

Abstract
Background and purpose:
3,4-methylenedioxymethamphetamine (MDMA) causes a persistent loss of dopaminergic cell bodies in the substantia nigra of mice. Current evidence indicates that such neurotoxicity is due to oxidative stress but the source of free radicals remains unknown. Inhibition of mitochondrial electron transport chain complexes by MDMA was assessed as a possible source.
Experimental approach:
Activities of mitochondrial complexes after MDMA were evaluated spectrophotometrically. In situ visualization of superoxide production in the striatum was assessed by ethidium fluorescence and striatal dopamine levels were determined by HPLC as an index of dopaminergic toxicity.
Key results:
3,4-methylenedioxymethamphetamine decreased mitochondrial complex I activity in the striatum of mice, an effect accompanied by an increased production of superoxide radicals and the inhibition of endogenous aconitase. α-Lipoic acid prevented superoxide generation and long-term toxicity independent of any effect on complex I inhibition. These effects of α-lipoic acid were also associated with a significant increase of striatal glutathione levels. The relevance of glutathione was supported by reducing striatal glutathione content with L-buthionine-(S,R)-sulfoximine, which exacerbated MDMA-induced dopamine deficits, effects suppressed by α-lipoic acid. The nitric oxide synthase inhibitor, NG-nitro-L-arginine, partially prevented MDMA-induced dopamine depletions, an effect reversed by L-arginine but not D-arginine. Finally, a direct relationship between mitochondrial complex I inhibition and long-term dopamine depletions was found in animals treated with MDMA in combination with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
Conclusions and implications:
Inhibition of mitochondrial complex I following MDMA could be the source of free radicals responsible for oxidative stress and the consequent neurotoxicity of this drug in mice.
This article is commented on by Moncada, pp. 217–219 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00706.x and to view related papers in this issue by Pravdic et al. and Kurz et al. visit http://dx.doi.org/10.1111/j.1476-5381.2010.00698.x and http://dx.doi.org/10.1111/j.1476-5381.2010.00656.x
Keywords: complex I, dopamine, lipoic acid, MDMA, mitochondria, oxidative stress
Introduction
Administration of the amphetamine derivative 3,4-methylenedioxymethamphetamine (MDMA, ecstasy) to mice produces persistent long-term effects in their brains which are considered to reflect neurodegenerative changes (Capela et al., 2009). Such neurotoxicity results in a decrease in the content of striatal dopamine and its main metabolites (Stone et al., 1987; Logan et al., 1988; O'Callaghan and Miller, 1994; Capela et al., 2009); the decline in L-tyrosine hydroxylase (TH) and dopamine transporter (DAT) immunostaining (Granado et al., 2008a); and increased markers of microglial and astrocytic activation in striatum, all providing anatomical correlation with dopaminergic deficits (Granado et al., 2008b). It is noteworthy that these latter authors were the first to show that MDMA in mice causes a persistent loss of dopaminergic cell bodies in the substantia nigra, indicating that MDMA neurotoxicity in this species is not restricted to the loss of neuronal 5-hydroxytryptaminergic terminals, as it is in rats (Granado et al., 2008b).
Recent evidence has revealed that striosomes are more vulnerable than the matrix to MDMA-induced loss of TH/DAT immunoreactive terminals (Granado et al., 2008a). This selective vulnerability has also been described for the Parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Iravani et al., 2005). Interestingly, there are other striking similarities between MDMA and MPTP toxicity. MPTP in mice is the most commonly studied animal model of Parkinson's disease (Przedborski and Vila, 2003). However, as found for MDMA, the dopaminergic neurotransmission system of the rat is essentially insensitive to the neurotoxic effects of systemically administered MPTP (Giovanni et al., 1994a, 1994b). Furthermore, DAT activity plays a key role in the neurotoxic process of both MPTP and MDMA as blockade of DAT by specific antagonists completely prevents both, MPTP- and MDMA-induced toxicity (Javitch et al., 1985; Bezard et al., 1999; O'Shea et al., 2001; Camarero et al., 2002). Moreover, reactive oxygen species (ROS) exert many of the toxic effects of MPTP and MDMA in mice (Przedborski and Vila, 2001, 2003; Capela et al., 2009), and transgenic mice with increased brain activity of copper/zinc superoxide dismutase (SOD1) are significantly more resistant to MPTP- or MDMA-induced dopaminergic toxicity than their non-transgenic littermates (Przedborski et al., 1992; Cadet et al., 1995).
It is well known that MPTP is metabolized into 1-methyl-4-phenylpyridinium (MPP+) which enters dopaminergic neurons by means of the DAT. Once inside, MPP+ impairs mitochondrial respiration by inhibiting complex I of the electron transport chain, which leads to an increased production of ROS responsible for oxidative stress (Nicklas et al., 1985; Mizuno et al., 1987; Przedborski and Vila, 2001, 2003). Current evidence indicates that neurotoxicity caused by MDMA in mice is also due to oxidative stress (Cadet et al., 1995; Jayanthi et al., 1999; Colado et al., 2001; Camarero et al., 2002; Sanchez et al., 2003); however, the source of free radicals remains largely unknown. Due to the similarities between MDMA and MPTP toxicity in mice we investigated whether MDMA administration to mice affects the activity of the mitochondrial transport chain complexes as a possible source of the ROS formed and its possible relationship with the long-term neurotoxic effects.
Methods
Animals and treatments
All animal care and experimental procedures were in accordance with the European Community Council Directive of 24 November 1986 (86/609/EEC) and approved by the Ethical Committee of the University of Navarra. Male Swiss-Webster mice (CD-1, Harlan Ibérica, Barcelona, Spain), 25–30 g weight, were housed in groups of five in constant conditions of humidity and temperature (22 ± 1°C) with a 12-h/12-h light-dark cycle (lights on at 07:00 h). Food and water were available ad libitum. The total number of mice used in the present work was 386 animals.
Study I: effect of MDMA on the activity of mitochondrial electron transport chain complexes
To study the effects of MDMA on the activities of mitochondrial electron transport chain complexes, mice were treated with saline or three increasing doses of MDMA (10, 20, 30 mg·kg−1 i.p. every 2 h) and were killed 1, 3, 6, 12 or 24 h after the last MDMA injection. The striatum was dissected free and was homogenized in buffer (20 mM HEPES pH 7.8) at a tissue concentration of 2 µg·µL−1. For aconitase activity measurements mice were treated with saline or MDMA and killed 1, 3 or 6 h later.
Study II: nitric oxide (NO) and MDMA-induced toxicity
Mice were pretreated with either saline or the non selective NO synthase (NOS) inhibitor NG-nitro-L-arginine (L-NNA; 10 mg·kg−1 i.p.) 30 min before MDMA administration, as described (Nowicki et al., 1991). The inhibition of NOS is stereospecific and is attenuated by high concentrations of L-arginine, which competes for binding to NOS, but not D-arginine, which does not selectively bind to NOS (Knowles et al., 1990). L-arginine or D-arginine were given at 300 mg·kg−1 i.p. 5 min before L-NNA pretreatment, as described (Matthews et al., 1997).
Study III: effect of α-lipoic acid (LA) on MDMA-induced toxicity
Mice were distributed into four groups which received: (1) vehicle/vehicle; (2) LA/vehicle; (3) vehicle/MDMA; and (4) LA/MDMA. Both LA (100 mg·kg−1 i.p. in sodium bicarbonate 7.5% w·v−1, pH 7.4) and vehicle were given twice daily for 2 days, and MDMA (10, 20, 30 mg·kg−1 i.p. 2 h apart) was given 30 min after the fourth dose of LA or vehicle. Animals were killed either 3 h later to visualize ROS production and to analyse complex I activity, or 7 days later to measure striatal dopamine content. LA dosage regimen was based on earlier work showing full protection of MDMA-induced 5-hydroxytryptaminergic deficits in rats (Aguirre et al., 1999).
Study IV: effect of L-buthionine-(S,R)-sulfoximine (BSO) on striatal glutathione (GSH) content and MDMA-induced toxicity
γ-Glutamylcysteine synthetase (γ-GCS) is the rate-limiting enzyme for GSH synthesis (Griffith, 1982). BSO is an irreversible inhibitor of γ-GCS and thereby depletes intracellular GSH. We used BSO to investigate the role of GSH and ROS in the mechanisms underlying MDMA toxicity.
A set of mice were distributed into four groups which received: (i) vehicle/vehicle; (ii) LA/vehicle; (iii) vehicle/BSO; and (iv) LA/BSO. The LA dosage regimen used was the same as described in Study III. BSO (3 mmol·kg−1 i.p., in sterile saline) was injected 24 h before the fourth injection of LA. Thirty min after the last dose of LA, mice were killed for GSH analysis. BSO treatment was chosen based on a previous report showing significant reductions in the striatal content of GSH without significantly affecting TH activity or dopamine concentrations (Andersen et al., 1996).
Another set of mice were treated identically as described above, and in addition they also received saline or MDMA (10, 20, 30 mg·kg−1 i.p. 2 h apart) 30 min after the last injection of LA. These animals were killed 7 days later, to measure dopamine content.
Study V: effect of MPTP on MDMA-induced toxicity
Mice were treated with a subtoxic (3 × 10 mg·kg−1 i.p. every 2 h) or a toxic (10, 20, 30 mg·kg−1 i.p. every 2 h) treatment of MDMA either alone or in combination with a subtoxic dosage regimen of MPTP (3 × 10 mg·kg−1 i.p. every 2 h). The dose of MPTP was chosen based upon previous studies showing that similar doses had little effect on striatal dopamine content or TH activity in the substantia nigra (Sonsalla and Heikkila, 1986; Albers et al., 1996). Striata were harvested 3 h or 7 days later to analyse complex I activity and to measure striatal dopamine content respectively.
In a final set of experiments, the toxic dosage regimen of MDMA was combined with a toxic MPTP treatment (3 × 20 mg·kg−1 i.p., every 2 h). Shortly after the third MDMA/MPTP injection, three mice begun to show signs of distress and suddenly died in the following 5–10 min. Because other animals in the MDMA/MPTP group started to show similar symptoms, the experiment was rapidly terminated and all remaining animals were killed humanely and promptly following the guidelines of our Institutional Animal Care and Use Committee.
Determination of activities of mitochondrial complexes
Mice were killed by decapitation 1, 3, 6, 12 and 24 h after the last MDMA injection. Striata were dissected free and homogenized in 20 mM HEPES buffer (pH 7.8) to get a final tissue concentration of 2 µg·µL−1. Samples were then freeze-thawed twice and 50 µL aliquots were transferred to 150 µL of the assay medium. Assays of all respiratory chain enzyme activities were carried out spectrophotometrically in a Multiskan Spectrum (Thermo, Electron Corporation, Finland) using standardized and reproducible methods. Complex I activity was determined as described by Ratner et al. (2009). Oxidation of nicotinamide adenine dinucleotide, reduced form (NADH) was followed at 340 nm using coenzyme Q1 as the electron acceptor. Complex II and complex II/III activities were measured as previously described (Klivenyi et al., 2004; Seo et al., 2008). Complex IV activity was determined by following cytochrome c oxidation, sensitive to potassium cyanide (KCN) inhibition, at 550 nm in a medium containing 20 mM HEPES (pH 7.8) supplemented with 0.025 mM reduced cytochrome c. Citrate synthase activity was determined as described by Aleardi et al. (2005).
Assay for aconitase activity
Aconitase activity was measured as described earlier (Cleren et al., 2005). Briefly, frozen-thawed mouse striatum samples were homogenized in 2 mM HEPES buffer (pH 7.8) and mixed with the reaction buffer (2 mM HEPES pH 7.8, 0.6 mM MgCl2, 0.5 mM NADP+, two units of isocitrate dehydrogenase, 1 µM rotenone, 10 µM CaCl2 and 1 mM citrate) in a 96-well plate. Aconitase activity was measured by following the appearance of NADPH at 340 nm (Gardner, 2002). The absorbance changes at 340 nm were followed for 10 min with a plate reader SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA).
In situ detection of superoxide radical (O2•−) production
In situ visualization of O2- production was assessed by hydroethidine histochemistry as previously described (Kim and Chan, 2002). Two and a half h after the last injection of MDMA, mice were injected i.p. with 200 µL of PBS containing 1 µg·µL−1 hydroethidine (Molecular Probes, Invitrogen, Carlsbad, CA, USA) and 1% DMSO. Brains were collected 30 min later and frozen on dry ice. Midbrain sections (25 µm thick) were mounted onto gelatin-coated glass slides, and examined for hydroethidine oxidation product, ethidium accumulation, by fluorescence microscopy (excitation, 510 nm; emission, 580 nm). Fluoresecence intensity was quantified using the image analysis software AnalySISD 5.0 (Soft Imaging System, Olympus, Münster, Germany).
Measurement of rectal temperature
Temperature measurement was performed using a TMP 812 thermometer, with digital readout (Panlab, Barcelona, Spain) and a lubricated YSI 451 rectal semi-flexible probe for mice. Each mouse was lightly restrained by hand, for approximately 10 s, while the probe was inserted approximately 2 cm into its rectum and a steady reading was obtained.
Determination of dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) in the striatum
Striatal concentrations of dopamine, DOPAC and HVA were determined by high performance liquid chromatography with electrochemical detection, as previously described (Goñi-allo et al., 2006).
GSH determination
Glutathione was measured by using the Bioxytech® GSH-400 assay Kit (OXIS, Foster City, CA, USA) according to the manufacturer's protocol.
Statistics
Temperatures were analysed by two-way anova for repeated measures. Treatment was used as the between subjects factor and time as the repeated measure. For the neurochemical analysis, differences in striatal dopamine concentrations, complex I or aconitase activities were analysed by one-way anova. Multiple pair wise comparisons were performed using the Student–Newman–Keuls test. Treatment differences were considered statistically significant at P < 0.05. Data analyses were performed using the Statistical Program for the Social Sciences (SPSS for Windows, 15.0; SPSS Inc., Chicago, IL, USA).
Materials
3,4-methylenedioxymethamphetamine-HCl was a gift from the ‘Servicio de Restricción de Estupefacientes’ (Spanish regulatory body on psychotropic drugs); The following reagents were purchased from Sigma (Madrid, Spain): dopamine, DOPAC, HVA, MPTP, KCN, β-NADH, 2,3-dimethoxy-5-methyl-6-(3-methyl-2-butenyl)-1,4-benzoquinone (coenzyme Q1), rotenone, 2,6-dichlorophenolindo phenol sodium salt, 4,4,4-trifluoro-1-(2-trienyl)-1,3-butadienone, 5,5′-dithiobis-(2-nitrobenzoic acid), oxaloacetic acid, LA, D- and L-arginine, and acetyl coenzyme A sodium salt; 1-buthionine-(S,R)-sulfoximine (BSO) and L-NNA were purchased from Tocris (Biogen Científica S.L., Madrid, Spain) and hydroethidine was from Invitrogen (Carlsbad, CA, USA); all other chemicals were from Merck (Darmstadt, Germany). Drug and receptor nomenclature follows Alexander et al. (2009).
Results
Effect of MDMA on the activity of mitochondrial complexes
In the first set of experiments, we analysed whether MDMA affects the activity of the mitochondrial complexes. As shown in Figure 1A, the administration of a toxic dosage regimen of MDMA (10, 20, 30 mg·kg−1 i.p. every 2 h) produced a significant decrease in mitochondrial complex I activity [F(5,53) = 3.696; P < 0.01]. Such changes are evident 1 h after drug administration and remain significantly below control values for up to 24 h later. However, no change was observed in the activity of any of the other mitochondrial complexes studied at any time (Table 1). No differences were found either in the activity of citrate synthase, a marker of the mitochondrial matrix, suggesting that decreased complex I activity was not due to differences in the amount of mitochondria present in the samples.
Figure 1.
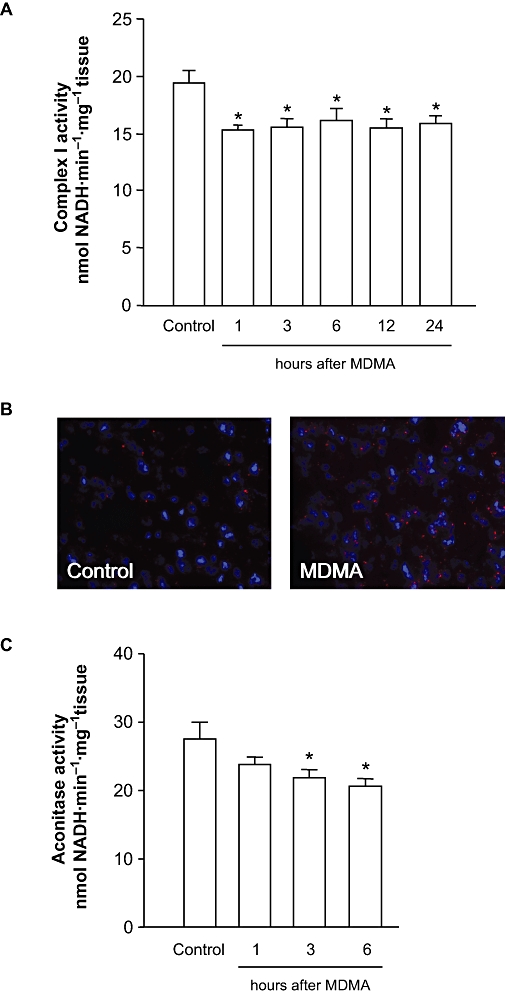
Effect of MDMA on mitochondrial complex I and aconitase activities and superoxide (O2-) production. (A) Mitochondrial complex I in the striatum of mice killed 1, 3, 6, 12 and 24 h after MDMA (10, 20, 30 mg·kg−1 i.p. every 2 h). (B) Representative photomicrographs showing fluorescent ethidium signals 3 h after saline or MDMA. Note that MDMA treatment resulted in a significant increased in oxidized hydroethidine signals compared with saline-treated mice. (C) Effect of MDMA on aconitase activity in mice killed 1, 3 and 6 h after MDMA. The results shown as mean ± SEM (n= 10; A and C). Differences from saline: *P < 0.05. MDMA, 3,4-methylenedioxymethamphetamine.
Table 1.
Effect of MDMA (10, 20, 30 mg·kg-1 i.p. every 2 h) on the activities of mitochondrial complexes II, II/III, IV and citrate synthase at various time points
| Complex II | Complex II/III | Complex IV | Citrate synthase | |
|---|---|---|---|---|
| Control | 13.28 ± 0.83 | 17.05 ± 0.51 | 6.01 ± 1.01 | 45.72 ± 1.19 |
| 1 h | 12.04 ± 0.61 | 16.97 ± 0.62 | 5.30 ± 0.58 | 43.93 ± 1.05 |
| 3 h | 11.67 ± 0.69 | 17.22 ± 0.49 | 6.33 ± 0.54 | 44.99 ± 1.05 |
| 6 h | 12.09 ± 0.76 | 17.15 ± 0.28 | 5.24 ± 0.51 | 45.41 ± 1.31 |
| 12 h | 12.49 ± 0.83 | 17.08 ± 0.54 | 5.03 ± 0.57 | 43.18 ± 1.68 |
| 24 h | 12.34 ± 0.82 | 16.75 ± 0.97 | 6.67 ± 0.56 | 45.73 ± 1.52 |
Results shown as mean ± SEM, n= 10, in DCPIP nmol·min−1·mg−1 tissue (complex II), cytochrome c nmol·min−1·mg−1 tissue (complex II/III), cytochrome c µmol·min−1·mg−1 tissue (complex IV) and DTNB nmol·min−1·mg−1 tissue (citrate synthase).
One-way anova revealed no significant differences between control and MDMA treatment at any time point.
DCPIP, 2,6-dichlorophenolindo phenol; DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); MDMA, 3,4-methylenedioxymethamphetamine.
In saline-injected mice, striatal O2- and O2--derived oxidant production, as assayed by ethidium fluorescence, was minimal (Figure 1B). In contrast, in MDMA-treated mice, striatal production of O2- or O2--derived oxidants shown by ethidium fluorescence was increased at 3 h after MDMA.
It has been previously reported that aconitase is sensitive to O2- production and several studies used aconitase as an intracellular indicator of superoxide formation (Patel et al., 1996; Liang et al., 2000; Li et al., 2001; Tretter et al., 2005), and has proven to be sensitive enough to reflect in situ ROS generation in mitochondria (Sipos et al., 2003). Under our experimental conditions, aconitase activity was significantly decreased 3 and 6 h after MDMA [F(3,77) = 3.361; P < 0.05] (Figure 1C).
Effect of NOS modulation on MDMA-induced depletion of striatal dopamine
We also examined whether pretreatment with a non-specific NOS inhibitor, L-NNA, could attenuate the depletion of striatal dopamine induced by MDMA (∼ 45% vs. control). We found that administration of L-NNA produced significant neuroprotection against dopamine loss (Figure 2). Furthermore we found that the administration of L-arginine (300 mg·kg−1 i.p. 0.5 h before MDMA) completely blocked the neuroprotective effect of L-NNA [F(7,65) = 10.78; P < 0.001], whereas D-arginine (at the same dose) had no significant effect on the protection afforded by L-NNA [F(7,62) = 7.444; P < 0.001].
Figure 2.
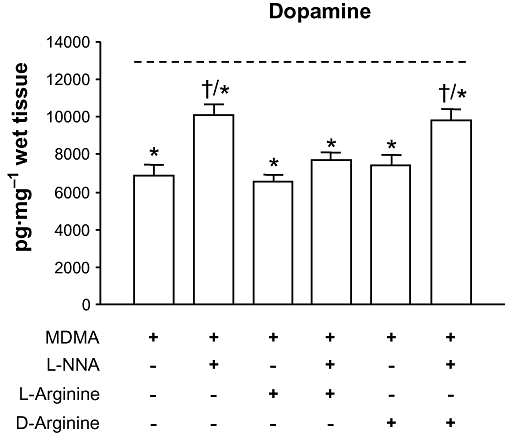
Effect of the NOS inhibitor L-NNA alone or in combination with D- or L-arginine on MDMA-induced striatal dopamine loss. L-NNA (10 mg·kg−1 i.p.) or saline were administered 30 min before MDMA (10, 20, 30 mg·kg−1 i.p. every 2 h), and D- or L-arginine (300 mg·kg−1 i.p.) were given 5 min before L-NNA or saline. Mice were killed 7 days later. L-arginine but not D-arginine reversed the protection afforded by L-NNA against MDMA-induced dopamine depletions. Discontinous line (----) represents dopamine levels in saline-treated mice. Results shown as means ± SEM (n= 8–12). *P < 0.05 different from saline; †P < 0.05 different from MDMA-treated mice. L-NNA, NG-nitro-L-arginine; MDMA, 3,4-methylenedioxymethamphetamine; NOS, nitric oxide synthase.
LA prevents MDMA-induced dopaminergic deficits but not mitochondrial complex I inhibition
Seven days after MDMA striatal concentrations of dopamine, DOPAC and HVA were decreased by 48%, 29% and 30% respectively. LA pretreatment afforded a complete protection against the loss of striatal dopamine and its metabolites caused by MDMA. One-way anova comparing the four groups of mice yielded the following results: dopamine: F(3,25) = 8.565; P < 0.001; DOPAC: F(3,25) = 6.649; P < 0.001 and HVA: F(3,25) = 4.151; P < 0.01] (Figure 3A).
Figure 3.
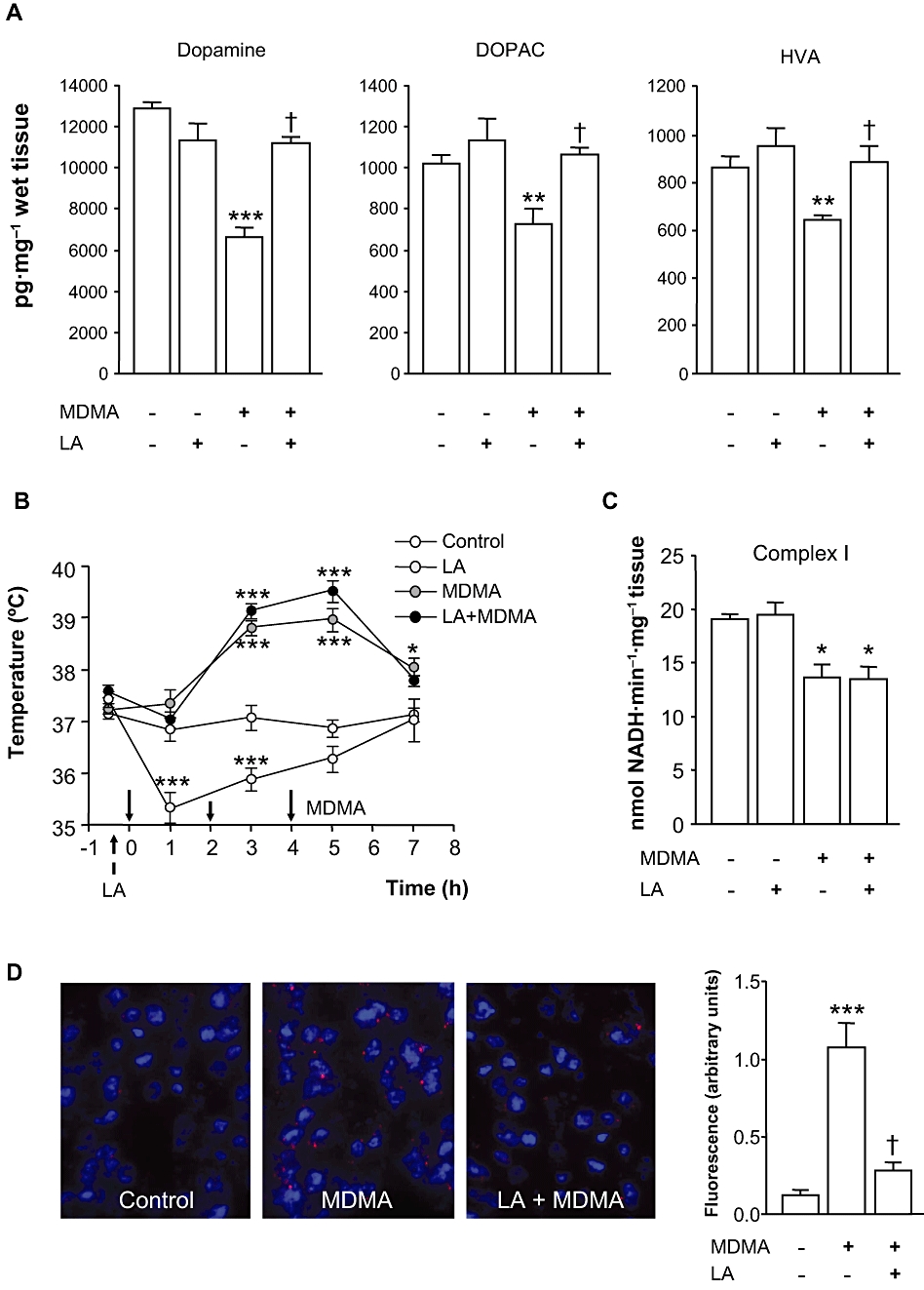
Effect of LA on MDMA-induced dopaminergic deficits, mitochondrial complex I inhibition and ROS production. LA (100 mg·kg−1 i.p.) or vehicle were given twice daily for 2 days, and MDMA (10, 20, 30 mg·kg−1 i.p. 2 h apart) was given 30 min after the fourth administration of LA or vehicle. (A) Striatal dopamine, DOPAC and HVA levels of mice 7 days after MDMA given alone or in combination with LA. Results shown as means ± SEM in pg·mg−1 wet tissue (n= 8–10). (B) Effect of LA on MDMA-induced acute hyperthermia. Arrows denote administration of LA or MDMA (solid arrows). Mice temperatures were recorded at baseline (t=–30 min) and 1 h after every injection of MDMA up to 7 h. Values are means ± SEM (n= 8–12 mice per group). (C) Complex I activity measured in the striata of mice 3 h after treatments. Results shown as means ± SEM (n= 8–12). (D) Effect of LA on MDMA-induced O2•− production. O2•− radicals were measured in animals treated with saline (control) or MDMA alone or in combination with LA. Note that LA decreased the ethidium signal evoked by MDMA. Results shown as means ± SEM (n= 3–4). In all panels: *P < 0.05, **P < 0.01 or ***P < 0.001 versus control group; †P < 0.01, different from MDMA-treated animals. One-way anova followed by Newman–Keuls test. DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; LA, α-lipoic acid; MDMA, 3,4-methylenedioxymethamphetamine; ROS, reactive oxygen species.
As expected, MDMA significantly increased the core body temperature of mice, while LA given alone caused hypothermia. The latter effect was not surprising as it has also been described in rats (Aguirre et al., 1999). Analysis of temperature curves using two-way anova for repeated measures revealed a significant interaction treatment × time [F(15,156) = 11.259, P < 0.001]. Single time point comparisons revealed no significant differences between mice treated with MDMA alone or in combination with LA, indicating that protection afforded by LA was independent of any effect on MDMA-induced hyperthermia (Figure 3B). We also analysed whether LA could have reversed the inhibition of complex I caused by MDMA. As shown in Figure 3C, complex I activity was significantly reduced in the groups of mice given MDMA alone or in combination with LA [F(3,33) = 9.177; P < 0.01].
As expected O2- and O2--derived oxidant production shown by the ethidium signal was increased in the striatum of MDMA-treated mice as compared with saline-treated animals. By contrast, the ethidium signal evoked by MDMA was markedly decreased in mice treated with the LA/MDMA combination, suggesting the inhibition of O2- production by LA (Figure 3D).
GSH depletion by BSO potentiates MDMA-induced toxicity: reversal by LA
To obtain further insight into the involvement of ROS in MDMA-induced dopamine depletions and LA afforded protection, we used the γ-GCS inhibitor BSO. First, we investigated the effects of BSO, LA and the LA/BSO combination on striatal GSH content. As shown in Figure 4A, BSO and LA caused opposite effects. While BSO significantly decreased the concentration of GSH in the striatum, LA not only increased it above control values but also restored the concentration of GSH in those animals receiving the LA/BSO combination [F(3,33) = 21.64; P < 0.01],
Figure 4.
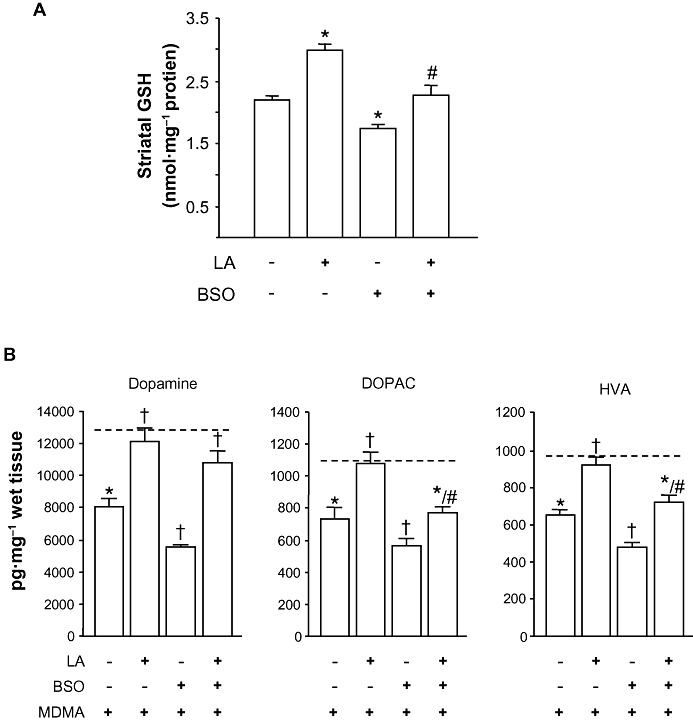
Effect of BSO on MDMA induced-toxicity. BSO (3 mmol·kg−1 i.p.) was given 24 h before the fourth injection of LA (4 × 100 mg·kg−1 i.p. 12 h apart). (A) Striatal GSH levels 24 h after BSO given alone or in combination with LA. Results shown as means ± SEM (n= 8–10). *P < 0.05 versus saline; #P < 0.05 versus BSO-treated animals. (B) Striatal dopamine, DOPAC and HVA levels of mice 7 days after administration of BSO or LA in combination with MDMA. Discontinous line (----) shows dopamine, DOPAC and HVA levels of saline-treated mice. Results shown as means ± SEM (n= 8–12). *P < 0.05, different from saline; †P < 0.05, different from MDMA-treated mice; #P < 0.05, different from BSO + MDMA treated animals. One-way anova followed by Newman–Keuls test. BSO, L-buthionine-(S,R)-sulfoximine; DOPAC, 3,4-dihydroxyphenylacetic acid; GSH, glutathione; LA, α-lipoic acid; MDMA, 3,4-methylenedioxymethamphetamine.
We next investigated the effect of BSO on MDMA-induced dopamine depletion and LA-afforded protection. As shown in Figure 4B, BSO exacerbated the reductions in the levels of secondary biomarkers of dopamine neurotoxicity (dopamine, DOPAC and HVA concentrations in the striatum 7 days after drug exposure). The effect of BSO on dopamine concentrations was completely reversed by LA [F(7,65) = 10.64; P < 0.001], and partially prevented in the case of DOPAC [F(7,65) = 15.54; P < 0.001] and HVA [F(7,65) = 11.23; P < 0.001]. BSO or LA given alone and their combination caused no significant effect on striatal dopamine concentrations (data not shown).
Effect of co-administration of MDMA and MPTP on striatal dopamine levels and mitochondrial complex I activity
Intraperitoneal administration of non-toxic doses of MDMA (3 × 10 mg·kg−1 every 2 h) or MPTP (3 × 10 mg·kg−1 every 2 h) produced no long-term effects in the dopamine content in the striatum (Figure 5A). In contrast, the co-administration of MDMA and MPTP resulted in a significant reduction of striatal dopamine concentrations [F(3,36) = 6.396; P < 0.05]. Consistent with these results, complex I activity remained unaltered 3 h after MDMA or MPTP, while the co-administration of both dopaminergic toxins resulted in a significant reduction in NADH oxidation rate (Figure 5B). Thus, one-way anova analysis revealed significant differences [F(3,36) = 6.666; P < 0.01].
Figure 5.
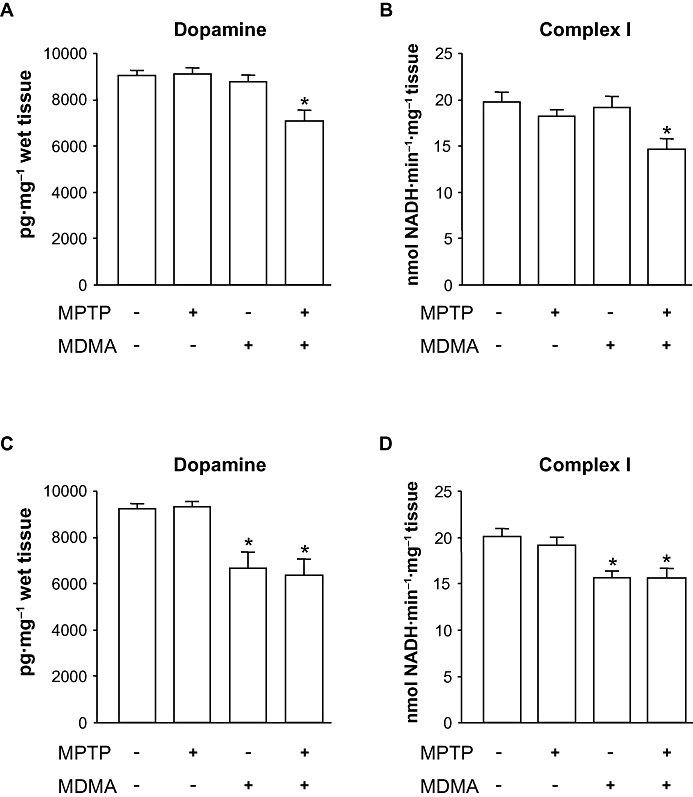
Effect of MPTP on MDMA-induced toxicity. (A) Striatal dopamine levels 7 days after the administration of MDMA (3 × 10 mg·kg−1 i.p. every 2 h) alone or in combination with MPTP (3 × 10 mg·kg−1 i.p. every 2 h). (B) mitochondrial complex I activity 3 h after the same drug treatments shown in panel A. (C) Striatal dopamine levels 7 days after the administration of MDMA (10, 20, 30 mg·kg−1 i.p. every 2 h) alone or in combination with MPTP (3 × 10 mg·kg−1 i.p. every 2 h). (D) mitochondrial complex I activity 3 h after the same drug treatments shown in panel C. Results shown as means ± SEM (n= 8–12). *P < 0.05 versus saline-treated mice. One-way anova followed by Newman–Keuls test. MDMA, 3,4-methylenedioxymethamphetamine; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
As co-administration of non-toxic treatments with MDMA and MPTP resulted in a significant decrease of striatal concentration of dopamine, we next tested whether a non-toxic dosage regimen of MPTP would exacerbate the long-term depletion of striatal dopamine caused by a toxic dosage regimen of MDMA. As it can be seen in Figure 5C, the administration of MDMA produced a significant long-term depletion of striatal dopamine content [F(3,38) = 9.934; P < 0.01] and its metabolites DOPAC and HVA (data not shown). The addition of MPTP, however, did not exacerbate the dopaminergic deficits caused by MDMA alone.
We also tested whether the MDMA/MPTP combination would result in a larger inhibition of mitochondrial complex I than that caused by MDMA alone. The results shown in Figure 5D indicate that while MDMA significantly reduced complex I activity [F(3,33) = 7.771; P < 0.01], such effects were not potentiated by the co-administration of non-toxic doses of MPTP. Citrate synthase activity used as a normalizing control did not vary significantly between groups (data not shown).
Discussion
In this study we provide new insight into the pathways involved in MDMA-induced dopaminergic deficits in mice. Our data demonstrate, for the first time, that MDMA inhibits mitochondrial complex I activity in the striatum of mice. As a consequence, there was a significant overproduction of O2- radicals, shown by ethidium fluorescence, resulting in a significant reduction of aconitase activity, possibly due to its oxidation. Consistent with these findings, the metabolic anti-oxidant LA afforded complete protection against MDMA-induced long-term loss of striatal dopamine content. LA not only abolished O2- radical production downstream complex I inhibition, but also increased striatal GSH content. Noteworthy, MDMA-induced neurotoxicity correlated negatively with striatal GSH levels. Moreover, NOS inhibition partially prevented the dopaminergic deficits caused by MDMA, an effect reversed by L-arginine but not by D-arginine. Finally, our data also showed a direct relationship between complex I inhibition and long-term dopamine depletions further suggesting that this effect could be, at least, partially responsible for the neurotoxicity produced by this amphetamine derivative in this animal species (Figure 6).
Figure 6.
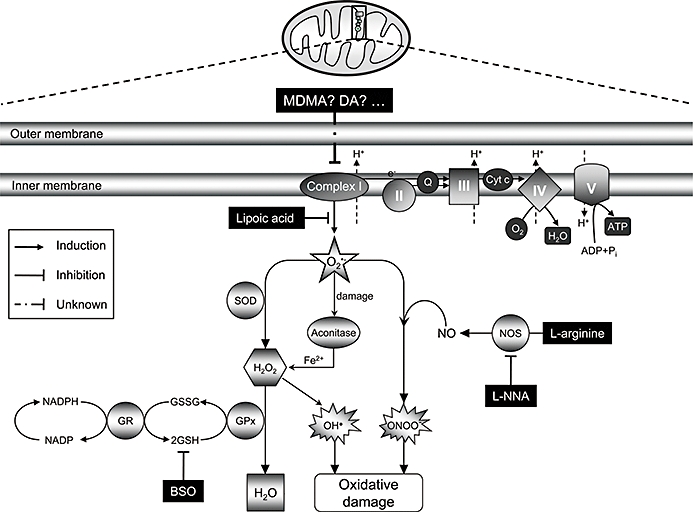
A hypothetical model of MDMA-induced dopaminergic toxicity in mice. The inhibition of mitochondrial complex I activity after systemic administration of MDMA promotes the generation of superoxide radicals (O2•−), which could lead to the formation of peroxynitrite (ONOO-), in the presence of NO, or to H2O2 by means of superoxide dismutase (SOD). The [4Fe-4S]2+ cluster of aconitase could then be inactivated by O2•−, peroxynitrite or H2O2 leading to the generation of Fe2+, which, together with H2O2, may increase the formation of hydroxyl radicals (OH•) by the Fenton reaction. Glutathione (GSH) is involved both as a non-enzymic, free radical scavenger for ROS and/or peroxynitrite and enzymically to inactivate H2O2. In summary, inhibition of mitochondrial complex I activity could be a plausible source of free radicals responsible for oxidative damage to dopamine neurons caused by MDMA in mice. Further studies are needed, however, to resolve which is/are the specific compound(s) responsible for such effects. GPx, gluthathione peroxidase; GR, glutathione reductase; MDMA, 3,4-methylenedioxymethamphetamine; ROS, reactive oxygen species.
Consistent with previous observations supporting the mitochondrion as a main player in neurodegenerative processes, our data revealed that one of the earlier events that take place in MDMA-induced neurotoxicity in mice is the inhibition of complex I of the mitochondrial electron transport chain. This effect appears to be specific, as MDMA failed to modify the activity of other complexes including complex II, II/III or IV.
It has been shown that increased ROS generation parallels the inhibition of complex I (Hasegawa et al., 1990; Ramsay and Singer, 1992; Liu et al., 2002; Perier et al., 2005). Also, inactivation of complex I to a similar extent to that found in our experiments (∼25%) resulted in a significant increase in superoxide production (Sipos et al., 2003). Furthermore, cell death caused by other complex I inhibitors has been specifically and quantitatively linked to ROS production (Barrientos and Moraes, 1999). Accordingly, we focused our work on the consequence of complex I inhibition, that is, increased ROS production. We used hydroethidine, which is selectively oxidized to ethidium by O2- (Bindokas et al., 1996), to visualize in situ the production of O2- after MDMA. Consistent with the inhibitory effect on mitochondrial complex I activity, MDMA caused the build-up of O2- radicals in the striatum of mice.
Aconitase, a Krebs cycle enzyme, is very sensitive to inhibition by O2- (Gardner et al., 1995) and has been used in several studies as a marker of its production (Bindokas et al., 1996; Patel et al., 1996; Kim and Chan, 2002). This enzyme is sensitive enough to reflect in situ ROS generation in mitochondria (Gardner, 2002; Sipos et al., 2003). In agreement with these reports, we found that aconitase activity was significantly decreased after MDMA treatment, further supporting the possible involvement of O2- in MDMA-induced dopamine toxicity. It is noteworthy that mitochondrial aconitase is not just a target but is also a source of ROS. The [4Fe-4S]2+ cluster of mitochondrial aconitase is oxidized by O2- leading to the generation of free Fe (II), which, together with H2O2, may increase the formation of hydroxyl radicals via the Fenton reaction. Because hydroxyl radicals were found to be the most efficient inhibitor of complex I among the ROS tested (Zhang et al., 1990), further ROS could be generated in a vicious cycle, which may exist between complex I inhibition and aconitase inactivation.
The [4Fe-4S]2+ cluster of aconitase is also inactivated by peroxynitrite or H2O2 (Gardner et al., 1995; Hausladen and Fridovich, 1996; Tretter and Adam-Vizi, 2000). Given this, toxicity after MDMA could derive from peroxynitrite that is formed by the diffusion-limited reaction of O2- with NO (Beckman et al., 1990; Ischiropoulos and al-Mehdi, 1995). Our data appear to support this notion as the non-selective NOS inhibitor L-NNA partially protected against the long-term dopamine-depleting effects of MDMA, an effect reversed by the NO precursor L-arginine, but not by D-arginine. These findings are also supported by previous reports showing that the neuronal NOS inhibitor, S-methyl-L-thiocitrulline provides significant neuroprotection against MDMA-induced long-term dopamine depletion in mice (Colado et al., 2001). Consistent with peroxynitrite involvement in MDMA neurotoxicity in mice are also the findings that mice genetically deficient in neuronal NOS and mice overexpressing human CuZn-SOD are less sensitive to MDMA toxicity compared with their wild-type counterparts (Cadet et al., 1995; Itzhak et al., 2004).
In order to further evaluate the relevance of ROS in MDMA-induced neurotoxicity, we also performed a pharmacological approximation using LA. We have previously demonstrated that LA fully prevents MDMA-induced 5-HT deficits in rats (Aguirre et al., 1999). In line with these findings we now show that it also completely prevented MDMA-induced dopamine depletions in mice. Protection afforded by LA was independent of any effect of LA on MDMA-induced hyperthermia or complex I inhibition. It is noteworthy that LA completely abolished O2- production when combined with MDMA, further suggesting that O2- derived ROS formation after complex I inhibiton by MDMA is a key step in the mechanisms underlying MDMA-induced dopamine toxicity. It has been recently reported that MDMA produces DNA single and double-strand breaks along with extensive mitochondrial oxidative damage (Frenzilli et al., 2007; Alves et al., 2009). Because LA potently inhibits peroxynitrite-mediated DNA strand breakage (Jia et al., 2009) and is also one of the major cofactors for a specific lipoyl dehydrogenase which catalyses the reduction of nitrated DNA (Chen et al., 2002), it may also specifically protect from MDMA-induced DNA damage.
Glutathione is considered to be the most prevalent and important intracellular non-protein thiolsulphydryl compound in mammalian cells (Bains and Shaw, 1997). A crucial role for GSH is as a free radical scavenger, particularly effective against the hydroxyl radical (Coyle and Puttfarcken, 1993). Because there are no known enzymatic defences against this radical species, the ability of GSH to non-enzymatically scavenge hydroxyl radicals provides a first line of anti-oxidant defence. In agreement with previous findings (Suh et al., 2004; Fujita et al., 2008; Petersen et al., 2008), our results demonstrate that LA significantly increased endogenous GSH levels in the striatum of mice. This effect may be particularly important in the case of MDMA toxicity, as hydroxyl radical formation has been demonstrated by measuring the conversion of salicylic acid to 2,3-dihydroxy benzoic acid in the striatum of MDMA-treated mice (Colado et al., 2001; Camarero et al., 2002).
Dismutation of O2- to H2O2 is catalysed by the action of Mn-SOD in the mitochondrion or by CuZn-SOD in the cytosol. H2O2 is then converted to water and oxygen by catalase and GSH peroxidase, which uses GSH as a proton donor. Although the activities of GSH peroxidase and GSH reductase in brain mitochondria are relatively low, these do nonetheless appear to play an important role in the elimination of H2O2 (Desagher et al., 1996; Dringen et al., 1999). This function appears to be also important in the case of MDMA as our data show that a small reduction in GSH levels, caused by BSO, was sufficient to potentiate MDMA-induced long-term dopamine depletions, effects that were reversed by previous LA administration. This evidence for a key protective role of GSH against MDMA-induced toxicity in mice is also supported by the study by Sanchez et al. (2003) who described a negative correlation between GSH peroxidase activity and the dopaminergic toxicity induced by MDMA.
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine represents the most important and most frequently used Parkinsonian toxin applied in animal models. MPTP delivered systemically to mice or non-human primates selectively damages dopaminergic neurons in the substantia nigra via inhibition of mitochondrial complex I (Beal, 2001; Przedborski and Vila, 2003). As MPTP-induced toxicity is associated with the inhibition of mitochondrial function we hypothesized that MPTP would synergize with MDMA and their combination would result in greater depletion of dopamine than that caused by either substance alone. In accordance with our assumption, sub-toxic dosage regimens of MDMA and MPTP alone had no effect on mitochondrial complex I activity; however, the combination of both resulted in a significant decrease in NADH oxidation rate. These results are consistent with the concentrations of dopamine found in the striatum 7 days later. We next studied whether the same MPTP treatment would exacerbate complex I inhibition and/or dopamine deficits caused by a toxic treatment of MDMA. Although we found a direct relationship between complex I activity and striatal dopamine content, in this case MPTP did not potentiate the effects of MDMA. Further experiments are warranted to clarify this issue. It is possible that MDMA and MPTP may compete for different mechanisms underlying dopamine toxicity such as the 5-HT transporter, the DAT, monoamine oxidase B (MAO-B) or the same mitochondrial complex I subunit (Leonardi and Azmitia, 1994; O'Shea et al., 2001; Przedborski and Vila, 2003; Alves et al., 2007).
Our results clearly demonstrate that systemic administration of MDMA to mice inhibits mitochondrial complex I activity; however, the exact mechanism(s) by which this may occur remain(s) to be elucidated. Intact mitochondria can accumulate dopamine, thereby enabling the interaction between dopamine and complex I (Brenner-lavie et al., 2008). As dopamine per se is capable of inhibiting complex I activity but not that of complex II, IV or V (Brenner-lavie et al., 2008, 2009), one could hypothesize that under conditions of increased availability of cytoplasmic dopamine or decreased metabolism of dopamine, the potential for dopamine-induced effects on mitochondria would increase. Such conditions are thought to exist following a toxic dosage regimen of MDMA, resulting in the redistribution of dopamine from vesicular storage to the cytoplasm (Sabol and Seiden, 1998). Moreover, MDMA is known to inhibit MAO-A activity (Leonardi and Azmitia, 1994), which would further increase the availability of cytoplasmic dopamine after MDMA within the dopaminergic terminal as MAO-A is predominantly expressed inside catecholaminergic neurons (Shih et al., 1999). It is also noteworthy that dopamine can be metabolized as the preferred substrate for MAO-B. Although this MAO isoform is mainly found inside 5-hydroxytryptaminergic terminals and glial cells (Shih et al., 1999), long-term dopamine depletions caused by MDMA was more pronounced in MAO-B deficient mice than in their wild-type counterparts (Fornai et al., 2001). Altogether these findings suggest that dopamine could be responsible for complex I inhibition after MDMA in mice; however, this hypothesis remains speculative and is the focus of ongoing experiments in the laboratory.
In summary, this study sheds light in the mechanisms underlying MDMA toxicity in mice. MDMA inhibition of complex I activity in the striatum results in the disruption of redox homeostasis, which may contribute to oxidative stress and final loss of dopamine-expressing cell bodies in the substantia nigra of this animal species. In addition, the inability of LA to reverse MDMA-induced inhibition of complex I, despite blocking the formation of O2-, suggests that complex I inhibition is an early event in MDMA neurotoxicity. In any case, these data raise a number of important issues which require further study.
Acknowledgments
The authors are grateful to Sandra Lizaso for technical assistance. They would also like to thank Ministerio de Educación y Ciencia for a fellowship to E.P. and I.H. is a fellow of the JAE-DOC program (CSIC). This work was supported by grants from the Ministerio de Ciencia e Innovación (SAF2008-05143-C03-03) to NA and SAF2008-05143-C03-01 and Plan Nacional sobre Drogas to J.J.
Glossary
Abbreviations:
- BSO
L-buthionine-(S,R)-sulfoximine
- DAT
dopamine transporter
- DOPAC
3,4-dihydroxyphenylacetic acid
- GSH
glutathione
- HVA
homovanillic acid
- L-NNA
NG-nitro-L-arginine
- LA
α-lipoic acid
- MDMA
3,4-methylenedioxymethamphetamine
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NADH
nicotinamide adenine dinucleotide, reduced form
- NOS
nitric oxide synthase
- O2•−
superoxide radical
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TH
L-tyrosine hydroxylase
Conflict of interest
None.
References
- Aguirre N, Barrionuevo M, Ramírez MJ, Del Río J, Lasheras B. Alpha-lipoic acid prevents 3,4-methylenedioxy-methamphetamine (MDMA)-induced neurotoxicity. Neuroreport. 1999;10:3675–3680. doi: 10.1097/00001756-199911260-00039. [DOI] [PubMed] [Google Scholar]
- Albers DS, Zeevalk GD, Sonsalla PK. Damage to dopaminergic nerve terminals in mice by combined treatment of intrastriatal malonate with systemic methamphetamine or MPTP. Brain Res. 1996;718:217–220. doi: 10.1016/0006-8993(96)00135-7. [DOI] [PubMed] [Google Scholar]
- Aleardi AM, Benard G, Augereau O, Malgat M, Talbot JC, Mazat JP, et al. Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J Bioenerg Biomembr. 2005;37:207–225. doi: 10.1007/s10863-005-6631-3. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves E, Summavielle T, Alves CJ, Custódio JB, Fernandes E, de Lourdes Bastos M, et al. Ecstasy-induced oxidative stress to adolescent rat brain mitochondria in vivo: influence of monoamine oxidase type A. Addict Biol. 2009;14:185–193. doi: 10.1111/j.1369-1600.2008.00143.x. [DOI] [PubMed] [Google Scholar]
- Alves E, Summavielle T, Alves CJ, Gomes-da-Silva J, Barata JC, Fernandes E, et al. Monoamine oxidase-B mediates ecstasy-induced neurotoxic effects to adolescent rat brain mitochondria. J Neurosci. 2007;27:10203–10210. doi: 10.1523/JNEUROSCI.2645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JK, Mo JQ, Hom DG, Lee FY, Harnish P, Hamill RW, et al. Effect of buthionine sulfoximine, a synthesis inhibitor of the antioxidant glutathione, on the murine nigrostriatal neurons. J Neurochem. 1996;67:2164–2171. doi: 10.1046/j.1471-4159.1996.67052164.x. [DOI] [PubMed] [Google Scholar]
- Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res Rev. 1997;25:335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- Beal MF. Experimental models of Parkinson's disease. Nat Rev Neurosci. 2001;2:325–334. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PM, Freeman BA. Apparent hydroxy radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1621–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Gross CE, Fournier MC, Dovero S, Bloch B, Jaber M. Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp Neurol. 1999;155:268–273. doi: 10.1006/exnr.1998.6995. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner-Lavie H, Klein E, Ben-Shachar D. Mitochondrial complex I as a novel target for intraneuronal DA: modulation of respiration in intact cells. Biochem Pharmacol. 2009;78:85–95. doi: 10.1016/j.bcp.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Brenner-Lavie H, Klein E, Zuk R, Gazawi H, Ljubuncic P, Ben-Shachar D. Dopamine modulates mitochondrial function in viable SH-SY5Y cells possibly via its interaction with complex I: relevance to dopamine pathology in schizophrenia. Biochim Biophys Acta. 2008;1777:173–185. doi: 10.1016/j.bbabio.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Ladenheim B, Hirata H, Rothman RB, Ali S, Carlson E, et al. Superoxide radicals mediate the biochemical effects of methylenedioxymethamphetamine (MDMA): evidence from using CuZn-superoxide dismutase transgenic mice. Synapse. 1995;21:169–176. doi: 10.1002/syn.890210210. [DOI] [PubMed] [Google Scholar]
- Camarero J, Sanchez V, O'shea E, Green AR, Colado MI. Studies, using in vivo microdialysis, on the effect of the dopamine uptake inhibitor GBR 12909 on 3,4-methylenedioxymethamphetamine (‘ecstasy’)-induced dopamine release and free radical formation in the mouse striatum. J Neurochem. 2002;81:961–972. doi: 10.1046/j.1471-4159.2002.00879.x. [DOI] [PubMed] [Google Scholar]
- Capela JP, Carmo H, Remião F, Bastos ML, Meisel A, Carvalho F. Molecular and cellular mechanisms of ecstasy-induced neurotoxicity: an overview. Mol Neurobiol. 2009;39:210–271. doi: 10.1007/s12035-009-8064-1. [DOI] [PubMed] [Google Scholar]
- Chen HJ, Chen YM, Chang CM. Lipoyl dehydrogenase catalyzes reduction of nitrated DNA and protein adducts using dihydrolipoic acid or ubiquinol as the cofactor. Chem Biol Interact. 2002;140:199–213. doi: 10.1016/s0009-2797(02)00019-4. [DOI] [PubMed] [Google Scholar]
- Cleren C, Starkov AA, Calingasan NY, Lorenzo BJ, Chen J, Beal MF. Promethazine protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Neurobiol Dis. 2005;20:701–708. doi: 10.1016/j.nbd.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Colado MI, Camarero J, Mechan AO, Sanchez V, Esteban B, Elliott JM, et al. A study of the mechanisms involved in the neurotoxic action of 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) on dopamine neurones in mouse brain. Br J Pharmacol. 2001;134:1711–1723. doi: 10.1038/sj.bjp.0704435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Desagher S, Glowinski J, Premont J. Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci. 1996;16:2553–2562. doi: 10.1523/JNEUROSCI.16-08-02553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Kussmaul L, Gutterer JM, Hirrlinger J, Hamprecht B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J Neurochem. 1999;72:2523–2530. doi: 10.1046/j.1471-4159.1999.0722523.x. [DOI] [PubMed] [Google Scholar]
- Fornai F, Giorgi FS, Gesi M, Chen K, Alessandri MG, Shih JC. Biochemical effects of the monoamine neurotoxins DSP-4 and MDMA in specific brain regions of MAO-B-deficient mice. Synapse. 2001;39:213–221. doi: 10.1002/1098-2396(20010301)39:3<213::AID-SYN1002>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Frenzilli G, Ferrucci M, Giorgi FS, Blandini F, Nigro M, Ruggieri S, et al. DNA fragmentation and oxidative stress in the hippocampal formation: a bridge between 3,4 and methylenedioxymethamphetamine (ecstasy) intake and long-lasting behavioral alterations. Behav Pharmacol. 2007;18:471–581. doi: 10.1097/FBP.0b013e3282d518aa. [DOI] [PubMed] [Google Scholar]
- Fujita H, Shiosaka M, Ogino T, Okimura Y, Utsumi T, Sato EF, et al. Alpha-lipoic acid suppresses 6-hydroxydopamine-induced ROS generation and apoptosis through the stimulation of glutathione synthesis but not by the expression of heme oxygenase-1. Brain Res. 2008;1206:1–12. doi: 10.1016/j.brainres.2008.01.081. [DOI] [PubMed] [Google Scholar]
- Gardner PR. Aconitase: sensitive target and measure of superoxide. Methods Enzymol. 2002;349:9–23. doi: 10.1016/s0076-6879(02)49317-2. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Raineri I, Epstein LB, White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Sieber BA, Heikkila RE, Sonsalla PK. Studies on species sensitivity to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Part 1: Systemic administration. J Pharmacol Exp Ther. 1994a;270:1000–1007. [PubMed] [Google Scholar]
- Giovanni A, Sonsalla PK, Heikkila RE. Studies on species sensitivity to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Part 2: central administration of 1-methyl-4-phenylpyridinium. J Pharmacol Exp Ther. 1994b;270:1008–1014. [PubMed] [Google Scholar]
- Goñi-Allo B, Ramos M, Hervias I, Lasheras B, Aguirre N. Studies on striatal neurotoxicity caused by the 3,4-methylenedioxymethamphetamine/malonate combination: implications for serotonin/dopamine interactions. J Psychopharmacol. 2006;20:245–256. doi: 10.1177/0269881106063264. [DOI] [PubMed] [Google Scholar]
- Granado N, Escobedo I, O'Shea E, Colado I, Moratalla R. Early loss of dopaminergic terminals in striosomes after MDMA administration to mice. Synapse. 2008a;62:80–84. doi: 10.1002/syn.20466. [DOI] [PubMed] [Google Scholar]
- Granado N, O'Shea E, Bove J, Vila M, Colado MI, Moratalla R. Persistent MDMA-induced dopaminergic neurotoxicity in the striatum and substantia nigra of mice. J Neurochem. 2008b;107:1102–1112. doi: 10.1111/j.1471-4159.2008.05705.x. [DOI] [PubMed] [Google Scholar]
- Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982;257:13704–13712. [PubMed] [Google Scholar]
- Hasegawa E, Takeshige K, Oishi T, Murai Y, Minakami S. 1-Mehtyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem Biophys Res Commun. 1990;170:1049–1055. doi: 10.1016/0006-291x(90)90498-c. [DOI] [PubMed] [Google Scholar]
- Hausladen A, Fridovich I. Measuring nitric oxide and superoxide: rate constants for aconitase reactivity. Methods Enzymol. 1996;269:37–41. doi: 10.1016/s0076-6879(96)69007-7. [DOI] [PubMed] [Google Scholar]
- Iravani MM, Syed E, Jackson MJ, Johnston LC, Smith LA, Jenner P. A modified MPTP treatment regime produces reproducible partial nigrostriatal lesions in common marmosets. Eur J Neurosci. 2005;21:841–854. doi: 10.1111/j.1460-9568.2005.03915.x. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, al-Mehdi AB. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-u. [DOI] [PubMed] [Google Scholar]
- Itzhak Y, Anderson KL, Ali SF. Differential response of nNOS knockout mice to MDMA (‘ecstasy’)- and methamphetamine-induced psychomotor sensitization and neurotoxicity. Ann NY Acad Sci. 2004;1025:119–128. doi: 10.1196/annals.1316.015. [DOI] [PubMed] [Google Scholar]
- Javitch JA, D'Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci U S A. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S, Ladenheim B, Andrews AM, Cadet JL. Overexpression of human copper/zinc superoxide dismutase in transgenic mice attenuates oxidative stress caused by methylenedioxymethamphetamine (ecstasy) Neuroscience. 1999;91:1379–1387. doi: 10.1016/s0306-4522(98)00698-8. [DOI] [PubMed] [Google Scholar]
- Jia Z, Zhu H, Vitto MJ, Misra BR, Li Y, Misra HP. Alpha-lipoic acid potently inhibits peroxynitrite-mediated DNA strand breakage and hydroxyl radical formation: implications for the neuroprotective effects of alpha-lipoic acid. Mol Cell Biochem. 2009;323:131–138. doi: 10.1007/s11010-008-9971-6. [DOI] [PubMed] [Google Scholar]
- Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;22:798–809. doi: 10.1097/00004647-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Starkov AA, Calingasan NY, Gardian G, Browne SE, Yang L, et al. Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. J Neurochem. 2004;88:1352–1360. doi: 10.1046/j.1471-4159.2003.02263.x. [DOI] [PubMed] [Google Scholar]
- Knowles RG, Palacios M, Palmer RMJ, Moncada S. Nitric oxide synthase in the brain. In: Moncada S, Higgs EA, editors. Nitric Oxide from L-Arginine: A Bioregulatory System. New York: Elsevier; 1990. pp. 139–146. [Google Scholar]
- Leonardi ET, Azmitia EC. MDMA (ecstasy) inhibition of MAO type A and type B: comparisons with fenfluramine and fluoxetine (Prozac) Neuropsychopharmacology. 1994;10:231–238. doi: 10.1038/npp.1994.26. [DOI] [PubMed] [Google Scholar]
- Li Q-Y, Pedersen C, Day BJ, Patel M. Dependence of excitotoxic neurodegeneration on mitochondrial aconitase inactivation. J Neurochem. 2001;78:746–755. doi: 10.1046/j.1471-4159.2001.00457.x. [DOI] [PubMed] [Google Scholar]
- Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000;101:563–570. doi: 10.1016/s0306-4522(00)00397-3. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Logan BJ, Laverty R, Sanderson WD, Yee YB. Differences between rats and mice in MDMA (methylenedioxymethylamphetamine) neurotoxicity. Eur J Pharmacol. 1988;152:227–234. doi: 10.1016/0014-2999(88)90717-0. [DOI] [PubMed] [Google Scholar]
- Matthews RT, Yang L, Beal MF. S-Methylthiocitrulline, a neuronal nitric oxide synthase inhibitor, protects against malonate and MPTP neurotoxicity. Exp Neurol. 1997;143:282–286. doi: 10.1006/exnr.1996.6406. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Sone N, Saitoh T. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J Neurochem. 1987;48:1787–1793. doi: 10.1111/j.1471-4159.1987.tb05737.x. [DOI] [PubMed] [Google Scholar]
- Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- Nowicki JP, Duval D, Poignet H, Scatton B. Nitric oxide mediates neuronal death after focal cerebral ischemia in the mouse. Eur J Pharmacol. 1991;204:339–340. doi: 10.1016/0014-2999(91)90862-k. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- O'Shea E, Esteban B, Camarero J, Green AR, Colado MI. Effect of GBR 12909 and fluoxetine on the acute and long term changes induced by MDMA (‘ecstasy’) on the 5-HT and dopamine concentrations in mouse brain. Neuropharmacology. 2001;40:65–74. doi: 10.1016/s0028-3908(00)00106-4. [DOI] [PubMed] [Google Scholar]
- Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- Perier C, Tieu K, Guégan C, Caspersen C, Jackson-Lewis V, Carelli V, et al. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci U S A. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SK, Moreau RF, Smith EJ, Hagen TM. Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity. IUBMB Life. 2008;60:362–367. doi: 10.1002/iub.40. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, et al. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci. 1992;12:1658–1667. doi: 10.1523/JNEUROSCI.12-05-01658.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Vila M. The last decade in Parkinson's disease research. Basic sciences. Adv Neurol. 2001;86:177–186. [PubMed] [Google Scholar]
- Przedborski S, Vila M. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: a tool to explore the pathogenesis of Parkinson's disease. Ann NY Acad Sci. 2003;991:189–198. [PubMed] [Google Scholar]
- Ramsay RR, Singer TP. Relation of superoxide generation and lipid peroxidation to the inhibition of NADH-Q oxidoreductase by rotenone, piericidin A, and MPP+ Biochem Biophys Res Commun. 1992;189:47–52. doi: 10.1016/0006-291x(92)91523-s. [DOI] [PubMed] [Google Scholar]
- Ratner V, Starkov A, Matsiukevich D, Polin RA, Ten VS. Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia-exposed mice. Am J Respir Cell Mol Biol. 2009;40:511–518. doi: 10.1165/rcmb.2008-0341RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabol KE, Seiden LS. Reserpine attenuates D-amphetamine and MDMA-induced transmitter release in vivo: a consideration of dose, core temperature and dopamine synthesis. Brain Res. 1998;806:69–78. doi: 10.1016/s0006-8993(98)00720-3. [DOI] [PubMed] [Google Scholar]
- Sanchez V, Camarero J, O'Shea E, Green AR, Colado MI. Differential effect of dietary selenium on the long-term neurotoxicity induced by MDMA in mice and rats. Neuropharmacology. 2003;44:449–461. doi: 10.1016/s0028-3908(02)00411-2. [DOI] [PubMed] [Google Scholar]
- Seo H, Kim W, Isacson O. Compensatory changes in the ubiquitin-proteasome system, brain-derived neurotrophic factor and mitochondrial complex II/III in YAC72 and R6/2 transgenic mice partially model Huntington's disease patients. Hum Mol Genet. 2008;17:3144–3153. doi: 10.1093/hmg/ddn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JC, Grimsby J, Chen K. Molecular biology of monoamine oxidase A and B: their role in the degradation of serotonin. In: Gothert HB, editor. Serotoninergic Neurons and 5-HT Receptors in the SNC. Berlin: Springer; 1999. pp. 655–670. [Google Scholar]
- Sipos I, Tretter L, Adam-Vizi V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem. 2003;84:112–118. doi: 10.1046/j.1471-4159.2003.01513.x. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Heikkila RE. The influence of dose and dosing interval on MPTP-induced dopaminergic neurotoxicity in mice. Eur J Pharmacol. 1986;129:339–345. doi: 10.1016/0014-2999(86)90444-9. [DOI] [PubMed] [Google Scholar]
- Stone DM, Hanson GR, Gibb JW. Differences in the central serotonergic effects of methylenedioxymethamphetamine (MDMA) in mice and rats. Neuropharmacology. 1987;26:1657–1661. doi: 10.1016/0028-3908(87)90017-7. [DOI] [PubMed] [Google Scholar]
- Suh JH, Wang H, Liu RM, Liu J, Hagen TM. (R)-alpha-lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: evidence for increased cysteine requirement for GSH synthesis. Arch Biochem Biophys. 2004;423:126–135. doi: 10.1016/j.abb.2003.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Liktor B, Adam-Vizi V. Dual effect of pyruvate in isolated nerve terminals: generation of reactive oxygen species and protection of aconitase. Neurochem Res. 2005;30:1331–1338. doi: 10.1007/s11064-005-8805-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem. 1990;265:16330–16336. [PubMed] [Google Scholar]