

Abstract
We investigated the frequency and function of mutations and increased copy number of the PIK3CA gene in lung cancers. PIK3CA mutations are one of the most common gene changes present in human cancers. We analyzed the mutational status of exons 9 and 20 and gene copy number of PIK3CA using 86 non–small cell lung cancer (NSCLC) cell lines, 43 small cell lung cancer (SCLC) cell lines, 3 extrapulmonary small cell cancer (ExPuSC) cell lines, and 691 resected NSCLC tumors and studied the relationship between PIK3CA alterations and mutational status of epidermal growth factor receptor (EGFR) signaling pathway genes (EGFR, KRAS, HER2, and BRAF). We also determined PIK3CA expression and activity and correlated the findings with effects on cell growth. We identified mutations in 4.7% of NSCLC cell lines and 1.6% of tumors of all major histologic types. Mutations in cell lines of small cell origin were limited to two ExPuSC cell lines. PIK3CA copy number gains were more frequent in squamous cell carcinoma (33.1%) than in adenocarcinoma (6.2%) or SCLC lines (4.7%). Mutational status of PIK3CA was not mutually exclusive to EGFR or KRAS. PIK3CA alterations were associated with increased phosphatidylinositol 3-kinase activity and phosphorylated Akt expression. RNA interference–mediated knockdown of PIK3CA inhibited colony formation of cell lines with PIK3CA mutations or gains but was not effective in PIK3CA wild-type cells. PIK3CA mutations or gains are present in a subset of lung cancers and are of functional importance.
Introduction
Most, if not all, of human malignancies are caused by somatic alterations within the cancer genome, leading to activation of oncogenes or inactivation of tumor suppressor genes, and many of the resultant changes target cell signaling pathways (1). Because of advances in sequencing technology, it is possible to perform exon resequencing for gene families involved in cellular signaling pathways, such as tyrosine kinases, tyrosine phosphatases, and phosphatidylinositol 3-kinases (PI3K; ref. 2). PI3Ks are heterodimeric lipid kinases composed of catalytic and regulatory subunits encoded by separate genes and subject to alternative splicing (3). The resultant proteins are critical regulators of growth, survival, and motility. PI3Ks are divided into three classes (I–III) according to their substrate preference and sequence homology (4). To date, class IA PI3Kα, consisting of catalytic subunit p110α and regulatory subunit p85α, is the only PI3K molecule found to have somatic mutations in human cancers; these occur predominantly in helical or kinase domains of its catalytic subunit encoded by the PIK3CA gene. Mutations of PIK3CA occur in many human epithelial cancers, resulting in PIK3CA being one of the two most commonly mutated oncogenes (along with KRAS) identified in human cancers (3, 5). However, individual types of epithelial cancers show great variability in their mutational rates, with high rates present in glioblastomas, gastric, hepatocellular, and breast cancers (5), whereas the rates described in non–small cell lung cancers (NSCLC) are relatively low (6, 7). To date, no information is available about small cell lung cancers (SCLC).
In addition to mutations, increased chromosomal copy number (by amplification or polysomy) is another method of oncogene activation (2). In general, increased copy number is frequently associated with increased mRNA expression (8, 9). A region of chromosome 3q (3q25-27), where PIK3CA (3q26) is located, is frequently amplified in lung cancers (10), especially squamous cell carcinomas (11, 12). However, the relationship between mutations and amplification of PIK3CA has not been studied comprehensively. Also, the functional effects of mutant or amplified PIK3CA in lung cancers are still unclear.
The PI3K/Akt pathway lies downstream of certain receptor tyrosine kinases, including epidermal growth factor receptor (EGFR). In addition to PIK3CA, other components of the pathway, including loss of the inhibitor PTEN or activating mutations of AKT, occur in certain cancers (13). In lung cancers, mutations have also been reported in several other genes involved in the EGFR signaling pathway, including EGFR, KRAS, HER2, and BRAF (14–16). As activation of EGFR family signaling is a mechanism to activate class IA PI3K (either directly or through RAS), we investigated the relationship between mutations of these genes and PIK3CA alterations.
As relatively little is known about the role of PI3K deregulation in lung cancer, we documented the frequency of mutations and amplifications in NSCLC tumors from four countries and in NSCLC and SCLC cell lines. We also studied the functional effects on PI3K deregulation in NSCLC cell lines.
Materials and Methods
Cell lines
We studied 86 NSCLC and 43 SCLC cell lines and three extrapulmonary small cell cancers (ExPuSC; ref. 17), most of which were established by the authors (18). We also investigated eight immortalized human bronchial epithelial cell lines (HBEC; refs. 19, 20). NCI-H3255 was obtained from Dr. Bruce Johnson (21). PC-9 (originally from Tokyo Medical University) was obtained from Dr. Bert Vogelstein. Calu-3 was purchased from the American Type Culture Collection.
Tumor samples
We previously reported on the mutational status of EGFR, KRAS, and HER2 in NSCLC (14, 15). Of the 617 samples used in that study, DNAs from 591 tumors were available for PIK3CA and BRAF mutational studies. In this study, we included a further 100 samples obtained from Okayama University. Thus, the total number of tumor samples available for mutational analyses of the five genes was 691. These tumors were from patients undergoing surgical resection from Japan (n = 323), Taiwan (n = 148), the United States (n = 150), or Australia (n = 70). Sufficient DNA was available from 356 of these tumors for PIK3CA gene copy number analysis. Corresponding nonmalignant adjacent tissues (n = 267) were also available. Institutional Review Board permission and informed consent were obtained at each collection site.
For array comparative genomic hybridization (CGH) analysis, microdissected DNA was available from a separate set of 40 fresh-frozen NSCLC obtained from Vancouver General Hospital.
DNA and RNA extraction
Genomic DNA was obtained from primary tumors and cell lines by standard phenol-chloroform (1:1) extraction followed by ethanol precipitation or by using DNeasy Tissue kit (QIAGEN). Total RNA was obtained from cell lines using RNeasy Plus Mini kit (QIAGEN).
Gene mutational analyses
The intron- based PCR primer sequences for PIK3CA (exons 9 and 20) with PCR product lengths of 273 and 359 bp, respectively, and PCR conditions are provided in Supplementary Data. Primers were designed to avoid amplification of a known PIK3CA pseudogene (22). All sequence variants were confirmed by independent PCR amplifications and sequenced in both directions. With one exception (U1171), all of the mutant cases had corresponding nonmalignant tissue DNA available and were of somatic origin.
BRAF mutational status (exons 11 and 15) was determined, as described in Supplementary Data.
PIK3CA copy number evaluation by real-time quantitative PCR
We studied PIK3CA copy number in all of the cell lines, 356 NSCLC tumors, and 267 corresponding nonmalignant adjacent tissues using real-time quantitative PCR, as described in Supplementary Data. Samples were analyzed in triplicate. Each amplification reaction was checked for the absence of nonspecific PCR products by melting curve analysis. PIK3CA copy number calculation was carried out using the comparative Ct method (23) after validating that the efficiencies of PCR reactions of both PIK3CA and COL8A1 were equal. Human genomic DNA (EMD Biosciences), which is made from a mixture of pooled human whole blood from six to eight individual male and female donors, was run in every assay as a calibrator sample. PIK3CA gene copy number in normal human genomic DNA was set as 2 and copy number of more than 4 in cell lines was considered as increased. For clinical samples, we reduced the cutoff value from 4 to 3 because contamination with nonmalignant cells is invariably present (estimated average per tumor = 50% tumor cells, 50% nonmalignant cells).
Tiling path array CGH
Array CGH was performed as previously described (24). Further details are provided in Supplementary Data.
PIK3CA mRNA expression
We analyzed PIK3CA mRNA expression levels in lung cancer cell lines as a part of RT2 Profiler Custom PCR Array (SuperArray Bioscience). After making cDNA from 1.0 μg total RNA using RT2 PCR Array First Strand kit (SuperArray Bioscience), quantitative PCR was performed with the Chromo4 PCR System (Bio-Rad Laboratories) using RT2 Real-Time SYBR Green PCR Master Mix (SuperArray Bioscience) according to the manufacturer's protocol. We chose three different housekeeping genes, β-actin (ACTB), glyceraldehyde-3-phosphate dehydrogenase, and hypoxanthine phosphoribosyltransferase 1, as internal controls, and the averages of their Ct values were used. We also analyzed the values of six HBECs (HBEC3KT, HBEC4KT, HBEC5KT, HBEC17KT, HBEC30KT, and HBEC34KT) for PIK3CA mRNA expression, and the tumor cell values were expressed relative to the mean of the six HBECs. PIK3CA mRNA relative value for each HBEC cell line was also calculated by comparison to the average value of the six HBECs. For data analysis, the comparative Ct method (23) was used.
PI3K activity assay
PI3K activity of lung cancer cell lines was measured using an ELISA kit (Echelon Biosciences)11 according to the manufacturer's protocol. PIP3 produced by one of PIK3CA wild-type cell line with normal copy number (NCI-H1299) was set as 100 (control), and others were compared with the control and expressed as relative values.
Western blot analysis
Preparation of total cell lysates and Western blotting were done as described previously (20). Primary antibodies used were rabbit polyclonal anti-PI3K p110α (Cell Signaling), rabbit polyclonal anti-Akt (Cell Signaling), rabbit polyclonal anti–phosphorylated Akt (pAkt; Ser473; Cell Signaling), rabbit polyclonal anti-PTEN (Cell Signaling), rabbit polyclonal anti–phosphorylated PTEN (Ser380; Cell Signaling), and mouse monoclonal anti-actin (Sigma-Aldrich) antibodies. Actin levels were used as a control for protein loading. Peroxidase-labeled antirabbit or antimouse antibodies (Amersham Pharmacia) were used as the second antibody.
RNA interference studies
RNA interference of PIK3CA was performed by two methods, using either small interfering RNA (siRNA) or small hairpin RNA (shRNA). For each technique, two individual sequences targeting different regions of the gene were used as described below.
Preparation and transfection of siRNAs
siRNAs targeting PIK3CA were designed through the RNAi Co. Ltd. website12 based on siDirect online software system (25), which can efficiently select siRNA sequences avoiding off-target effects. siRNA sequences targeting two different regions of PIK3CA are provided in Supplementary Data. The siRNA sequences against Tax (the human leukemia virus gene) were reported previously (26). The siRNAs were chemically synthesized by Dharmacon. Cells were transfected with 5 nmol/L of siRNA using HiPerFect Transfection Reagent (QIAGEN) according to the manufacturer's protocol. Control cells were treated with HiPerFect alone or with Tax siRNA. Cells were grown and harvested 72 h after the transfection for additional analyses.
Preparation and transfection of shRNAs
HuSH 29mer shRNA constructs against PIK3CA (TR310428) gene-specific shRNA expression plasmids, along with control shRNA plasmids, including the original pRS vector (TR20003), were purchased from OriGene. We used the following shRNAs: pRSPIK3CA_1 (TI341705) and pRSPIK3CA_3 (TI341707). To produce viral-containing medium, 293T cells were transiently transfected with shRNA vectors together with pVpack-VSVG and pVpack-GP vectors (Stratagene). Transfection assays were performed as described previously (20). Two days after incubation, drug selection was initiated with 2 μg/mL of puromycin.
Colony formation assays
The in vitro growth characteristics were tested by colony formation assays. siRNA-transfected cells were harvested 72 h after the transfection and colony formation assays were performed. For shRNA experiments, shRNA-transfected cells were selected appropriately by antibiotics, and the colony formation assays were performed. For liquid colony formation assays, 500 viable cells were plated onto six-well plates in triplicate. Cells were cultured in RPMI 1640 supplemented with 5% serum, and colonies were counted 14 d later after staining with 0.5% methylene blue. For soft agar growth assays, 2,000 viable cells were suspended and plated in 0.33% agar in appropriate medium with 10% fetal bovine serum and layered over a 0.50% agar base in six-well plates in triplicate. After 2 wk, the number of microscopically visible colonies (>50 cells) was counted.
Statistical analyses
All statistical analyses in this study were performed using GraphPad Prism 4 software (GraphPad Software).
Results
PIK3CA mutations in lung cancer cell lines and tumors
We analyzed PIK3CA mutations in exons 9 and 20 in lung cancer cell lines and tumors (Supplementary Fig. S1; Tables 1 and 2) because 80% of the reported mutations of PIK3CA are clustered in these two exons (27). We identified PIK3CA mutations in NSCLC cell lines (4 of 86, 4.7%) and tumors (11 of 691, 1.6%; Table 1). PIK3CA mutations were distributed among all the major histologic subtypes (Table 1 and Supplementary Tables S1 and S2). Mutations in cancer cell lines of small cell origin were limited to two of three ExPuSC cell lines (Table 1 and Supplementary Table S2). All of these mutations were missense mutations. One primary tumor (U1171) had two missense mutations and one of these (C1622T, S541F) was a novel sequence variant (Supplementary Fig. S1). Whereas mutations at M1043 position have been previously described (27), an A3127C (M1043L) mutation, detected in tumor J-OK398, was also a novel sequence variant (Supplementary Fig. S1). The mutations in NSCLC were more frequent in exon 9 (3 of 4 in cell lines and 7 of 11 in tumors) than in exon 20, whereas both mutations in ExPuSC cell lines were in exon 20.
Table 1.
Summary of PIK3CA mutations in lung cancers
| Exon | Amino acid change | Functional domain | NSCLC cell lines (n = 86) | NSCLC tumors (n = 691) | SCLC cell lines (n = 43) | ExPuSC cell lines (n = 3) | Total no. mutations |
|---|---|---|---|---|---|---|---|
| 9 | S541F | Helical | 0 | 1* | 0 | 0 | 1 |
| 9 | E542K | Helical | 0 | 2* | 0 | 0 | 2 |
| 9 | E545K | Helical | 2 | 5 | 0 | 0 | 7 |
| 9 | Q546K | Helical | 1 | 0 | 0 | 0 | 1 |
| 20 | M1043L | Kinase | 0 | 1 | 0 | 0 | 1 |
| 20 | H1047R | Kinase | 1 | 1 | 0 | 1 | 3 |
| 20 | H1047L | Kinase | 0 | 2 | 0 | 1 | 3 |
| Total cell lines/tumors (%) | 4 (4.7%) | 11* (1.6%) | 0 (0%) | 2 (66.7%) |
One NSCLC tumor had both S541F and E542K mutations. For histologic subtype data of cell lines, see Table 2. Mutations in the 691 NSCLC tumors occurred in the following histologic subtypes: 5 of 249 (2.0%) squamous cell tumors, 5 of 400 (1.3%) adenocarcinomas, and 1 of 42 (2.4%) other NSCLC tumors.
Table 2.
PIK3CA mutation or copy number gain in lung cancer cell lines and tumors
| Samples | No. samples with the following factor in PIK3CA | |||
|---|---|---|---|---|
| Mutations | Copy number gain | Both mutations and gain | Either mutations or gain | |
| NSCLC cell lines (n = 86) | 4 (4.7%) | 8 (9.3%) | 1 (1.2%) | 11 (12.8%) |
| Squamous cell lines (n = 14) | 1 (7.1%) | 5 (35.7%) | 1 (7.1%) | 5 (35.7%) |
| Adenocarcinoma cell lines (n = 51) | 1 (2.0%) | 3 (5.9%) | 0 (0%) | 4 (7.8%) |
| Other NSCLC cell lines (n = 21) | 2 (9.5%) | 0 (0%) | 0 (0%) | 2 (9.5%) |
| NSCLC tumors (n = 356) | 11 (3.1%)* | 61 (17.1%) | 4(1.1%) | 68 (19.1%) |
| Squamous cell tumors (n = 139) | 5 (3.6%) | 46 (33.1%) | 3 (2.2%) | 48 (34.5%) |
| Adenocarcinomas (n = 195) | 5 (2.6%) | 12 (6.2%) | 0 (0%) | 17 (8.7%) |
| Other NSCLC tumors (n = 22) | 1 (4.5%) | 3 (13.6%) | 1 (4.5%) | 3 (13.6%) |
| SCLC cell lines (n = 43) | 0 (0%) | 2 (4.7%) | 0 (0%) | 2 (4.7%) |
| ExPuSC cell lines (n = 3) | 2 (66.7%) | 0 (0%) | 0 (0%) | 2 (66.7%) |
PIK3CA mutations were detected in 11 of 691 NSCLC tumors (1.6%), as shown in Table 1. However, as only 356 of 691 tumors were analyzed for both mutations and gene copy number of PIK3CA (all 11 mutant tumors were represented in this subset), the frequency of mutations in this subset increased to 3.1%.
The frequencies of mutations in these four genes in 86 NSCLC cell lines were as follows: EGFR (n = 10, 11.6%), KRAS (n = 26, 30.2%), HER2 (n = 1, 1.2%), BRAF (n = 5, 5.8%). There were no mutations in these four genes in 35 SCLC cell lines. We confirmed that the mutational status of EGFR, KRAS, HER2, and BRAF in NSCLC tumors and cell lines were mutually exclusive. However, we found that one cell line and three clinical samples with PIK3CA mutations also had KRAS or EGFR mutations (Supplementary Table S3). Thus, whereas mutations in the other EGFR pathway genes (EGFR, KRAS, HER2, and BRAF) are mutually exclusive, mutations of PIK3CA are not mutually exclusive to these genes.
Relationship of mutations and clinicopathologic features
A summary of the relationship between mutational status of NSCLC tumors and clinicopathologic features is presented in Supplementary Table S4 (individual tumor information is presented in Supplementary Table S1). As indicated by our data and several previous reports (14, 15), EGFR, KRAS, and HER2 mutations target specific histology, gender, smoking status, and ethnic subgroups. Our data confirm these findings and extend them to BRAF mutations (although the data are limited). However, PIK3CA mutations do not show association with any of these clinicopathologic features.
PIK3CA copy number evaluation in lung cancer cell lines and tumors
We evaluated PIK3CA copy number by real-time quantitative PCR (Fig. 1). Increased PIK3CA copy number was detected in eight (9.3%) of NSCLC cell lines, two (4.7%) of SCLC cell lines, none of three ExPuSC lines, and in 61 (17.1%) of NSCLC tumors. Increased PIK3CA copy number was significantly more frequent in squamous cell carcinomas (5 of 14 cell lines, 35.7%, and 46 of 139 tumors, 33.1%) than in adenocarcinomas (3 of 51 cell lines, 5.9%, and 12 of 195 tumors, 6.2%; Fig. 1; Supplementary Tables S1 and S2), P = 0.009 and P < 0.0001, respectively. Adenocarcinomas with PIK3CA copy gain occasionally had other gene mutations (one of three in cell lines and 7 of 12 in primary tumors; see Supplementary Tables S1-S3), whereas squamous cell carcinomas with PIK3CA copy gains tended to have other gene mutations less frequently (a single tumor with copy gain also had a KRAS mutation).
Figure 1.
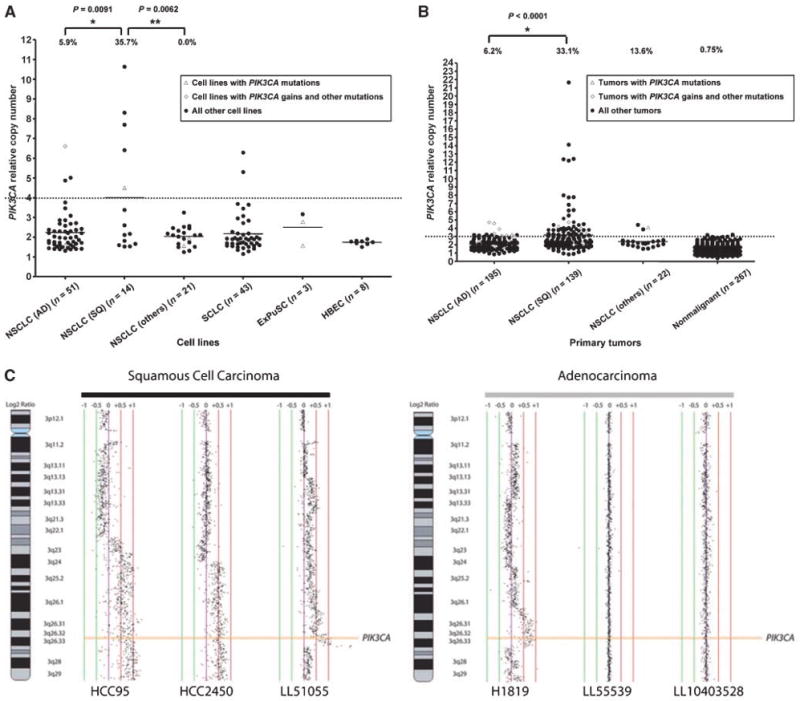
PIK3CA gene copy number in lung cancer cell lines (A) and primary tumors (B). Array CGH. Representative array CGH profiles at the PIK3CA gene locus for squamous cell carcinoma and adenocarcinoma (C). A and B, PIK3CA copy number was expressed as relative values compared with pooled human genomic DNA from multiple healthy donors. A, copy number in lung cancer cell lines. Open triangles indicate PIK3CA mutant cell lines, and open diamonds indicate PIK3CA copy number gain cell lines with mutations in other EGFR pathway genes. B, copy number in primary lung tumors. Corresponding adjacent nonmalignant tissues (n = 267) were also available. Copy numbers more than 4 (in cell lines) or 3 (in tumors) were considered as increased. We reduced the cutoff value to determine gains from 4 (for cell lines) to 3 for tumors to compensate for contamination with nonmalignant tissue in tumor samples (estimated average = 50% tumor cells, 50% nonmalignant cells). Fisher's exact test (cell lines) or χ2 test (primary tumors) were used to determine significance. C, normalized log2 signal intensity ratios were plotted using SeeGH software. Vertical lines denote log2 signal ratios from −1 to +1 with copy number increases to the right (red lines) and decreases to the left (green lines) of 0 (purple line). Each black dot represents a single bacterial artificial chromosome (BAC) clone. The region containing PIK3CA is shaded orange. For squamous cell carcinomas, two cell lines (HCC95 and HCC2450) displaying long segment PIK3CA gain/amplification and a clinical tumor specimen (LL51055) demonstrating focal amplification are depicted. For adenocarcinomas, one cell line, NCI-H1819 (H1819), with long segment high-level gain is depicted along with two clinical tumor specimens with diploid copy number for PIK3CA.
In both tumors and cell lines, copy gain values were usually modest (<5 copies per cell). With one exception (adenocarcinoma line NCI-H3255, copy number 6.61, also having an EGFR mutation), all the NSCLC cell lines and tumors having high-level gains (>5 copies per cell) were squamous cell cancers.
We also examined for geographic differences between East Asian and other ethnicities (mainly Caucasian) in PIK3CA copy number. Although not statistically significant, East Asian tumors tended to have more cases with increased PIK3CA copy number in both adenocarcinoma and squamous cell carcinoma compared with other ethnicities. PIK3CA copy gains were more frequent in squamous cell carcinomas than in adenocarcinomas in both ethnicities (41.2% of squamous cell carcinomas and 9.5% of adenocarcinomas in East Asians and 25.4% of squamous cell carcinomas and 1.3% of adenocarcinomas in other ethnicities).
Table 2 indicates the frequencies of either PIK3CA mutation or copy gains in lung cancers. PIK3CA alterations were more frequent in squamous cell carcinoma than in adenocarcinoma.
To verify the PIK3CA copy number results obtained by qPCR, we also examined a subset of cell lines and a separate cohort of tumors by array CGH. Figure 1C shows representative results for PIK3CA amplification. We obtained similar results of PIK3CA copy number by both techniques in cell lines HCC95, HCC2450, and NCI-H1819 (also see Supplementary Tables S2 and S5). CGH data confirmed that PIK3CA copy gains are more frequent in squamous cell carcinomas than in adenocarcinomas from North America (Supplementary Table S6).
PIK3CA mRNA expression in lung cancer cell lines
PIK3CA mRNA relative expression in lung cancer cell lines with PIK3CA mutation or increased copy number and other mutations was compared with that of wild-type cell lines (Fig. 2A). Wild-type cell lines indicate cell lines without mutations of EGFR, KRAS, HER2, BRAF, or PIK3CA. PIK3CA mRNA expression was significantly increased in cell lines with PIK3CA copy gains (cPIK3CA) or EGFR mutations (mEGFR) compared with wild-type cell lines. Eight of 9 cPIK3CA and 8 of 10 mEGFR cell lines had values more than double the mean values for all tumor lines, whereas lines with mutations other than mEGFR (including mPIK3CA lines without copy gains) had similar mRNA expression levels. Highly expressed PIK3CA mRNA was observed only in cPIK3CA or mEGFR cell lines.
Figure 2.

PIK3CA mRNA expression (A) and PI3K activity (B) in lung cancer cell lines. A, PIK3CA mRNA expression was compared among cell lines having different features, such as PIK3CA alterations or mutations of other genes involved in the EGFR signaling pathway. PIK3CA mRNA expression was expressed as a relative value compared with the mean of values of six HBEC cell lines. PIK3CA mRNA expression in PIK3CA gain or EGFR mutant cell lines was significantly increased compared with that of wild-type cell lines. However, PIK3CA mutant lines do not express increased mRNA levels. Horizontal bars indicate mean values. The Kruskal-Wallis test with Dunn's multiple comparison test was used to determine significance. B, equal amounts of protein (500 μg) were immunoprecipitated with anti-PI3K p85 antibody. The PI3K activity of each precipitate was then measured by ELISA and expressed as the relative production of phosphatidylinositol 3,4,5-triphosphate (PIP3) in each sample using a commercially available ELISA kit. PIP3 produced by NCI-H1299 cells (being wild-type for EGFR, KRAS, HER2, BRAF or PIK3CA, PIK3CA copy number 2.45) was set as 100 (control), and others were compared with this value. Each assay was done in triplicate. Mean values of PI3K activity in PIK3CA mutant cell lines were significantly higher than those of wild-type cell lines. Two cell lines having mutations of EGFR or HER2 also had relatively high PI3K activity, indicating that mutant HER2 or EGFR activates PI3K, which is downstream of these genes. Error bars indicate SD. The Mann-Whitney U test was used to determine significance.
PI3K activity and protein expression profile in PI3K/Akt pathway in lung cancer cell lines
We measured PI3K activity by competitive ELISA in a subset of 16 cell lines (Fig. 2B). We tested all available mPIK3CA cell lines and representative examples of cell lines with other molecular alterations. The mean value of PI3K activity in all six mPIK3CA cell lines was significantly increased compared with that of five PIK3CA wild-type cell lines (i.e., wild-type cell lines here means cell lines without mutations of any of the five tested genes). One cPIK3CA cell line, one mEGFR cell line, and one mHER2 cell line had increased kinase activity, whereas mKRAS or mBRAF cell lines had no increased kinase activity. Thus, PI3K activity was increased in lines having PIK3CA mutations or increased copy number or by mutations in genes upstream of PIK3CA.
We also correlated genotypic changes of PIK3CA with protein expression of PI3K pathway genes AKT and PTEN (Fig. 3). PIK3CA mutations and gains were strongly correlated with expression of activated pAkt (Ser473). Phosphorylated PTEN (Ser380), which counteracts PIK3CA (28), is expressed by most of lung cancer cell lines.
Figure 3.

Protein expression profiles of PI3K/Akt pathway genes in lung cancer cell lines. Whole-cell lysates were subjected to Western blotting using the antibodies indicated in the left-hand column. Total PIK3CA is expressed in all lung cancer cell lines to varying degrees. However, pAkt, which is downstream of PIK3CA, is expressed in all the PIK3CA mutant and PIK3CA high copy number cell lines. pAkt was also detected in EGFR mutant and HER2 mutant cell lines but was not detected in KRAS mutant or BRAF mutant cell lines. Of interest, high expression of pAkt is present in some SCLC lines lacking known EGFR pathway alterations. PTEN, which counters PI3K activity, is phosphorylated (and thus inactivated) at position Ser380 in most of the lung cancer cell lines.
RNA interference of PIK3CA
To investigate the phenotypic effects of PIK3CA mutations and gains, we performed RNA interference–mediated knockdown of mPIK3CA, cPIK3CA, and wild-type cell lines (Fig. 4). As detailed in the legend of Fig. 4, using both transient (siRNA) or stable (shRNA) techniques, expression of both PIK3CA and its downstream effector molecule AKT could be down-regulated in both mPIK3CA and cPIK3CA cell lines (only the data of representative siRNA experiments are shown). RNA interference also inhibited PIK3CA expression in a wild-type line having a near diploid gene copy number (NCI-H226). However, this cell line was the only line that did not express pAKT in its unaltered state.
Figure 4.
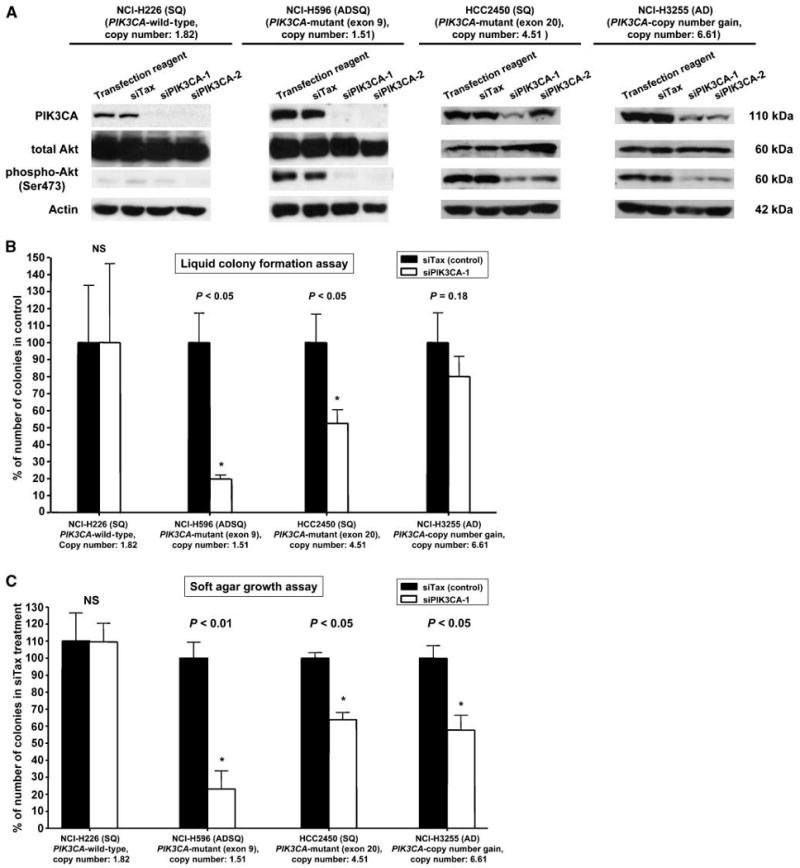
RNA interference of PIK3CA by either siRNA or shRNA. A, knockdown of PIK3CA was performed using two transient siRNAs and by two stable shRNAs. (Note the data of shRNA experiments are not shown because of the limitation of the space). We tested lung cancer cell lines having wild-type PIK3CA (NCI-H226) or lines having mutant (NCI-H596), high copy number (HCC2814, NCI-H3255, and HCC95), or both (HCC2450). All cell lines expressed PIK3CA protein and pAkt (except for NCI-H226). Knockdown experiments decreased or abolished PIK3CA and pAkt expression in all lines to varying degrees. (Note the data for HCC95 and HCC2814 are not shown because of the limitation of the space.) B and C, colony formation assays in liquid (B) or soft agar media (C). Knockdown of PIK3CA by transient siPIK3CA-1 or stable pRSPIK3CA-1 significantly inhibited anchorage-dependent (B) and anchorage-independent (C) colony formation in mutant cell line NCI-H596. In addition, there was a downward trend (although not always significant) in mutant and high copy number line HCC2450 and high copy number lines NCI-H3255, HCC2814, and HCC95. However, in wild-type line NCI-H226 (having near diploid copy number), there was no effect on growth. Note the data of shRNA experiments are not shown. Error bars indicate SD. The unpaired t test (Student's t test) was used to determine the significance.
As illustrated in Fig. 4, RNA interference–mediated knockdown of PIK3CA inhibited colony formation in both anchorage-dependent and anchorage-independent conditions in both mPIK3CA and cPIK3CA cell lines but had no effect on the growth of the wild-type cell line with near diploid copy number (NCI-H226). Of the lines tested, one had a mutation in exon 9 and one in exon 20. With minor discrepancies, the results of siRNA and shRNA interference were complementary.
Discussion
The PI3K/Akt pathway is one of the important regulators of mammalian cell proliferation and survival, because several components of this pathway are deregulated in many cancers (13). We performed a large-scale mutational and copy number gain analysis of lung cancer cell lines and primary tumors followed by functional analyses. Consistent with earlier smaller-scale analyses by others (7, 27), we found a relatively low percentage of PIK3CA mutations in lung cancers compared with several other epithelial cancers. Whereas we did not find PIK3CA mutations in SCLC cell lines of pulmonary origin, we found mutations in two ExPuSC cell lines. To date, mutations in the EGFR pathway genes that are well studied (EGFR, BRAF, KRAS, HER2, and PIK3CA) have not been reported in SCLC.
Previously, we reported that mutations of EGFR, KRAS, and HER2 were mutually exclusive, indicating that at least one activating mutation in the EGFR-RAS-RAF signaling pathway is sufficient for the pathogenesis of many lung cancers (15). In this study, we added BRAF to the mutual exclusion list. However, the mutational status of PIK3CA was not mutually exclusive to EGFR or KRAS, which is similar to previous reports (6, 29, 30). Whereas the other genes targeted adenocarcinomas, PIK3CA mutations targeted different subpopulations of NSCLC.
PI3K may be activated by receptor kinases and Ras, which in turn activates Akt. However, whereas the PI3K and EGFR signaling pathways closely interact, PI3K signaling has additional activators and downstream targets (4). Our findings of dual mutations in PI3KCA and genes within the direct EGFR signaling pathways are consistent with these observations.
We found that PIK3CA copy number gains in cell lines and tumors were more frequent in squamous cell carcinomas than in adenocarcinomas, which is consistent with previous smaller studies (11, 12). Array CGH analyses of a subset of our cell lines and an independent smaller tumor set confirmed the qPCR data. Adenocarcinomas with PIK3CA gains tended to have other gene mutations, suggesting that PIK3CA gain may not be enough for the pathogenesis of adenocarcinoma. However, most of squamous cell carcinomas with PIK3CA gains had no other alterations in the genes studied, indicating that PIK3CA may play a pivotal role in pathogenesis of squamous cell cancers. High-level PIK3CA gains (copy number, >5) were exclusively present in squamous cell carcinomas. PIK3CA mutations and copy number gains occurred independently of each other, and either molecular event may be sufficient to drive the cell toward tumorigenesis.
We profiled PIK3CA mRNA expression, PI3K activity and protein expression status in PI3K/Akt pathway to see if PIK3CA alterations have a biological effect. Cell lines with PIK3CA gains had increased mRNA expression, as did lines with EGFR activating mutations, as previously reported (31, 32). Somewhat surprisingly, PIK3CA mutant cell lines did not have high mRNA expression.
However, cell lines having either PIK3CA mutations or gains (or both) were associated with increased PI3K activity and increased expression of pAkt protein (but not with increased total PIK3CA protein) when compared with wild-type cell lines or immortalized bronchial epithelial cells. EGFR and HER2 mutant cell lines had the same effects, whereas KRAS and BRAF mutant cell lines did not have these effects. According to previous reports, mutant PIK3CA protein causes a gain of enzymatic function and induces oncogenic transformation when expressed in primary chicken embryo fibroblasts and in NIH 3T3 cells (33, 34). Samuels and colleagues also reported that mutant PIK3CA promotes cell growth and invasion of human cancer cells (35). Expression of PTEN, a negative regulator of the PI3K/Akt pathway, was present (as total and phosphoprotein) in most of lung cancer cell lines irrespective of their mutational or copy number status.
To determine whether alterations in PIK3CA contribute toward the oncogenic phenotype, we used RNA interference, both transient (siRNA) and stable (shRNA). Results of both knockdown techniques were similar and showed reduction of PIK3CA and pAkt proteins in mutant and high copy number cell lines accompanied by decreased anchorage-dependent and independent growth. However, knockdown of PIK3CA protein in a wild-type cell line lacking detectable pAkt had no effect on cell growth. These results confirm that mutations or copy number gains in squamous lung cancer cells confer a growth advantage and that inhibition of pAkt may be a better indicator of cellular effect (and perhaps of therapeutic efficacy) than inhibition of PIK3CA protein.
The finding that PI3K, a key regulator of cell growth, metabolism, and survival, is frequently activated in cancers has stimulated widespread interest in identifying potent and selective inhibitors of PI3K isoforms (36). Whereas several potential therapeutic agents have been identified, most do not show isoform selectivity. Recently, the structure of a human p110α/p85α complex was clarified, and the location of the common mutations at specific interfaces identified (37). These new insights into the structure of PIK3CA offer insights into the design of specific inhibitors.
In conclusion, PIK3CA copy number gains occur at much higher frequencies in lung cancers than do activating mutations and that the gains target squamous cell carcinomas. As these alterations confer a growth advantage to the cancer cells, targeting the PI3K/Akt pathway is a potential therapeutic option for squamous cell lung cancers. It is of particular interest to target squamous cell carcinomas, the commonest form of lung cancer in ever smokers (38), as most recent trials of targeted agents for lung cancer have focused on adenocarcinomas.
Supplementary Material
Supplemental Figure 1
Supplemental table 1. PIK3CA mutations and copy number in lung cancers (n = 356)
Supplemental table 2. PIK3CA mutations and copy number in lung cancer cell lines
Supplemental Table 3. Mutational status of EGFR, HER2, KRAS and BRAF genes in PIK3CA-mutant or -high copy number lung cancers
Supplemental Table 4. Characteristics of mutations in PIK3CA and other 4 genes (EGFR, KRAS, HER2 and BRAF)
Supplemental table 5. PIK3CA Array CGH Data for 30 NSCLC Cell Lines
Supplemental table 6. PIK3CA Array CGH Data for 40 Fresh-Frozen NSCLC Tumors
Acknowledgments
Grant support: Specialized Program of Research Excellence in Lung Cancer grant P50CA70907, National Cancer Institute.
We thank Dr. Mamoru Ouchida for providing advice and encouragement.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 2.Weir B, Zhao X, Meyerson M. Somatic alterations in the human cancer genome. Cancer Cell. 2004;6:433–8. doi: 10.1016/j.ccr.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006;94:455–9. doi: 10.1038/sj.bjc.6602970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 5.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 6.Kawano O, Sasaki H, Endo K, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006;54:209–15. doi: 10.1016/j.lungcan.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Lee JW, Soung YH, Kim SY, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–80. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- 8.Hyman E, Kauraniemi P, Hautaniemi S, et al. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res. 2002;62:6240–5. [PubMed] [Google Scholar]
- 9.Heidenblad M, Lindgren D, Veltman JA, et al. Microarray analyses reveal strong influence of DNA copy number alterations on the transcriptional patterns in pancreatic cancer: implications for the interpretation of genomic amplifications. Oncogene. 2005;24:1794–801. doi: 10.1038/sj.onc.1208383. [DOI] [PubMed] [Google Scholar]
- 10.Balsara BR, Testa JR. Chromosomal imbalances in human lung cancer. Oncogene. 2002;21:6877–83. doi: 10.1038/sj.onc.1205836. [DOI] [PubMed] [Google Scholar]
- 11.Massion PP, Kuo WL, Stokoe D, et al. Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization: implications of the phosphatidylinositol 3-kinase pathway. Cancer Res. 2002;62:3636–40. [PubMed] [Google Scholar]
- 12.Garnis C, Lockwood WW, Vucic E, et al. High resolution analysis of non-small cell lung cancer cell lines by whole genome tiling path array CGH. Int J Cancer. 2006;118:1556–64. doi: 10.1002/ijc.21491. [DOI] [PubMed] [Google Scholar]
- 13.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 14.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–46. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 15.Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–6. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 16.Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res. 2002;62:7001–3. [PubMed] [Google Scholar]
- 17.Galanis E, Frytak S, Lloyd RV. Extrapulmonary small cell carcinoma. Cancer. 1997;79:1729–36. doi: 10.1002/(sici)1097-0142(19970501)79:9<1729::aid-cncr14>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Phelps RM, Johnson BE, Ihde DC, et al. NCI-Navy Medical Oncology Branch cell line data base. J Cell Biochem Suppl. 1996;24:32–91. doi: 10.1002/jcb.240630505. [DOI] [PubMed] [Google Scholar]
- 19.Ramirez RD, Sheridan S, Girard L, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 20.Sato M, Vaughan MB, Girard L, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–28. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 21.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 22.Qiu W, Schonleben F, Li X, et al. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:1441–6. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Lockwood WW, Coe BP, Williams AC, MacAulay C, Lam WL. Whole genome tiling path array CGH analysis of segmental copy number alterations in cervical cancer cell lines. Int J Cancer. 2007;120:436–43. doi: 10.1002/ijc.22335. [DOI] [PubMed] [Google Scholar]
- 25.Naito Y, Yamada T, Ui-Tei K, Morishita S, Saigo K. siDirect: highly effective, target-specific siRNA design software for mammalian RNA interference. Nucleic Acids Res. 2004;32:W124–9. doi: 10.1093/nar/gkh442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma UN, Surabhi RM, Schmaltieg A, Becerra C, Gaynor RB. Small interfering RNAs directed against β-catenin inhibit the in vitro and in vivo growth of colon cancer cells. Clin Cancer Res. 2003;9:1291–300. [PubMed] [Google Scholar]
- 27.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 28.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–8. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Endoh H, Yatabe Y, Kosaka T, Kuwano H, Mitsudomi T. PTEN and PIK3CA expression is associated with prolonged survival after gefitinib treatment in EGFR-mutated lung cancer patients. J Thorac Oncol. 2006;1:629–34. [PubMed] [Google Scholar]
- 30.Marks JL, McLellan MD, Zakowski MF, et al. Mutational analysis of EGFR and related signaling pathway genes in lung Adenocarcinomas identifies a novel somatic kinase domain mutation in FGFR4. PLoS ONE. 2007;2:e426. doi: 10.1371/journal.pone.0000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angulo B, Suarez-Gauthier A, Lopez-Rios F, et al. Expression signatures in lung cancer reveal a profile for EGFR-mutant tumours and identify selective PIK3CA overexpression by gene amplification. J Pathol. 2008;214:347–56. doi: 10.1002/path.2267. [DOI] [PubMed] [Google Scholar]
- 32.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 33.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–7. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikenoue T, Kanai F, Hikiba Y, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–7. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- 35.Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–73. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 36.Knight ZA, Shokat KM. Chemically targeting the PI3K family. Biochem Soc Trans. 2007;35:245–9. doi: 10.1042/BST0350245. [DOI] [PubMed] [Google Scholar]
- 37.Huang CH, Mandelker D, Schmidt-Kittler O, et al. The structure of a human p110α/p85α complex elucidates the effects of oncogenic PI3Kα mutations. Science. 2007;318:1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 38.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers-a different disease. Nat Rev Cancer. 2007;7:778–90. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Supplemental table 1. PIK3CA mutations and copy number in lung cancers (n = 356)
Supplemental table 2. PIK3CA mutations and copy number in lung cancer cell lines
Supplemental Table 3. Mutational status of EGFR, HER2, KRAS and BRAF genes in PIK3CA-mutant or -high copy number lung cancers
Supplemental Table 4. Characteristics of mutations in PIK3CA and other 4 genes (EGFR, KRAS, HER2 and BRAF)
Supplemental table 5. PIK3CA Array CGH Data for 30 NSCLC Cell Lines
Supplemental table 6. PIK3CA Array CGH Data for 40 Fresh-Frozen NSCLC Tumors