

Abstract
The family of G protein-coupled receptors (GPCRs) constitutes the largest class of signalling receptors in the human genome, controlling vast physiological responses and are the target of many drugs. After activation, GPCRs are rapidly desensitized by phosphorylation and β-arrestin binding. Most classic GPCRs are internalized through a clathrin, dynamin and β-arrestin-dependent pathway and then recycled back to the cell surface or sorted to lysosomes for degradation. Given the vast number and diversity of GPCRs, different mechanisms are likely to exist to precisely regulate the magnitude, duration and spatial aspects of receptor signalling. The G protein-coupled protease-activated receptors (PARs) provide elegant examples of GPCRs that are regulated by distinct desensitization and endocytic sorting mechanisms, processes that are critically important for the spatial and temporal fidelity of PAR signalling. PARs are irreversibly activated through proteolytic cleavage and transmit cellular responses to extracellular proteases. Activated PAR1 internalizes through a clathrin- and dynamin-dependent pathway independent of β-arrestins. Interestingly, PAR1 is basally ubiquitinated and deubiquitinated after activation and traffics from endosomes to lysosomes independent of ubiquitination. In contrast, β-arrestins mediate activated PAR2 internalization and function as scaffolds that promote signalling from endocytic vesicles. Moreover, activated PAR2 is modified with ubiquitin, which facilitates lysosomal degradation. Activated PARs also adopt distinct active conformations that signal to diverse effectors and are likely regulated by different mechanisms. Thus, the identification of the molecular machinery important for PAR signal regulation will enable the development of new strategies to manipulate receptor signalling and will provide novel targets for the development of drugs.
Keywords: arrestin, caveolae, clathrin, endocytosis, GPCR, G protein, thrombin, protease, ubiquitin, trafficking
The family of protease-activated receptors (PARs) is comprised of four members: PAR1, PAR2, PAR3 and PAR4 (Alexander et al., 2008). PARs are G protein-coupled receptors (GPCRs) that are uniquely activated by proteolysis. PARs are expressed predominantly in vascular, immune and epithelial cells, astrocytes and neurons and transmit cellular responses to coagulant proteases as well as other proteases expressed in distinct tissues (Coughlin, 2005; Russo et al., 2009b). PARs are critical mediators of haemostasis, thrombosis, inflammation, and have been implicated in cancer progression, making this receptor class an important drug target. Several recent studies suggest that different agonists, proteases and synthetic peptide ligands, elicit distinct signalling responses through the activation of the same PAR. The observation that different ligands acting at the same receptor stimulate distinct signalling responses is best characterized for GPCRs and is a process termed ‘functional selectivity’ or ‘biased agonism’ (Urban et al., 2007). The mechanism responsible for agonist-induced biased agonism at PARs appears to involve stabilization of distinct active receptor conformations that may be facilitated by receptor compartmentalization in plasma membrane microdomains.
The ability of different agonists to distinctly activate PAR signalling presumably by stabilization of distinct active receptor conformations raises the question of whether a single or multiple processes of signal termination exist for distinctly activated PARs. Similar to other GPCRs, activated PARs are rapidly desensitized by phosphorylation and β-arrestin binding. In addition to rapid desensitization, PAR trafficking is crucial for the temporal and spatial control of receptor signalling. The mechanisms responsible for endocytic trafficking of PARs remain poorly understood. Moreover, the efficiency with which PARs are degraded makes this receptor class an excellent model system to investigate the molecular basis of GPCR lysosomal degradation. PAR2, like many GPCRs, is modified with ubiquitin, which facilitates lysosomal trafficking through the endosomal-sorting complex required for transport (ESCRT) pathway (Jacob et al., 2005; Hasdemir et al., 2007). We recently discovered that activated PAR1 traffics from endosomes to lysosomes independent of ubiquitination and the ubiquitin-binding components of the ESCRT machinery (Gullapalli et al., 2006; Wolfe et al., 2007). The molecular basis of the novel ubiquitin-independent lysosomal sorting of PAR1 as well as the identities of the adaptor molecules and processes that control endocytic trafficking remains largely unknown. Here, we discuss the mechanisms of PAR activation, signalling, desensitization, resensitization, endocytic trafficking and crosstalk with other receptors.
PAR activation and signalling
Activation of PARs occurs through an irreversible proteolytic mechanism. Proteases bind to and cleave the extracellular N-terminal domain of PARs at specific sites to unmask a new N-terminus that acts as a tethered ligand that binds to the receptor and triggers intracellular signalling (Figure 1) (Vu et al., 1991a,b;). Synthetic peptides that mimic the first six amino acids of the newly formed N-terminus can activate PARs independent of protease and receptor cleavage (Scarborough et al., 1992; Chen et al., 1994; Lerner et al., 1996), except for PAR3, which is unresponsive to synthetic peptide agonists (Nakanishi-Matsui et al., 2000). After protease cleavage, the N-terminal 41-amino-acid domain of PAR1 is released into the extracellular milieu and may exert some biological activity, but this remains controversial. Recent work suggests that the released N-terminal region of PAR1 containing an N-terminal hydrophobic domain and dubbed ‘Parstatin’, inhibits vascular endothelial cell growth factor (VEGF) and fibroblast growth factor-induced angiogenesis in vitro and in vivo by regulating intracellular signalling events (Zania et al., 2009). Previous studies have also reported effects of the released N-terminus of PAR1 at the platelet cell surface that appears to modulate platelet function (Furman et al., 2000a,b;). The mechanism by which the cleaved-off N-terminal domain of PAR1 exerts its effects on cells remains unclear and whether the released N-terminal extracellular regions of other PARs also have biological activities has not been determined.
Figure 1.
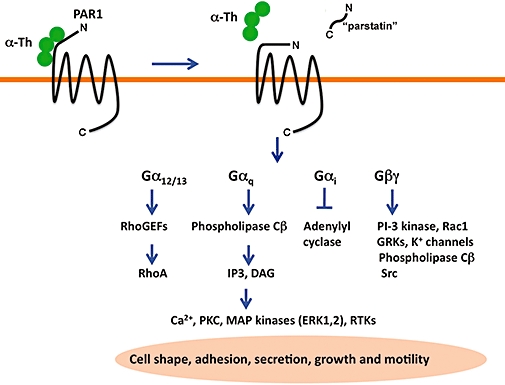
Activation and signalling by protease-activated receptor-1 (PAR1). PAR1 is a seven transmembrane G protein-coupled receptor that is irreversibly proteolytically activated by thrombin. Thrombin binds to and cleaves the N-terminus of PAR1, generating a new N-terminal domain that binds intramolecularly to trigger transmembrane signalling. Recent studies suggest that the cleaved N-terminal domain of PAR1 is released and exhibits biological activity in certain settings and has been termed ‘parstatin’. Activated PAR1 couples to multiple heterotrimeric G protein subtypes including G12/13, Gq and Gi and activates a variety of signalling effectors important for inducing cell shape changes, adhesion molecule expression, secretion of vasoactive factors, cellular growth and motility. The mechanisms that specify PAR1 coupling to distinct G protein subtypes is not known.
The ligand activation of PARs is likely to induce conformational changes within the transmembrane helices that expose receptor cytoplasmic surfaces important for interaction with the α subunits of heterotrimeric G proteins at the inner leaflet of the plasma membrane (Oldham and Hamm, 2007; 2008;). The activation of PARs is thought to occur through peptide ligand interactions with residues residing in the second extracellular loop (Gerszten et al., 1994), unlike most classic GPCRs where ligand binding occurs in a pocket formed by the transmembrane helices. New high-resolution structures of four different GPCRs reveal considerable divergence in the second extracellular loops as well as in the ligand-binding pocket and intracellular loops (Hanson et al., 2009; Rosenbaum et al., 2009), suggesting distinct mechanisms of GPCR activation and signal transduction. Once activated, GPCRs function as guanine nucleotide exchange factors and promote exchange of GDP for GTP on the α subunit leading to βγ subunit dissociation. Both the GTP bound α subunit and βγ subunits signal to various effectors to promote diverse cellular responses (Figure 1) (Oldham and Hamm, 2007; 2008;). Activated PAR1 and PAR2 couple to multiple heterotrimeric G-protein subtypes including Gi, Gq and G12/13 (Coughlin, 2005; Russo et al., 2009b). PAR4 couples to Gq and G12/13 activation, whereas PAR3 was previously thought not to signal autonomously (Nakanishi-Matsui et al., 2000). However, new studies indicate that thrombin activation of natively expressed PAR3 can elicit Rho- and Ca2+-dependent release of ATP from lung epithelial A549 cells, a cell type that does not appear to express either PAR1 or PAR4 (Seminario-Vidal et al., 2009).
Activated PARs also interact with various adaptor proteins that facilitate signal transduction independent of heterotrimeric G protein coupling. The best-characterized G protein-independent effectors for GPCRs are the β-arrestins, multifunctional adaptor proteins (Table 1) (Lefkowitz and Shenoy, 2005). There are four members of the arrestin family including visual arrestin (arrestin 1) and X-arrestin (arrestin 4), which are expressed primarily in retinal rods or cones, and the highly abundant, ubiquitously expressed β-arrestin 1 and 2 (also known as arrestin 2 and 3). Arrestins are comprised of distinct N and C structural domains linked by a 12-residue polar core that engages receptor-associated phosphates (Hirsch et al., 1999; Gurevich and Gurevich, 2006). Interestingly, the Vps26 protein, a component of the retromer complex important for retrograde trafficking of cargo from endosomes to the trans-Golgi network (Bonifacino and Hurley, 2008), has a structure surprisingly similar to β-arrestins, despite limited primary amino acid sequence homology (Shi et al., 2006). Strikingly, Vps26 appears to be more closely related to α-arrestins, a new family of arrestin-like proteins discovered in yeast; humans appear to have six α-arrestin proteins (Alvarez, 2008). The α-arrestins are predicted to harbour distinct N and C domains and to resemble β-arrestins and Vps26 structurally, but unlike these molecules α-arrestins contain proline-sequence (PY) motifs that can bind to WW domains of E3 ubiquitin ligases to promote ubiquitination of cargo proteins (Lin et al., 2008; Nikko et al., 2008). Thus far, there are no reported studies that have addressed the function of α-arrestin proteins in the regulation of mammalian GPCR signalling and trafficking.
Table 1.
Agonist, signalling and shut-off mechanisms of protease-activated receptors (PARs)
| Receptor | Tethered ligand | Activating proteases | Signalling effectors | Signal termination |
|---|---|---|---|---|
| PAR1 | SFLLRN | Thrombin TF-VIIa-Xa or Xa APC-EPCR Trypsin Plasmin MMP-1 Granzyme 1 | GqGiG12/13Hsp90 Creatine kinase Zyxin | Phosphorylation β-Arrestins Internalization Degradation |
| PAR2 | SLIGKV | Trypsin Tryptase TF-VIIa TF-VIIa-Xa Matriptase (MT-SP1) Bacterial gingipains Kallikreins Granzyme A | GqGiG12/13β-Arrestins Jab1 | Phosphorylation β-Arrestins |
| PAR3 | TFRGAP | Thrombin | Gq | ? |
| PAR4 | GYGQV | Thrombin Trypsin TF-VIIa-Xa Plasmin Cathepsin G Bacterial ginginpains Killikreins MASP-1 | GqG12/13 | Internalization |
The multifaceted β-arrestins control the magnitude and duration of GPCR-mediated G protein signalling as well as signalling to non-G protein effectors by functioning as scaffolds that form discrete signalling complexes with certain GPCRs (Lefkowitz and Shenoy, 2005; Moore et al., 2007). β-Arrestins recruit c-Src to some activated GPCRs to facilitate activation of extracellular signal-regulated kinases 1 and 2 (ERK1, 2). In addition, β-arrestins co-internalize with activated GPCRs and function as scaffolds that bring components of the ERK1, 2 and JNK3 signalling modules together, a process that is regulated by agonist stimulation. Unlike other PARs, activated PAR2 is the only protease-activated receptor shown to bind and co-internalize with β-arrestins, which facilitates ERK1, 2 signalling from endocytic vesicles to mediate changes in the actin cytoskeleton and cell migration (Defea et al., 2000; Ge et al., 2004; Stalheim et al., 2005). In addition to β-arrestins, PAR2 has been reported to bind to the Jun activating binding protein-1 (Jab1), a protein that stabilizes complexes of c-Jun or Jun D with the transcription factor activator protein 1 DNA binding sites (Luo et al., 2006). Jab1 was identified using PAR2 in a yeast two-hybrid screen, and has been reported to mediate PAR2-induced activation of c-Jun and gene transcription (Table 1). Previous studies using the PAR1 cytoplasmic tail in a yeast two-hybrid cDNA library screen led to the identification of several binding partners including heat shock protein 90 (Hsp90) and creatine kinase (Table 1). PAR1 interacts directly with creatine kinase and Hsp90, which appears to modulate activated receptor stimulation of RhoA signalling and cytoskeleton rearrangements in neuronal systems (Mahajan et al., 2000; Pai et al., 2001). A recent study suggests that the PAR1 cytoplasmic tail domain interacts directly with zyxin, a LIM domain containing protein, which signals to the actin cytoskeleton independent of RhoA activation (Han et al., 2009). There are no known reports of G protein-independent effectors that mediate signalling by activated PAR3 or PAR4.
PAR desensitization and resensitization
In the classic paradigm, activated GPCRs are rapidly desensitized and uncoupled from G protein signalling by phosphorylation and β-arrestin binding. GPCR phosphorylation occurs predominantly on serine and threonine residues within the cytoplasmic tail and third intracellular loop but rarely on tyrosine residues. G protein-coupled receptor kinases (GRKs) mediate phosphorylation of activated GPCRs, which increases receptor affinity towards β-arrestins and thereby prevents further receptor–G protein coupling and consequent signalling (Lohse et al., 1990; Krupnick and Benovic, 1998). The second messenger kinases, protein kinase A and protein kinase C (PKC), can also phosphorylate and desensitize GPCRs regardless of the state of receptor activation, through a process that does not involve β-arrestin recruitment to the receptor (Pitcher et al., 1992). In addition, phosphorylation is not absolutely required for agonist-dependent β-arrestin binding and desensitization of certain activated GPCRs (Mukherjee et al., 2002; Jala et al., 2005). Moreover, some GRKs can uncouple GPCRs from G protein signalling independent of receptor phosphorylation (Ferguson, 2007). Recent work further indicates that GPCRs undergo site-specific phosphorylation that confers important functions and signalling outcomes in distinct cellular contexts (Tobin et al., 2008). Taken together, many studies suggest that GPCR signalling is regulated by multiple complex pathways that is likely important for distinct biological functions.
Despite the irreversible proteolytic mechanism of PAR activation that results in the generation of a tethered ligand that cannot diffuse away, signalling by the receptor is rapidly shut off. Previous studies suggest that each activated PAR1 generates a defined amount of second messenger response, and then shuts off, at least in terms of Gq signalling (Ishii et al., 1993). Phosphorylation of activated PAR1 appears to be important for rapid uncoupling from G-protein signalling as overexpression of either GRK3 or GRK5 enhances PAR1 phosphorylation and markedly inhibits signalling (Figure 2) (Ishii et al., 1994; Tiruppathi et al., 2000). A PAR1 mutant in which all serines and threonines in the cytoplasmic tail are converted to alanines is neither extensively phosphorylated nor inhibited by GRK3 overexpression in multiple cell types. Interestingly, Paing et al. previously showed that β-arrestin 1 is more effective than β-arrestin 2 at desensitizing PAR1 signalling, through a pathway that occurs independent of receptor phosphorylation and internalization (Figure 1) (Paing et al., 2002; Chen et al., 2004). Moreover, β-arrestin 1 R169E and β-arrestin 2 R170E mutants that bind to activated GPCRs with high affinity independent of receptor phosphorylation were equally effective at promoting desensitization of both PAR1 wild type and a phosphorylation-defective mutant. Thus, desensitization of PAR1 signalling is regulated by multiple independent mechanisms including phosphorylation of the cytoplasmic tail and the binding of β-arrestin 1, which occurs independent of receptor phosphorylation.
Figure 2.
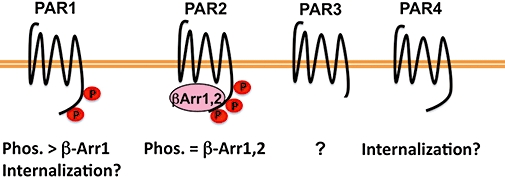
Protease-activated receptor (PAR) desensitization mechanisms. The mechanisms responsible for desensitization of PARs are varied and poorly understood. Phosphorylation of activated by PAR1 by GRK3 and GRK5 is the predominant mediator of PAR1 uncoupling from G protein signalling at least to Gq. β-Arrestin 1, but not β-arrestin 2, also contributes to PAR1 desensitization through a mechanism that does not involve receptor phosphorylation or internalization. The contribution of internalization to the uncoupling of PAR1 from G proteins signalling is not clearly understood. In contrast, activated PAR2 is robustly phosphorylated by protein kinase C and other kinases, binds both β-arrestin 1 and β-arrestin 2 and is rapidly uncoupled from G protein signalling at the cell surface. The mechanisms responsible for desensitization of PAR3 and PAR4 are largely unknown; however, internalization may contribute to the termination of PAR4 signalling.
A role for phosphorylation in desensitization of activated PAR2 signalling has recently been reported (Ricks and Trejo, 2009). Previous studies showed that pharmacological inhibitors of PKC enhance PAR2-mediated calcium responses in transformed rat kidney epithelial (KNRK) cells and Berkeley rat intestinal (hBRIE 380) cells, suggesting a role for phosphorylation in PAR2 regulation (Bohm et al., 1996). We recently demonstrated that PAR2 activation caused a rapid and robust increase in phosphorylation of PAR2 wild type, but not a mutant receptor in which all serines and threonines in the cytoplasmic tail were converted to alanines, suggesting that the major sites of PAR2 phosphorylation occur within the cytoplasmic tail (Figure 2) (Ricks and Trejo, 2009). Similar to other GPCRs, phosphorylation of PAR2 occurred on multiple redundant sites, any of which were sufficient for receptor desensitization or internalization. In addition, a phosphorylation-defective PAR2 mutant failed to recruit either β-arrestin 1 or β-arrestin 2 and desensitization was markedly impaired compared with wild type receptor (Figure 2). These findings are consistent with previous work showing that β-arrestins are essential for activated PAR2 desensitization and internalization (Dery et al., 1999; Defea et al., 2000; Stalheim et al., 2005). However, internalization of phosphorylation-defective PAR2 mutant proceeded through a dynamin-dependent but clathrin- and β-arrestin-independent pathway in both a constitutive and agonist-dependent manner (Ricks and Trejo, 2009). We interpret these findings to suggest that mutations of serines and threonines within the PAR2 cytoplasmic tail promote constitutive internalization by either diminishing basal receptor phosphorylation and/or altering a distinct receptor conformation important for retention at the plasma membrane. Thus, unlike PAR1, β-arrestins mediate activated PAR2 desensitization, presumably through phosphorylation, internalization and signalling to downstream effectors. PAR4, a low-affinity receptor for thrombin, promotes sustained signalling responses and does not appear to undergo agonist-promoted phosphorylation when overexpressed in Rat1 fibroblasts (Shapiro et al., 2000), despite the presence of multiple serines and threonines residues within its cytoplasmic tail. Activated PAR4 also displays a slowed rate of internalization, through a poorly understood process. The cytoplasmic tail of PAR3 is considerably shorter than the cytoplasmic tail domains of other PARs, and the regulatory mechanisms responsible for termination of PAR3 signalling have not been determined (Figure 2).
The proteolytic activation of PARs is unique among GPCRs, and distinct mechanisms mediate the disposal of irreversibly activated receptors and replenish the cell surface with uncleaved receptor critical for cellular resensitization. The redistribution of uncleaved PARs from an intracellular pool to the cell surface permits rapid recovery of protease signalling independent of de novo receptor synthesis in certain cell types and is critical for cellular resensitization (Hein et al., 1994; Bohm et al., 1996). This ensures that endothelial cells, fibroblasts and other cell types that are exposed to proteases repeatedly recover signalling responses in a timely manner. The mechanisms responsible for replenishing the cell surface with uncleaved PAR1 and PAR2 appear to be distinct. In endothelial cells and fibroblasts, unactivated PAR1 is delivered to the cell surface and cycles constitutively between the plasma membrane and an early endosomal recycling compartment (Paing et al., 2006), forming a cytoplasmic pool that is protected from protease cleavage. Although PAR1 internalization is dependent on clathrin and dynamin, it occurs independent of β-arrestins (Trejo et al., 2000; Paing et al., 2002; Chen et al., 2004). Instead, the clathrin adaptor protein complex-2 (AP-2) binds directly to a tyrosine-based motif in the cytoplasmic tail of PAR1 and is essential for constitutive internalization and cellular recovery following thrombin exposure (Paing et al., 2006). The expression of a PAR1 tyrosine mutant or depletion of AP-2 by RNA interference leads to significant inhibition of PAR1 constitutive internalization, loss of intracellular uncleaved PAR1, and failure of endothelial cells and other cell types to regain thrombin responsiveness. In contrast, human platelets, which presumably respond to thrombin only once, the majority of PAR1 is retained on the cell surface (Molino et al., 1997). Thus, platelets lack an internal pool of protected receptors, as recovery of thrombin responsiveness is not a physiological requirement of this cell type.
In contrast to PAR1, full cellular resensitization to PAR2 signalling involves mobilization of naïve receptor from the Golgi apparatus as well as de novo receptor synthesis (Bohm et al., 1996). In KNRK and hBRIE 380 epithelial cells, initial resensitization to trypsin, a potent activator of PAR2 signalling, remained intact after incubation with brefeldin A, an inhibitor of Golgi protein transport, or cycloheximide, an inhibitor of de novo protein synthesis, suggesting that initial resensitization depends upon the existence of a reserve receptor pool (Bohm et al., 1996). However, subsequent challenge with agonist required both intact Golgi function and de novo synthesis for full PAR2 responsiveness. A more recent study identified the type I transmembrane protein p24a as an important mediator of PAR2 transport from the Golgi apparatus to the cell surface and cellular resensitization (Luo et al., 2007). P24a is a member of the p24 family of proteins that facilitates protein transport through the Golgi network and binds to the second extracellular loop of PAR2 retaining it at the Golgi until receptor activation causes receptor dissociation and exocytic transport, a process that involves activation of the small GTPase, ADP-ribosylation factor-1 (ARF1). In addition to ARF1, the small GTPase rab11a, a key regulator of perinuclear, plasma membrane and Golgi endosomal recycling, has also been implicated in PAR2 exocytic trafficking and cellular resensitization (Roosterman et al., 2003). The mechanisms that regulate PAR3 and PAR4 trafficking and cellular resensitization have not been determined.
Endocytic trafficking of PARs
The internalization, recycling and lysosomal sorting of PARs are crucial for the spatial and temporal regulation of receptor signalling. Unactivated PAR1 cycles tonically between the cell surface and endosomes and is important for cellular resensitization (Paing et al., 2006), whereas activated PAR1 internalization and lysosomal degradation is critical for signal termination (Trejo et al., 1998; Trejo and Coughlin, 1999). In contrast, activated PAR2 internalization is required for signal propagation from endocytic vesicles (Defea et al., 2000; Stalheim et al., 2005). The underlying mechanistic basis for these processes is just beginning to emerge and has been reported in recent studies as discussed below.
The transport of GPCRs within the endocytic pathway is initiated through a highly regulated process that leads to deformation of select regions of the plasma membrane (Marchese et al., 2008). Clathrin-coated pits formed at the plasma membrane are largely responsible for recruitment and internalization of most GPCRs. Clathrin-coated pits assemble at sites enriched in phosphatidylinositol (4,5)-bisphosphate, and involve the recruitment of clathrin, adaptor proteins and many other regulatory proteins that coordinate the formation and invagination of clathrin-coated vesicles (Edeling et al., 2006). The recruitment of cargo to clathrin-coated pits is mediated by clathrin adaptors that recognize phosphorylated or ubiquitinated cargo or short linear sorting sequences residing within the cytoplasmic domains of cargo proteins. Most activated and phosphorylated GPCRs are recognized by β-arrestins, which facilitate receptor internalization through clathrin-coated pits. The binding of β-arrestins to GPCRs induces a conformational change in β-arrestins exposing C-terminal sequences that bind to clathrin and the β2-adaptin subunit of the clathrin adaptor protein AP-2 (Goodman et al., 1996; Laporte et al., 1999).
Similar to most classic GPCRs, activated PAR2 is robustly phosphorylated and recruited to clathrin-coated pits by binding to β-arrestins (Defea et al., 2000; Stalheim et al., 2005; Ricks and Trejo, 2009). Moreover, activated PAR2 is phosphorylated on multiple serine and threonine residues positioned at the end of the cytoplasmic tail (Ricks and Trejo, 2009), which increases its affinity for β-arrestins and facilitates PAR2–β-arrestin complex co-internalization and colocalization on endocytic vesicles. Unlike most classic GPCRs, activated and internalized PAR2 does not recycle, but instead remains associated with β-arrestins on endocytic vesicles and promotes sustained ERK1, 2 signalling in the cytoplasm (Defea et al., 2000; Stalheim et al., 2005).
PAR1 displays both constitutive and agonist-promoted internalization, which proceeds through a clathrin- and dynamin-dependent pathway independent of β-arrestins (Figure 3) (Trejo et al., 2000; Paing et al., 2002; 2006;). We previously showed that the clathrin adaptor AP-2 is essential for constitutive PAR1 internalization (Paing et al., 2006; Wolfe et al., 2007). In addition, the post-translational modification of PAR1 with ubiquitin specifies a distinct clathrin adaptor requirement that mediates activated receptor internalization through clathrin-coated pits independent of AP-2 and β-arrestins (Wolfe et al., 2007). Ubiquitin is a 76-amino-acid polypeptide that is covalently linked to lysine residues of substrate proteins by ubiquitin ligases and removed by deubiquitinating enzymes. A role for ubiquitination in GPCR internalization was first demonstrated for the yeast Saccharomyces cerevisiae G protein-coupled Ste2 and Ste3 receptors (Hicke and Riezman, 1996; Roth and Davis, 1996). The ubiquitination of PAR1 is likely a highly dynamic and reversible process, and, the receptor probably exists in both ubiquitinated and deubiquitinated forms at steady state. Remarkably, the major sites of PAR1 ubiquitination occur within the cytoplasmic tail at lysines residues, which are located within the distal tyrosine-based motif, an important binding site for the µ2 subunit of the AP-2 complex that mediates constitutive receptor internalization (Figure 3) (Paing et al., 2006; Wolfe et al., 2007). Thus, PAR1 ubiquitination may preclude AP-2 binding to negatively regulate constitutive receptor internalization. Consistent with this model, an ubiquitin-deficient PAR1‘0K’ mutant displays an enhanced rate of constitutive internalization and the fusion of an ubiquitin moiety to the cytoplasmic tail of PAR1‘0K’ mutant attenuated constitutive internalization. Thus, modification of PAR1 by ubiquitination switches the trafficking phenotype and the endocytic machinery required to internalize activated receptor. In addition to PAR1, several other mammalian GPCRs internalize through clathrin-coated pits independent of β-arrestins (Wolfe and Trejo, 2007) and raise the possibility that similar internalization mechanisms exist for other mammalian GPCRs.
Figure 3.
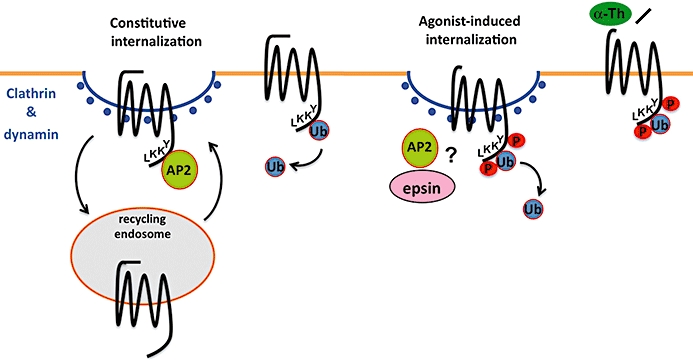
Constitutive and activated protease-activated receptor-1 (PAR1) internalization are regulated by distinct mechanisms. Unactivated PAR1 cycles constitutively between the cell surface and an intracellular compartment, which generates an internal pool of uncleaved receptor that functions to rapidly resensitize cells after protease cleavage. In contrast, activated PAR1 is phosphorylated, internalized and sorted directly to lysosomes for degradation, a process critical for termination of activated receptor signalling. Both constitutive and activated PAR1 internalization occur through a clathrin- and dynamin-dependent pathway that is independent of β-arrestins. Rather than β-arrestins, the clathrin adaptor (protein) AP-2 binds directly to a tyrosine-based motif at the C-terminus of PAR1 to mediate constitutive internalization. Interestingly, PAR1 is basally ubiquitinated and ubiquitination appears to negatively regulate PAR1 constitutive internalization, perhaps by impeding AP-2 binding. In contrast, ubiquitination of activated PAR1 specifies a distinct clathrin-dependent internalization pathway that is independent of AP-2. However, both AP-2 and epsin, a clathrin adaptor that binds ubiquitin, mediate activated PAR1 internalization through clathrin-coated pits but precisely how this occurs remains to be determined. Moreover, deubiquitination of PAR1 occurs after activation through mechanisms that remain poorly understood.
Once internalized, GPCRs are sorted within an endosomal tubulo-vesicular compartment to either a recycling or lysosomal degradation pathway. Sorting of GPCRs to a recycling pathway occurs through a default pathway similar to bulk membrane flow or through a regulated process (Hanyaloglu and von Zastrow, 2008). Trafficking of internalized receptor from endosomes to lysosomes is a major pathway for GPCR degradation or down-regulation. The regulatory mechanisms that control degradation of GPCRs by internalization and lysosomal sorting involve adaptor protein recognition of either structural determinants within the receptor cytoplasmic tail and/or post-translational modification with ubiquitin (Marchese et al., 2008).
Several mammalian GPCRs harbour tyrosine-based motifs within their cytoplasmic tails that may function as binding sites for endocytic AP complexes to facilitate membrane trafficking (Marchese et al., 2008). In addition to their endocytic function, tyrosine-based motifs have been implicated in targeting transmembrane proteins to lysosomes. The lysosomal targeting tyrosine-based motifs are often found close to the end of the transmembrane domain. The heterotetrameric AP complexes bind directly to tyrosine-based motifs and have distinct functions in membrane trafficking (Bonifacino and Traub, 2003). AP-2 is abundant at the plasma membrane and mediates endocytosis. AP-1 localizes predominantly to the trans-Golgi network, whereas AP-3 associates with early-endosome-associated tubules and facilitates endosome-to-lysosome trafficking. AP complexes are composed of α, β, µ and σ subunits; the µ subunit directly binds to tyrosine-based motifs. We previously found that a proximal PAR1 cytoplasmic tail tyrosine-based motif localized near the end of the seventh transmembrane domain facilitates lysosomal degradation (Paing et al., 2004), but whether this involves recognition by distinct AP complexes has not been determined. Moreover, lysosomal sorting of the galanin R1 receptor and M2 muscarinic receptor has also been shown to require a cytoplasmic localized tyrosine-based motif (Goldman and Nathanson, 1994; Xia et al., 2008), but how this occurs mechanistically is not known.
The sorting of ubiquitinated cargo from early endosomes to lysosomes involves transit through an endosomal compartment that contains intralumenal vesicles (ILVs) and is termed multivesicular bodies (MVBs). MVBs (also known as late endosomes) fuse with lysosomes, resulting in the degradation of ILVs containing lipids by lipases and proteins by proteases. The ubiquitin-dependent ESCRT machinery is comprised of a complex network of proteins that function coordinately to sort ubiquitinated cargo to a degradative pathway and was discovered in yeast S. cerevisiae (Piper and Katzmann, 2007). The ESCRT machinery is conserved in mammalian cells, although mammalian cells have a greater diversity and specialization in endocytic sorting pathways. The ESCRT machinery is comprised of three distinct complexes that recognize ubiquitinated cargo and prevent their recycling or retrograde trafficking and has the capacity to deform the endosomal membrane allowing cargo to be sorted into ILVs of MVBs. The hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) (also known as ESCRT-0) is recruited to endosomes via its FYVE domain, which binds to phosphatidylinositol-3-phosphate, a lipid enriched on endosomal membranes. HRS binds directly to ubiquitinated cargo on endosomes and to tumour suppressor gene 101 (Tsg101), a component of ESCRT-I. ESCRT-II and ESCRT-III complexes are then recruited sequentially to endosomes and coordinate the recruitment and sorting of ubiquitinated cargo to MVBs. Cargo is then deubiquitinated before entry into ILVs of MVBs. The activity of vacuolar protein sorting protein-4, an AAA-ATPase that catalyses the disassembly of the ESCRT components, is critical for ESCRT function.
Several mammalian GPCRs are directly modified with ubiquitin and sorted to lysosomes through an ESCRT-dependent pathway (Marchese et al., 2008). Activated PAR2 is internalized via a β-arrestin, clathrin and dynamin-dependent pathway (Defea et al., 2000; Stalheim et al., 2005; Ricks and Trejo, 2009), and is directly ubiquitinated by the RING-finger ubiquitin ligase c-Cbl and sorted to lysosomes for degradation (Jacob et al., 2005). Activated PAR2 requires not only ubiquitination but also HRS, a component of the ESCRT machinery, for lysosomal degradation and not for receptor internalization (Figure 4) (Jacob et al., 2005; Hasdemir et al., 2007). In addition, overexpression of the catalytically inactive deubiquitinating enzymes AMSH and UBPY increase the steady state amount of ubiquitinated PAR2 and slowed agonist-triggered lysosomal degradation (Hasdemir et al., 2009). Although PAR2 is considered a class B GPCR as defined by its ability to form stable complexes with β-arrestins, rather than transient interactions like class A receptors, the role of β-arrestin ubiquitination and its ability to recruit E3 ubiquitin ligases to the regulation of PAR2 ubiquitination is not known (Drake et al., 2006). However, not all GPCRs that sort rapidly and efficiently to lysosomes require direct ubiquitination (Marchese et al., 2008). We previously found that activation of wild type and ubiquitin-deficient PAR1 expressed in HeLa cells or Rat1 fibroblasts caused a similar extent of receptor degradation, suggesting that PAR1 ubiquitination is not required for lysosomal degradation (Figure 4) (Wolfe et al., 2007). Moreover, activated PAR1 sorts from endosomes to lysosomes independent of HRS and Tsg101 (Gullapalli et al., 2006), components of the ubiquitin-binding ESCRT machinery. These findings strongly suggest that deubiquitinated rather than ubiquitinated PAR1 transits through the endosomal–lysosomal system independent of the ESCRT proteins HRS and Tsg101. However, the sorting of PAR1 from early endosomes to lysosomes at an early stage in the pathway is dependent on sorting nexin-1 (SNX1) (Figure 4) (Wang et al., 2002; Gullapalli et al., 2006). SNX1 associates with the cytosolic face of endosomes and contains a phox homology (PX) domain important for binding to endosomal-enriched phosphoinositides. The C-terminal BAR domain of SNX1 forms banana-shaped dimers with different degrees of curvature that sense or induce membrane curvature and is thought to drive tubule formation by preferentially interacting with the narrow diameter of membrane tubules (Carlton et al., 2004; Cullen, 2008). Thus, SNX1 facilitates the pinching off of endosomal tubules through a process that involves other as-yet unidentified endocytic proteins. The identities of other endocytic adaptor proteins that mediate ubiquitin-independent lysosomal sorting of PAR1 and other GPCRs remain to be determined.
Figure 4.
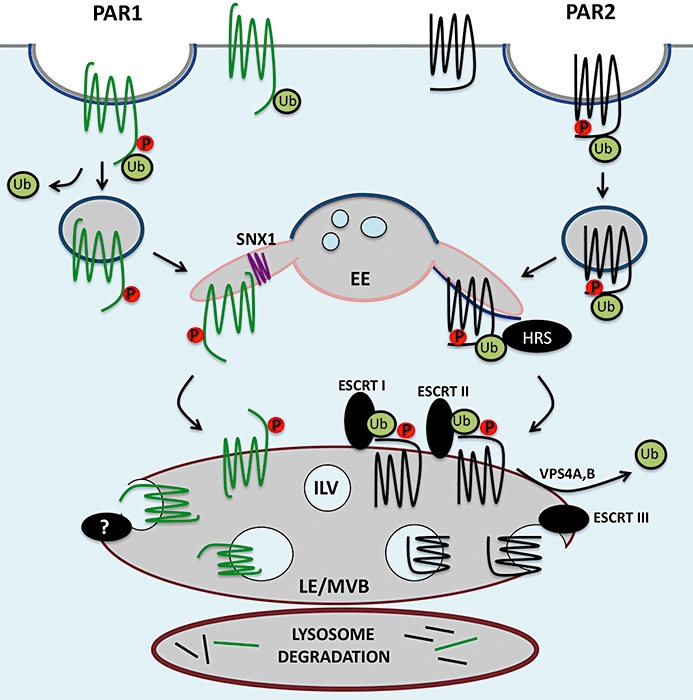
Ubiquitin-dependent and -independent lysosomal trafficking of protease-activated receptors (PARs). Activated PAR2 is phosphorylated, binds β-arrestins and is then recruited to clathrin-coated pits and internalized from the cell surface. In contrast, internalization of activated PAR1 occurs via a phosphorylation, clathrin- and dynamin-dependent pathway independent of β-arrestins. Clathrin-coated pits pinch off to form vesicles, which fuse with early endosomes (EE). Internalized PARs are then sorted through endocytic compartments to lysosomes and degraded. Most ubiquitinated cargo are sorted to lysosomes through the well-characterized ubiquitin-dependent endosomal-sorting complex required for transport (ESCRT) pathway. Activated PAR2 is ubiquitinated and requires hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) and vacuolar protein sorting protein-4 (Vps4) for lysosomal sorting. Remarkably, activated PAR1 is delivered to lysosomes independent of ubiquitination and the ESCRT machinery – HRS, tumour suppressor gene 101 and Vps4. Trafficking of PAR1 to lysosomes, however, requires its C-terminal tyrosine-based motif and is mediated by sorting nexin 1 (SNX1). The mechanism by which activated PAR1 is sorted to intralumenal vesicles (ILVs) of late endosomes (LE)/multivesicular bodies (MVBs) is not known.
Agonist selective PAR signalling
‘Functional selectivity’ or ‘biased agonism’ is a process by which distinct ligands acting on the same receptor can elicit different signalling responses by stabilizing distinct active receptor conformations and has been best characterized for GPCRs (Urban et al., 2007). In many cases, differences in GPCR signalling have been observed in studies comparing synthetic ligands to natural ligands. Indeed, previous studies suggest that distinct cellular responses could be evoked by PAR1 when activated proteolytically by its tethered ligand versus activation by untethered ‘free’ synthetic peptide agonists (Blackhart et al., 2000). McLaughlin et al. demonstrated that thrombin activation of PAR1 in human endothelial cells favours activation of G12/13 signalling and induction of endothelial barrier permeability over Gq or Gi signalling (McLaughlin et al., 2005). In contrast, the activation of PAR1 by synthetic peptide agonists SFLLRN or TFLLRNPNDK caused preferential coupling to Gq-triggered increases in Ca2+ mobilization rather than G12/13 signalling, suggesting that tethered versus ‘free’ peptide ligands can distinctly activate PAR signalling. Consistent with this idea, the activation of PAR2 by distinct tethered versus ‘free’ peptide agonist also appears to cause biased receptor signalling. The mutant SLAAA peptide sequence stimulates PAR2 induction of Ca2+ mobilization when tethered to the receptor but failed to elicit signalling responses when presented to cells as a soluble ligand (Al-Ani et al., 2004). In new work, Ramachandran et al. showed that although the soluble SLAAA peptide ligand could not trigger Ca2+ mobilization, it retained the capacity to activate ERK1, 2 signalling comparable to native agonist peptide SLIGKV (Ramachandran et al., 2009), suggesting that the agonist peptide SLAAA agonist induces biased signalling.
The unique proteolytic cleavage mechanism of PAR activation that occurs at a defined site in the N-terminus and results in the generation of a specific tethered ligand sequence, suggested that different proteases are unlikely to function as biased agonists for PAR signalling. However, several recent studies suggest that distinct proteases can stabilize different active PAR conformations to elicit distinct cellular responses that may be facilitated by compartmentalization in plasma membrane microdomains (Figure 5) (Russo et al., 2009b). Several proteases have been reported to cleave and activate PARs including serine, cysteine and metalloproteases that either function as soluble enzymes or require membrane-associated cofactors that facilitate membrane localization and/or allosterically modulate protease activity (Table 1) (Russo et al., 2009b). Thrombin, the main effector protease of the coagulation cascade, promotes pro-inflammatory responses and disrupts endothelial barrier permeability through the activation of PAR1 (Komarova et al., 2007). In contrast, activated protein C (APC), an anticoagulant protease, elicits anti-inflammatory responses and promotes endothelial barrier stabilization through PAR1 signalling (Feistritzer and Riewald, 2005; Finigan et al., 2005). APC is generated on the endothelial cell surface via activation of protein C by the thrombin–thrombomodulin complex. APC bound to endothelial protein C receptor (EPCR) cleaves and inactivates coagulation factors Va and VIIa diminishing thrombin generation and induces cellular responses through the activation of PAR1 (Riewald et al., 2002; Mosnier et al., 2007). These findings raise the question of how two different proteases can activate the same receptor to elicit opposite cellular responses.
Figure 5.
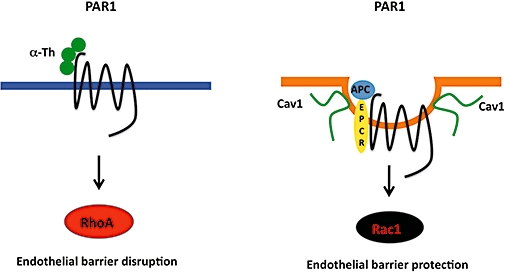
Protease-selective protease-activated receptor-1 (PAR1) activation and signalling is mediated by compartmentalization in caveolae. Thrombin efficiently binds to and cleaves PAR1 to elicit preferential activation of G12/13 signalling and Rho activation in endothelial cells that results in endothelial barrier disruption. In contrast, the activated protein C (APC) cofactor endothelial protein C receptor, PAR1 and signalling effectors sequester in caveolae, a subtype of lipid raft, and promote Rac1 activation and endothelial barrier stabilization.
Our recent work demonstrates that compartmentalization of PAR1 in caveolae is essential for APC-dependent cytoprotective signalling in endothelial cells (Figure 5) (Russo et al., 2009a). We hypothesized that if the difference between thrombin and APC signalling were due to efficiency in PAR1 cleavage and activation we would expect to observe quantitative, but not qualitative differences in signalling responses. Thus, to determine whether this was indeed the case we examined the ability of thrombin and APC to activate RhoA and Rac1, small GTPases that differentially regulate endothelial barrier permeability (Komarova et al., 2007). We found that thrombin caused robust RhoA signalling but not Rac1 activation, whereas APC stimulated a marked increase in Rac1 activation but not RhoA signalling (Russo et al., 2009a), consistent with the opposing functions of these proteases on endothelial barrier integrity. Moreover, one mechanism that contributes to the differential activation of PAR1 by thrombin versus APC involves localization of the receptor to caveolae, a subtype of lipid rafts (Figure 5). Caveolae are enriched in cholesterol and caveolin-1, a structural protein essential for caveolae formation in endothelial cells (Razani et al., 2001). The APC cofactor EPCR, PAR1 and Gi partition into lipid rafts and cofractionate with caveolin-1 on sucrose gradients (Bae et al., 2007a,b;), suggesting that these molecules exist as a preassembled complex poised to signal after APC generation. Several reports indicate that APC's proteolytic activity is required for PAR1-dependent cytoprotective signalling and that APC cleaves the N-terminus of PAR1 (Riewald et al., 2002; Ludeman et al., 2004; Mosnier et al., 2004); however, the precise cleavage site of PAR1 that confers cytoprotective signalling by APC remains to be determined. Together these studies suggest that localization of PAR1 and EPCR with specific G protein signalling effectors stabilizes a distinct active receptor conformation that favours endothelial barrier protective signalling. Interestingly, caveolae are also required for the activation of PAR2 by the tissue factor-coagulant protease factor VIIa complex but not by synthetic peptide agonists (Awasthi et al., 2008); however, it was not determined whether the different agonists caused biased signalling.
In human platelets, collagen initiates activation of matrix metalloprotease-1 (MMP-1) on the platelet surface; MMP-1 then cleaves and activates platelet PAR1 (Trivedi et al., 2009). Interestingly, MMP-1 cleaves the N-terminus of PAR1 at a different site from that previously reported for thrombin, but surprisingly MMP-1 signals comparably to thrombin to induce platelet shape change. However, previous studies showed that MMP-1 and thrombin differentially activated gene transcription through PAR1 activation in endothelial cells to promote angiogenesis (Blackburn and Brinckerhoff, 2008), suggesting that the two proteases elicit distinct cellular signalling responses through the activation of the same receptor. Other metalloproteases such as disintegrin and metalloprotease 17 (ADAM17)/TACE have been shown to cleave PAR1 at sites that are unproductive and render PAR1 unresponsive to thrombin signalling (Ludeman et al., 2004). Plasmin, a serine protease generated in plasma by urokinase and tissue plasminogen activator cleavage of plasminogen, cleaves PAR1 at multiple sites, which either activates or incapacitates the receptor depending on the cleavage site (Kuliopulos et al., 1999). In astrocytes, plasmin functions as an endogenous activator of PAR1 and increases Ca2+ mobilization, which potentiates N-methyl-d-aspartate receptor function in hippocampal neurons (Mannaioni et al., 2008). Thus, it remains to be determined whether proteases such as MMPs and/or plasmin cleave and activate PAR1, which results in the stabilization of an active receptor conformation that is able to induce selective signalling responses that are distinct from thrombin.
PAR crosstalk with other receptors
Although typically proteolytic cleavage leads to activation of the same receptor, there is evidence of crosstalk between different PARs. In murine platelets and transfected COS-7 cells, human PAR3 binds to thrombin with high affinity but does not appear to signal (Nakanishi-Matsui et al., 2000). Instead, PAR3 localizes thrombin to facilitate activation of PAR4, a receptor with low affinity for thrombin, to elicit platelet activation (Nakanishi-Matsui et al., 2000). In addition, PAR3 dimerizes with PAR1 and consequently potentiates thrombin signalling in endothelial cells, suggesting that PAR3 functions as an allosteric modulator of PAR1 signalling in certain cell types (McLaughlin et al., 2007). Another type of PAR crosstalk occurs in endothelial cells, where the tethered ligand domain of signalling defective cleaved PAR1 containing a mutation in a critical proline residue in the second extracellular loop, transactivates PAR2 (O'Brien et al., 2000). In addition, during progression of sepsis, a severe inflammatory condition, activated PAR1 switches from endothelial-disruptive signalling to protective signalling via transactivation of PAR2, a receptor whose expression is up-regulated in endothelial cells during sepsis (Kaneider et al., 2007).
In addition to other PARs, PAR1 has been shown to modulate signalling of other GPCRs including the sphingosine 1-phosphate (S1P) receptor and platelet-activating factor receptor (PAFR). Interestingly, the endothelial barrier protective effects elicited by APC activation of PAR1 involve crosstalk with S1P1 receptor, but not the S1P3 receptor (Feistritzer and Riewald, 2005). The sphingolipid S1P is generated intracellularly by sphingosine kinases 1 and 2 (SPK1) and depletion of SPK1 or S1P1 receptor expression by RNAi resulted in the loss of APC-induced barrier protective signalling, a process that requires expression of PAR1 (Feistritzer and Riewald, 2005; Russo et al., 2009a). These findings indicate that S1P is an important mediator of endothelial barrier integrity. In contrast, dendritic cell signalling by PAR1 induces S1P activation of the S1P3 receptor to promote inflammation in decompensated innate immune response (Niessen et al., 2008). In melanoma, PAR1 expression is important for basal PAF production and PAFR expression, which mediates induction of the melanoma adhesion molecule via activation of the cAMP-responsive element-binding protein and tumour progression (Melnikova et al., 2009).
In addition to S1P and PAF, activation of PARs stimulates production and release of a variety of cytokines, chemokines and growth factors including interleukin (IL)-6, IL-8, VEGF, platelet-derived growth factor and modulates the activation of integrins to elicit various cellular responses (Figure 1) (Coughlin and Camerer, 2003; Coughlin, 2005). Transactivation of epidermal growth factor receptors and ErbB2 by PAR1 and PAR2 through activation of ADAMs or MMPs, which in turn releases membrane-anchored ligands such as heparin-binding epidermal growth factor and transforming growth factor-α or through intracellular signalling pathways have been reported in both normal and transformed cells (Daub et al., 1996; Prenzel et al., 1999; Darmoul et al., 2004; Melnikova et al., 2009). In recent work, PAR2 has been demonstrated to signal synergistically with Toll-like receptor 4 (TLR4) (Chi et al., 2001; Rallabhandi et al., 2008). The Toll-like receptors are type I transmembrane glycoproteins that mediate innate immune responses. The coexpression of TLR4 complex with PAR2 caused synergistic signalling to inflammatory responses that was distinct from PAR2 or TLR4 signalling alone and required specific TLR4 adaptor components (Rallabhandi et al., 2008). Interestingly, unlike other signalling crosstalk the PAR2 and TLR4 receptor cooperativity appears to be dependent on physical interaction. This is similar to that reported for receptor-activity-modifying proteins (RAMPs), which are type I transmembrane proteins that interact with many GPCRs to modulate receptor signalling and trafficking (Sexton et al., 2009), although RAMPs have not been reported to interact with PARs.
Conclusions
PARs are expressed in a variety of cell types and mediate critical cellular responses important for haemostasis, thrombosis, and inflammatory and proliferative responses associated with tissue injury (Coughlin, 2005). With the exception of coagulant proteases, the specific proteases responsible for activation of PARs expressed in tissues other than the vasculature remain poorly characterized. Several recent studies provide compelling evidence that activation of PARs by different proteases or in complex with other receptors elicit distinct signalling outcomes and raise the possibility that distinct active receptor conformations are differentially regulated. In addition to desensitization, the internalization, recycling and lysosomal sorting of PARs is crucial for the spatial and temporal regulation of receptor signalling and appropriate cellular responses. However, the mechanisms responsible for regulation of PAR signalling when distinctly activated by different proteases or in complex with other receptors remain poorly understood. Indeed, activation of PAR1 by APC causes minimal receptor internalization and lysosomal degradation (Schuepbach et al., 2008; Russo et al., 2009a), a process critical for termination of thrombin-activated PAR1 signalling (Trejo et al., 1998). The mechanisms responsible for APC-activated PAR1 desensitization are not known. The finding that PARs can be differentially activated by distinct ligands also provides new important therapeutic opportunities. Moreover, as alterations in PAR trafficking can promote changes in the magnitude, duration and localization of receptor signalling, it is critical to understand the processes that control sorting of these receptors through the endocytic pathway. An understanding of the mechanisms by which specific adaptor proteins and processes regulate ubiquitin-dependent and -independent PAR lysosomal sorting will enable us to develop new strategies to manipulate receptor signalling and provide novel targets for the development of drugs that can be used in the prevention and treatment of a wide range of human diseases and cancer progression. This is particularly important because there are currently no available drugs that selectively block PAR signalling although a new PAR1 antagonist compound based on the natural product himbacine is currently undergoing Phase III clinical trials for acute coronary syndrome (Chackalamannil et al., 2008). Finally, the development of other drugs such as allosteric modulators that could promote PAR internalization independent of cellular signalling or cause sustained signalling without receptor down-regulation could also be potentially useful in certain disease scenarios.
Acknowledgments
The work on PARs in the author's laboratory was funded by an American Heart Association Established Investigator Award and National Institutes of Health Grants HL073328 and GM090689. UJK Soh is supported by an American Heart Association Postdoctoral Fellowship Award.
Glossary
Abbreviations:
- AP-2
adaptor protein complex-2
- APC
activated protein C
- EPCR
endothelial protein C receptor
- ERK1, 2
extracellular signal-regulated kinases 1 and 2
- ESCRT
endosomal-sorting complex required for transport
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- HRS
hepatocyte growth factor-regulated tyrosine kinase substrate
- ILVs
intraluminal vesicles
- MVBs
multivesicular bodies
- PAR
protease-activated receptor
- PKC
protein kinase C
- Tsg101
tumour suppressor gene 101
- VEGF
vascular endothelial growth factor
Conflict of interest
The authors state no conflict of interest.
References
- Al-Ani B, Hansen KK, Hollenberg MD. Proteinase-activated receptor-2: key role of amino-terminal dipeptide residues of the tethered ligand for receptor activation. Mol Pharm. 2004;65:149–156. doi: 10.1124/mol.65.1.149. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez CE. On the origins of arrestin and rhodopsin. BMC Evol Biol. 2008;8:222. doi: 10.1186/1471-2148-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi V, Mandal SK, Papanna V, Rao LVM, Pendurthi UR. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler Thromb Vasc Biol. 2008;27:1447–1455. doi: 10.1161/ATVBAHA.107.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor-1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007a;110:3909–3916. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JS, Yang L, Rezaie AR. Receptors of the protein C activation and activated protein C signaling pathways are colocalized in lipid rafts of endothelial cells. Proc Natl Acad Sci USA. 2007b;104:2867–2872. doi: 10.1073/pnas.0611493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn JS, Brinckerhoff CE. Matrix metalloproteinase-1 and thrombin differentially activate gene expression in endothelial cells via PAR-1 and promote angiogenesis. Am J Pathol. 2008;173:1736–1746. doi: 10.2353/ajpath.2008.080512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackhart BD, Ruslim-Litrus L, Lu CC, Alves VL, Teng W, Scarborough RM, et al. Extracellular mutations of protease-activated receptor-1 result in differential activation by thrombin and thrombin receptor agonist peptide. Mol Pharm. 2000;58:1178–1187. doi: 10.1124/mol.58.6.1178. [DOI] [PubMed] [Google Scholar]
- Bohm SK, Khitin LM, Grady EF, Aponte G, Payan DG, Bunnett NW. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem. 1996;271:22003–22016. doi: 10.1074/jbc.271.36.22003. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton J, Bujny M, Peter BJ, Oorschot VMJ, Rutherford A, Meller H, et al. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high curvature membranes and 3-phosphoinositides. Curr Biol. 2004;14:1791–1800. doi: 10.1016/j.cub.2004.09.077. [DOI] [PubMed] [Google Scholar]
- Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, et al. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- Chen CH, Paing MM, Trejo J. Termination of protease-activated receptor-1 signaling by β-arrestins is independent of receptor phosphorylation. J Biol Chem. 2004;279:10020–10031. doi: 10.1074/jbc.M310590200. [DOI] [PubMed] [Google Scholar]
- Chen J, Ishii M, Wang L, Ishii K, Coughlin SR. Thrombin receptor activation: confirmation of the intramolecular tethered liganding hypothesis and discovery of an alternative intermolecular liganding mode. J Biol Chem. 1994;269:16041–16045. [PubMed] [Google Scholar]
- Chi L, Li Y, Stehno-Bittel L, Gao J, Morrison DC, Stechschulte DJ, et al. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J Interferon Cytokine Res. 2001;21:231–240. doi: 10.1089/107999001750169871. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111:25–27. doi: 10.1172/JCI17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen PJ. Endosomal sorting and signalling: an emerging role for sorting nexins. Nat Rev Mol Cell Biol. 2008;7:574–582. doi: 10.1038/nrm2427. [DOI] [PubMed] [Google Scholar]
- Darmoul D, Gratio V, Devaud H, Labrurthe M. Protease-activated receptor-2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor transactivation. J Biol Chem. 2004;279:20297–20934. doi: 10.1074/jbc.M401430200. [DOI] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- DeFea KA, Zalevski J, Thoma MS, Dery O, Mullins RD, Bunnett NW. β-Arrestin-dependent endocytosis of proteinase-activated receptor-2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dery O, Thoma MS, Wong H, Grady EF, Bunnett NW. Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein. J Biol Chem. 1999;274:18524–18535. doi: 10.1074/jbc.274.26.18524. [DOI] [PubMed] [Google Scholar]
- Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circ Res. 2006;99:570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- Edeling MA, Smith C, Owen D. Life of a clathrin coat: insights from clathrin and AP structures. Nat Mol Cell Biol. 2006;7:32–44. doi: 10.1038/nrm1786. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine-1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- Ferguson SS. Phosphorylation-independent attenuation of GPCR signalling. Trends Pharmacol Sci. 2007;28:173–179. doi: 10.1016/j.tips.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, et al. Activated protein C mediates novel lung endothelial barrier enhancement. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- Furman MI, Krueger LA, Frelinger AL, 3rd, Barnard MR, Mascelli MA, Nakada MT, et al. GPIIb-IIIa antagonist-induced reduction in platelet surface factor V/Va binding and phosphatidylserine expression in whole blood. Thromb Haemost. 2000a;84:492–498. [PubMed] [Google Scholar]
- Furman MI, Nurden P, Berndt MC, Nurden AT, Benoit SE, Barnard MR, et al. The cleaved peptide of PAR1 results in a redistribution of the platelet surface GPIb-IX-V complex to the surface-connected canalicular system. Thromb Haemost. 2000b;84:897–903. [PubMed] [Google Scholar]
- Ge L, Shenoy SK, Lefkowitz RJ, DeFea K. Constitutive protease-activated-receptor-2 mediated migration of MDA-MB-231 breast cancer cells requires both β-arrestin-1 and 2. J Biol Chem. 2004;279:55419–55424. doi: 10.1074/jbc.M410312200. [DOI] [PubMed] [Google Scholar]
- Gerszten RE, Chen J, Ishii M, Ishii K, Wang L, Nanevicz T, et al. The thrombin receptor's specificity for agonist peptide is defined by its extracellular surface. Nature. 1994;368:648–651. doi: 10.1038/368648a0. [DOI] [PubMed] [Google Scholar]
- Goldman PS, Nathanson NM. Differential Role of the carboxyl-terminal tyrosine in down-regulation and sequestration of the m2 muscarinic acetylcholine receptor. J Biol Chem. 1994;269:15640–15645. [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the Beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Gullapalli A, Wolfe BL, Griffin CT, Magnuson T, Trejo J. An essential role for SNX1 in lysosomal sorting of protease-activated receptor-1: evidence for retromer, Hrs and Tsg101 independent functions of sorting nexins. Mol Biol Cell. 2006;17:1228–1238. doi: 10.1091/mbc.E05-09-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Liu G, Profirovic J, Niu J, Voyno-Yasenetskaya T. Zyxin is involved in thrombin signaling via interaction with PAR-1 receptor. FASEB J. 2009;12:4193–4206. doi: 10.1096/fj.09-131862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Stevens RC. Discovery of new GPCR biology: one receptor structure at a time. Structure. 2009;17:8–14. doi: 10.1016/j.str.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Hasdemir B, Bunnett NW, Cottrell GS. Hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J Biol Chem. 2007;282:29646–29657. doi: 10.1074/jbc.M702974200. [DOI] [PubMed] [Google Scholar]
- Hasdemir B, Murphy JE, Cottrell GS, Bunnett NW. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J Biol Chem. 2009;284:28453–28466. doi: 10.1074/jbc.M109.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein L, Ishii K, Coughlin SR, Kobilka BK. Intracellular targeting and trafficking of thrombin receptors: a novel mechanism for resensitization of a G protein-coupled receptor. J Biol Chem. 1994;269:27719–27726. [PubMed] [Google Scholar]
- Hicke L, Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich V, Sigler PA. The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Ishii K, Hein L, Kobilka B, Coughlin SR. Kinetics of thrombin receptor cleavage on intact cells: relation to signaling. J Biol Chem. 1993;268:9780–9786. [PubMed] [Google Scholar]
- Ishii K, Chen J, Ishii M, Koch WJ, Freedman NJ, Lefkowitz RJ, et al. Inhibition of thrombin receptor signaling by a G protein-coupled receptor kinase. Functional specificity among G protein-coupled receptor kinases. J Biol Chem. 1994;269:1125–1130. [PubMed] [Google Scholar]
- Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, Bunnett NW. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem. 2005;280:16076–16087. doi: 10.1074/jbc.M500109200. [DOI] [PubMed] [Google Scholar]
- Jala VR, Shao WH, Haribabu B. Phosphorylation-independent beta-arrestin translocation and internalization of leukotriene B4 receptors. J Biol Chem. 2005;280:4880–4887. doi: 10.1074/jbc.M409821200. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;12:1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova YA, Mehta D, Malik AB. Dual regulation of endothelial junctional permeability. Sci STKE. 2007;2007:re8. doi: 10.1126/stke.4122007re8. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- Kuliopulos A, Covic L, Seely SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;38:4572–4585. doi: 10.1021/bi9824792. [DOI] [PubMed] [Google Scholar]
- Laporte S, Oakley RH, Zhang J, Holt JA, Ferguson SSG, Caron MG, et al. The β2-adrenergic receptor/β-arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Lerner DJ, Chen M, Tram T, Coughlin SR. Agonist recognition by proteinase-activated receptor 2 and thrombin receptor. Importance of extracellular loop interactions for receptor function. J Biol Chem. 1996;271:13943–13947. [PubMed] [Google Scholar]
- Lin CH, MacGurn JA, Chu T, Stefan CJ, Emr SD. Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell. 2008;135:714–725. doi: 10.1016/j.cell.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Lohse M, Benovic J, Codina J, Caron M, Lefkowitz R. β-arrestin: a protein that regulates B-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Ludeman MJ, Zheng YW, Ishii K, Coughlin SR. Regulated shedding of PAR1 N-terminal exodomain from endothelial cells. J Biol Chem. 2004;279:18592–18599. doi: 10.1074/jbc.M310836200. [DOI] [PubMed] [Google Scholar]
- Luo W, Wang Y, Hanck T, Stricker R, Reiser G. Jab1, a novel protease-activated receptor-2 (PAR-2)-interacting protein, is involved in PAR-2-induced activation of activated protein-1. J Biol Chem. 2006;281:7927–7936. doi: 10.1074/jbc.M510784200. [DOI] [PubMed] [Google Scholar]
- Luo W, Wang Y, Reiser G. p24A, a type I transmembrane protein, controls ARF1-dependent resensitization of protease-activated receptor-2 by influence on receptor trafficking. J Biol Chem. 2007;282:30246–30255. doi: 10.1074/jbc.M703205200. [DOI] [PubMed] [Google Scholar]
- McLaughlin JN, Shen L, Holinstat M, Brooks JD, DiBenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem. 2005;280:25048–25059. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci USA. 2007;104:5662–5667. doi: 10.1073/pnas.0700763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan VB, Pai KS, Lau A, Cunningham DD. Creatine kinase, an ATP-generating enzyme, is required for thrombin receptor signaling to the cytoskeleton. Proc Natl Acad Sci USA. 2000;97:12062–12067. doi: 10.1073/pnas.97.22.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaioni G, Orr AG, Hamill CE, Yuan H, Pedone KH, McCoy KL, et al. Plasmin potentiates synaptic N-methly-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J Biol Chem. 2008;283:20600–20611. doi: 10.1074/jbc.M803015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Paing MM, Temple BRS, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmcol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikova VO, Balasubramanian K, Villares GJ, Dobroff AS, Zigler M, Wang H, et al. Crosstalk between protease-activated receptor 1 and platelet-activating factor receptor regulates melanoma cell adhesion molecule (MCAM/MUC18) expression and melanoma metastasis. J Biol Chem. 2009;284:28845–28855. doi: 10.1074/jbc.M109.042150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molino M, Bainton DF, Hoxie JA, Coughlin SR, Brass LF. Thrombin receptors on human platelets. Initial localization and subsequent redistribution during platelet activation. J Biol Chem. 1997;272:6011–6017. doi: 10.1074/jbc.272.9.6011. [DOI] [PubMed] [Google Scholar]
- Moore CAC, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:19.11–19.32. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104:1740–1744. doi: 10.1182/blood-2004-01-0110. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Gurevich VV, Preninger A, Hamm HE, Bader M-F, Fazleabas AT, et al. Aspartic acid 564 in the third cytoplasmic loop of the luteinizing hormone/choriogonadotropin receptor is crucial for phosphorylation-independent interaction with arrestin2. J Biol Chem. 2002;277:17916–17927. doi: 10.1074/jbc.M110479200. [DOI] [PubMed] [Google Scholar]
- Nakanishi-Matsui M, Zheng YW, Weiss EJ, Sulciner D, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, et al. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008;452:654–658. doi: 10.1038/nature06663. [DOI] [PubMed] [Google Scholar]
- Nikko E, Sullivan JA, Pelham HR. Arrestin-like proteins mediate ubiquitination and endocytosis of the yeast metal transporter Smf1. EMBO Rep. 2008;9:1216–1221. doi: 10.1038/embor.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, et al. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275:13502–13509. doi: 10.1074/jbc.275.18.13502. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. How do receptors activate G proteins? Adv Protein Chem. 2007;74:67–93. doi: 10.1016/S0065-3233(07)74002-0. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Pai KS, Mahajan VB, Lau A, Cunningham DD. Thrombin receptor signaling to cytoskeleton requires Hsp90. J Biol Chem. 2001;276:32642–32647. doi: 10.1074/jbc.M104212200. [DOI] [PubMed] [Google Scholar]
- Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. β-arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J Biol Chem. 2002;277:1292–1300. doi: 10.1074/jbc.M109160200. [DOI] [PubMed] [Google Scholar]
- Paing MM, Temple BRS, Trejo J. A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1: multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J Biol Chem. 2004;279:21938–21947. doi: 10.1074/jbc.M401672200. [DOI] [PubMed] [Google Scholar]
- Paing MM, Johnston CA, Siderovski DP, Trejo J. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol Cell Biol. 2006;28:3221–3242. doi: 10.1128/MCB.26.8.3231-3242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Katzmann DJ. Biogenesis and function of multivesicular bodies. Annu Rev Cell Dev Biol. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher J, Lohse MJ, Codina J, Caron MG, Lefkowitz RJ. Desensitization of the isolated beta 2-adrenergic receptor by beta-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry. 1992;31:3193–3197. doi: 10.1021/bi00127a021. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Rallabhandi P, Nhu QM, Toshchakov VY, Piao W, Medvedev AE, Hollenberg MD, et al. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J Biol Chem. 2008;283:24314–24325. doi: 10.1074/jbc.M804800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol. 2009;76:791–801. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Ricks T, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem. 2009;284:34444–34457. doi: 10.1074/jbc.M109.048942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor-1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- Roosterman D, Schmidlin F, Bunnett NW. Rab5a and rab11a mediate agonist-induced trafficking of protease-activated receptor-2. Am J Physiol Cell Physiol. 2003;284:C1319–C1329. doi: 10.1152/ajpcell.00540.2002. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Davis NG. Ubiquitination of the yeast a-factor receptor. J Cell Biol. 1996;134:661–674. doi: 10.1083/jcb.134.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A, Soh JK, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci USA. 2009a;106:6393–6397. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A, Soh UJ, Trejo J. Proteases display biased agonism at protease-activated receptors: location matters! Mol Interv. 2009b;9:87–96. doi: 10.1124/mi.9.2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarborough RM, Naughton MA, Teng W, Hung DT, Rose J, Vu TK, et al. Tethered ligand agonist peptides. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J Biol Chem. 1992;267:13146–13149. [PubMed] [Google Scholar]
- Schuepbach RA, Feistritzer C, Brass LF, Riewald M. Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood. 2008;111:2667–2673. doi: 10.1182/blood-2007-09-113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminario-Vidal L, Kreda S, Jones L, O'Neal W, Trejo J, Boucher RC, et al. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of Rho- and Ca2+-dependent signaling pathways. J Biol Chem. 2009;284:20638–20648. doi: 10.1074/jbc.M109.004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton PM, Poyner DR, Simms J, Christopoulos A, Hay DL. Modulating receptor function through RAMPs: can they represent drug targets in themselves? Drug Discov Today. 2009;14:413–419. doi: 10.1016/j.drudis.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. 2000;275:25216–25221. doi: 10.1074/jbc.M004589200. [DOI] [PubMed] [Google Scholar]
- Shi H, Rojas R, Bonifacino JS, Hurley JH. The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nat Struct Mol Biol. 2006;13:540–548. doi: 10.1038/nsmb1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, et al. Multiple independent functions of arrestins in regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharm. 2005;67:1–10. doi: 10.1124/mol.104.006072. [DOI] [PubMed] [Google Scholar]
- Tiruppathi C, Yan W, Sandoval R, Naqvi T, Pronin AN, Benovic JL, et al. G protein-coupled receptor kinase-5 regulates thrombin-activated signaling in endothelial cells. Proc Natl Acad Sci USA. 2000;97:7440–7445. doi: 10.1073/pnas.97.13.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AB, Butcher AJ, Kong KC. Location, location, location … site specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;8:413–420. doi: 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo J, Coughlin SR. The cytoplasmic tails of protease-activated receptor-1 and substance P receptor specify sorting to lysosomes vs recycling. J Biol Chem. 1999;274:2216–2224. doi: 10.1074/jbc.274.4.2216. [DOI] [PubMed] [Google Scholar]
- Trejo J, Hammes SR, Coughlin SR. Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc Natl Acad Sci USA. 1998;95:13698–13702. doi: 10.1073/pnas.95.23.13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo J, Altschuler Y, Fu H-W, Mostov KE, Coughlin SR. Protease-activated receptor downregulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. J Biol Chem. 2000;275:31255–31265. doi: 10.1074/jbc.M003770200. [DOI] [PubMed] [Google Scholar]
- Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K, et al. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991a;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Vu TK, Wheaton VI, Hung DT, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991b;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhou Y, Szabo K, Haft CR, Trejo J. Down-regulation of protease-activated receptor-1 is regulated by sorting nexin 1. Mol Biol Cell. 2002;13:1965–1976. doi: 10.1091/mbc.E01-11-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe BL, Marchese A, Trejo J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J Cell Biol. 2007;177:905–916. doi: 10.1083/jcb.200610154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe BL, Trejo J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic. 2007;8:1–9. doi: 10.1111/j.1600-0854.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- Xia S, Dun XP, Hu PS, Kjaer S, Zheng K, Qian Y, et al. Postendocytotic traffic of the galanin R1 receptor: a lysosomal signal motif on the cytoplasmic terminus. Proc Natl Acad Sci USA. 2008;105:5609–5613. doi: 10.1073/pnas.0801456105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zania P, Gourni D, Aplin AC, Nicosia RF, Flordellis CS, Maragoudakis ME, et al. Parstatin, the cleaved peptide on proteinase-activated receptor 1 activation, is a potent inhibitor of angiogenesis. J Pharmacol Exp Ther. 2009;328:378–389. doi: 10.1124/jpet.108.145664. [DOI] [PubMed] [Google Scholar]