

SUMMARY
Multiple angiogenesis inhibitors have been therapeutically validated in preclinical cancer models, and several in clinical trials. Here we report that angiogenesis inhibitors targeting the VEGF pathway demonstrate antitumor effects in mouse models of pancreatic neuroendocrine carcinoma and glioblastoma but concomitantly elicit tumor adaptation and progression to stages of greater malignancy, with heightened invasiveness and in some cases increased lymphatic and distant metastasis. Increased invasiveness is also seen by genetic ablation of the Vegf-A gene in both models, substantiating the results of the pharmacological inhibitors. The realization that potent angiogenesis inhibition can alter the natural history of tumors by increasing invasion and metastasis warrants clinical investigation, as the prospect has important implications for the development of enduring antiangiogenic therapies.
SIGNIFICANCE.
Angiogenesis inhibitors targeting the VEGF signaling pathway have proven to be efficacious in preclinical cancer models and in clinical trials. While antitumoral effects and survival benefit are often evident, relapse to progressive tumor growth typically ensues, reflecting multiple mechanisms of adaptation to antiangiogenic therapies. Our findings further implicate angiogenesis inhibition as a driving force in tumor progression to stages of greater malignancy, reflected in heightened invasion into surrounding tissue and in some cases increased lymphatic and distant metastasis. Thus, antiangiogenic therapy that effectively inhibits neovascularization and produces antitumor effects and survival benefit can additionally alter the phenotype of tumors by increasing invasion and metastasis. This realization motivates clinical studies to confirm and potentially target this insidious consequence of antiangiogenic therapies.
INTRODUCTION
Judah Folkman's long-standing vision of angiogenesis as a therapeutic target (Folkman, 1971) has been increasingly validated in both traditional transplant tumor models and genetically engineered mouse models of cancer, beginning in the mid-1990s and continuing to the present (Parangi et al., 1996; Shaked et al., 2005). The “angiogenic switch” during tumor progression (Hanahan and Folkman, 1996) is increasingly recognized as constituting a rate-limiting secondary event in tumorigenesis (Hanahan and Weinberg, 2000) that can be effectively targeted as a successful therapeutic approach to treat cancer; clinically, proof of concept has begun with the recent regulatory approvals of three antiangiogenic therapies targeting the VEGF/VEGFR2 pathway in certain types of cancer (Folkman, 2007; Folkman and Ingber, 1992; Kerbel, 2000). Notably, like most systemic therapies, these drugs have not produced enduring efficacy in terms of either tumor shrinkage or dormancy (stable disease) or long-term survival; rather, the common result is delayed time to progression following a period of clinical benefit, which is suggestive of an emergent resistance to the antiangiogenic therapy (Bergers and Hanahan, 2008; Kerbel et al., 2001; Miller et al., 2005).
Recently, experimental evidence has been developed in support of this proposition: VEGF receptor inhibition in a mouse model of pancreatic islet cancer reveals an initial response with vascular dropout and tumor stasis, but tumors then adapt and begin regrowing via a process referred to as “evasive resistance,” based on the observed upregulation of alternative proangiogenic signals that include functional involvement of fibroblast growth factor (FGF) ligands (Casanovas et al., 2005). Additionally, it was noted that the relapsing tumors appear to be more invasive. Other studies have also associated increased invasiveness with impaired angiogenesis in the context of genetic ablation of the hypoxia response and/or the VEGF/VEGFR pathways (Blouw et al., 2007; Du et al., 2008; Pennacchietti et al., 2003).
RESULTS
Increased Invasiveness in Response to a Specific VEGFR2 Inhibitor
To further extend our previous study of resistance to abrogation of VEGF signaling with a function-blocking antibody against VEGFR2 (DC101) in the RIP1-Tag2 model of pancreatic neuroendocrine cancer (PNET) (Casanovas et al., 2005), we sought to focus on the initial onset of malignant progression to invasive islet carcinoma. Tumor-bearing immunocompromised RIP1-Tag2 mice treated with DC101 for a relatively brief period of 1 week had reduced tumor vasculature and volume compared to control age-matched untreated animals, as described previously (Figure 1A and data not shown; Casanovas et al., 2005). Nevertheless, histological analysis showed a significantly more invasive phenotype after 1 week of treatment, an effect that was exacerbated when therapy was maintained for 4 continuous weeks. These more aggressive tumors had wide fronts of invasion, which prominently intermingled with the surrounding acinar tissue, whereas the majority of control tumors were predominantly encapsulated or microinvasive (Figure 1A, top panels). This aggravated invasive phenotype was readily revealed by fluorescent immunostaining for a marker of cancer cells, the SV40 T antigen oncoprotein (Figure 1A, middle panels). Congruently, immunostaining for the tumor collagenous capsule with anti-collagen type I antibody revealed the anti-VEGFR2-treated tumors to have a much thinner capsule, with areas of breakage or its complete absence (data not shown).
Figure 1. Increased Invasive Phenotype after Anti-VEGFR2 Therapy.

Histological analysis and quantification of tumor invasion in anti-VEGFR2 antibody (DC101)-treated or control immunocompromised tumor-bearing RIP1-Tag2 animals are presented.
(A) Histological images of tumors from control, 1-week, and 4-week treatment arms are shown in hematoxylin and eosin staining (H&E; top row), indicating prominent invasive fronts in the treated animals (black arrowheads); high-magnification images of immunofluorescence staining for SV40 T antigen (middle row), where T indicates tumor and Ac indicates surrounding acinar tissue; and immunodetection of the vasculature with anti-CD31 antibody (bottom row), where a central view of the tumor parenchyma is shown.
(B) Quantification of tumor invasiveness represented as the percentage of encapsulated islet tumors (IT), microinvasive carcinomas (IC1), and fully invasive carcinomas (IC2) for the three different treatments. Both anti-VEGFR2 treatments show a statistically significant decrease in the percentage of IT and a significant increase in IC2 tumors (**p < 0.01 by Kruskal-Wallis test).
(C) Quantification of tumor invasiveness in animals treated for only 1 week with anti-VEGFR2 antibody and then maintained without therapy for 1, 2, or 3 more weeks. Percentage of IT (white bars), IC1 (gray bars), and IC2 (black bars) is shown per treatment and time point. All treatment groups showed statistical differences by Mann-Whitney test (p < 0.01) when compared to the control group at the point when treatment was started. Age-matched treated versus control statistical analysis is shown for each time point. Note that no animals in the control group survived to the 16 week time point.
*p < 0.05, **p < 0.01 by Mann-Whitney test.
Error bars indicate ± SD.
In order to quantify the degree of invasion, the frequency of invasive lesions was scored in short-term (1 week) and long-term (4 weeks) VEGFR2-blocking antibody-treated RIP1-Tag2 animals using a three-grade nomenclature of noninvasive encapsulated islet tumors (IT), microinvasive carcinomas (IC1), and widely invasive carcinomas (IC2) as described previously (Lopez and Hanahan, 2002). While control untreated animals showed a 6% incidence of highly invasive tumors (IC2), treatment with anti-VEGFR2 antibody for 1 week increased the IC2 tumor incidence to 54%, and furthermore, treatment for 4 continuous weeks with this antibody led to an incidence of 62.5% (Figure 1B). Thus, the event of increased tumor invasiveness was already evident and significant following 1 week of antiangiogenic treatment, and the effect persisted and increased during longer-term continuous treatment for 4 weeks. Notably, in some tumors from long-term anti-VEGFR2-treated animals, tumor cells could be observed invading not only into the surrounding acinar tissue but also into the vasculature (see Figure S1 available online); such “vasoinvasion” has been used as an indicator of poor prognosis in some human tumor types (Offerhaus et al., 1991; Waggoner, 2003).
Persistence of the Invasive Phenotype after Cessation of Anti-VEGFR2 Treatment
We next sought to investigate the reversibility of this heightened invasive phenotype in the presence of angiogenesis inhibition by assessing its persistence when the angiogenic blockade was lifted. Thus, 1-week anti-VEGFR2-treated animals were then maintained for an additional 1, 2, or 3 weeks without treatment, and tumor invasiveness was determined at each time point. Again, the proportion of invasive tumors was increased after 1-week treatment, and strikingly, invasiveness remained augmented after termination of therapy, with a 10-fold higher incidence of widely invasive carcinomas persisting 1, 2, and 3 weeks after cessation of treatment (Figure 1C). It is noteworthy that the treated animals survived until 16 weeks of age due to the beneficial effect of the 1-week therapy (which produced stable disease for that period), whereas control untreated animals survived only until 15 weeks of age (Figure S2). Thus, in order to rule out the possibility that this increased invasion was due to an increased life span of the animals and not to the inferred drug-related effect, we also performed an age-matched comparison of the tumor invasion incidence in treated animals versus controls at each defined time point. Again, all anti-VEGFR2 treatment groups also exhibited statistically significant differences when compared to their respective age-matched control groups (Figure 1C).
Thus, the augmentation of tumor invasiveness is a rapidly occurring event that develops within 1 week of initiating anti-VEGFR2 therapy, producing a condition of increased malignancy that persists even when the treatment is terminated.
Increased Invasiveness in Tumors Carrying a Genetic Deletion of Vegf-A
To substantiate the hypothesis that this augmented invasive and malignant phenotype is a result of impairing the VEGF/VEGFR2 proangiogenic signaling pathway, we asked whether a genetic disruption of this pathway would produce a similar adaptive response. To do so, we analyzed RIP1-Tag2 mice carrying a tumor cell-specific deletion of the Vegf-A gene, by means of the Cre-loxP system. We have previously shown that RIP1-Tag2/Cre;Vegf-Afl/fl mice (β-VEGF-KO) exhibit a dramatic suppression of angiogenesis and impairment of tumor development and growth (Inoue et al., 2002). Detailed histological observation of the borders of β-VEGF-KO lesions revealed the tumors to be much more irregular and invasive, encroaching into adjacent exocrine tissue, as compared to tumors in RIP1-Tag2/Cre;Vegf-A+/+ mice (β-VEGF-WT), a result that was further confirmed by positive immunostaining for insulin, a marker of these islet b cell tumors (Figure 2A). Consistent with previous observations (Inoue et al., 2002), β-VEGF-KO lesions had remarkably fewer blood vessels compared with their β-VEGF-WT counterparts, and their invasive fronts had a microvessel density similar to that of normal exocrine tissue, suggestive of a possible invasive co-option of the neighboring normal vasculature in the exocrine pancreas (Figures 2A and 2B). When tumor invasiveness was quantified in both genotypes, encapsulated tumors and both grades of invasive lesions were observed in the control β-VEGF-WT mice, whereas, remarkably, no noninvasive tumors were observed in the β-VEGF-KO mice (Figure 2C). Instead, all tumors were invasive to varying degrees, even in the smaller lesions. Taken together, these results recapitulate the effect of the anti-VEGFR2 blocking antibody and thus confirm the specificity of the heightened invasiveness caused by the specific disruption of the VEGF/VEGFR signaling axis.
Figure 2. Increased Tumor Invasion after Tumor-Specific Vegf-A Gene Deletion.

(A) Top row: H&E staining of lesions in RIP1-Tag2/Cre;Vegf-A+/+ (β-VEGF-WT; left) and RIP1-Tag2/Cre;Vegf-Afl/fl (β-VEGF-KO; middle) mice. Middle row: insulin immunostaining reveals the tumor cells in serial sections to the H&E images shown in the top row. Bottom row: CD31 vessel staining (red) and DAPI nuclear staining (blue) of tumors in a β-VEGF-WT mouse and a β-VEGF-KO mouse. Images at right show a higher magnification of images in the middle column. (B) Microvessel area (MVA) at the invasive front was equivalent to that of exocrine tissue in β-VEGF-KO tumors. MVA of six tumors from four β-VEGF-WT and β-VEGF-KO animals was determined in the tumor “border” as the band-like region 50 μm inside the tumor perimeter (gray bars), the surrounding exocrine tissue (“Exo”; white bars), and the tumor “core” (black bars). MVA values were standardized to those of exocrine tissue distant from the tumors in each section. *p < 0.05. (C) Quantification of invasiveness represented by the frequency of each tumor grade in 27 tumors from 5 β-VEGF-KO mice (gray bars) versus 75 tumors from 5 β-VEGF-WT mice (black bars) at 13 weeks of age. *p < 0.05, **p < 0.01 by Mann-Whitney test.
Error bars indicate ± SD.
Increased Metastasis in Response to VEGFR2 Blockade
To further determine the consequences of this augmented invasiveness, we next sought to determine whether anti-VEGFR2 antiangiogenic therapy was associated with a higher incidence of tumor dissemination and metastasis formation in immuno-competent RIP1-Tag2 mice. Recognizing the rapid time course to end-stage disease in this model, we started the treatment when solid tumors begin to appear (at 10 weeks of age) and continued it for 10 days. The treatment was then discontinued, and the animals were maintained until 16 weeks of age, when they were sacrificed along with age-matched untreated control animals. Gross morphology of surgically removed pancreata revealed that anti-VEGFR2-treated animals more frequently contained enlarged and hemorrhagic peripancreatic lymph nodes (LNs). The presence of tumor metastases in these lymph nodes was determined by hematoxylin and eosin (H&E) staining and corroborated by immunohistochemical staining for the tumor-specific SV40 T antigen oncoprotein (Figure 3A). Quantification of the incidence of macroscopic LN metastasis in both treatment groups revealed that the incidence of LN metastasis was 4-fold higher in treated animals than in controls, with a relative risk of 4.3 [1.5-12.4] (95% confidence interval) (Figure 3B).
Figure 3. Increased Incidence of Lymph Node and Liver Metastasis in Anti-VEGFR2-Treated Animals.

Histological analysis of lymph node (LN) and liver metastasis (Met) in RIP1-Tag2 animals treated with anti-VEGFR2 for 10 days starting at 10 weeks of age and then left untreated until 16 weeks of age.
(A) LN and Met observed by histological H&E staining of tissue sections from anti-VEGFR2-treated animals appear as enlarged hemorrhaging LNs infiltrated with tumor cells and a small tumor nodule in the liver parenchyma (left panels). Immunohistochemical staining for the tumor marker SV40 T antigen (brown) reveals the presence of tumor cells infiltrated into a LN or in the midst of the liver parenchyma (right panels).
(B) Top: quantification of the incidence of animals with microscopic liver micrometastasis and macroscopic LN metastasis in the control (gray bars) and anti-VEGFR2-treated (black bars) treatment arms. Bottom: contingency table relating the number and percentage of animals in each treatment/metastasis case. Treated animals show a statistically significant increase in the incidence of liver micrometastasis and LN metastasis by the chi-square test (*p < 0.05; **p < 0.01).
(C) Quantification of the number of microscopic liver metastasis in the anti-VEGFR2 treated (black bars) and control (gray bars) treatment arms. Treated animals show a statistically significant increase in the number of liver micrometastases per animal by the Mann-Whitney test (*p < 0.05). Error bars indicate ± SEM.
We next investigated the possible occurrence of distant metastasis, focusing on the liver, which is a prevalent site for human pancreatic metastases. Microscopic liver metastases were detected by H&E staining and further confirmed by tumor-specific immunostaining for the T antigen oncoprotein, revealing microscopic tumor nodules in the liver parenchyma (Figure 3A). In this case, the incidence of animals with liver micrometastases was also 2-fold higher in the treated animals than in controls, with a relative risk of 2.0 [1.1-3.7] (95% confidence interval) (Figure 3B). Furthermore, the average number of metastatic lesions in the liver was also significantly increased (2.7-fold) in the treated animals compared to controls (Figure 3C). We infer that death due to primary tumor burden precluded the development of macrometastatic lesions.
A Multitargeted Antiangiogenic Kinase Inhibitor also Induces Increased Invasiveness
We further sought to determine whether these surprising adaptive responses occurred in the context of treatment with other angiogenesis inhibitors. We chose to investigate a more potent inhibitor of tumor angiogenesis, sunitinib (Sutent, Pfizer Inc.), which effectively inhibits both VEGFR and PDGFR signaling, a combination that we previously demonstrated to have potent efficacy and added benefit over singular inhibition of VEGFR, by targeting both endothelial cells and their supporting pericytes (Bergers et al., 2003; Pietras and Hanahan, 2005). Consistent with these results, continuous sunitinib treatment of tumor-bearing RIP1-Tag2 mice was markedly efficacious, producing a significant survival benefit (Figure 4A) and a 65% decrease in tumor burden after 5 weeks of treatment when compared to age-matched control animals (Figure 4B). Histological analysis of animals treated for 5 weeks starting at 10 weeks of age showed potent inhibition of angiogenesis and disruption of the preexisting tumor vasculature, with concomitant exacerbated invasive and more aggressive tumor phenotype (Figure 5A). Fluorescent immunostaining for T antigen further revealed widespread tumor infiltration, with apparent dissemination of tumor cell clusters far from the primary tumor masses (Figure 5A; Figure S3). Quantification of this increased invasiveness demonstrated that sunitinib treatment for 5 weeks more than doubled the incidence of highly invasive tumors (IC2) compared to control animals (Figure 5B). Notably, the tumors in sunitinib-treated mice, while graded as IC2, were often remarkable for the aggressive character of their invasiveness to a degree of malignancy rarely seen in untreated tumors (Figure S3).
Figure 4. Increased Life Span and Tumor Reduction in Sunitinib-Treated RIP1-Tag2 Animals.
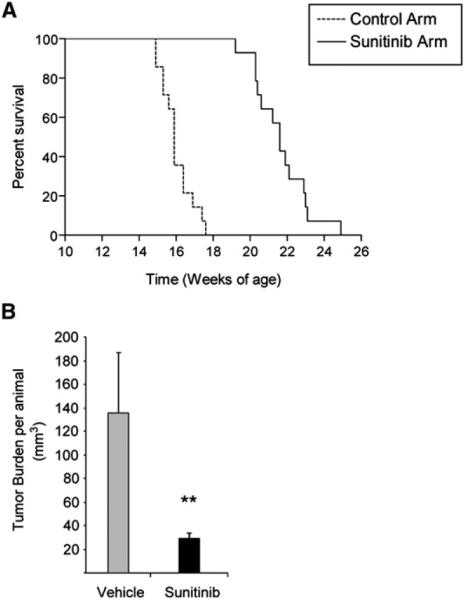
(A) Kaplan-Meier survival curves in tumor-bearing RIP1-Tag2 mice (12 weeks) treated continuously with vehicle control or sunitinib starting at 12 weeks. While vehicle-treated mice showed a median life span of 15.2 weeks, mice receiving continuous sunitinib treatment demonstrated a survival benefit of 7 additional weeks. (Kaplan-Meier statistic: p < 0.01.)
(B) Total tumor burden analysis in 5-week treatment trials with sunitinib or vehicle control starting at 10 weeks of age. Gray bar shows tumor burden of vehicle-treated RIP1-Tag2 animals, and black bar shows the statistically significant tumor volume reduction of efficacious sunitinib therapy. Each treatment cohort involved a minimum of ten animals per group. **p < 0.01 by Mann-Whitney test. Error bars indicate ± SEM.
Figure 5. Increased Tumor Invasion Evoked by Treatment with a Multitargeted Angiogenic Kinase Inhibitor.

Histological analysis and quantification of tumor invasion in RIP1-Tag2 mice treated for 5 weeks with sunitinib compared to control vehicle-treated mice are shown.
(A) Top row: H&E staining of control- and sunitinib-treated tumors showing the invasive front extensively intercalating into the surrounding tissue (dashed white line), producing isolated islands of normal exocrine tissue inside treated tumors (white arrow). Middle row: immunofluorescence staining for SV40 T antigen showing the widely invasive phenotype in treated tumors, with several fronts invading into the surrounding tissue. Bottom row: visualization of blood vessels by intravenous perfusion of FITC-conjugated tomato lectin (green), revealing the effect of the antiangiogenic treatment on the tumor vasculature.
(B)Quantification of the percentage of IT, IC1, and IC2 tumors in vehicle- versus sunitinib-treated animals. Sunitinib treatment shows statistically significant decrease in IT and increase in the highly invasive grade of tumor (IC2) by Mann-Whitney test (*p < 0.01; **p < 0.01). Error bars indicate ± SD.
(C) Left panels: histological analysis by H&E staining of tissue sections of lymph node and liver metastasis from sunitinib-treated animals. Right panels: confirmation of metastasis is shown by immunofluorescence staining for the tumor marker T antigen (red). The images give evidence of tumor cells infiltrated into the lymph node or the liver parenchyma.
(D) Top: quantification of the proportion of animals with liver and lymph node (LN) micrometastasis in the vehicle-treated (gray bars) and sunitinib-treated (black bars) arms. Bottom: contingency table listing the number and percentage of animals in each treatment/metastasis case. Treated animals show a 3.5-fold increase in the incidence of liver micrometastasis by the chi-square test (*p < 0.05).
(E) Quantification of the number of liver micrometastases in the vehicle-treated (gray bars) and sunitinib-treated (black bars) arms. The number of liver micro-metastases was 3.7-fold increased in the sunitinib-treated group, which showed a clear but not statistically significant trend compared to control animals. Error bars indicate ± SEM.
Increased Metastasis in Sunitinib-Treated PNET Tumors
To further investigate the adaptive response to potent angiogenesis inhibition with sunitinib, peripancreatic LNs were assessed for the presence of metastatic lesions by H&E and by T antigen immunostaining of tissue sections from treated animals (Figure 5C). Quantitative analysis revealed a similar incidence of animals with LN metastasis in the sunitinib-treated versus control vehicle-treated groups (Figure 5D). In contrast to the lack of effect on lymphatic metastasis in sunitinib-treated mice, hematogenous dissemination was prevalent and arguably more aggressive. H&E staining confirmed by immunostaining for the T antigen oncoprotein consistently showed more abundant and larger liver micrometastases in the sunitinib-treated animals than in controls (Figure 5C, bottom row). The incidence of liver micrometastases was significantly increased by 3.5-fold in treated animals (χ2, p < 0.05), with a relative risk of 3.5 (95% confidence interval 0.8-14.3) (Figure 5D). When quantified, the number of liver micrometastases per animal was also increased by 3.7-fold in the sunitinib-treated animal group, which showed a clear but not statistical significant tendency when compared with control animals (Figure 5E). Thus, pancreatic islet tumors treated with a multitargeted kinase inhibitor also adapt by increasing their metastatic phenotype.
VEGF Inhibitors also Evoke Increased Invasiveness in Orthotopic Glioblastoma
These provocative observations in a mouse model of PNET raised the question of generality to other tumor types. As a first step, we asked whether this adaptive response of heightened malignancy in response to genetic or pharmacological angiogenesis inhibition occurred in a second tumor type, glioblastoma multiforme (GBM), by utilizing an orthotopic mouse model of genetically engineered transformed mouse astrocytes (Blouw et al., 2003; Du et al., 2008) characterized by development of aggressive and locally invasive glioblastomas that phenocopy the major characteristics of human glioblastomas (Berger and Wilson, 1999). Thus, the effects of a VEGFR-selective kinase inhibitor (SU10944, Pfizer Inc.) or the multitarget VEGFR kinase inhibitor sunitinib were assessed along with GBM cells genetically deficient in Vegf-A gene expression (VEGF-KO GBMs) (Blouw et al., 2003). In all three situations in which VEGF activity was either pharmacologically or genetically impaired, the tumor vasculature appeared thinner when compared to that of control tumors (Figure 6A). The VEGF-KO GBMs produced a survival advantage, consistent with our previous report (Blouw et al., 2003), whereas the kinase inhibitors produced minimal (SU10944) or modest (sunitinib) effects on tumor growth and animal survival (Figure 6B), consistent with a clinical study wherein a VEGFR kinase inhibitor transiently normalized the tumor vessels and reduced side effects such as edema but had little impact on GBM growth or patient survival (Batchelor et al., 2007). The most notable consequence of all three conditions of impaired VEGFR signaling in our mouse model was alterations in the invasive phenotype of the GBM, in the form of a more highly invasive and qualitatively distinct tumor phenotype, with tumor cells dispersed throughout the brain in close proximity to resident normal blood vessels (perivascular tumor invasion), which contrasted with the less prominent single-cell infiltrative pattern that was typical in the control GBM tumors (Figure 6A). The relative prevalence of the perivascular invasive mode was assessed by counting the invading tumor cells in proximity to vessels and by grading the extent of perivascular invasion (which accounts for both single vascular-proximal tumor cells as well as focal clumps of tumor cells clustered around vessels). In all three cases, a significant increase in perivascular tumor cell invasion was observed, but the invasive effects were less pronounced in the case of both chemical VEGFR inhibitors (SU10944 and sunitinib) than in VEGF-KO GBM (Figure 6C). The differing degrees of invasion could be explained by the fact that VEGF-KO GBM cells were forced to grow from the time of their inoculation without a cancer cell-supplied source of VEGF, whereas WT GBM tumors were subjected to VEGF blockade only after having formed and grown to a substantive degree.
Figure 6. Effects of VEGFR-Selective Kinase Inhibitors on an Orthotopic Mouse Model of Glioblastoma Multiforme.

(A) Top row: immunohistological analysis of glioblastoma multiforme (GBM) with fluorescent SV40 T antigen staining (red) on whole brain sections counterstained with DAPI (blue). Control wild-type (WT) GBMs appear less invasive than WT GBMs treated with SU10944 or sunitinib, or VEGF-KO GBMs (purple arrowheads). Bottom row: high-magnification images of fluorescent detection of tumor cells (anti-T antigen antibody; red) and the perfused vasculature (FITC-lectin, green). WT GBM cells infiltrate as single cells into the brain parenchyma without associating with blood vessels (white arrowheads) or invade alongside blood vessels in the brain (perivascular invasion; yellow arrowheads). GBMs treated with either SU10944 or sunitinib as well as VEGF-KO GBMs are more invasive and predominantly migrate along blood vessels (yellow arrowheads).
(B) Kaplan-Meier survival curves of WT GBM-bearing mice untreated or treated with a relatively selective VEGFR inhibitor (SU10944) or a multitargeted VEGFR inhibitor (sunitinib) starting 3 days after inoculation. The effects on survival observed with SU10944 and sunitinib treatment were minimal or modest, respectively, and were not statistically significant.
(C) Top: grading of perivascular invasion was performed on a scale from 1 to 4, where 1 indicates minimal distant spread of tumor cells and 4 indicates substantial and marked distant spread; this parameter indicated a significant increase in perivascular invasion in response to both pharmacological treatments and in the VEGF-KO GBMs (**p < 0.01). Bottom: quantification of perivascular invasive mode by immunohistochemical analysis on tumor sections. Perivascular invasive cells, counted as the number of cells in the tumor periphery tightly associated with vessels, were significantly increased by both antiangiogenic small-molecule treatments and the genetic ablation of VEGF-KO GBM cells (**p < 0.01). Error bars indicate ± SD.
Thus, genetically or pharmacologically impaired VEGF signaling in an orthotopic model of GBM also evokes a more invasive and aggressive phenotype, distinct in form and yet analogous in essence to that observed in pancreatic neuroendocrine tumors subjected to similar genetic or pharmacological VEGF blockade.
Hypoxia Is Implicated in the Adaptive Response
To gain insight into one possible molecular mechanism for the increased invasion and subsequent dissemination, we sought to determine whether hypoxia developed in the treated tumors concomitant with invasion. While in control untreated animals, most of the tumors showed little or no hypoxia as revealed by pimonidazole adduct staining, both β-VEGF-KO RIP1-Tag2 animals and animals treated with anti-VEGFR2 antibody or sunitinib displayed multiple tumors with distinct regions of intense hypoxia (Figure 7A). Moreover, when the incidence of hypoxic pimonidazole-positive tumors was quantified, both anti-VEGFR2 and sunitinib treatments showed a significant increase in hypoxic tumors (Figure 7B). Furthermore, intense hypoxia was evident in the disseminated liver micrometastases of the sunitinib-treated animals together with lack of vascularity inside the metastatic lesions, while in control animals, the micrometastases were vascularized and showed no hypoxia (Figures 7Ag-7Ai). Thus, the hyperinvasive and metastatic lesions that develop in the PNET model treated with a potent angiogenesis inhibitor exhibit marked hypoxia, suggestive of a role for the hypoxia response system in their manifestation.
Figure 7. Antiangiogenic Treatment also Provokes Hypoxia in Tumors and Liver Micrometastases.

(A) Hypoxia in islet tumors was detected by immunofluorescence staining of pimonidazole adducts in sections of pancreas (Aa-Af) or liver (Ag-Ai) from control untreated animals (Aa and Ag), animals receiving short-term (Ab) or long-term (Ac) anti-VEGFR2 treatment, β-VEGF-WT (Ad) and β-VEGF-KO (Ae) islet tumors, and sunitinib-treated animals (Af, Ah, and Ai). (Aa)-(Ag) show pimonidazole immunodetection (green) with blood vessel CD31 staining (red); (Ah) shows pimonidazole immunodetection (green) and T antigen onco-protein (red); (Ai) shows blood vessel MECA32 staining (green) and T antigen oncoprotein (red). All images show nuclei counterstained with DAPI (blue).
(B) Quantitation of the incidence of hypoxic tumors was performed in long-term anti-VEGFR2-treated and sunitinib-treated animals and plotted as the percentage of pimonidazole-positive tumors per animal compared to control animals. **p < 0.01 by Mann-Whitney test. Error bars indicate ± SEM.
DISCUSSION
Proinvasive consequences of antiangiogenic therapy had been described previously as a collateral to the antitumor response in glioblastoma multiforme and were attributed to vessel co-option by cancer cells, but with uncertain significance for therapeutic efficacy (Kunkel et al., 2001; Rubenstein et al., 2000). Recently, heightened invasion has been further implicated as a response to antiangiogenic therapy in another model of orthotopic GBM (Gomez-Manzano et al., 2008), consistent with these previous studies and our observations. More recently, we reported that abrogation of VEGF signaling with a function-blocking antibody against VEGFR2 elicits, after a period of transitory response, a bimodal resistance reaction with evident revascularization and increased invasiveness; while the revascularization was demonstrably important in the development of resistance (Casanovas et al., 2005), the significance of the heightened invasiveness was unclear. Here we demonstrate in two distinct engineered mouse models of cancer (PNET and GBM) that therapeutically efficacious antiangiogenic therapy can elicit an adaptive-evasive response involving an augmented invasive phenotype and, in some cases, increased dissemination and the emergence of distant metastasis (Figure 8).
Figure 8. Adaptive-Evasive Responses by Tumors to Antiangiogenic Therapies.

Schematic summary of adaptive responses to VEGF/VEGFR inhibitors (and likely other angiogenesis inhibitors) that elicit “evasive resistance.” Tumors respond to VEGF/VEGFR pathway inhibition with tumor stasis or regression and a loss of blood vessels, but mechanisms of evasive resistance to the antiangiogenic treatment are then induced that can variously enable revascularization via alternative proangiogenic signals, increased local invasiveness, and/or enhanced distant metastasis.
Interestingly, PNET development in the context of genetic ablation of the Vegf-A gene produced small pancreatic lesions with increased invasiveness but without evident metastasis (data not shown), in contrast to the case of VEGF pathway inhibition. We suspect that the metastasis response is somehow enabled by the initial presence of an angiogenic vasculature in established solid tumors containing hyperproliferative cancer cells that, when disrupted, fosters metastatic dissemination. In contrast, the small avascular lesions populated by VEGF-KO cancer cells may have failed to acquire other necessary capabilities for productive dissemination. Regardless, the preclinical trials in the PNET model unambiguously demonstrate the enhancement of invasion and metastasis consequent to pharmacological angiogenesis inhibition.
In the PNET model, the multitargeted inhibitor sunitinib consistently demonstrated significantly better efficacy than the VEGFR2-specific antibody, in regard to both tumor shrinkage and augmented survival benefit. But in turn, sunitinib evidently elicited a more highly invasive adaptation, with some of the most aggressively invasive tumors observed heretofore in this model. Thus, the more effective the VEGF/angiogenesis inhibition, the more pronounced the adaptive progression to heightened and altered invasiveness. Interestingly, while both angiogenesis inhibitors enhanced liver metastasis, sunitinib did not increase lymphatic metastasis, in contrast to the anti-VEGFR2 antibody. This difference could be due to the differential specificity of inhibition by the two drugs: sunitinib potently blocks not only VEGFR2 and the PDGFRs but also lymphatic vessel-related VEGFR3 (Faivre et al., 2007; Roskoski, 2007). An attractive hypothesis is that the specific blockade of VEGFR3 signaling by sunitinib serves to alter the structure or permeability of lymphatic vessels or lymph nodes, thereby impeding formation of LN metastasis.
An intriguing paradox involving these results is raised by previous reports in other model systems, in which VEGF-driven angiogenesis was found to positively correlate with increased tumor invasiveness (Skobe et al., 1997) or metastasis (Warren et al., 1995). In contrast, our results as well as those presented in the accompanying study by Ebos et al. (2009) in this issue of Cancer Cell support the opposite conclusion, raising the possibility that both induction and suppression of tumor angiogenesis can exert proinvasive/prometastatic effects. We envision that the two effects, while related, may have distinct physiological bases. For example, tumors that express very high levels of VEGF (as in Skobe et al. and Warren et al.) may produce a tumor vasculature with excessive sprouting, poor pericyte coverage, and collectively impaired vascular integrity. Notably, Xian et al. (2006) demonstrated that vessels with poor pericyte coverage are prone to increased metastasis and that overexpression of VEGF can drive intense angiogenesis with low pericyte coverage and a hyperpermeable vasculature, which can be more permissive for tumor cell intravasation and dissemination. On the other hand, antiangiogenic therapy demonstrably disturbs the tumor vasculature and in some cases (e.g., with sunitinib) also disrupts pericyte coverage, and may in addition be indirectly affecting the tumor cells by inducing a more invasive phenotype in response to hypoxia, leading to increased intravasation and metastatic dissemination.
How might tumors become more invasive during antiangiogenic therapy? One possibility is that tumors may elevate the activity of a preexisting invasion program that was not previously the driving force of expansive tumor growth, given the capability for angiogenesis. Alternatively, some tumors may switch on an invasive growth program distinct from that arising spontaneously during unperturbed tumor development and progression, as is evidently the case for GBMs, in which antiangiogenic therapy induced a phenotypic change from single-cell infiltration to migration of cell clusters along normal blood vessels. These observations suggest that the invasive growth program induced in response to therapy may be qualitatively different than the pathway used in normal tumor progression.
In this regard, genetic studies have implicated the hypoxia/HIF-1α pathway as an instigator of invasion and metastasis (Du et al., 2008; Pennacchietti et al., 2003). Interestingly in the GBM model, hypoxia via consequent HIF-1 activation induced angiogenesis and more infiltrative tumor cell behavior, while inhibition of HIF-1 and consequent failure to respond to hypoxia led to a blockade of angiogenesis and an exacerbated proinvasive phenotype. Due to the degree of invasion and the close proximity of invading tumor cells to blood vessels, these tumors are less hypoxic (Blouw et al., 2003; Du et al., 2008), despite the implication that hypoxia in the primary tumor drove their switch to a hyperinvasive condition. In concordance with the GBM results, we document herein the concomitant triggering of hypoxia with increased invasion in response to antiangiogenic therapy in the PNET model. While the concurrent initiation of hypoxia and invasion could be a circumstantial coincidence, a body of published evidence linking hypoxia and invasion (Cairns et al., 2001; Pennacchietti et al., 2003; Young and Hill, 1990) suggests that the hypoxia response system could be involved in regulating invasion in this tumor type as well. Additionally, other therapy-induced mechanisms could contribute to the phenotypic progression of malignancy, such as activation or upregulation of extracellular proteases (e.g., matrix metalloproteinases and cathepsins), latent signaling circuits (e.g., c-Met), or triggering of epithelial-to-mesenchymal transition programs, each of which can demonstrably lead to more invasive and/or metastatic phenotypes and has been linked to the hypoxia response in particular model systems (Higgins et al., 2007; Munoz-Najar et al., 2006; Pennacchietti et al., 2003). The possible contributions of the hypoxia response system and other factors to adaptation and emergent resistance to angiogenesis inhibitors remain to be determined and are an important topic for future research.
Translational Considerations
It must be emphasized that the three VEGF pathway inhibitors currently approved for certain cancer types (variously including colorectal, renal, breast, and hepatocellular), and likely others (both new drugs and new indications) to follow, are markedly altering the standard of care for these cancers. Despite their transitory efficacy, angiogenesis inhibitors present important therapeutic options that constitute improvements in terms of both clinical benefit and reduced toxicity compared to conventional agents used to treat these diseases. Recognizing both their benefits and their limitations, a pertinent question at hand is that of the broader applicability of our findings to antiangiogenic therapies aimed at other organ-specific cancers, and in particular their extrapolation to the outcomes of antiangiogenic therapies in cancer patients. Notably, in the accompanying paper by Ebos et al. (2009), one of these approved angiogenesis inhibitors, sunitinib, is shown to facilitate metastatic dissemination of both human breast cancer cells and syngeneic melanoma in mice following either orthotopic or intravenous inoculation; a second approved drug, sorafenib, evidently has similar effects. Even brief week-long antiangiogenic treatment elicited increased metastasis, much as we observed following a similar week-long treatment with the anti-VEGFR2 antibody in the PNET model. Collectively, the two studies document increased invasion and/or metastasis in four distinct mouse models of organ-specific cancer and begin to establish a mechanistic pattern of increased invasion and metastasis as an adaptive response to antiangiogenic therapy, one that seems likely to be operative in certain human tumors.
In regard to the relevance of our findings in mouse models of neuroendocrine tumors and glioblastoma to antiangiogenic therapies for the cognate human cancers, there are currently insufficient data about the responses of human pancreatic neuroendocrine tumors due to both their rarity and the limited number of patients that have been treated in clinical trials with angiogenesis inhibitors. In the case of glioblastoma, there is growing experience with antiangiogenic therapies, specifically the VEGF ligand-trapping antibody bevacizumab (Fischer et al., 2008; Narayana et al., 2009; Norden et al., 2008) and the VEGFR1/2/3 kinase inhibitor AZD2171 (Batchelor et al., 2007). In both cases, there seems to be a proinvasive adaptation to antiangiogenic therapy, as suggested by magnetic resonance imaging in a subset of GBM patients that developed multifocal recurrence of tumors during the course of therapy with anti-VEGF (bevacizumab) (Fischer et al., 2008; Narayana et al., 2009; Norden et al., 2008) or AZD2171 (Batchelor et al., 2007). Much as in the mouse GBM model, we infer that the human glioblastoma cells could be co-opting blood vessels by invading along them into the surrounding normal brain tissue, thereby achieving vascular/nutrient/oxygen sufficiency while consequently promoting an invasive dispersion phenotype (perivascular tumor invasion). Thus, there is reason to predict clinical relevance of our collective observations.
How then could the improvement of survival in many clinical trials in spite of increased invasiveness and metastasis be explained? The increased progression-free survival (PFS) could likely reflect the impairment of primary tumor growth, which, while initially efficacious, is short lived, with the onset of multiple forms of evasive resistance that would inevitably produce a return to progressive disease. This tumor relapse would compromise this initial benefit such that the prolongation of PFS and even overall survival (OS) would not be particularly robust, as is the case in many clinical trials. For example, recent results from two randomized clinical trials with bevacizumab and taxanes for the treatment of metastatic breast cancer (the E2100 study and the AVADO study) demonstrated small benefits in PFS but not an OS benefit, and our results provide a possible explanation for this disconnect.
Indeed, this undesirable therapy-triggered change in the natural history of the treated tumors is convergent with emerging examples involving other targeted therapy-driven changes, such as the case of trastuzumab (Herceptin) therapy in HER2-positive breast cancer patients. Although effective in its antitumor and prosurvival effects (13 months increased life span), trastuzumab therapy changes the course of disease progression in these patients to one with a markedly increased incidence of brain metastasis (Bendell et al., 2003; Clayton et al., 2004; Miller et al., 2003; Weil et al., 2005), which is otherwise rare in untreated patients. The companion study by Ebos et al. (2009) documents increased metastasis in multiple organs, including the brain, in response to the angiogenesis inhibitor sunitinib in a mouse model of breast cancer, illustrating that convergence.
Another clinically relevant question raised by our results is whether concomitant chemotherapy in some tumor types (e.g., in all currently approved indications for bevacizumab) could alter or even abrogate this increased malignant phenotype. Both preclinical and clinical trials investigating the effects of standard-of-care and more recent (e.g., metronomic) chemotherapy regimens on proinvasive and prometastatic responses to anti-angiogenic therapies are warranted.
Certainly, the characteristics and consequences of the exacerbated invasive-metastatic phenotype that develops in response to treatment with angiogenesis inhibitors motivate critical assessment in the clinical setting. Moreover, further preclinical studies are warranted to elucidate the mechanisms of this adaptive-evasive resistance, so as to design and test the potential of mechanism-based combination therapies aimed at impeding this insidious consequence of singular antiangiogenic therapy.
EXPERIMENTAL PROCEDURES
Animal Models
RIP1-Tag2 (Hanahan, 1985), immunocompromised RIP1-Tag2;Rag1-KO (Casanovas et al., 2005), and RIP1-Tag2/Cre;Vegf-Afl/fl mice (Inoue et al., 2002) have been described previously. The orthotopic mouse GBM models utilizing intracranial injection of transformed mouse astrocytes of wild-type or Vegf-A-loxP mice have also been described previously (Blouw et al., 2003; Du et al., 2008). Animal housing, handling, and all procedures involving mice were approved by the University of California, San Francisco (UCSF), Catalan Institute of Oncology, and Osaka Medical Center for Cancer and Cardiovascular Diseases institutional committees that approve and oversee research involving vertebrate animals, and all experiments were performed according to each country's guidelines governing animal care in the USA, Spain, and Japan, respectively.
Therapeutic Trials
RIP1-Tag2 mice were treated starting at 10 or 12 weeks of age as indicated, and immunocompromised RIP1-Tag2;Rag1-KO mice were used for long-term DC101 treatment to avoid immune reaction against the therapeutic antibody. The following antiangiogenic regimens were used: (1) 1 mg/mouse twice per week of anti-VEGFR2 blocking antibody (DC101) purified from a hybridoma culture (American Type Culture Collection) and its control purified rat IgG (Jackson ImmunoResearch) as described previously (Casanovas et al., 2005) administered intraperitoneally (i.p.). (2) 40 mg/kg/day sunitinib L-malate (Sutent, Pfizer Inc.) and its control carboxymethylcellulose vehicle formulation, administered daily by oral gavage for 5 weeks. Tumor volume in the RIP1-Tag2 animals was measured as described previously (Inoue et al., 2002). For therapeutic trials in the GBM model, FVB/N Rag1-KO mice were injected intracranially with 25,000 cells and treated with 150 mg/kg/day SU10944 or 40 mg/kg/day sunitinib L-malate administered by oral gavage starting 3 days after inoculation until moribund (average 13.5 days in both groups).
Histopathological Analyses
Pancreata and livers were either OCT embedded and frozen or fixed in buffered formalin and embedded in paraffin. Antibodies used for specific tissue immunostaining included rat monoclonal anti-mouse CD31 (clone MEC13.3, 1:50, BD Pharmingen), rabbit polyclonal serum anti-large T antigen (1:10,000, prepared in the Hanahan laboratory), polyclonal guinea pig anti-insulin (A0564, DAKO), and rat anti-MECA32 (550563, BD Pharmingen). For immunohistochemistry, the EnVision system of labeled polymer-HRP anti-rabbit IgG (Dako-Cytomation) was used as a secondary antibody. For immunofluorescence detection, Alexa Fluor 488 goat anti-rat IgG and Alexa Fluor 546 goat anti-rabbit IgG secondary antibodies were used at a dilution of 1:200. For nuclear counterstaining, tissue sections were either mounted with Vectashield with DAPI (Vector Laboratories) or stained with TO-PRO-3 iodide used at a 1:1000 dilution and then mounted with Vectashield without DAPI.
Systemic perfusion of fluoresceinated lectin to visualize the functional blood vasculature was performed as described previously (Inoue et al., 2002). Briefly, 0.1 mg of fluorescein-labeled tomato lectin (Vector Laboratories) was injected intravenously in a total volume of 0.1 ml and allowed to circulate for 5-10 min, followed by heart perfusion with neutral buffered formalin and tissue sample preparation for OCT-embedded frozen sectioning.
For microvessel area (MVA) quantification, the ratio of CD31-stained area to total area was calculated in ten randomly chosen circular spots (50 μm diameter) using Lumina Vision image analysis software (Mitani Corporation). The MVA of the region was expressed as the average ratio of these ten spots.
Hypoxia detection was performed 1 hr after i.p. injection of 60 mg/kg pimonidazole using a Hypoxyprobe Plus kit (Natural Pharmacia International Inc.) following the manufacturer's instructions.
Invasion and Metastasis Analysis
Invasion grading in the RIP1-Tag2 model was performed as described previously (Lopez and Hanahan, 2002) from five H&E-stained sections per animal from a minimum of five mice per treatment group. The incidence of LN/liver metastasis was determined by scoring for presence or absence in each animal and counting the number of liver micrometastases in five sections per animal from a minimum of ten animals per treatment group. In the GBM model, perivascular invasion was quantified by counting the number of invasive cells/clusters associated with a vessel at the tumor edge (20x field) in double-stained frozen sections with large T antigen and CD31 from 3-10 sections per 2-4 different tumor samples per group. Perivascular invasion grading of tumors was determined by staining tumor cells with an antibody for SV40 large T antigen on whole-brain sections, and 5-8 tumors from each group were graded from 1 to 4, where 1 indicates minimal distant spread of tumor cells and 4 indicates substantial and marked distant spread.
Statistical Analysis
All results were evaluated using the SPSS statistical software package. Due to the small sample size in each analysis and the fact that not all of the data were normally distributed, nonparametric statistical tests were used in each case (Kruskal-Wallis, Mann-Whitney, or chi-square test).
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge J.G. Christensen of Pfizer for advice and encouragement; Pfizer for generous gifts of SU10944 and sunitinib; and X. Hernando (Casanovas laboratory), C. Guinto and E. Drori (Hanahan laboratory), and K. Lu and B. Kaplan (Bergers laboratory) for excellent technical support. This work was funded by research grants SAF2006-00590, SAF2007-60955, RTICC-RD2006-0092, and SGR727 from MICINN and AGAUR (Spain); grants from the US National Cancer Institute to G.B. and D.H.; a grant from the William K. Bowes, Jr. Foundation to D.H.; and a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science to M.I. D.H. is a American Cancer Society Research Professor, O.C. is a Ramon y Cajal fellow of the Spanish Ministry of Education, G.B. holds the Neill H. and Linda S. Brownstein Chair in Brain Tumor Research at UCSF, and M.P.-R. is a recipient of an IDIBELL fellowship.
D.H. serves on a strategic advisory panel for oncology at Pfizer Inc.
Footnotes
SUPPLEMENTAL DATA The Supplemental Data include three figures and can be found with this article online at http://www.cancercell.org/supplemental/S1535-6108(09)00034-8.
REFERENCES
- Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendell JC, Domchek SM, Burstein HJ, Harris L, Younger J, Kuter I, Bunnell C, Rue M, Gelman R, Winer E. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer. 2003;97:2972–2977. doi: 10.1002/cncr.11436. [DOI] [PubMed] [Google Scholar]
- Berger MS, Wilson CB. The Gliomas. W.B. Saunders Company; Philadelphia: 1999. [Google Scholar]
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, Johnson RS, Bergers G. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 2003;4:133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- Blouw B, Haase VH, Song H, Bergers G, Johnson RS. Loss of vascular endothelial growth factor expression reduces vascularization, but not growth, of tumors lacking the Von Hippel-Lindau tumor suppressor gene. Oncogene. 2007;26:4531–4540. doi: 10.1038/sj.onc.1210249. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Kalliomaki T, Hill RP. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001;61:8903–8908. [PubMed] [Google Scholar]
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Clayton AJ, Danson S, Jolly S, Ryder WD, Burt PA, Stewart AL, Wilkinson PM, Welch RS, Magee B, Wilson G, et al. Incidence of cerebral metastases in patients treated with trastuzumab for metastatic breast cancer. Br. J. Cancer. 2004;91:639–643. doi: 10.1038/sj.bjc.6601970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos JML, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 2007;6:734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- Fischer I, Cunliffe C, Bollo R, Raza S, Monoky D, Chiriboga L, Parker E, Golfinos J, Kelly P, Knopp E, et al. High-grade glioma before and after treatment with radiation and Avastin: initial observations. Neuro-oncol. 2008;10:700–708. doi: 10.1215/15228517-2008-042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- Folkman J, Ingber D. Inhibition of angiogenesis. Semin. Cancer Biol. 1992;3:89–96. [PubMed] [Google Scholar]
- Gomez-Manzano C, Holash J, Fueyo J, Xu J, Conrad CA, Aldape KD, de Groot JF, Bekele BN, Yung WK. VEGF Trap induces anti-glioma effect at different stages of disease. Neuro Oncol. 2008;10:940–945. doi: 10.1215/15228517-2008-061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Hager JH, Ferrara N, Gerber HP, Hanahan D. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell. 2002;1:193–202. doi: 10.1016/s1535-6108(02)00031-4. [DOI] [PubMed] [Google Scholar]
- Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis. 2000;21:505–515. doi: 10.1093/carcin/21.3.505. [DOI] [PubMed] [Google Scholar]
- Kerbel RS, Yu J, Tran J, Man S, Viloria-Petit A, Klement G, Coomber BL, Rak J. Possible mechanisms of acquired resistance to anti-angiogenic drugs: implications for the use of combination therapy approaches. Cancer Metastasis Rev. 2001;20:79–86. doi: 10.1023/a:1013172910858. [DOI] [PubMed] [Google Scholar]
- Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, Westphal M, Lamszus K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61:6624–6628. [PubMed] [Google Scholar]
- Lopez T, Hanahan D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumor-igenesis. Cancer Cell. 2002;1:339–353. doi: 10.1016/s1535-6108(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Miller KD, Weathers T, Haney LG, Timmerman R, Dickler M, Shen J, Sledge GW., Jr. Occult central nervous system involvement in patients with metastatic breast cancer: prevalence, predictive factors and impact on overall survival. Ann. Oncol. 2003;14:1072–1077. doi: 10.1093/annonc/mdg300. [DOI] [PubMed] [Google Scholar]
- Miller KD, Sweeney CJ, Sledge GW., Jr. Can tumor angiogenesis be inhibited without resistance? EXS. 2005;94:95–112. doi: 10.1007/3-7643-7311-3_7. [DOI] [PubMed] [Google Scholar]
- Munoz-Najar UM, Neurath KM, Vumbaca F, Claffey KP. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene. 2006;25:2379–2392. doi: 10.1038/sj.onc.1209273. [DOI] [PubMed] [Google Scholar]
- Narayana A, Kelly P, Golfinos J, Parker E, Johnson G, Knopp E, Zagzag D, Fischer I, Raza S, Medabalmi P, et al. Antiangiogenic therapy using bevacizumab in recurrent high-grade glioma: impact on local control and patient survival. J. Neurosurg. 2009;110:173–180. doi: 10.3171/2008.4.17492. [DOI] [PubMed] [Google Scholar]
- Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R, Ross GL, Ciampa AS, Ebbeling LG, Levy B, Drappatz J, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779–787. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- Offerhaus GJ, Giardiello FM, Bruijn JA, Stijnen T, Molyvas EN, Fleuren GJ. The value of immunohistochemistry for collagen IV expression in colorectal carcinomas. Cancer. 1991;67:99–105. doi: 10.1002/1097-0142(19910101)67:1<99::aid-cncr2820670119>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Parangi S, O'Reilly M, Christofori G, Holmgren L, Grosfeld J, Folkman J, Hanahan D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc. Natl. Acad. Sci. USA. 1996;93:2002–2007. doi: 10.1073/pnas.93.5.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Pietras K, Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J. Clin. Oncol. 2005;23:939–952. doi: 10.1200/JCO.2005.07.093. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 2007;356:323–328. doi: 10.1016/j.bbrc.2007.02.156. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306–314. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked Y, Bertolini F, Man S, Rogers MS, Cervi D, Foutz T, Rawn K, Voskas D, Dumont DJ, Ben-David Y, et al. Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis; Implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell. 2005;7:101–111. doi: 10.1016/j.ccr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Skobe M, Rockwell P, Goldstein N, Vosseler S, Fusenig NE. Halting angiogenesis suppresses carcinoma cell invasion. Nat. Med. 1997;3:1222–1227. doi: 10.1038/nm1197-1222. [DOI] [PubMed] [Google Scholar]
- Waggoner SE. Cervical cancer. Lancet. 2003;361:2217–2225. doi: 10.1016/S0140-6736(03)13778-6. [DOI] [PubMed] [Google Scholar]
- Warren RS, Yuan H, Matli MR, Gillett NA, Ferrara N. Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis. J. Clin. Invest. 1995;95:1789–1797. doi: 10.1172/JCI117857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS. Breast cancer metastasis to the central nervous system. Am. J. Pathol. 2005;167:913–920. doi: 10.1016/S0002-9440(10)61180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian X, Hakansson J, Stahlberg A, Lindblom P, Betsholtz C, Gerhardt H, Semb H. Pericytes limit tumor cell metastasis. J. Clin. Invest. 2006;116:642–651. doi: 10.1172/JCI25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SD, Hill RP. Effects of reoxygenation on cells from hypoxic regions of solid tumors: anticancer drug sensitivity and metastatic potential. J. Natl. Cancer Inst. 1990;82:371–380. doi: 10.1093/jnci/82.5.371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.