

Abstract
Background and purpose:
The volatile anaesthetic isoflurane protects the heart from ischaemia and reperfusion (I/R) injury when applied at the onset of reperfusion [anaesthetic postconditioning (APoC)]. However, the mechanism of APoC-mediated protection is unknown. In this study, we examined the effect of APoC on mitochondrial bioenergetics, mitochondrial matrix pH (pHm) and cytosolic pH (pHi), and intracellular Ca2+.
Experimental approach:
Cardiac mitochondria from Wistar rats were isolated after in vivo I/R with or without APoC (1.4%-vol isoflurane, 1 minimum alveolar concentration), and mitochondrial permeability transition pore (mPTP) opening, mitochondrial membrane potential (ΔΨm), and oxygen consumption were assessed. In isolated cardiomyocytes and isolated mitochondria I/R injury was produced in vitro, with or without APoC (0.5 mM isoflurane). Intracellular Ca2+, pHm, pHi and ΔΨm were monitored with SNARF-1, TMRE and fluo-4, respectively. Myocyte survival was assessed when APoC was induced at pH 7.4 and 7.8. In isolated mitochondria oxygen consumption and ATP synthesis were measured.
Key results:
In vivo APoC protected against mPTP opening, slowed mitochondrial respiration and depolarized mitochondria. APoC decreased the number of hypercontracted cardiomyocytes at pH 7.4, but not at pH 7.8. APoC attenuated intracellular Ca2+ accumulation, maintained lower pHm, and preserved ΔΨm during reoxygenation. Isoflurane did not affect the regulation of cytosolic pH. In mitochondria, APoC preserved ATP production rate and respiration.
Conclusions and implications:
At reperfusion, APoC inhibited mitochondrial respiration, depolarized mitochondria and acidified pHm. These events may lead to inhibition of mPTP opening and, consequently, to preserved ΔΨm and ATP synthesis. This reduces intracellular Ca2+ overload and cell death.
This article is commented on by Moncada, pp. 217–219 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00706.x and to view related papers in this issue by Puerta et al. and Kurz et al. visit http://dx.doi.org/10.1111/j.1476-5381.2010.00663.x and http://dx.doi.org/10.1111/j.1476-5381.2010.00656.x
Keywords: anaesthetic-induced postconditioning, cardioprotection, volatile anaesthetics, isoflurane, mitochondria
Introduction
Numerous studies have shown that volatile anaesthetics can protect myocardium not only when applied before a harmful ischaemic event [anaesthetic preconditioning (APC)], but also when applied at the beginning of reperfusion. This phenomenon is known as anaesthetic postconditioning (APoC) (Chen et al., 2008; He et al., 2008; Huhn et al., 2008; Lee et al., 2008). Cardioprotective mechanisms produced by APoC were shown to involve activation of phosphoinositide 3-kinase, extracellular regulated kinases 1 and 2 (ERK1 and ERK1), the 70-kDa ribosomal protein S6 kinase, endothelial nitric oxide synthase (eNOS), mitochondrial KATP channels and inhibition of glycogen synthase kinase 3-β (Tanaka et al., 2002; Chiari et al., 2005; Krolikowski et al., 2005; Weihrauch et al., 2005; Pagel et al., 2006). However, it is unlikely that stimulation of pro-survival signalling pathways occurs rapidly enough to prevent damage resulting from the initial injury during reperfusion. Recently, attention has focused on mitochondria as a target of cardioprotection by volatile anaesthetics (Ljubkovic et al., 2007; Pravdic et al., 2009), and it is feasible that immediate effects of these agents on mitochondria could contribute to protection at the onset of reperfusion.
Reperfusion of ischaemic myocardium with an acidotic buffer during the first minutes of reperfusion, as occurring during ischaemic postconditioning, exerts cardioprotection (Cohen et al., 2007). Acidosis is known to suppress mitochondrial permeability transition pore (mPTP) opening, prevent the development of hypercontracture and reduce calpain-dependent proteolysis (Piper et al., 2004; Cohen et al., 2007; Inserte et al., 2009). In order to maintain normal Ca2+ homeostasis, the ability of mitochondria to resume ATP synthesis after hypoxia needs to be preserved (Piper et al., 2004). Furthermore, inhibition of contractile mechanisms and mPTP opening induced by acidosis is crucial for effective postconditioning (Cohen et al., 2008). The effect of volatile anaesthetics such as isoflurane during reperfusion on mitochondrial function and bioenergetics, mitochondrial and cytosolic pH and intracellular Ca2+ is not known.
Therefore, in this investigation we tested the hypothesis that APoC-induced protection against hypoxia and reoxygenation (H/R) injury is dependent on changes in mitochondrial bioenergetics, including pHm, during reoxygenation. Our results showed that APoC triggered a significant protection of mitochondrial function along with depolarization of mitochondrial membrane potential and acidification of the mitochondrial matrix.
Methods
Animals
All procedures that involved the use of animals were approved by the Institutional Animal Use and Care Committee of the Medical College of Wisconsin (Milwaukee, Wisconsin), and all experiments were conducted in accordance with US National Institutes of Health standards (NIH Publication 95-23, revised 1996).
In vivo myocardial ischaemia and reperfusion injury and APoC with isoflurane
Myocardial infarction was performed on male Wistar rats (280–420 g, corresponding to an age of 5–9 weeks) as previously described (Ludwig et al., 2004). Animals were anaesthetized with thiobutabarbital sodium (100–150 mg·kg−1; Inactin, Sigma-Aldrich, St. Louis, MO, USA), tracheotomy was performed and the trachea cannulated. Rats were then ventilated with a rodent ventilator (Harvard Apparatus 683, South Natick, MA, USA), with positive end-expiratory pressure using an air-oxygen mixture. After a left thoracotomy was performed in the fifth intercostal space, the pericardium was opened. Myocardial ischaemia was induced by occluding the left descending coronary artery, and reperfusion was initiated by loosening the suture. Occlusion of the coronary artery for 30 min was followed by 15 min of reperfusion (n= 8 per group). The short reoxygenation protocol was choosen to study the acute changes in mitochondrial function following APoC (Paillard et al., 2009). Isoflurane postconditioning group received isoflurane (Baxter, Deerfield, IL, USA) at 1.0 minimum alveolar concentration (corresponding to 1.4%), 3 min before and 2 min during the beginning of reoxygenation using a vaporizer (Ohio Medical Products 100F, Madison, WI, USA). Control rats remained without isoflurane treatment, and sham rats underwent tracheal intubation and thoracotomy with no coronary artery occlusion throughout the experiment. After 15 min of reoxygenation, left ventricular area at risk (left ventricle below the left coronary artery occlusion) was excised and mitochondria were isolated as described in the further discussion (preparation of mitochondria).
Cardiac myocyte isolation
Ventricular myocytes were obtained by enzymatic dissociation with 0.5 mg·mL−1 collagenase type II (Invitrogen, Carlsberg, CA, USA) and 0.25 mg·mL−1 protease type XIV (Sigma-Aldrich, St. Louis, MO, USA), as previously described (Aizawa et al., 2004; Marinovic et al., 2005). After isolation, the myocytes were resuspended and stored in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered Tyrode solution (in mM: 132 NaCl, 10 HEPES, 5 d-glucose, 4.8 KCl, 1 CaCl2 and 1.2 MgCl2, pH 7.4 adjusted with 1 M NaOH) at 20–22°C for 1 h, and used for experiments within 5 h. All cells used in this study were rod shaped in appearance, had well-defined striations and did not spontaneously contract.
Laser scanning confocal microscopy
Isolated cardiomyocytes were studied in a hypoxic chamber (RC-21B, Warner Instruments, Hamden, CT, USA) mounted on the stage of a confocal microscope (Nikon Eclipse TE2000-U; Nikon, Tokyo, Japan) with a 40×/1.3 oil-immersion and 10×/0.45 dry objectives (Nikon). Data were analyzed using MetaMorph 6.2 software (Universal Imaging, West Chester, PA, USA), and NIH ImageJ software 1.41 (National Institutes of Health, Bethesda, MD, USA).
H/R injury of cardiomyocytes
The protocol for H/R was modified from a previously described model (O'Brien et al., 2008) that included hypoxia, acidosis, lactate accumulation, hyperkalaemia and substrate depletion. Cells were placed in a hypoxic chamber covered with a glass coverslip, which was sealed with high vacuum grease (Dow Corning Corporation, Midland, MI, USA) and perfused under controlled conditions. For baseline measurements, myocytes were superfused with normoxic Tyrode solution containing (in mM): 132 NaCl, 10 HEPES, 5 d-glucose, 4.8 KCl, 1 CaCl2 and 1.2 MgCl2 (pH 7.4 if not otherwise stated; equilibrated with room air; PO2 199.1 ± 14.1 mmHg). The same solution was used during reoxygenation. Hypoxia was produced by superfusing myocytes with HEPES-buffered glucose-free Tyrode solution containing (in mM): 118 NaCl, 10 HEPES, 8 KCl, 1 CaCl2, 1.2 MgCl2, 1 Na2HPO4 and 10 mM Na-L-lactate (pH 6.8; previously gassed with 100% N2 for 20 min; PO2 26.5 ± 2.8 mmHg). Decrease in PO2 was ∼85% which is comparable with other models of simulated ischaemia (Maddaford et al., 1999; Lu et al., 2005). To diminish contamination with room air, all solutions were placed in glass syringes and delivered to a recording chamber using a polyethylene air-tight tubing system. The PO2 of normal and hypoxic Tyrode solutions was measured with a gas analyzer (Radiometer ABL 505, Radiometer, Copenhagen, Denmark).
Experimental protocol for APoC in isolated cardiomyocytes
In the time control group, myocytes were perfused with normoxic Tyrode solution for 5 min. Ischaemia was simulated by switching perfusion to hypoxic Tyrode solution for 20 min. To simulate reperfusion, the perfusate was switched back to normoxic Tyrode solution for 35 min. Isoflurane was included in the perfusion solution for the first 5 min of simulated reoxygenation (APoC group). Lactate (as sodium lactate) concentrations were fixed at 20 mM to mimic conditions of cardiac ischaemia. An identical amount of NaCl was omitted in order to maintain constant osmolarity. Isoflurane was dissolved in Tyrode solution by sonication, to a final concentration of 0.75 ± 0.1 mM. Anaesthetic concentrations were measured by gas chromatography (Gas chromatograph GC-8A; Shimadzu, Kyoto, Japan). All experiments using cardiac myocytes were performed at room temperature.
Assessment of cellular viability
Assay of cellular viability was performed after 35 min of reoxygenation, as previously described (Faghihi et al., 2008). During the first 5 min of reoxygenation a perfusate at either pH 7.4 or 7.8 was used. Cell death was defined based on morphological characteristics. Rounded cells (ratio of diameters <2) were considered dead. The percentage of hypercontracted cells was calculated from the cell count at the beginning and at the end of reoxygenation. The location of the cells was monitored using a labelled grid to ensure counting of the same cardiomyocytes.
Measurement of mitochondrial pH
The pHm was measured in single cardiomyocytes using the dual emission, pH sensitive indicator, 5-(and-6)-carboxy SNARF-1 acetoxymethyl ester (SNARF-1; 5 µM; Invitrogen). Cells were loaded with SNARF-1 at 4°C for 30 min. Loaded cells were then washed with dye-free Tyrode solution at room temperature for additional 30 min to remove extracellular dye and to allow complete de-esterification of the dye. This loading procedure allowed SNARF-1 to distribute preferentially to mitochondria. To assure mitochondrial loading of SNARF-1, we examined colocalization of SNARF-1 with mitochondrial flavoproteins before each experiment. Measurements were performed using dual emission ratiometric analysis on a fluorescence microscope. Excitation was performed at 543 nm by a green HeNe laser, and the emitted fluorescence was simultaneously recorded at 650 and 590 nm. The ratio of emitted fluorescence at 650 and 590 nm was calculated and converted to pHm values (Buckler and Vaughan-Jones, 1990). A calibration curve was made by exposing the cells to a depolarizing high K+ buffer (140 mM KCl, 1.0 mM MgCl2, 5 mM d-glucose, 10 mM HEPES, in the presence of 10 µM nigericin) at pH 6.5, 7.5, and 8.5 (Salvi et al., 2002). The pH calibration data were averaged from 20 different cells.
Measurement of intracellular pH
Measurements of intracellular pH (pHi) of isolated cardiomyocytes were performed using SNARF-1 as described previously (Ch'en et al., 2008). Myocytes were loaded with SNARF-1 acetoxymethyl ester (5 µM) for 10 min at room temperature, followed by 10 min washout with dye-free solution (Figure 5A). Sodium hydrogen exchanger (NHE) activity was assessed after myocytes were exposed to an acid load consisting of 10 mM NH4Cl for 3 min (Vaughan-jones et al., 2009). In addition, experiments were conducted in sodium-free solution by replacing sodium with 132 mM N-methyl-D-glucamine. The effect of isoflurane on carbonic anhydrase activity was performed and calculated as described previously (Ch'en et al., 2008). To determine whether isoflurane induces pH changes due to increased base efflux through Cl- OH- and Cl- HCO3- exchangers, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS; 0.5 mM; Sigma-Aldrich) was used as inhibitor (Ch'en et al., 2008). The effect of isoflurane on metabolically-induced pH changes was examined in glucose-free HEPES-buffered Tyrode solution in the presence of 10 mM 2-deoxy-D-glucose.
Figure 5.
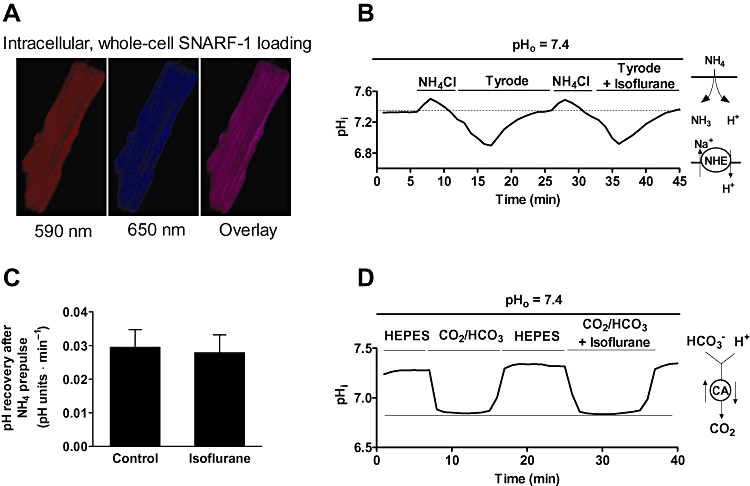
Isoflurane does not modulate NHE exchanger and carbonic anhydrase activities. (A) Representative images showing whole-cell loading of SNARF-1. (B) Representative trace of pHi recovery from the cardiomyocyte following exposure to NH4Cl (10 mM) in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered Tyrode solution, pHo 7.4. The trace demonstrates that after two consecutive NH4Cl pulses, pHi recovery from an intracellular acid load was not different in the presence of isoflurane. (C) Summary for recovery of pHi from NH4Cl -induced acidosis in the presence and subsequent absence of 0.5 mM isoflurane. (D) Effect of isoflurane on carbonic anhydrase activity following intracellular acid loading induced by application of 5% CO2/HCO-3. The magnitude of acidosis was not different in the presence of isoflurane. Data are presented as means ± standard deviation. Traces are representative from at least five measurements. SNARF-1, 5-(and-6)-carboxy SNARF-1, acetoxymethyl ester; NHE, Na+-H+ exchanger, CA, carbonic anhydrase.
Measurement of intracellular Ca2+
Changes in intracellular Ca2+ were monitored with the Ca2+ sensitive indicator fluo-4 AM. Myocytes were incubated with fluo-4 AM (2 µM; Invitrogen) at room temperature for 20 min. Following incubation, the myocytes were superfused with dye-free Tyrode solution for additional 15 min at room temperature to remove extracellular dye and to allow complete de-esterification of the dye. Fluo-4 was excited with the 488 nm line of an argon laser, and the emitted fluorescence was collected at 520 nm. Changes in intracellular Ca2+ were expressed as a percent relative to the baseline (F/F0× 100), where F is the measured fluo-4 fluorescence and F0 is the baseline fluo-4 fluorescence at the beginning of each experiment.
Monitoring of mitochondrial membrane potential in cardiomyocytes
Isolated cells were incubated with the mitochondrial membrane potential indicator tetramethylrhodamine ethyl ester (TMRE, 50 nM; Invitrogen) for 30 min at room temperature. TMRE (30 nM) was also included in the superfusing solution throughout the experiments. TMRE fluorescence intensity was recorded at 590 nm (excitation at 543 nm with green HeNe laser), and the changes in mitochondrial membrane potential (ΔΨm) were monitored by measuring the mean and standard deviation intensity of TMRE fluorescence. Accumulation of the dye into mitochondria causes a bright signal emitted from mitochondria and very dark signal over the cytosol, providing a high standard deviation of TMRE fluorescence. Depolarization causes more evenly distributed signal over the cell and decrease in standard deviation of the fluorescence signal. The ratio of mean/standard deviation was calculated as a measure of changes in ΔΨm (Brennan et al., 2006).
Preparation of cardiac mitochondria
Mitochondria were isolated from male Wistar rat hearts following 30 min ischaemia and 15 min reoxygenation or from untreated rats (Ljubkovic et al., 2007). The atrium and right ventricle were removed, and left ventricle was minced in isolation buffer [in mM: 50 sucrose, 200 mannitol, 5 KH2PO4, 1 EGTA, 5 3-(N-morpholino)propanesulfonic acid and 0.1% bovine serum albumin, pH 7.3)]. The tissue was homogenized twice for 5 s with T 25 disperser (IKA-Werke, Staufen, Germany), and the mitochondria isolated by differential centrifugation. The homogenate was first centrifuged at 8000 g for 10 min; the pellet was removed, resuspended in the isolation buffer using a Potter-Elvehjem homogenizer, and centrifuged at 750 g for 10 min. Finally, the supernatant was centrifuged at 8000 g for 10 min to pellet the mitochondria. The final mitochondrial pellet was resuspended in isolation buffer without EGTA, stored on ice and used for experiments within 4 h. Protein concentration was determined by a modified Lowry assay kit (Bio-Rad, Hercules, CA, USA).
Recording of mPTP opening
We used the amount of Ca2+ required to trigger massive mitochondrial ΔΨm depolarization as an index of mPTP opening (Pravdic et al., 2009). ΔΨm was measured using rhodamine 123 (50 nM, Invitrogen). Recordings were performed with a spectrofluorometer (QM-8, PTI Technologies, Birmingham, NY, USA) operating at excitation and emission wavelengths set at 503 and 527 nm, respectively. Isolated mitochondria (0.5 mg·mL−1) were suspended in experimental buffer containing (in mM): 220 sucrose, 10 KH2PO4, 10 HEPES, 5 pyruvate, 5 malate, pH 7.4. Mitochondria were exposed to incremental amounts of CaCl2 every 60 s until ΔΨm abruptly decreased indicating mPTP opening.
Monitoring of mitochondrial membrane potential (ΔΨm) in isolated mitochondria
ΔΨm was monitored during state 2 respiration in respiration buffer containing mitochondrial suspension (0.5 mg·mL−1) and pyruvate/malate (5 mM each) as substrates (Ljubkovic et al., 2007). Rhodamine 123 (50 nM) fluorescence was monitored with emission and excitation wavelengths set to 503 nm and 527 nm, respectively. After equilibrium was reached, maximal depolarization was induced by addition of 1 µM carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP), a mitochondrial uncoupler. ΔΨm at state 2 was expressed as percent of rhodamine 123 fluorescence relative to the fluorescence recorded in the presence of FCCP.
Measurement of mitochondrial oxygen consumption
Mitochondrial oxygen consumption was measured with an oxygen electrode (Hansatech Instruments, Norfolk, UK) at 30°C in mitochondrial respiration buffer (in mM: 125 KCl, 5 K2HPO4, 20 3-(N-morpholino)propanesulfonic acid, 2.5 EGTA, 1 mM Na4P2O7 and 0.1% bovine serum albumin, pH 7.4) containing mitochondria at a final concentration of 1 mg protein·mL−1. State 2 of respiration was initiated with 5 mM pyruvate and 5 mM malate or 10 mM succinate and 2 µM rotenone as substrates. The ADP-stimulated respiration (state 3) was measured in the presence of 250 mM ADP, and state 4 respiration was monitored after all ADP was consumed.
In vitro H/R injury of isolated mitochondria
The protocol for H/R was adopted from the previously described model (Ljubkovic et al., 2007). After state 3 respiration was initiated, mitochondria were allowed to consume all oxygen in the chamber. Hypoxia was reached within 5 min as mitochondria consumed all available oxygen in the chamber. The oxygen concentration detectable within the closed chamber containing the mitochondrial suspension was zero throughout the hypoxic interval (30 min). After the hypoxic interval, the chamber containing the mitochondrial suspension was opened to room air to initiate a 15 min reoxygenation period. Oxygen consumption in the chamber was measured again as described in the previous section for oxygen consumption measurements. The respiratory control ratio (RCR), defined as the ratio of oxygen consumption during state 3 to state 4 (a measure of the coupling of ATP synthesis to respiration) was calculated to evaluate the degree of mitochondrial damage before and after exposure to 30 min hypoxia and 15 min reoxygenation.
Experimental protocol for APoC and APC in isolated cardiac mitochondria
To test APoC on isolated mitochondria, isoflurane (0.5 mM) dispersed in dimethyl sulfoxide was added to the mitochondria at the beginning of reoxygenation. Dimethyl sulfoxide without isoflurane was added to control mitochondria. To test the effect of APC on mitochondrial respiration isolated mitochondria were exposed to 0.5 mM isoflurane before anoxia was initiated. Isoflurane was then removed by an additional centrifugation for 10 min at 6000× g. Control mitochondria were incubated with dimethyl sulfoxide, and centrifuged after incubation. Isoflurane concentrations were measured in experimental buffers by gas chromatography.
Measurement of mitochondrial ATP synthesis
Isolated mitochondria were exposed to 30 min hypoxia by saturating the solution with nitrogen. Isoflurane (0.5 mM) was added to the mitochondria at the beginning of 15 min reoxygenation to produce APoC. After 15 min reoxygenation by exposure to room air, isolated mitochondrial ATP synthesis was determined via a chemiluminescence-based method utilizing firefly luciferase and luciferin (Invitrogen). Reaction solutions contained respiration buffer, 0.2 µM diadenosine pentaphosphate, 30 µM ADP (made ATP free by hexokinase treatment), 0.2 mg/mL mitochondria, 0.1 mg/mL luciferin, and 1.25 µg/mL luciferase. The reaction was initiated by the addition of 5 mM pyruvate and 5 mM malate. The blank was obtained by the measurements of luminescence in the absence of substrates. Chemiluminescence was measured in a Modulus luminometer (Turner Biosystems, Sunnyvale, CA, USA) at room temperature for 100 s. The standard curve was obtained with defined ATP concentrations, from which the rate of mitochondrial ATP production was calculated. To test the effect of APC on ATP synthesis, isolated mitochondria were preconditioned as described in the section for experimental protocol for APC.
Statistical analysis
Statistical analyses were performed with Origin 7 (OriginLab, Northampton, MA, USA) and GraphPad 4.03 softwares (GraphPad Sofware Inc., San Diego, CA, USA). Data are presented as means ± SD, with the sample size (n). One-way analysis of variance was used for statistical evaluation of differences between groups, with Bonferroni's post hoc test. Analysis of variance for repeated measures with Tukey's post hoc test was used for temporal comparisons in any given group. Student's t-test was used wherever appropriate. All P values were two-tailed. A P value of 0.05 or less was accepted as significantly different.
Nomenclature
Our receptor and ion channel nomenclature conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2009).
Results
Hemodynamics
Heart rate and mean arterial blood pressure were comparable among groups at baseline (Table 1). There were no significant differences in hemodynamics between ischaemia and reoxygenation and APoC groups during and after coronary artery occlusion.
Table 1.
Systemic hemodynamics
| Groups |
Baseline |
30 min CAO |
15 min reperfusion |
|||
|---|---|---|---|---|---|---|
| HR (bpm) | MAP (mmHg) | HR (bpm) | MAP (mmHg) | HR (bpm) | MAP (mmHg) | |
| Sham | 383 ± 44 | 106 ± 15 | 370 ± 39 | 110 ± 19 | 352 ± 34 | 107 ± 18 |
| Control | 368 ± 38 | 116 ± 17 | 347 ± 28 | 127 ± 10 | 317 ± 46 | 116 ± 20 |
| APoC | 344 ± 31 | 119 ± 9 | 350 ± 35 | 126 ± 12 | 322 ± 37 | 121 ± 22 |
Values are means ± standard deviation; n= 8.
HR, heart rate in beats per minute (bpm); MAP, mean arterial blood pressure; CAO, coronary artery occlusion; ApoC, isoflurane postconditioning.
*P < 0.05 versus baseline.
APoC in vivo involves modulation of oxidative phosphorylation and ΔΨm
To investigate the effect of APoC on mitochondrial oxidative phosphorylation we isolated mitochondria from hearts after ischaemia and reperfusion (I/R) injury. Hearts subjected to sham surgery had highly coupled mitochondria (RCR averaged 6.6 ± 1.3 with complex I and 3.3 ± 0.3 with complex II substrates, respectively), with respiratory rate in the presence of ATP (state 3) at 118 ± 6 nmol O2·min·mg protein−1 with complex I and 163 ± 10 nmol O2·min·mg·protein−1 with complex II substrates, respectively. Following 30 min ischaemia and 15 min reperfusion, in mitochondria from control rats state 3 respiration was significantly decreased in comparison with that in sham operated rats (Table 2). After APoC, in mitochondria with pyruvate/malate, but not with succinate/rotenone as substrates state 2 and state 3 were further decreased compared with control mitochondria (Table 2).
Table 2.
Oxydative phosphorylation after in vivo APoC
| Sham | Control | APoC | |
|---|---|---|---|
| Complex I | |||
| State 2 | 14 ± 1 | 10 ± 1* | 8 ± 1*# |
| State 3 | 118 ± 6 | 46 ± 7* | 36 ± 7*# |
| State 4 | 24 ± 5 | 14 ± 3* | 12 ± 2* |
| RCR | 6.6 ± 1.3 | 3.4 ± 0.6* | 3.2 ± 0.1* |
| Complex II | |||
| State 2 | 42 ± 12 | 27 ± 3* | 25 ± 4* |
| State 3 | 163 ± 10 | 75 ± 6* | 68 ± 11* |
| State 4 | 50 ± 6 | 31 ± 6* | 33 ± 11* |
| RCR | 3.3 ± 0.3 | 2.5 ± 0.3* | 2.2 ± 0.4* |
Mitochondrial respiration was measured with pyruvate/malate (5 mM each) as substrates for complex I and succinate/rotenone (10 mM/2 µM) as substrates for complex II. Rates are expressed in nmol O2·min·mg mitochondrial protein−1.
Values are means ± standard deviation; n= 6.
P < 0.05 versus respective Sham,
P < 0.05 versus respective control.
RCR, respiratory control ratio (state 3/state 4); ApoC, isoflurane postconditioning.
Monitoring rhodamine 123 fluorescence revealed a significant degree of depolarization in ΔΨm of control mitochondria after I/R injury with pyruvate/malate as substrates(P < 0.05; Figure 1A and B). In APoC group, ΔΨm was further decreased as compared with control mitochondria (P < 0.05; Figure 1B).
Figure 1.
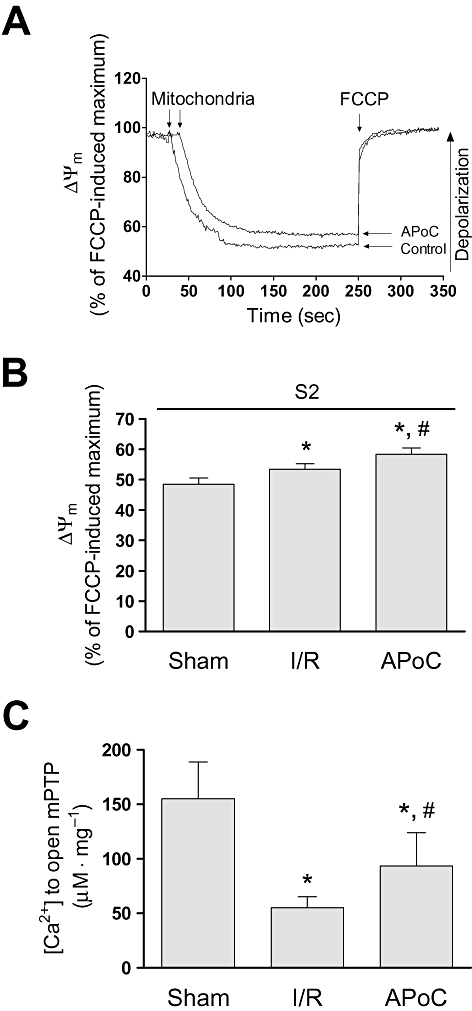
Isoflurane postconditioning in vivo protects mitochondria from ischaemia and reperfusion (I/R) injury. (A) Original recordings of ΔΨm in mitochondria from control and APoC hearts in the presence of pyruvate/malate. (B) Summarized data showing ΔΨm relative to maximum depolarization. (C) The amount of Ca2+ necessary to open the mPTP in sham, control and APoC mitochondria. Values are means ± standard deviation. *P < 0.05 versus sham, #P < 0.05 versus control, n= 9. ΔΨm, mitochondrial membrane potential; mPTP, mitochondrial permeability transition pore; FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; S2, state 2 respiration; APoC, isoflurane postconditioning.
In vivo APoC delays opening of mPTP
We investigated whether the cardioprotective effects of APoC could in part be caused by protection against mPTP opening after I/R injury. Isolated mitochondria were subjected to Ca2+ overload. In sham group, the concentration of Ca2+ necessary to trigger mPTP opening was 155 ± 34 µM CaCl2 mg protein−1 (n= 6). In control mitochondria, after 30 min ischaemia and 15 min reoxygenation, this concentration was markedly reduced (n= 6, P < 0.05; Figure 1C). In APoC group, the amount of Ca2+ required for mPTP opening was significantly increased, compared with control group, indicating that APoC delayed mPTP opening (Figure 1C).
In vitro APoC protects the cardiomyocytes from H/R injury in a pH dependent manner
Figure 2A shows representative images of cardiomyocyte hypercontracture during reoxygenation following hypoxia. In the time control group, normoxic Tyrode solution did not cause significant cell hypercontracture (92 ± 4% hypercontracted cells compared with the baseline) at pH 7.4. Cells exposed to H/R started to contract during reoxygenation, which resulted in hypercontracture, indicating cell death (Figure 2A). Treatment of cardiomyocytes with isoflurane at the beginning of reoxygenation reduced the proportion of hypercontracted cells at pH 7.4. When cardiomyocytes were exposed to isoflurane at pH 7.8, the APoC-induced protection was lost. The number of hypercontracted cells in H/R groups was not different when the first five minutes of reperfusion occured either at pH 7.4 or 7.8. These experiments demonstrate that protection elicited by APoC was dependent on pH. These results are summarized in Figure 2B, as cell survival (n= 6, 50–70 cells were counted per experiment in each group, P < 0.05).
Figure 2.
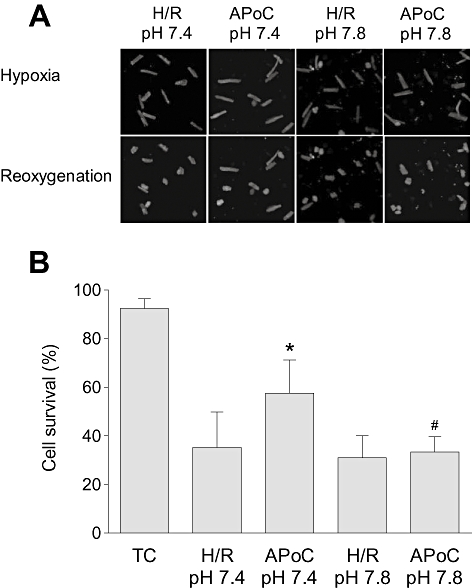
Isoflurane postconditioning protects isolated cardiomyocytes from H/R injury. (A) Representative images of isolated cardiomyocyte changes during H/R. During reoxygenation after hypoxia cardiomyocytes became spherical, hypercontracted and exhibited membrane blebs. APoC protected cardiomyocytes against H/R injury at pH 7.4, but the protection was lost when APoC was performed at pH 7.8. (B) Summary graph showing the percentage of cell survival after 35 min of reoxygenation. Data are means ± standard deviation; *P < 0.05 versus time control; #P < 0.05 versus ApoC; n= 5. TC, time control; H/R, hypoxia and reoxygenation; APoC, isoflurane postconditioning.
APoC attenuates intracellular Ca2+ accumulation during reoxygenation
To investigate the effect of APoC on intracellular Ca2+ accumulation, isolated cardiomyocytes were incubated with the fluorescent indicator fluo-4 (Figure 3A). Figure 3B summarizes the results of H/R on intracellular Ca2+ levels. In control cardiomyocytes, 60 min of perfusion with normoxic Tyrode solution did not change fluo-4 fluorescence (96 ± 4% from baseline value, n= 17/group; Figure 3A). Perfusion of cardiomyocytes with hypoxic Tyrode solution for 20 min elicited an increase in fluo-4 fluorescence, indicating an increase in intracellular Ca2+ (142 ± 12% of prehypoxic levels, P < 0.05) (O'Brien et al., 2008). There was no significant difference in fluo-4 fluorescence between control and APoC groups during the period of hypoxia. However, as displayed in Figure 3B, during early reoxygenation intracellular Ca2+ decreased to prehypoxic values, but then again increased throughout reoxygenation. In the APoC group, the increase in fluo-4 fluorescence after 60 min reoxygenation was attenuated from 136 ± 11% in control to 108 ± 6% in the APoC group (P < 0.05).
Figure 3.
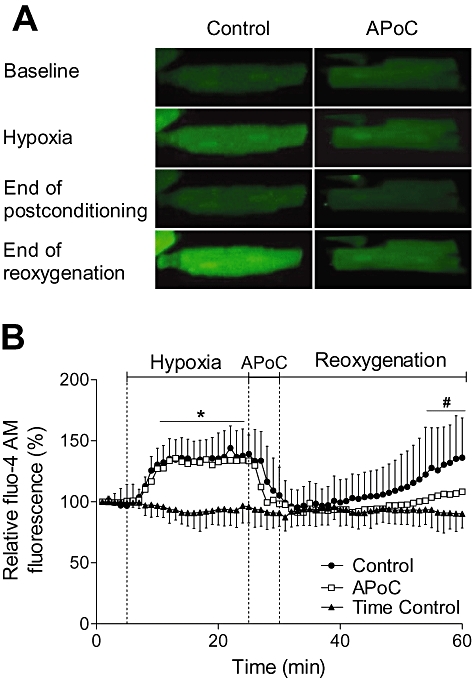
APoC decreases cytosolic Ca2+ overload in cardiomyocytes during reoxygenation. (A) Representative image of isolated cardiomyocytes loaded with fluo-4, fluorescent indicator for intracellular Ca2+. Under hypoxic conditions intracellular Ca2+ increased relative to pre-hypoxic values and then recovered at reoxygenation. During reoxygenation, Ca2+ remained low at early reoxygenation, but then increased. APoC decreased cytosolic Ca2+ overload during reoxygenation. (B) Summarized data for intracellular Ca2+ during hypoxia and reoxygenation. Standard deviations (SDs) are omitted for clarity in APoC group. Values are means ± SD; *P < 0.05 versus time control; #P < 0.05 versus control; n= 17. APoC, isoflurane postconditioning.
APoC prevents rapid recovery of pHm during reoxygenation
The pHm during H/R was monitored in isolated cardiomyocytes loaded with SNARF-1 (5 µM). SNARF-1 was taken up preferentially in mitochondria, which was confirmed by colocalization with mitochondrial flavoproteins (Figure 4A). Figure 4B illustrates the pHm changes during H/R. The exposure of cardiomyocytes to hypoxic Tyrode solution (pH 6.8) significantly decreased pHm, as shown by a reduction of the SNARF-1 ratio. There was no difference in pHm in control and APoC groups at the end of hypoxia. During reoxygenation pHm returned to pre-hypoxic baseline levels. However, APoC produced a lower pHm during the postconditioning period, and the time required for recovery of pHm to pre-hypoxic values increased to 18.6 ± 2.6 min compared with 12.4 ± 4.5 min in control cells (Figure 4B, n= 20 per group, P < 0.05).
Figure 4.
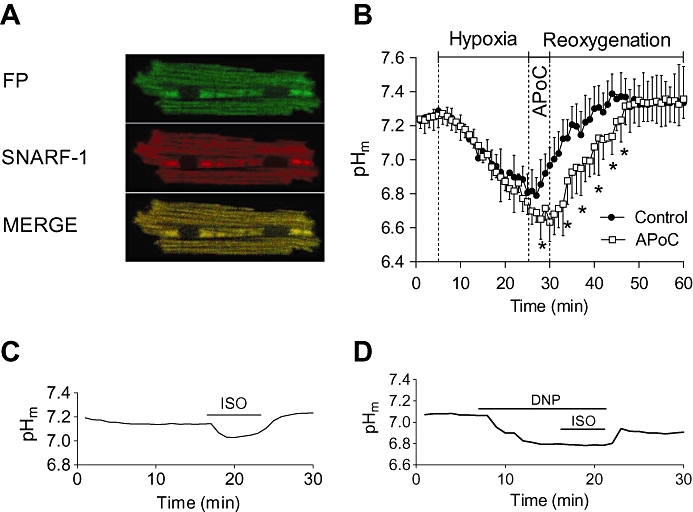
APoC prevents rapid pHm recovery in cardiomyocytes at reoxygenation. (A) Representative images showing selective mitochondrial loading of SNARF-1. SNARF-1 colocalizes with mitochondrial flavoproteins (FP). (B) pHm decreased during hypoxia as shown by a decrease in SNARF-1 ratio. Upon reoxygenation, pHm quickly recoverd to baseline levels. Treatment of cardiomyocytes with isoflurane at the beginning of reoxygenation preserved acidic intracellular pHm during reoxygenation as compared with the control group. (C) The panel shows representative time lapse of baseline pHm recording. Baseline pHm was reduced upon addition of isoflurane into superfusing solution, and this effect was reversible after anaesthetic washout. (D) Administration of the mitochondrial uncoupler 2-4-dinitrophenol (DNP) prevented any further effect of isoflurane on pHm. Data are presented as means ± standard deviation, *P < 0.05 versus APoC; n= 20. APoC, isoflurane postconditioning; SNARF-1, 5-(and-6)-carboxy SNARF-1, acetoxymethyl ester.
When cardiomyocytes were exposed to 0.5 mM isoflurane in normoxic Tyrode solution, a decrease in baseline pHm occurred compared with control cells (0.10 ± 0.02 pH units, P < 0.05, n= 25 Figure 4C). However, treatment of cardiomyocytes with mitochondrial uncouplers, 2-4-dinitrophenol (50 µM) and carbonylcyanide-p-trifluoromethoxyphenylhydrazone (1 µM, data not shown) (Abad et al., 2004) prevented any additional effect of isoflurane (Figure 4D), suggesting that isoflurane indeed elicited an effect at the level of mitochondria.
Isoflurane does not affect NHE, carbonic anhydrase, chloride-mediated base extrusion transporters, and glycolysis-mediated generation of lactic acid in rat ventricular cardiomyocytes
Isoflurane-induced regulation of pHi may contribute to changes of pHm (Gursahani and Schaefer, 2004). Further, activity of the NHE reduces intracellular Ca2+ overload, and decreases pHi (Murphy and Steenbergen, 2008). Therefore we tested whether isoflurane modified these mechanisms involved in pHi regulation. To examine the effect of isoflurane on NHE we induced intracellular acidosis with 10 mM NH4Cl in HEPES-buffered Tyrode solution. In the absence of CO2/HCO3- buffer recovery from intracellular acidosis is mediated by NHE alone. As shown in the representative trace in Figure 5B, pHi recovery in the presence and absence of isoflurane was not changed, indicating that isoflurane did not change activity of the NHE. The rates of pH recovery are summarized in Figure 5C (n= 18). Carbonic anhydrase was proposed to mediate fast pH changes, and to buffer H+ with HCO3-. Therefore, we examined whether isoflurane-induced pH changes were secondary to an inhibition of carbonic anhydrase activity. When switching the superfusate from HEPES-buffered to CO2/HCO3--buffered solution at the same extracellular pH (7.4), the initial fall and following recovery in pHi were not different in the presence or absence of isoflurane. This suggests that carbonic anhydrase activity was not changed after exposure of the myocyte to isoflurane. Further, no effect of isoflurane on Cl-/HCO3- and Cl-/OH- exchangers and glycolysis-induced pH changes were found (data not shown).
APoC preserves ΔΨm during reoxygenation in rat ventricular cardiomyocytes
The effect of APoC on ΔΨm was investigated by incubating isolated cardiomyocytes with a positively charged mitochondrial potentiometric dye, TMRE. As shown in Figure 6A, hypoxia caused a decrease in the standard deviation of the TMRE signal, indicating the ΔΨm was falling. There was no significant difference in ΔΨm during hypoxia between groups. As shown by the standard deviation of the TMRE signal, throughout reoxygenation ΔΨm was significantly greater in the APoC group (P < 0.05; Figure 6B).
Figure 6.
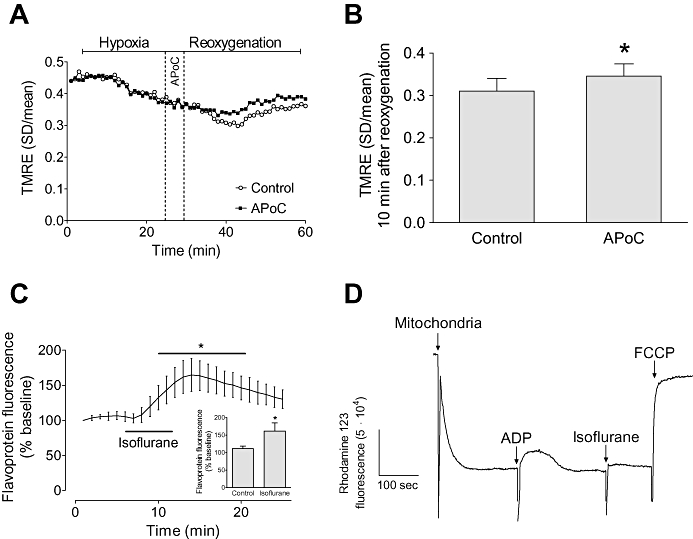
APoC preserves ΔΨm in isolated myocytes throughout reoxygenation. (A) Representative recording of ΔΨm changes during H/R. Hypoxia caused a significant decrease in the standard deviation of TMRE fluorescence indicating mitochondrial depolarization whereas reoxygenation caused an increase in the standard deviation indicating recovery of ΔΨm. Treatment of cardiomyocytes with isoflurane at the beginning of reoxygenation preserved ΔΨm as compared with control cells. (B) Summary data for control and APoC groups at 10 min of reoxygenation. (C) Effect of isoflurane on mitochondrial flavoprotein fluorescence, and summary data (inset). (D) Representative trace of ΔΨm in isolated mitochondria. Isoflurane induced slight depolarization of ΔΨm. Data are means ± standard deviation; n= 15; n= 6 in flavoprotein and isolated mitochondria experiments. *P < 0.05 versus control. APoC, isoflurane postconditioning, TMRE, tetramethylrhodamine ethyl ester. FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone.
When cardiomyocytes were exposed to 0.5 mM isoflurane for 5 min under normoxic conditions, isoflurane significantly increased mitochondrial flavoprotein fluorescence, indicating that isoflurane depolarized mitochondria (Figure 6C). However, after 5 min we did not see any significant effect of isoflurane on TMRE fluorescence (not shown). This might be related to the short exposure to the anaesthetic and/or artifacts produced by redistribution of the dye from the mitochondria to the cytosolic compartment (Ljubkovic et al., 2007). To confirm that observed changes in flavoprotein fluorescence were related to mitochondrial depolarization, we measured ΔΨm in isolated mitochondria. Direct exposure of mitochondria to isoflurane caused slight depolarization of ΔΨm (Figure 6D).
APoC but not APC preserves oxidative phosphorylation and ATP production in isolated mitochondria
Typical chart recordings of oxygen consumption in rat mitochondria subjected to H/R after isolation are depicted in Figure 7A and summarized in Figure 7B and C. After reoxygenation, ADP-stimulated oxygen consumption (state 3 respiration) in isolated mitochondria was better preserved in APoC treated than in control mitochondria (n= 5, P < 0.05; Figure 7B). As a consequence, the RCR was better preserved in the APoC group (P < 0.05). Interestingly, application of isoflurane to mitochondria before hypoxia, followed by isoflurane removal with mitochondrial centrifugation (APC), did not preserve respiration.
Figure 7.
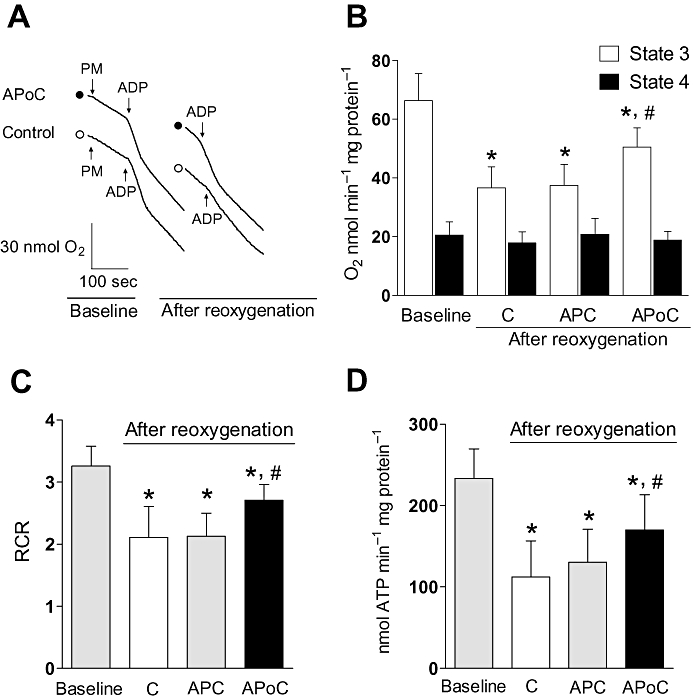
In vitro APoC protects isolated mitochondria from H/R injury. (A) Typical recordings showing respiration under baseline condition and after reoxygenation in control and isoflurane-postconditioned mitochondria. Oxygen consumption was initiated by addition of pyruvate/malate (PM), accelerated by addition of ADP (state 3 respiration), and decelerated after all ADP was consumed (state 4 respiration). Following H/R, state 3 respiration was preserved in APoC-treated mitochondria. (B) After H/R, state 3 respiration decreased in all groups; however, state 3 respiration in APoC-treated mitochondria was better preserved as compared with control and preconditioned mitochondria. (C) Respiratory control ratio (RCR) was better maintained in APoC treated mitochondria, whereas preconditioning did not improve RCR. (D) The rate of ATP production was decelerated after reoxygenation. However, ATP synthesis in APoC-treated mitochondria was better preserved as compared with control and preconditioned mitochondria. Values are mean ± SD. *P < 0.05 versus baseline or APoC; #P < 0.05 versus control or APC. C, control; APoC, isoflurane postconditioning; APC, isoflurane preconditioning; open circle, control; solid circle, isoflurane postconditioning, n= 7 per group.
Monitoring the rate of ATP production revealed that ATP production in both control and APoC treated mitochondria decreased after reoxygenation (Figure 7D). ATP generation was maintained at higher levels in APoC-treated, than in control mitochondria, agreeing with results obtained by oxidative phosphorylation measurements. In contrast to the effect of APoC, ATP synthesis in isoflurane-preconditioned (APC) mitochondria was not preserved after H/R. Thus APoC, but not APC prevented the decline in ATP synthesis in post-hypoxic mitochondria.
Discussion and conclusions
In this investigation, we examined the direct effect that isoflurane has on mitochondria, particularly at the time of reoxygenation. This involves primarily mitochondrial depolarization and lowering of the matrix pH, which in turn may explain the outcomes, such as preserved bioenergetics, delayed permeability transition pore opening, and, ultimately, better survival. In vitro postconditioning, but not preconditioning of isolated mitochondria protected them from ischaemic injury and preserved oxidative phosphorylation and ATP production. Thus, an immediate and intracellular signalling pathway-independent action on mitochondrial function is critical for the cardioprotective effects provided by APoC.
The cardioprotective effect of applying several short periods of ischaemia or hypoxia at the beginning of reperfusion (ischaemic postconditioning, IPoC) originally described by Zhao et al. (2003) has been verified in many models (Zhao et al., 2003; Kin et al., 2004; Yang et al., 2004; 2005; Sun et al., 2005). Application of volatile anaesthetics immediately during early reperfusion has similar protective actions, and several elements of the prosurvival signalling pathways have been proposed to be involved (Chiari et al., 2005; Krolikowski et al., 2005; Weihrauch et al., 2005; Pagel et al., 2006; Pagel, 2008). The actions of volatile anaesthetics that contribute to cardioprotection during early reperfusion are mediated by phosphoinositide 3-kinase, ERK1 and ERK2, the 70-kDa ribosomal protein S6 kinase, eNOS, glycogen synthase kinase 3-β, mitochondrial KATP channels and protein kinase C (Chiari et al., 2005; Weihrauch et al., 2005; Pagel et al., 2006; Venkatapuram et al., 2006; Pagel, 2008). Prosurvival kinases are known to target several mitochondrial sites, particularly the mPTP (Chiari et al., 2005; Krolikowski et al., 2005; Weihrauch et al., 2005; Pagel et al., 2006; Pagel, 2008). Furthermore, phosphoinositide 3-kinase activates phosphoinositide-dependent kinase 1 and Akt (protein kinase B), leading to inhibition of pro-apoptotic proteins such as p53, Bad and Bax, which stimulate the mitochondrial apoptosis pathway including cytochrome c release (Pagel, 2008). Isoflurane administered immediately before or immediately at reperfusion reduced cytochrome c release from mitochondria and decreased the number of TUNEL-positive cells, as shown previously in our laboratory (Weihrauch et al., 2005). Postconditioning-induced activation of prosurvival signalling pathways may take several minutes, but the first minutes of reoxygenation are essential for salvage of ischaemic myocardium (Piper et al., 2004; Darling et al., 2005). Therefore, the aim of this investigation was to examine the immediate action of isoflurane on mitochondrial and cellular function necessary for cardioprotection.
Gradual or intermittent reperfusion may delay recovery of pH during reperfusion, and this delay may be beneficial (Cohen et al., 2007). Interestingly, a slightly acidotic pH at reperfusion has been shown to be crucial for protection against I/R injury (Cohen et al., 2007; Inserte et al., 2008). At reperfusion, mPTP opening occurs and it is delayed at lower pH values (Crompton, 1999; Kim et al., 2006; Javadov and Karmazyn, 2007). Two minutes of reperfusion at pH 6.9 exhibited the same cardioprotection as IPoC (Cohen et al., 2007). These authors suggested that the acidotic environment at the beginning of reperfusion is necessary to block mPTP opening until signalling pathways are activated (Cohen et al., 2008; Inserte et al., 2008; 2009;). Reperfusion with a mildly alkaline perfusate abolished the protective effects of postconditioning, but the protection was restored by cyclosporin A, drug that maintains the mPTP in a relatively closed state. It has been unclear whether volatile anaesthetics may also affect the pHm during reperfusion. We found that, after exposure to isoflurane, an acidotic pHm is maintained and mPTP opening delayed. Prevention of mPTP formation stabilizes ΔΨm, thereby maintaining electron flux and oxidative phosphorylation (Crompton, 1999). The mechanism for lower pHm may not be related to anaerobic metabolism during reperfusion as isoflurane also decreased pHm under normoxic conditions. The fact that isoflurane did not further decrease pHm in the presence of mitochondrial uncoupler 2,4-dinitrophenol, which is known to induce only slight changes in cytosolic pH (Buckler and Vaughan-Jones, 1998) suggested that isoflurane elicited an effect on the pH at the level of mitochondria. Isoflurane has been shown to mildly inhibit complex I of the respiratory chain and uncouple mitochondria (Hanley et al., 2002; Ljubkovic et al., 2007). We confirmed that complex I plays a role by measuring oxidative phosphorylation in isolated mitochondria after in vivo APoC, in contrast to findings after ischaemic postconditioning (Paillard et al., 2009). These effects of isoflurane on mitochondrial bioenergetics could lower the pH in the mitochondrial matrix, either by reduced H+ pumping by complex I or increased influx of H+ due to mitochondrial uncoupling (Abad et al., 2004; Brennan et al., 2006; Dlaskova et al., 2008). Although changes in the cytosolic pH may also influence mitochondrial pH (Gursahani and Schaefer, 2004) we excluded this possibility by ruling out an effect of isoflurane on major regulators of cytosolic pH: NHE, carbonic anydrase, Cl- HCO3- and Cl- OH- exchangers, and glycolysis.
Elevated levels of intracellular Ca2+ contribute to induction of cell death during H/R (Moens et al., 2005; Murphy and Steenbergen, 2008). It is therefore important that intracellular Ca2+ after exposure of cardiomyocytes to H/R injury was reduced in APoC-treated relative to untreated cardiomyocytes. This decrease in intracellular Ca2+ is in agreement with previous findings that IPoC reduces cardiomyocyte death and attenuates intracellular Ca2+ overload during reoxygenation (Sun et al., 2005). Sevoflurane administered after ischaemia also attenuated intracellular Ca2+ loading in isolated guinea pig hearts (Varadarajan et al., 2002). Halothane reduced oscillations of intracellular Ca2+ and protected cardiomyocytes against reoxygenation-induced hypercontracture (Siegmund et al., 1997). Preservation of mitochondrial function, as observed in the present investigation, may enable the cell to provide sufficient ATP necessary for Ca2+ homeostasis via sarcoplasmic reticulum and sarcolemmal Ca2+-ATPases (Igarashi-Saito et al., 1998; Murphy and Steenbergen, 2008). Our observation that mitochondria exhibit preserved oxidative phosphorylation after APoC could explain the lower accumulation of intracellular Ca2+ in cardiomyocytes treated with isoflurane. Isoflurane could also inhibit Ca2+ uptake into mitochondria by reducing membrane potential and hence uniporter activity (Ljubkovic et al., 2007). This would indirectly reduce mPTP opening. In addition, inhibition of extrusion of H+ from the cardiomyocytes by NHE may affect intracellular Ca2+ levels (Murphy and Steenbergen, 2008). Our data, however, showed that isoflurane did not alter NHE-mediated H+ fluxes through the plasma membrane, indicating that this mechanism does not influence intracellular Ca2+ in APoC.
To test whether the isoflurane-induced protection of mitochondria in APoC was due to a direct effect of isoflurane on mitochondrial function, we subjected isolated mitochondria to either an APoC or APC protocol. Mitochondrial RCR and rate of ATP synthesis after H/R were preserved in APoC, but not in APC mitochondria. These findings indicate that for protection of isolated mitochondria the presence of isoflurane is required at the time of reperfusion. Cytosolic components and post-translational protein modifications are not required for protection of mitochondria in APoC, but may be needed for APC (Pravdic et al., 2009). This further supports our hypothesis that the direct effect of isoflurane on mitochondria plays a crucial role for protection within the first minutes of reperfusion in APoC. Interestingly, after in vivo APoC, the electron transport chain was mildly inhibited at the level of complex I. The fact that this effect was not observed when isolated mitochondria were subjected to APoC could be due to the lack of a cytosolic component required for this effect. Alternatively, better preserved bioenergetics may overcome the inhibitory effect of isoflurane on respiration in isolated mitochondria after APoC. A beneficial effect of complex I inhibition has been shown previously with a reversible complex I inhibitor amobarbital and with rotenone (Lesnefsky et al., 2004; Chen et al., 2006). Transnitrosation with nitrosoglutathione also inhibits complex I and protects against I/R injury (Burwell et al., 2006). Moreover, hearts from mice with a heterozygous mutation in the complex I NDUSF4 subunit are resistant to cardioprotection by ischaemic preconditioning and by S-nitroso-2-mercaptopropionyl glycine (Nadtochiy et al., 2009). Inhibition of complex I in I/R leads to decreased production of reactive oxygen species (ROS), particularly due to reverse electron transport, whereas mitochondrial oxidative phosphorylation is maintained through complex II (Heinen et al., 2007; Shiva et al., 2007). Here, inhibition of complex I was associated with mildly reduced ΔΨm and blunted recovery of matrix pH at reoxygenation. Both, lower pH and ΔΨm, decrease ROS production, an important inducer of mPTP opening (Selivanov et al., 2008).
In conclusion, the present investigation demonstrates that APoC produces a significant protection of mitochondrial function. In contrast to findings after ischaemic postconditioning, APoC-induced cardioprotection involves modulation of ΔΨm and oxidative phosphorylation. A mildly acidotic pHm due to a mildly reduced oxidative phosphorylation and/or depolarization at the onset of reperfusion may be responsible for prevention of opening of mPTP and preservation of mitochondrial bioenergetics immediately during reperfusion. These mechanisms lead to attenuation of intracellular Ca2+ accumulation and an increase in cell survival.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants P01GM066730 and HL034708 (to ZJB) and HL054820 (to DCW), Bethesda, Maryland and Advancing a Healthier Wisconsin Program (to MB). We thank Chiaki Kwok, B.S., Research Technologist, for technical assistance in isolation of mitochondria and David A. Schwabe, B.S., Research Technologist, for assistance in rat surgical procedures. We also thank Mary B. Ziebell, Research Technologist, for isoflurane measurements and Terri L. Misorski, A.A.S., Program Coordinator, for editorial assistance (all from Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, Wisconsin).
Glossary
Abbreviations:
- APC
anaesthetic-induced preconditioning
- APoC
anaesthetic-induced postconditioning
- H/R
hypoxia and reoxygenation
- IPoC
ischaemic postconditioning
- I/R
ischaemia and reperfusion
- mPTP
mitochondrial permeability transition pore
- pHi
cytosolic pH
- pHm
mitochondrial matrix pH
- ΔΨm
mitochondrial membrane potential
Conflicts of interest
The authors state no conflict of interests.
References
- Abad MFC, Di Benedetto G, Magalhaes PJ, Filippin L, Pozzan T. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J Biol Chem. 2004;279:11521–11529. doi: 10.1074/jbc.M306766200. [DOI] [PubMed] [Google Scholar]
- Aizawa K, Turner LA, Weihrauch D, Bosnjak ZJ, Kwok WM. Protein kinase C-epsilon primes the cardiac sarcolemmal adenosine triphosphate-sensitive potassium channel to modulation by isoflurane. Anesthesiology. 2004;101:381–389. doi: 10.1097/00000542-200408000-00019. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan JP, Berry RG, Baghai M, Duchen MR, Shattock MJ. FCCP is cardioprotective at concentrations that cause mitochondrial oxidation without detectable depolarisation. Cardiovasc Res. 2006;72:322–330. doi: 10.1016/j.cardiores.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Application of a new pH-sensitive fluoroprobe (carboxy-SNARF-1) for intracellular pH measurement in small, isolated cells. Pflugers Arch. 1990;417:234–239. doi: 10.1007/BF00370705. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. 1998;513:819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'en FF, Villafuerte FC, Swietach P, Cobden PM, Vaughan-Jones RD. S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br J Pharmacol. 2008;153:972–982. doi: 10.1038/sj.bjp.0707667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- Chen HT, Yang CX, Li H, Zhang CJ, Wen XJ, Zhou J, et al. Cardioprotection of sevoflurane postconditioning by activating extracellular signal-regulated kinase 1/2 in isolated rat hearts. Acta Pharmacol Sin. 2008;29:931–941. doi: 10.1111/j.1745-7254.2008.00824.x. [DOI] [PubMed] [Google Scholar]
- Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology. 2005;102:102–109. doi: 10.1097/00000542-200501000-00018. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation. 2007;115:1895–1903. doi: 10.1161/CIRCULATIONAHA.106.675710. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning's success. Basic Res Cardiol. 2008;103:464–471. doi: 10.1007/s00395-008-0737-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Darling CE, Jiang R, Maynard M, Whittaker P, Vinten-Johansen J, Przyklenk K. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am J Physiol Heart Circ Physiol. 2005;289:H1618–H1626. doi: 10.1152/ajpheart.00055.2005. [DOI] [PubMed] [Google Scholar]
- Dlaskova A, Hlavata L, Jezek J, Jezek P. Mitochondrial Complex I superoxide production is attenuated by uncoupling. Int J Biochem Cell Biol. 2008;40:2098–2109. doi: 10.1016/j.biocel.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Faghihi M, Sukhodub A, Jovanovic S, Jovanovic A. Mg2+ protects adult beating cardiomyocytes against ischaemia. Int J Mol Med. 2008;21:69–73. [PMC free article] [PubMed] [Google Scholar]
- Gursahani HI, Schaefer S. Acidification reduces mitochondrial calcium uptake in rat cardiac mitochondria. Am J Physiol Heart Circ Physiol. 2004;287:H2659–H2665. doi: 10.1152/ajpheart.00344.2004. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH : ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687–693. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Zhang FJ, Wang SP, Chen G, Chen CC, Yan M. Postconditioning of sevoflurane and propofol is associated with mitochondrial permeability transition pore. J Zhejiang Univ Sci B. 2008;9:100–108. doi: 10.1631/jzus.B0710586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen A, Aldakkak M, Stowe DF, Rhodes SS, Riess ML, Varadarajan SG, et al. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am J Physiol Heart Circ Physiol. 2007;293:H1400–H1407. doi: 10.1152/ajpheart.00198.2007. [DOI] [PubMed] [Google Scholar]
- Huhn R, Heinen A, Weber NC, Hollmann MW, Schlack W, Preckel B. Hyperglycaemia blocks sevoflurane-induced postconditioning in the rat heart in vivo: cardioprotection can be restored by blocking the mitochondrial permeability transition pore. Br J Anaesth. 2008;100:465–471. doi: 10.1093/bja/aen022. [DOI] [PubMed] [Google Scholar]
- Igarashi-Saito K, Tsutsui H, Yamamoto S, Takahashi M, Kinugawa S, Tagawa H, et al. Role of SR Ca2+-ATPase in contractile dysfunction of myocytes in tachycardia-induced heart failure. Am J Physiol. 1998;275:H31–H40. doi: 10.1152/ajpheart.1998.275.1.H31. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Hernando V, Abellan A, Ruiz-Meana M, Rodriguez-Sinovas A, et al. Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage. Cardiovasc Res. 2008;77:782–790. doi: 10.1093/cvr/cvm082. [DOI] [PubMed] [Google Scholar]
- Inserte J, Barba I, Hernando V, Garcia-Dorado D. Delayed recovery of intracellular acidosis during reperfusion prevents calpain activation and determines protection in postconditioned myocardium. Cardiovasc Res. 2009;81:116–122. doi: 10.1093/cvr/cvn260. [DOI] [PubMed] [Google Scholar]
- Javadov S, Karmazyn M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol Biochem. 2007;20:1–22. doi: 10.1159/000103747. [DOI] [PubMed] [Google Scholar]
- Kim J-S, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, et al. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res. 2004;62:74–85. doi: 10.1016/j.cardiores.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, Kersten JR, Pagel PS. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: the role of mitochondrial KATP channels. Anesth Analg. 2005;101:1590–1596. doi: 10.1213/01.ANE.0000181288.13549.28. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055–1062. doi: 10.1097/ALN.0b013e3181730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, Bosnjak ZJ, et al. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–C1590. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- Lu J, Zang WJ, Yu XJ, Chen LN, Zhang CH, Jia B. Effects of ischaemia-mimetic factors on isolated rat ventricular myocytes. Exp Physiol. 2005;90:497–505. doi: 10.1113/expphysiol.2004.029421. [DOI] [PubMed] [Google Scholar]
- Ludwig LM, Weihrauch D, Kersten JR, Pagel PS, Warltier DC. Protein kinase C translocation and Src protein tyrosine kinase activation mediate isoflurane-induced preconditioning in vivo: potential downstream targets of mitochondrial adenosine triphosphate-sensitive potassium channels and reactive oxygen species. Anesthesiology. 2004;100:532–539. doi: 10.1097/00000542-200403000-00011. [DOI] [PubMed] [Google Scholar]
- Maddaford TG, Hurtado C, Sobrattee S, Czubryt MP, Pierce GN. A model of low-flow ischemia and reperfusion in single, beating adult cardiomyocytes. Am J Physiol. 1999;277:H788–H798. doi: 10.1152/ajpheart.1999.277.2.H788. [DOI] [PubMed] [Google Scholar]
- Marinovic J, Bosnjak ZJ, Stadnicka A. Preconditioning by isoflurane induces lasting sensitization of the cardiac sarcolemmal adenosine triphosphate-sensitive potassium channel by a protein kinase C-delta-mediated mechanism. Anesthesiology. 2005;103:540–547. doi: 10.1097/00000542-200509000-00017. [DOI] [PubMed] [Google Scholar]
- Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int J Cardiol. 2005;100:179–190. doi: 10.1016/j.ijcard.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy SM, Burwell LS, Ingraham CA, Spencer CM, Friedman AE, Pinkert CA, et al. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. J Mol Cell Cardiol. 2009;46:960–968. doi: 10.1016/j.yjmcc.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JD, Ferguson JH, Howlett SE. Effects of ischemia and reperfusion on isolated ventricular myocytes from young adult and aged Fischer 344 rat hearts. Am J Physiol Heart Circ Physiol. 2008;294:H2174–H2183. doi: 10.1152/ajpheart.00058.2008. [DOI] [PubMed] [Google Scholar]
- Pagel PS. Postconditioning by volatile anesthetics: salvaging ischemic myocardium at reperfusion by activation of prosurvival signaling. J Cardiothorac Vasc Anesth. 2008;22:753–765. doi: 10.1053/j.jvca.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Pagel PS, Krolikowski JG, Neff DA, Weihrauch D, Bienengraeber M, Kersten JR, et al. Inhibition of glycogen synthase kinase enhances isoflurane-induced protection against myocardial infarction during early reperfusion in vivo. Anesth Analg. 2006;102:1348–1354. doi: 10.1213/01.ane.0000202379.61338.37. [DOI] [PubMed] [Google Scholar]
- Paillard M, Gomez L, Augeul L, Loufouat J, Lesnefsky EJ, Ovize M. Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential. J Mol Cell Cardiol. 2009;46:902–909. doi: 10.1016/j.yjmcc.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Piper HM, Abdallah Y, Schafer C. The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res. 2004;61:365–371. doi: 10.1016/j.cardiores.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Pravdic D, Sedlic F, Mio Y, Vladic N, Bienengraeber M, Bosnjak ZJ. Anesthetic-induced preconditioning delays opening of mitochondrial permeability transition pore via protein Kinase C-epsilon-mediated pathway. Anesthesiology. 2009;111:267–274. doi: 10.1097/ALN.0b013e3181a91957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi A, Quillan JM, Sadee W. Monitoring intracellular pH changes in response to osmotic stress and membrane transport activity using 5-chloromethylfluorescein. AAPS PharmSci. 2002;4:1–8. doi: 10.1208/ps040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selivanov VA, Zeak JA, Roca J, Cascante M, Trucco M, Votyakova TV. The role of external and matrix pH in mitochondrial reactive oxygen species generation. J Biol Chem. 2008;283:29292–29300. doi: 10.1074/jbc.M801019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund B, Schlack W, Ladilov YV, Balser C, Piper HM. Halothane protects cardiomyocytes against reoxygenation-induced hypercontracture. Circulation. 1997;96:4372–4379. doi: 10.1161/01.cir.96.12.4372. [DOI] [PubMed] [Google Scholar]
- Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, et al. Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload. Am J Physiol Heart Circ Physiol. 2005;288:H1900–H1908. doi: 10.1152/ajpheart.01244.2003. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Weihrauch D, Kehl F, Ludwig LM, LaDisa JF, Jr, Kersten JR, et al. Mechanism of preconditioning by isoflurane in rabbits: a direct role for reactive oxygen species. Anesthesiology. 2002;97:1485–1490. doi: 10.1097/00000542-200212000-00021. [DOI] [PubMed] [Google Scholar]
- Varadarajan SG, An J, Novalija E, Stowe DF. Sevoflurane before or after ischemia improves contractile and metabolic function while reducing myoplasmic Ca(2+) loading in intact hearts. Anesthesiology. 2002;96:125–133. doi: 10.1097/00000542-200201000-00025. [DOI] [PubMed] [Google Scholar]
- Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46:318–331. doi: 10.1016/j.yjmcc.2008.10.024. [DOI] [PubMed] [Google Scholar]
- Venkatapuram S, Wang C, Krolikowski JG, Weihrauch D, Kersten JR, Warltier DC, et al. Inhibition of apoptotic protein p53 lowers the threshold of isoflurane-induced cardioprotection during early reperfusion in rabbits. Anesth Analg. 2006;103:1400–1405. doi: 10.1213/01.ane.0000240903.63832.d8e. [DOI] [PubMed] [Google Scholar]
- Weihrauch D, Krolikowski JG, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS. Morphine enhances isoflurane-induced postconditioning against myocardial infarction: the role of phosphatidylinositol-3-kinase and opioid receptors in rabbits. Anesth Analg. 2005;101:942–949. doi: 10.1213/01.ane.0000171931.08371.a2. [DOI] [PubMed] [Google Scholar]
- Yang XM, Philipp S, Downey JM, Cohen MV. Postconditioning's protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation. Basic Res Cardiol. 2005;100:57–63. doi: 10.1007/s00395-004-0498-4. [DOI] [PubMed] [Google Scholar]
- Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol. 2004;44:1103–1110. doi: 10.1016/j.jacc.2004.05.060. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]