogenic stress in primary cells.
More recently, Bin1 was found to be essential for a default pathway of senescence that is also triggered by oncogenes in primary cells. Specifically, Bin1 was identified in a large-scale screen for genes that are required by the Ras effector kinase B-Raf to induce senescence in primary human fibroblasts or melanocytes [123]. This study screened ~28,000 genes by an siRNA-based method, identifying Bin1 in a set of 17 genes that also included p53 and other tumor suppressor genes known to be critical mediators of cell cycle arrest and senescence. This study has important implications for unraveling the precise suppressor functions of Bin1, insofar as other genes identified in the screen might be expected to participate in common or overlapping default pathways of senescence triggered by oncogene stress. In this context, it is interesting to note that the screen also identified SmarcB1, which encodes the SWI/SNF family transcriptional co-factor Ini1/SNF5 that binds Myc and mediates it transactivation activity [124]. Similar to its role in default pathways of apoptosis, the essential role revealed for Bin1 in oncogene-induced senescence is essential in primary cells where cell polarity signaling is enabled and helps restrict cancer at its earliest stages.
Bin1 in muscle development: potential integration of stress and polarity signaling roles
The finding that Bin1 is needed for default pathways of apoptosis or senescence under conditions of oncogenic stress may prompt a re-interpretation of earlier studies of the role of Bin1 in myogenesis, obtained originally in models where differentiation is triggered by growth factor deprival. In mouse C2C12 myoblasts, a commonly accepted model of muscle terminal differentiation, inhibiting Bin1 expression prevents upregulation of the cell cycle kinase inhibitor p21WAF and entry to terminal cell cycle arrest, which in turn prevents cell fusion events that form myotubes [30, 31, 95]. Moreover, a genetic suppressor screen for genes that block C2C12 differentiation identified only Bin1 and the retinoblastoma protein gene Rb [25], the loss of which produces a muscle phenotype [125]. The notion that Bin1 might participate in Rb-mediated cell cycle arrest has received an additional line of support recently [100]. Given its high levels of expression in skeletal muscle, these findings have tended to encourage the interpretation that Bin1 is crucial for terminal cell differentiation in muscle. However, more recent observations in Bin1 null mice call this interpretation into question since no defect in skeletal muscle is evident [52]. Thus, one interpretation of the findings of C2C12 studies which are consistent with other findings is that Bin1 may participate in cell fate decisions that are triggered by a growth-related stress, in this case by growth factor deprival [31].
This interpretation does not discount other evidence that Bin1 has a non-redundant function in muscle [95], and in fact, a recent genetic study indicates that germ-line mutations in human BIN1 cause a rare myopathy known as centronuclear myopathy [126]. Myopathies are marked by muscle weakness and this particular disease is associated with abnormal centralization of nuclei in muscle fibers formed by cell-cell fusion during myoblast differentation. Specifically, in three families exhibiting autosomal recessive inheritance, five individuals were found to harbor homozygous mutations in BIN1, all of which were implicated in disrupting recruitment or tubulation activity at membranes [126]. While these defects were interpreted to underlie nuclei mislocalization and muscle function, the impact of the mutations on polarity signaling was not considered, which could conceivably be relevant to the subcellular movement and positioning of nuclei in muscle fibers.
This study noted no cases of cancer among the five affected individuals, however, all but one died in infancy or at an early age, largely precluding an assessment of the effects of BIN1 alteration on cancer. In multicellular organisms, germ line and somatic alterations differ in their phenotypic consequences. Germ line mutations in suppressor genes can cause developmental abnormalities, but even if this is not the case, such mutations may not manifest their effects on cancer until later in adulthood (as studies of Bin1 in the mouse would suggest [86]). Thus, for practical purposes, early deaths associated with germ line mutations in BIN1 would seem to prevent an evaluation of their effect on cancer. In contrast, as discussed above, numerous studies have documented somatic alterations in Bin1 expression or splicing in cancer that are associated with poor outcomes and/or deficiencies in suppressor activities. Moreover, as discussed below, genetic ablation studies in the mouse establish that Bin1 loss-of-function is sufficient to cause cancer and to drive cancer progression [85, 86]. Thus, functions of Bin1 in muscle and cancer suppression may be non-redundant and distinct. Nevertheless, in considering findings from C2C12 myoblasts, one can re-interpret the results in a manner that is consistent with an emerging theme for Bin1 as an essential mediator of apoptosis and senescence pathways that help restrict the cancerous effects of ‘rogue’ oncogenic signals.
Bin1 and Bin3 have essential functions in cancer suppression
Four amphiphysin-like genes – the ubiquitous Bin1 and Bin3 and the tissue-specific Amph I and Bin2 – representing a subset of the BAR adapter family in mammals can be defined on the basis of close structural relatedness and biochemical interactions. Deletion of Bin1 in mice causes developmentally lethality, but analysis of a conditional mutant has permitted investigations of cancer susceptibility using mosaic or tissue-specific strategies to delete the gene under circumstances where viability is maintained [52, 85, 86]. While the consequences of Bin2 deletion have not been described, inactivation of Bin3 or Amph I does not affect viability or fecundity, permitting a direct evaluation of the essential functions of these genes [127, 128]. Figure 4 summarizes the phenotypes observed in knockout mice lacking Bin1, Amph I, or Bin3.
Figure 4. Phenotypes caused by deletion of amphiphysin-like genes in the mouse.
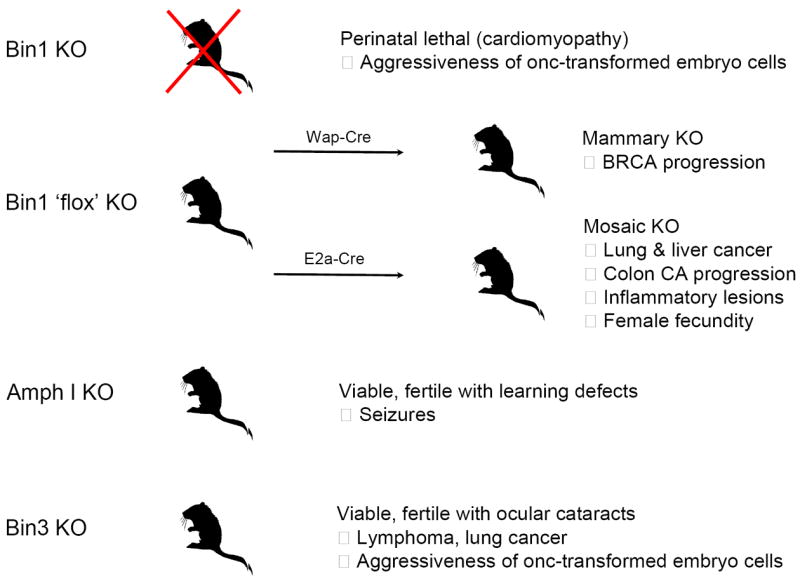
See text for details. Deletion of the amphiphysin-like Bin2 gene in the mouse has yet to be reported.
Bin1 deletion in the mouse produces a perinatal lethality associated with the development of cardiomyopathy in the embryo [52]. While precluding studies of cancer susceptibility, this phenotype was interesting in light of the evidence of a cardiomyopathy susceptibility locus in humans that maps to chromosome 2q14-22 where the Bin1 gene can be found [129]. Extensive investigations of mutant embryo cells revealed no apparent defects in endocytosis, phagocytosis, actin organization, proliferation, or survival [52]. Thus, in contrast to its homologs in yeast [1], Bin1 seems to be non-essential for these processes. While Bin1 is highly expressed in the CNS and in skeletal muscle, where it has been implicated in myoblast differentiation [25, 31, 95], no histological deficiencies are apparent in the brain or skeletal muscle, however, sluggish movement is apparent in neonates consistent with a muscle or neuromuscular deficiency [52]. In brain, Bin1 deletion associates with a concomitant loss of Amph I protein, arguing that Amph I is unstable in the absence of the ability to heterodimerize with Bin1 [Amph II] proteins in that tissue. This effect exaggerates the loss of amphiphysin-like function in brain without any obvious impact on development. In contrast, cardiac hypertrophy was evident in Bin1 null animals where the myofibrils of ventricular cardiomyocytes were severely disorganized. Interestingly, immunohistochemical analysis revealed that Bin1 proteins were predominantly cytoplasmic in skeletal muscle cells but predominantly nuclear in cardiac muscle cells [52], suggesting that nuclear interactions might contribute distally to the formation of a proper myofibril architecture in cardiomyocytes. While it is conceivable that compensary mechanisms act to limit effects of Bin1 loss in the brain or skeletal muscle, no upregulation of the amphiphysin-like genes Amph I, Bin2, or Bin3 was observed in the mutant embryo tissues. Although some studies have argued that Bin1 is essential for endocytosis or phagocytosis [93, 130], evaluations in nullizygous cells do not support this conclusion; the inconsistent findings obtained in the earlier studies may reflect the promiscuous and weakly controlled dominant inhibitory strategies (e.g. SH3 domain overexpression). More recent investigations in yeast and mammalian cells suggest a role in vesicle trafficking processes but where essential sites of function have not yet been established [60, 131]. However, in general, genetic knockout studies of Amph I and Bin1 in mice tend to reinforce the conclusions of Drosophila and fission yeast studies arguing that amphiphysin-like functions are non-essential for endocytotic processes.
More recently, a conditional cre-loxP strategy was developed to directly test the hypothesis that Bin1 suppresses cancer in tissue-specific or mosaic mutant settings where cardiac development and viability were preserved [85, 86]. Based upon the frequent attenuations of Bin1 expression found in human breast carcinomas [24], the effects of mammary gland-specific deletion were investigated on the initiation and progression of breast cancer [85]. Bin1 loss delayed the explosive outgrowth and later involution of the glandular ductal network that occurs during pregnancy, however, this effect was not associated with increased susceptibility to breast cancer during the lifetime of a parous female. In contrast, when breast cancer was initiated by treatment with the classical ras-activating carcinogen DMBA, Bin1 loss strongly accentuated the formation of poorly differentiated tumors characterized by low tubule formation, high mitotic index, and a high degree of nuclear pleomorphism. Analysis of epithelial tumor cell populations indicated that Bin1 loss provided significant benefits to proliferation, survival, and invasive capacity, in support of similar correlations in human breast carcinoma [101]. These effects were specific, because in parallel experiments Bin1 loss did not accentuate progression of tumors initiated by an overexpressed c-myc transgene, which on its own produced poorly differentiated and aggressive tumors [85]. Together, these findings extend the notion of some functional overlap between Bin1 loss and c-myc activation in breast cancer, given that ras activation can cooperate with either event to drive breast tumor progression [86, 132]. Clearly, this overlap is partial, because Bin1 loss does not fully phenocopy c-myc activation. Thus, it can be inferred that the extent of the overlap relates only to a subset of functions that are specifically germane to malignant progression in cooperation with ras . Here it is worth noting that while deregulating c-myc expression is sufficient to prevent cell cycle exit and to drive tumorigenesis, while all human cancers deregulate c-myc some also overexpress it, implying that overexpression confers additional benefits in cancer. With this in mind, a model was proposed in which Bin1 loss partly or fully phenocopies the benefits gained by c-myc overexpression in cooperation with ras activation [85].
A subsequent study conducted in mosaic mice extended the evidence that Bin1 is essential to suppress cancer during aging [86]. Mosaic mice were generated in animals where Cre recombinase is expressed only very early in development in oocytes and preimplantation embryos including at the one-cell stage zygote [133]. Loss of Bin1 was associated with an increased incidence of inflammation and the development of premalignant and malignant lesions in a variety of tissues during aging. By the age of 18-20 months, mosaic null mice displayed an increased incidence of myocarditis, an inflammatory condition of the heart, and pancreatitis, a known risk factor for pancreatic cancer. Additionally, mosaic mice displayed a significantly increased incidence of prostatitis or prostate hyperplasia, atypia or intraepithelial neoplasia [86]. These findings were notable, given the frequent loss of heterozygosity and expression of Bin1 documented in human cases of metastatic prostate cancer [23]. None of these features were apparent in control animals of a similar age. These findings suggested that by promoting inflammation, which contributes to the genesis and progression of many age-associated epithelial cancers, Bin1 attenuation might contribute to cancers in the prostate, pancreas, or other tissues if appropriate initiating lesions are present. More dramatically, >50% of mosaic null animals examined displayed frank carcinomas of the lung or liver by 18 months of age [86]. In support of a negative modifier role in progression, in young mice where colon carcinoma was initiated by exposure to a carcinogen, Bin1 loss was associated with increased tumor invasiveness. One other notable phenotype was an extended period of fecundity in female mosaic null mice, which retained reproductive capability to the unusually old age of 17 months [86]. Elderly age is the top risk factor for cancer, but few genes that modify cancer incidence during aging are known. Thus, one important implication of this study was the identification of Bin1 as an important modifier of cancer susceptibility during aging. Along with the broad evidence that its attenuation occurs often in human malignancy [2, 21-24, 29, 85, 91, 101-103], these findings establish directly that Bin1 has an essential and non-redundant function in cancer suppression, perhaps including by restricting inflammation.
Amph I is expressed specifically in the central nervous system (CNS) and mice lacking this gene exhibit increased susceptibility to epileptic-like seizures upon reaching adulthood. Amph I null mice also show learning deficiencies in support of a role in brain function [128]. However, Amph I deletion has little effect on synaptic vesicle recycling [128]. This finding is inconsistent with others based on dominant negative strategies to disrupt Amph I function, for example, by overexpressing the Amph I SH3 domain [134], possibly reflecting the pleiotropic and non-specific effects of dominant negative mutants which can arise (especially with promiscuous binding domains such as the SH3 domain). More recently, syndaptin I was defined as a physiologically relevant partner to dynamin in synaptic vesicle endocytosis and synaptic transmission rather than Amph-I [135]. Thus, while the learning deficiencies and increased seizures seen in Amph I null mice point to important essential functions in the CNS the molecular basis for these effects remain somewhat unclear. As mentioned above, Amph I acts as a paraneoplastic autoimmune antigen in rare occult cases of lung and breast cancer, suggesting that aberrant expression of Amph I in these cancers might contribute a target for immune surveillance during cancer development. In support of some link, remission of neurological symptoms has been documented to occur in patients after tumor excision and therapy [136]. Prompted by evidence of Amph I-Bin1 interaction, the role of Bin1 in suppressing breast and lung cancer, and the role of Bin1 in supporting tumoral immune surveillance (discussed below), it may be interesting to compare the susceptibility of Amph I null mice to breast or lung carcinogenesis to assess their propensity for tumoral immune escape [137].
Bin3 deletion causes ocular cataracts and increased lymphomas during aging [127]. In young animals, the lens is profoundly affected with vacuoles arising in cortical fibers and a near total loss of F-actin occuring in lens fiber cells but not lens epithelial cells. Young animals up to one year of age displayed no other overt phenotypes. However, reminiscent of the observations in Bin1 mosaic mice, in aging animals loss of Bin3 was associated with an increased incidence of lymphoma formation. This finding is of clinical interest, because the human Bin3 gene is located at chromosome 8p21.3 within a region of frequent deletions in non-Hodgkin’s lymphoma and various epithelial tumors where a tumor suppressor gene has yet to be defined [138-140]. High-resolution chromosomal deletion analyses have center on a ~1 Mb region that includes Bin3 among 10 other genes (Fig. 5). While several TRAIL family receptors within this region were originally considered good suppressor candidates in non-Hodgkins lymphoma [138], they have been ruled out as relevant recently whereas loss of heterozygosity of Bin3 has been confirmed (J. Martinez-Climent, pers. comm.). In younger Bin3 null mice that are treated with carcinogens, an increase is observed in the incidence of lung cancer was observed. Cellular analyses suggested that Bin3 was dispensable for normal cell proliferation, phagocytosis, cytosolic actin organization, or susceptibility to oncogenic transformation. In contrast, Bin3 deletion was found to increase the proliferation and motility of primary cells transformed by the SV40 T antigen and Ras oncogenes [127]. Together, the findings indicate that Bin3 has an essential function in cancer suppression.
Figure 5. Bin3 maps to a hotspot for deletions at chromosome 8p21.3 in non-Hodgkin’s lymphoma and other tumors.
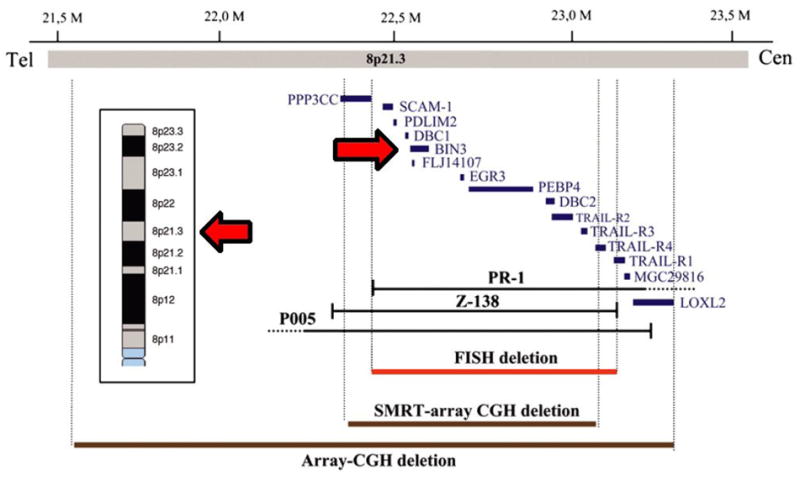
Figure is adapted from Rubio-Moscardo et al. [34].
Bin1 supports tumoral immune surveillance via indoleamine 2,3-dioxygenase (IDO)
Tumor graft studies of oncogenically transformed primary epithelial cells revealed unexpectedly that in addition to its cell-intrinsic suppressive effects Bin1 also exerts a powerful support to T cell-mediated immune surveillance [27]. Specifically, it was found that primary keratinocytes co-transformed with c-Myc+Ras (MR KECs) formed more aggressive tumors in the absence of Bin1 but only if the mouse host was competent for T cell immunity. In syngeneic animals, MR KECs expressing Bin1 formed only small, indolent nodules, whereas MR KECs lacking Bin1 formed large aggressive tumors that grew to an average size of >30-fold larger over the same period. This difference depende upon T cell immunity in the host animal, because Bin1 loss in the transformed cells conferred no advantage to tumor growth in either immunocompromised nude mice that are deficient for T cell function or in syngeneic mice that were depleted of CD4+CD8+ T cells [27]. Thus, Bin1 restricted the formation of epithelial tumors in a cell-extrinsic manner that was mediated by an ability to promote T cell-mediated anti-tumor immunity. Mechanistic investigations traced this effect to transcriptional repression of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO). In support of the hypothesis that IDO upregulation mediates the benefits of Bin1 loss to tumor outgrowth, pharmacological inhibition of IDO with 1-methyl-tryptophan (1MT) specifically blocks the in vivo growth of Bin1−/− MR KECs and this effect is dependent upon intact T cell immunity in the host [27]. Thus, Bin1 loss facilitated immune escape due to increased expression of IDO, a potent T cell suppressor that is frequently overexpressed in human tumors where it contributes to immune tolerance [141-143].
This finding that IDO is under the genetic control of Bin1 is important, because while immune escape is a fundamental trait of cancer and a critical feature of progression, relatively little is known about how it develops [144]. Since Bin1 supports immune surveillance by restricting IDO, a positive selection would exist for cells that have attenuated Bin1 and elevated IDO as one means to promote immune escape and progression. This connection does not rely upon cell transformation insofar as the same regulatory relationship was seen in primary monocytes where IDO is normally expressed [137]. IDO dysregulation caused by Bin1 loss is mediated by the increased activity of STAT1 and NF-κB, a finding that is interesting in light of the evidence that Bin1 may influence the nuclear localization efficiency of these transcription factors [27, 145]. IDO expression also appears to be elevated as a result of Bin3 loss (J. DuHadaway and G.C.P., unpublished observations). Thus, IDO may represent a generalized integration point for BAR adapter signaling by Bin1 or Bin3. Together, these findings have provided a major stimulus in the evaluation of IDO inhibitors as potential anti-tumor agents, as reviewed elsewhere [28, 144, 146, 147], and they have prompted further genetic investigations of the role of IDO in cancer and the its relationship to the cancer suppressor functions encoded by Bin1 and Bin3.
Future Perspectives
With the growing use of transgenic mouse models for cancer genetics studies, it is becoming clear that genes that modify oncogenesis may have strong effects on dormancy versus progression in cancer, a gateway that in humans would be expected to strongly affect clinical outcomes. Thus, further studies of Bin1 and Bin3 and other BAR adapter proteins and their critical signaling pathways in cancer may lead to insights into how cell polarity signals can govern cell proliferation survival, motility, and immune response, and how these signaling pathways break down during tumorigenesis and malignant progression. Signaling pathways that mediate cell polarity and immune escape represent hot emerging areas of signal transduction research in cancer, and the link revealed between these processes through Bin1 studies is an especially interesting development. In any case, given that the purposes of basic research is to develop new hypotheses, work on BAR adapter pathways in cancer seem like to provide valuable jumping-off points to new insights into the cellular pathophysiology of cancer and its treatment.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ren G, Vajjhala P, Lee JS, Winsor B, Munn AL. The BAR domain proteins: molding membranes in fission, fusion, and phagy. Microbiol Mol Biol Rev. 2006;70:37–120. doi: 10.1128/MMBR.70.1.37-120.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakamuro D, Elliott K, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumor suppressor. Nature Genet. 1996;14:69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 3.Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2003;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 4.Habermann B. The BAR domain family of proteins: a case of bending and binding? EMBO Rep. 2004;5:250–255. doi: 10.1038/sj.embor.7400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Pilecka I, Banach-Orlowska M, Miaczynska M. Nuclear functions of endocytic proteins. Eur J Cell Biol. 2007;86:533–547. doi: 10.1016/j.ejcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Wechsler-Reya R, Elliott K, Herlyn M, Prendergast GC. The putative tumor suppressor BIN1 is a short-lived nuclear phosphoprotein whose localization is altered in malignant cells. Cancer Res. 1997;57:3258–3263. [PubMed] [Google Scholar]
- 8.Elliott K, Sakamuro D, Basu A, Du W, Wunner W, Staller P, Gaubatz S, Zhang H, Prochownik E, Eilers M, Prendergast GC. Bin1 functionally interacts with Myc in cells and inhibits cell proliferation by multiple mechanisms. Oncogene. 1999;18:3564–3573. doi: 10.1038/sj.onc.1202670. [DOI] [PubMed] [Google Scholar]
- 9.Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- 10.Casal E, Federici L, Zhang W, Fernandez-Recio J, Priego EM, Miguel RN, DuHadaway JB, Prendergast GC, Luisi BF, Laue ED. The crystal structure of the BAR domain from human Bin1/amphiphysin II and its implications for molecular recognition. Biochemistry. 2006;45:12917–12928. doi: 10.1021/bi060717k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson JC, Legg JA, Machesky LM. Bar domain proteins: a role in tubulation, scission and actin assembly in clathrin-mediated endocytosis. Trends Cell Biol. 2006;16:493–498. doi: 10.1016/j.tcb.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Itoh T, De Camilli P. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim Biophys Acta. 2006;1761:897–912. doi: 10.1016/j.bbalip.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 13.Chitu V, Stanley ER. Pombe Cdc15 homology (PCH) proteins: coordinators of membrane-cytoskeletal interactions. Trends Cell Biol. 2007;17:145–156. doi: 10.1016/j.tcb.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Butler MH, David C, Ochoa G-C, Freyberg Z, Daniell L, Grabs D, Cremona O, De Camilli P. Amphiphysin II (SH3P9; BIN1), a member of the amphiphysin/RVS family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of Ravier in brain and around T tubules in skeletal muscle. J Cell Biol. 1997;137:1355–1367. doi: 10.1083/jcb.137.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsutsui K, Maeda Y, Tsutsui K, Seki S, Tokunaga A. cDNA cloning of a novel amphiphysin isoform and tissue-specific expression of its multiple splice variants. Biochem Biophys Res Comm. 1997;236:178–183. doi: 10.1006/bbrc.1997.6927. [DOI] [PubMed] [Google Scholar]
- 16.Kadlec L, Pendergast A-M. The amphiphysin-like protein 1 (ALP1) interacts functionally with the cABL tyrosine kinase and may play a role in cytoskeletal regulation. Proc Natl Acad Sci U S A. 1997;94:12390–12395. doi: 10.1073/pnas.94.23.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sparks AB, Hoffman NG, McConnell SJ, Fowlkes DM, Kay BK. Cloning of ligand targets: systematic isolation of SH3 domain-containing proteins. Nat Biotech. 1996;14:741–744. doi: 10.1038/nbt0696-741. [DOI] [PubMed] [Google Scholar]
- 18.Wechsler DS, Dang CV. Opposite orientations of DNA bending by c-Myc and Max. Proc Natl Acad Sci USA. 1992;89:7635–7639. doi: 10.1073/pnas.89.16.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DuHadaway JB, Sakamuro D, Ewert DL, Prendergast GC. Bin1 mediates apoptosis by c-Myc in transformed primary cells. Cancer Res. 2001;16:3151–3156. [PubMed] [Google Scholar]
- 20.Elliott K, Ge K, Du W, Prendergast GC. The c-Myc-interacting adapter protein Bin1 activates a caspase-independent cell death program. Oncogene. 2000;19:4669–4684. doi: 10.1038/sj.onc.1203681. [DOI] [PubMed] [Google Scholar]
- 21.Galderisi U, Di Bernardo G, Cipollaro M, Jori FP, Piegari E, Cascino A, Peluso G, Melone MAB. Induction of apoptosis and differentiation in neuroblastoma and astrocytoma cells by the overexpression of Bin1, a novel Myc interacting protein. J Cell Biochem. 1999;74:313–322. [PubMed] [Google Scholar]
- 22.Ge K, DuHadaway J, Du W, Herlyn M, Rodeck U, Prendergast GC. Mechanism for elimination of a tumor suppressor: aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci USA. 1999;96:9689–9694. doi: 10.1073/pnas.96.17.9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge K, Minhas F, DuHadaway J, Mao N-C, Wilson D, Sakamuro D, Buccafusca R, Nelson P, Malkowicz SB, Tomaszewski JT, Prendergast GC. Loss of heterozygosity and tumor suppressor activity of Bin1 in prostate carcinoma. Int J Cancer. 2000;86:155–161. doi: 10.1002/(sici)1097-0215(20000415)86:2<155::aid-ijc2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 24.Ge K, DuHadaway J, Sakamuro D, Wechsler-Reya R, Reynolds C, Prendergast GC. Losses of the tumor suppressor Bin1 in breast carcinoma are frequent and reflect deficits in a programmed cell death capacity. Int J Cancer. 2000;85:376–383. [PubMed] [Google Scholar]
- 25.Li FQ, Coonrod A, Horwitz M. Selection of a dominant negative retinoblastoma protein (RB) inhibiting satellite myoblast differentiation implies an indirect interaction between MyoD and RB. Mol Cell Biol. 2000;20:5129–5139. doi: 10.1128/mcb.20.14.5129-5139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pineda-Lucena A, Ho CS, Mao DY, Sheng Y, Laister RC, Muhandiram R, Lu Y, Seet BT, Katz S, Szyperski T, Penn LZ, Arrowsmith CH. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J Mol Biol. 2005;351:182–194. doi: 10.1016/j.jmb.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 27.Muller AJ, DuHadaway JB, Sutanto-Ward E, Donover PS, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunomodulatory target of the tumor suppressor gene Bin1, potentiates cancer chemotherapy. Nature Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 28.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Targeted deletion of the suppressor gene Bin1/Amphiphysin2 enhances the malignant character of transformed cells. Cancer Biol Ther. 2004;3:1236–1242. doi: 10.4161/cbt.3.12.1232. [DOI] [PubMed] [Google Scholar]
- 29.Tajiri T, Liu X, Thompson PM, Tanaka S, Suita S, Zhao H, Maris JM, Prendergast GC, Hogarty MD. Expression of a MYCN-interacting isoform of the tumor suppressor BIN1 is reduced in neuroblastomas with unfavorable biological features. Clin Cancer Res. 2003;9:3345–3355. [PubMed] [Google Scholar]
- 30.Mao NC, Steingrimsson E, DuHadaway J, Wasserman W, Ruiz JC, Copeland NG, Jenkins NA, Prendergast GC. The murine Bin1 gene functions early in myogenesis and defines a new region of synteny between mouse chromosome 18 and human chromosome 2. Genomics. 1999;56:51–58. doi: 10.1006/geno.1998.5709. [DOI] [PubMed] [Google Scholar]
- 31.Wechsler-Reya R, Elliott K, Prendergast GC. A role for the putative tumor suppressor Bin1 in muscle cell differentiation. Mol Cell Biol. 1998;18:566–575. doi: 10.1128/mcb.18.1.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dydensborg AB, Teller IC, Basora N, Groulx J-F, Auclair J, Francoeur C, Paré F, Herring E, Gauthier R, Prendergast GC, Jean D, Beaulieu J-F. Evidence for differential functions of the integrins a6Aß4 and a6Bß4 in human intestinal normal and adenocarcinoma cells. Gastroenterology accepted. 2008 [Google Scholar]
- 33.Routhier EL, Burn TC, Abbaszade I, Summers M, Albright CF, Prendergast GC. Human Bin3 complements the F-actin localization defects caused by loss of Hob3p, the fission yeast homolog of Rvs161p. J Biol Chem. 2001;276:2167–21677. doi: 10.1074/jbc.M101096200. [DOI] [PubMed] [Google Scholar]
- 34.Ramalingam A, Duhadaway JB, Sutanto-Ward E, Wang Y, Dinchuk J, Huang M, Donover PS, Boulden J, McNally LM, Soler AP, Muller AJ, Duncan MK, Prendergast GC. Bin3 deletion causes cataracts and increased susceptibility to lymphoma during aging. Cancer Res. 2008;68:1683–1690. doi: 10.1158/0008-5472.CAN-07-6072. [DOI] [PubMed] [Google Scholar]
- 35.Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P, Heim J. SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics. 2001;71:222–234. doi: 10.1006/geno.2000.6378. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, Youle RJ, Wang HG. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol. 2005;25:9369–9382. doi: 10.1128/MCB.25.21.9369-9382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–1039. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Modregger J, Schmidt AA, Ritter B, Huttner WB, Plomann M. Characterization of Endophilin B1b, a brain-specific membrane-associated lysophosphatidic acid acyl transferase with properties distinct from endophilin A1. J Biol Chem. 2003;278:4160–4167. doi: 10.1074/jbc.M208568200. [DOI] [PubMed] [Google Scholar]
- 39.Mitsuuchi Y, Johnson SW, Sonoda G, Tanno S, Golemis EA, Testa JR. Identification of a chromosome 3p14.3-21.1 gene, APPL, encoding an adaptor molecule that interacts with the oncoprotein-serine/threonine kinase AKT2. Oncogene. 1999;18:4891–4898. doi: 10.1038/sj.onc.1203080. [DOI] [PubMed] [Google Scholar]
- 40.Yang L, Lin HK, Altuwaijri S, Xie S, Wang L, Chang C. APPL suppresses androgen receptor transactivation via potentiating Akt activity. J Biol Chem. 2003;278:16820–16827. doi: 10.1074/jbc.M213163200. [DOI] [PubMed] [Google Scholar]
- 41.Kovacs EM, Makar RS, Gertler FB. Tuba stimulates intracellular N-WASP-dependent actin assembly. J Cell Sci. 2006;119:2715–2726. doi: 10.1242/jcs.03005. [DOI] [PubMed] [Google Scholar]
- 42.Salazar MA, Kwiatkowski AV, Pellegrini L, Cestra G, Butler MH, Rossman KL, Serna DM, Sondek J, Gertler FB, De Camilli P. Tuba: A novel protein containing Bin/Amphiphysin/Rvs (BAR) and Dbl homology domains links dynamin to regulation of the actin cytoskeleton. J Biol Chem. 2003 doi: 10.1074/jbc.M308104200. [DOI] [PubMed] [Google Scholar]
- 43.Parks WT, Frank DB, Huff C, Renfrew Haft C, Martin J, Meng X, de Caestecker MP, McNally JG, Reddi A, Taylor SI, Roberts AB, Wang T, Lechleider RJ. Sorting nexin 6, a novel SNX, interacts with the transforming growth factor-beta family of receptor serine-threonine kinases. J Biol Chem. 2001;276:19332–19339. doi: 10.1074/jbc.M100606200. [DOI] [PubMed] [Google Scholar]
- 44.Borkhardt A, Bojesen S, Haas OA, Fuchs U, Bartelheimer D, Loncarevic IF, Bohle RM, Harbott J, Repp R, Jaeger U, Viehmann S, Henn T, Korth P, Scharr D, Lampert F. The human GRAF gene is fused to MLL in a unique t(5;11)(q31;q23) and both alleles are disrupted in three cases of myelodysplastic syndrome/acute myeloid leukemia with a deletion 5q. Proc Natl Acad Sci U S A. 2000;97:9168–9173. doi: 10.1073/pnas.150079597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ge K, Prendergast GC. Bin2, a functionally nonredundant member of the BAR adaptor gene family. Genomics. 2000;67:210–220. doi: 10.1006/geno.2000.6216. [DOI] [PubMed] [Google Scholar]
- 46.Antoine JC, Absi L, Honnorat J, Boulesteix JM, de Brouker T, Vial C, Butler M, De Camilli P, Michel D. Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol. 1999;56:172–177. doi: 10.1001/archneur.56.2.172. [DOI] [PubMed] [Google Scholar]
- 47.Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol. 1996;16:3169–3178. doi: 10.1128/mcb.16.6.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siesser PM, Hanks SK. The signaling and biological implications of FAK overexpression in cancer. Clin Cancer Res. 2006;12:3233–3237. doi: 10.1158/1078-0432.CCR-06-0456. [DOI] [PubMed] [Google Scholar]
- 49.Mon NN, Ito S, Senga T, Hamaguchi M. FAK signaling in neoplastic disorders: a linkage between inflammation and cancer. Ann N Y Acad Sci. 2006;1086:199–212. doi: 10.1196/annals.1377.019. [DOI] [PubMed] [Google Scholar]
- 50.Ishibashi Y, Maita H, Yano M, Koike N, Tamai K, Ariga H, Iguchi-Ariga SM. Pim-1 translocates sorting nexin 6/TRAF4-associated factor 2 from cytoplasm to nucleus. FEBS Lett. 2001;506:33–38. doi: 10.1016/s0014-5793(01)02881-2. [DOI] [PubMed] [Google Scholar]
- 51.Chen JL, Limnander A, Rothman PB. Pim-1 and Pim-2 kinases are required for efficient pre-B-cell transformation by v-Abl oncogene. Blood. 2008;111:1677–1685. doi: 10.1182/blood-2007-04-083808. [DOI] [PubMed] [Google Scholar]
- 52.Muller AJ, Baker JF, DuHadaway JB, Ge K, Farmer G, Donover PS, Meade R, Reid C, Grzanna R, Roach AH, Shah N, Soler AP, Prendergast GC. Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol Cell Biol. 2003;23:4295–4306. doi: 10.1128/MCB.23.12.4295-4306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crouzet M, Urdaci M, Dulau L, Aigle M. Yeast mutant affected for viability upon nutrient starvation: characterization and cloning of the RVS161 gene. Yeast. 1991;7:727–743. doi: 10.1002/yea.320070708. [DOI] [PubMed] [Google Scholar]
- 54.Bauer F, Urdaci M, Aigle M, Crouzet M. Alteration of a yeast SH3 protein leads to conditional viability with defects in cytoskeletal and budding patterns. Mol Cell Biol. 1993;13:5070–5084. doi: 10.1128/mcb.13.8.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munn AL, Stevenson BJ, Geli MI, Riezman H. end5, end6, and end7: mutations that cause actin delocalization and block the internalization step of endocytosis in Saccharomyces cerevisiae. Mol Biol Cell. 1995;6:1721–1742. doi: 10.1091/mbc.6.12.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friesen H, Colwill K, Robertson K, Schub O, Andrews B. Interaction of the Saccharomyces cerevisiae cortical actin patch protein Rvs167p with proteins involved in ER to Golgi vesicle trafficking. Genetics. 2005;170:555–568. doi: 10.1534/genetics.104.040063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Germann M, Swain E, Bergman L, Nickels JT., Jr Characterizing the sphingolipid signaling pathway that remediates defects associated with loss of the yeast amphiphysin-like orthologs, Rvs161p and Rvs167p. J Biol Chem. 2005;280:4270–4278. doi: 10.1074/jbc.M412454200. [DOI] [PubMed] [Google Scholar]
- 58.Navarro P, Durrens P, Aigle M. Protein-protein interaction between the RVS161 and RVS167 gene products of Saccharomyces cerevisiae. Biochim Biophys Acta. 1997;1343:187–192. doi: 10.1016/s0167-4838(97)00108-8. [DOI] [PubMed] [Google Scholar]
- 59.Lombardi R, Riezman H. Rvs161p and Rvs167p, the two yeast amphiphysin homologs, function together in vivo. J Biol Chem. 2001;276:6016–6022. doi: 10.1074/jbc.M008735200. [DOI] [PubMed] [Google Scholar]
- 60.Friesen H, Humphries C, Ho Y, Schub O, Colwill K, Andrews B. Characterization of the yeast amphiphysins Rvs161p and Rvs167p reveals roles for the Rvs heterodimer in vivo. Mol Biol Cell. 2006;17:1306–1321. doi: 10.1091/mbc.E05-06-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friesen H, Murphy K, Breitkreutz A, Tyers M, Andrews B. Regulation of the yeast amphiphysin homologue Rvs167p by phosphorylation. Mol Biol Cell. 2003;14:3027–3040. doi: 10.1091/mbc.E02-09-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee J, Colwill K, Aneuliunas V, Tennyson c, Moore L, Ho Y, Andrews B. Interaction of yeast Rvs167 and Pho85 cyclin-dependent kinase complexes may link the cell cycle to the actin cytoskeleton. Curr Biol. 1998;8:1310–1321. doi: 10.1016/s0960-9822(07)00561-1. [DOI] [PubMed] [Google Scholar]
- 63.Floyd SR, Porrom EB, Slepnev VI, Ochoa GC, Tsai LH, De Camilli P. Amphiphysin 1 binds the cyclin-dependent kinase (cdk) 5 regulatory subunit p35 and is phosphorylated by cdk5 and cdc2. J Biol Chem. 2001;276:8104–8110. doi: 10.1074/jbc.M008932200. [DOI] [PubMed] [Google Scholar]
- 64.Huang D, Patrick G, Moffat J, Tsai LH, Andrews B. Mammalian cdk5 is a functional homologue of the budding yeast Pho85. Proc Natl Acad Sci USA. 1999;96:14445–14450. doi: 10.1073/pnas.96.25.14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, Hansra G, McClure SJ, Sarcevic B, Boadle RA, Larsen MR, Cousin MA, Robinson PJ. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol. 2003;5:701–710. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- 66.Smith DS, Tsai LH. Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol. 2002;12:28–36. doi: 10.1016/s0962-8924(01)02181-x. [DOI] [PubMed] [Google Scholar]
- 67.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 68.Sivadon P, Bauer F, Aigle M, Crouzet M. Actin cytoskeleton and budding pattern are altered in the yeast rvs161 mutant: the Rvs161 protein shares common domains with the brain protein amphiphysin. Mol Gen Genet. 1995;246:485–495. doi: 10.1007/BF00290452. [DOI] [PubMed] [Google Scholar]
- 69.Breton AM, Aigle M. Genetic and functional relationship between Rvsp, myosin and actin in Saccharomyces cerevisiae. Curr Genet. 1998;34:280–286. doi: 10.1007/s002940050397. [DOI] [PubMed] [Google Scholar]
- 70.Balguerie A, Sivadon P, Bonneu M, Aigle M. Rvs167p, the budding yeast homolog of amphiphysin, colocalizes with actin patches. J Cell Sci. 1999;112:2529–2537. doi: 10.1242/jcs.112.15.2529. [DOI] [PubMed] [Google Scholar]
- 71.Brizzio V, Gammie AE, Rose MD. Rvs161p interacts with Fus2p to promote cell fusion in Saccharomyces cerevisiae. J Cell Biol. 1998;141:567–584. doi: 10.1083/jcb.141.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drees BL, Sundin B, Brazeau E, Caviston JP, Chen GC, Guo W, Kozminski KG, Lau MW, Moskow JJ, Tong A, Schenkman LR, McKenzie Ar, Brennwald P, Longtine M, Bi E, Chan C, Novick P, Boone C, Pringle JR, Davis TN, Fields S, Drubin DG. A protein interaction map for cell polarity development. J Cell Biol. 2001;154:549–571. doi: 10.1083/jcb.200104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Routhier EL, Donover PS, Prendergast GC. hob1+, the homolog of Bin1 in fission yeast, is dispensable for endocytosis but required for the response to starvation or genotoxic stress. Oncogene. 2003;22:637–648. doi: 10.1038/sj.onc.1206162. [DOI] [PubMed] [Google Scholar]
- 74.Huang A, Fuchs D, Widner B, Glover C, Henderson DC, Allen-Mersh TG. Serum tryptophan decrease correlates with immune activation and impaired quality of life in colorectal cancer. Br J Cancer. 2002;86:1691–1696. doi: 10.1038/sj.bjc.6600336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramalingam A, Prendergast GC. Bin1 homolog hob1 supports a rad6-set1 pathway of transcriptional repression in fission yeast. Cell Cycle. 2007;6:1655–1662. doi: 10.4161/cc.6.13.4413. [DOI] [PubMed] [Google Scholar]
- 76.Ramalingam A, Farmer GE, Stamato TD, Prendergast GC. Bin1 interacts with and restrains the DNA end-binding protein complex Ku. Cell Cycle. 2007;6:1914–1918. doi: 10.4161/cc.6.15.4514. [DOI] [PubMed] [Google Scholar]
- 77.Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR, 3rd, Menssen A, Hermeking H. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle. 2007;6:205–217. doi: 10.4161/cc.6.2.3742. [DOI] [PubMed] [Google Scholar]
- 79.Coll PM, Rincon SA, Izquierdo RA, Perez P. Hob3p, the fission yast ortholog of human Bin3, localizes Cdc42p to the division site and regulates cytokinesis. EMBO J. 2007;26:1865–1877. doi: 10.1038/sj.emboj.7601641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rincon S, Coll PM, Perez P. Spatial regulation of Cdc42 during cytokinesis. Cell Cycle. 2007;6:1687–1691. doi: 10.4161/cc.6.14.4481. [DOI] [PubMed] [Google Scholar]
- 81.Razzaq A, Robinson IM, McMahon HT, Skepper JN, Su Y, Zelhof AC, Jackson AP, Gay NJ, O’Kane CJ. Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila. Genes Dev. 2001;15:2967–2979. doi: 10.1101/gad.207801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zelhof AC, Bao H, Hardy RW, Razzaq A, Zhang B, Doe CQ. Drosophila Amphiphysin is implicated in protein localization and membrane morphogenesis but not in synaptic vesicle endocytosis. Development. 2001;128:5005–5015. doi: 10.1242/dev.128.24.5005. [DOI] [PubMed] [Google Scholar]
- 83.Humbert P, Russell S, Richardson H. Dlg, Scribble and Lgl in cell polarity, cell proliferation and cancer. Bioessays. 2003;25:542–553. doi: 10.1002/bies.10286. [DOI] [PubMed] [Google Scholar]
- 84.Pagliarini RA, Xu T. A genetic screen in drosophila for metastatic behavior. Science. 2003;302:1227–1231. doi: 10.1126/science.1088474. [DOI] [PubMed] [Google Scholar]
- 85.Chang M, Boulden J, Sutanto-Ward E, Duhadaway JB, Soler AP, Muller AJ, Prendergast GC. Bin1 ablation in mammary gland delays tissue remodeling and drives cancer progression. Cancer Res. 2007;67:100–107. doi: 10.1158/0008-5472.CAN-06-2742. [DOI] [PubMed] [Google Scholar]
- 86.Chang M, Boulden J, Katz JB, Wang L, Meyer TJ, Soler AP, Muller AJ, Prendergast GC. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 2007;67:7605–7612. doi: 10.1158/0008-5472.CAN-07-1100. [DOI] [PubMed] [Google Scholar]
- 87.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 88.Kajiho H, Saito K, Tsujita K, Kontani K, Araki Y, Kurosu H, Katada T. RIN3 a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J Cell Sci. 2003;116:4159–4168. doi: 10.1242/jcs.00718. [DOI] [PubMed] [Google Scholar]
- 89.Hirst M, Ho C, Sabourin L, Rudnicki M, Penn L, Sadowski I. A two-hybrid system for transactivator bait proteins. Proc Natl Acad Sci U S A. 2001;98:8726–8731. doi: 10.1073/pnas.141413598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.EntrezBin1, http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSe arch=274&ordinalpos=1&itool=EntrezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum
- 91.DuHadaway JB, Lynch FJ, Brisbay S, Bueso-Ramos C, Troncoso P, McDonnell T, Prendergast GC. Immunohistochemical analysis of Bin1/Amphiphysin II in human tissues: Diverse sites of nuclear expression and losses in prostate cancer. J Cell Biochem. 2003;88:635–642. doi: 10.1002/jcb.10380. [DOI] [PubMed] [Google Scholar]
- 92.Wechsler-Reya R, Sakamuro D, Zhang J, Duhadaway J, Prendergast GC. Structural analysis of the human BIN1 gene: evidence for tissue-specific transcriptional regulation and alternate RNA splicing. J Biol Chem. 1997;272:31453–31458. doi: 10.1074/jbc.272.50.31453. [DOI] [PubMed] [Google Scholar]
- 93.Gold ES, Morrissette NS, Underhill DM, Guo J, Bassetti M, Aderem A. Amphiphysin IIm, a novel amphiphysin II isoform, is required for macrophage phagocytosis. Immunity. 2000;12:285–292. doi: 10.1016/s1074-7613(00)80181-8. [DOI] [PubMed] [Google Scholar]
- 94.Ramjaun AR, Micheva KD, Bouchelet I, McPherson PS. Identification and characterization of a nerve terminal-enriched amphiphysin isoform. J Biol Chem. 1997;272:16700–16706. doi: 10.1074/jbc.272.26.16700. [DOI] [PubMed] [Google Scholar]
- 95.Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, De Camilli P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–1196. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- 96.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu Q, Lee C. Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res. 2003;31:5635–5643. doi: 10.1093/nar/gkg786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roy M, Xu Q, Lee C. Evidence that public database records for many cancer-associated genes reflect a splice form found in tumors and lack normal splice forms. Nucleic Acids Res. 2005;33:5026–5033. doi: 10.1093/nar/gki792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nesbit CE, Grove LE, Yin X, Prochownik EV. Differential apoptotic behaviors of c-myc, N-myc, and L-myc oncoproteins. Cell Growth Diff. 1998;9:731–741. [PubMed] [Google Scholar]
- 100.Kinney EL, Tanida S, Rodrigue AA, Johnson JK, Tompkins VS, Sakamuro D. Adenovirus E1A oncoprotein liberates c-Myc activity to promote cell proliferation through abating Bin1 expression via an Rb/E2F1-dependent mechanism. J Cell Physiol. 2008 doi: 10.1002/jcp.21437. [DOI] [PubMed] [Google Scholar]
- 101.Ghaneie A, Zemba-Palko V, Itoh H, Itoh K, Sakamuro D, Nakamura S, Soler AP, Prendergast GC. Bin1 attenuation in breast cancer is correlated to nodal metastasis and reduced survival. Cancer Biol Ther. 2007;6 doi: 10.4161/cbt.6.2.3587. [DOI] [PubMed] [Google Scholar]
- 102.Hogarty MD, Liu X, Thompson PM, White PS, Sulman EP, Maris JM, Brodeur GM. BIN1 inhibits colony formation and induces apoptosis in neuroblastoma cell lines with MYCN amplification. Med Pediatr Oncol. 2000;35:559–562. doi: 10.1002/1096-911x(20001201)35:6<559::aid-mpo14>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 103.Huang H, Colella S, Kurrer M, Yonekawa Y, Kleihues P, Ohgaki H. Gene expression profiling of low-grade diffuse astrocytomas by cDNA arrays. Cancer Res. 2000;60:6868–6874. [PubMed] [Google Scholar]
- 104.Cher ML, Bova GS, Moore DH, Small EJ, Carroll PR, Pin SS, Epstein JI, Isaacs WB, Jensen RH. Genetic alterations in untreated metastases and androgen-independent prostate cancer detected by comparative genomic hybridization and allelotyping. Cancer Res. 1996;56:3091–3102. [PubMed] [Google Scholar]
- 105.DuHadaway JB, Du W, Liu A-X, Baker J, Donover PS, Sharp DM, Muller AJ, Prendergast GC. Transformation selective apoptosis by farnesyltransferase inhibitors requires Bin1. Oncogene. 2003;22:3578–3588. doi: 10.1038/sj.onc.1206481. [DOI] [PubMed] [Google Scholar]
- 106.McCarthy NJ, Whyte MKB, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC, de The H. PML induces a novel caspase-independent death process. Nat Genet. 1998;20:259–265. doi: 10.1038/3068. [DOI] [PubMed] [Google Scholar]
- 108.Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, Pandolfi PP. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20:266–272. doi: 10.1038/3073. [DOI] [PubMed] [Google Scholar]
- 109.Partanen JI, Nieminen AI, Makela TP, Klefstrom J. Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc Natl Acad Sci U S A. 2007;104:14694–14699. doi: 10.1073/pnas.0704677104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baas AF, Smit L, Clevers H. LKB1 tumor suppressor protein: PARtaker in cell polarity. Trends Cell Biol. 2004;14:312–319. doi: 10.1016/j.tcb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 111.Frisch SM. E1a induces the expression of epithelial characteristics. J Cell Biol. 1994;127:1085–1096. doi: 10.1083/jcb.127.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frisch SM. The epithelial cell default-phenotype hypothesis and its implications for cancer. Bioessays. 1997;19:705–709. doi: 10.1002/bies.950190811. [DOI] [PubMed] [Google Scholar]
- 113.Grooteclaes ML, Frisch SM. Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene. 2000;19:3823–3828. doi: 10.1038/sj.onc.1203721. [DOI] [PubMed] [Google Scholar]
- 114.Shiio Y, Donohoe S, Yi EC, Goodlett DR, Aebersold R, Eisenman RN. Quantitative proteomic analysis of Myc oncoprotein function. EMBO J. 2002;21:5088–5096. doi: 10.1093/emboj/cdf525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Telfer JF, Urquhart J, Crouch DH. Suppression of MEK/ERK signalling by Myc: role of Bin-1. Cell Signal. 2005;17:701–708. doi: 10.1016/j.cellsig.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 116.Prendergast GC. Mechanisms of apoptosis by c-Myc. Oncogene. 1999;18:2966–2986. doi: 10.1038/sj.onc.1202727. [DOI] [PubMed] [Google Scholar]
- 117.Soucie EL, Annis MG, Sedivy J, Filmus J, Leber B, Andrews DW, Penn LZ. Myc potentiates apoptosis by stimulating Bax activity at the mitochondria. Mol Cell Biol. 2001;21:4725–4736. doi: 10.1128/MCB.21.14.4725-4736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nieminen AI, Partanen JI, Hau A, Klefstrom J. c-Myc primed mitochondria determine cellular sensitivity to TRAIL-induced apoptosis. EMBO J. 2007;26:1055–1067. doi: 10.1038/sj.emboj.7601551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Janicke RU, Lee FH, Porter AG. Nuclear c-Myc plays an important role in the cytotoxicity of tumor necrosis factor alpha in tumor cells. Mol Cell Biol. 1994;14:5661–5670. doi: 10.1128/mcb.14.9.5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Klefstrom J, Arighi E, Littlewood T, Jaattela M, Saksela E, Evan GI, Alitalo K. Induction of TNF-sensitive cellular phenotype by c-Myc involves p53 and impaired NF-kappaB activation. EMBO J. 1997;16:7382–7392. doi: 10.1093/emboj/16.24.7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Klefstrom J, Vastrik I, Saksela E, Valle J, Eilers M, Alitalo K. c-Myc induces cellular susceptibility to the cytotoxic action of TNF-alpha. EMBO J. 1994;13:5442–5450. doi: 10.1002/j.1460-2075.1994.tb06879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Prendergast GC. Farnesyltransferase inhibitors: antineoplastic mechanism and clinical prospects. Curr Opin Cell Biol. 2000;12:166–173. doi: 10.1016/s0955-0674(99)00072-1. [DOI] [PubMed] [Google Scholar]
- 123.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet. 1999;22:102–105. doi: 10.1038/8811. [DOI] [PubMed] [Google Scholar]
- 125.Novitch BG, Mulligan GJ, Jacks T, Lassar AB. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol. 1996;135:441–456. doi: 10.1083/jcb.135.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nicot AS, Toussaint A, Tosch V, Kretz C, Wallgren-Pettersson C, Iwarsson E, Kingston H, Garnier JM, Biancalana V, Oldfors A, Mandel JL, Laporte J. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39:1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 127.Ramalingam A, Duhadaway JB, Sutanto-Ward E, Wang Y, Dinchuk J, Huang M, Donover PS, Boulden J, McNally LM, Soler AP, Muller AJ, Duncan MK, Prendergast GC. Bin3 deletion causes cataracts and elevates the incidence of lymphomas during aging. Cancer Res. 2008 doi: 10.1158/0008-5472.CAN-07-6072. in press. [DOI] [PubMed] [Google Scholar]
- 128.Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, Flavell R, Picciotto MR, Ryan TA, Cremona O, De Camilli P. Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron. 2002;33:789–804. doi: 10.1016/s0896-6273(02)00601-3. [DOI] [PubMed] [Google Scholar]
- 129.Jung M, Poepping I, Perrot A, Ellmer AE, Wienker TF, Dietz R, Reis A, Osterziel KJ. Investigation of a family with autosomal dominant dilated cardiomyopathy defines a novel locus on chromosome 2q14-q22. Am J Hum Genet. 1999;65:1068–1077. doi: 10.1086/302580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Owen DJ, Wigge P, Vallis Y, Moore JD, Evans PR, McMahon HT. Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J. 1998;17:5273–5285. doi: 10.1093/emboj/17.18.5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Leprince C, Le Scolan E, Meunier B, Fraisier V, Brandon N, De Gunzburg J, Camonis J. Sorting nexin 4 and amphiphysin 2, a new partnership between endocytosis and intracellular trafficking. J Cell Sci. 2003;116:1937–1948. doi: 10.1242/jcs.00403. [DOI] [PubMed] [Google Scholar]
- 132.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 133.Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wigge P, Vallis Y, McMahon HT. Inhibition of receptor-mediated endoctosis by the amphiphysin SH3 domain. Curr Biol. 1997;7:554–560. doi: 10.1016/s0960-9822(06)00254-5. [DOI] [PubMed] [Google Scholar]
- 135.Anggono V, Robinson PJ. Syndapin I and endophilin I bind overlapping proline-rich regions of dynamin I: role in synaptic vesicle endocytosis. J Neurochem. 2007;102:931–943. doi: 10.1111/j.1471-4159.2007.04574.x. [DOI] [PubMed] [Google Scholar]
- 136.De Camilli P, Thomas A, Cofiell R, Folli F, Lichte B, Piccolo G, Meinck HM, Austoni M, Fassetta G, Bottazzo G, et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of Stiff-Man syndrome with breast cancer. J Exp Med. 1993;178:2219–2223. doi: 10.1084/jem.178.6.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Prendergast GC. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008 doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 138.Rubio-Moscardo F, Blesa D, Mestre C, Siebert R, Balasas T, Benito A, Rosenwald A, Climent J, Martinez JI, Schilhabel M, Karran EL, Gesk S, Esteller M, deLeeuw R, Staudt LM, Fernandez-Luna JL, Pinkel D, Dyer MJ, Martinez-Climent JA. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood. 2005;106:3214–3222. doi: 10.1182/blood-2005-05-2013. [DOI] [PubMed] [Google Scholar]
- 139.Chang BL, Liu W, Sun J, Dimitrov L, Li T, Turner AR, Zheng SL, Isaacs WB, Xu J. Integration of somatic deletion analysis of prostate cancers and germline linkage analysis of prostate cancer families reveals two small consensus regions for prostate cancer genes at 8p. Cancer Res. 2007;67:4098–4103. doi: 10.1158/0008-5472.CAN-06-4570. [DOI] [PubMed] [Google Scholar]
- 140.Ye H, Pungpravat N, Huang BL, Muzio LL, Mariggio MA, Chen Z, Wong DT, Zhou X. Genomic assessments of the frequent loss of heterozygosity region on 8p21.3 approximately p22 in head and neck squamous cell carcinoma. Cancer Genet Cytogenet. 2007;176:100–106. doi: 10.1016/j.cancergencyto.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141..
- 142.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van Den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 143.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in immune suppression and cancer. Curr Cancer Drug Targets. 2007;7:31–40. doi: 10.2174/156800907780006896. [DOI] [PubMed] [Google Scholar]
- 145.Bild AH, Turkson J, Jove R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. EMBO J. 2002;21:3255–3263. doi: 10.1093/emboj/cdf351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Muller AJ, Prendergast GC. Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res. 2005;65:8065–8068. doi: 10.1158/0008-5472.CAN-05-2213. [DOI] [PubMed] [Google Scholar]
- 147.Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–625. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.