

Abstract
Background and purpose:
β-Amyloid peptide (Aβ) is implicated in the pathogenesis of Alzheimer's disease by initiating a cascade of events from mitochondrial dysfunction to neuronal death. The metabolic enhancer piracetam has been shown to improve mitochondrial dysfunction following brain aging and experimentally induced oxidative stress.
Experimental approach:
We used cell lines (PC12 and HEK cells) and murine dissociated brain cells. The protective effects of piracetam in vitro and ex vivo on Aβ-induced impairment of mitochondrial function (as mitochondrial membrane potential and ATP production), on secretion of soluble Aβ and on neurite outgrowth in PC12 cells were investigated.
Key results:
Piracetam improves mitochondrial function of PC12 cells and acutely dissociated brain cells from young NMRI mice following exposure to extracellular Aβ1-42. Similar protective effects against Aβ1-42 were observed in dissociated brain cells from aged NMRI mice, or mice transgenic for mutant human amyloid precursor protein (APP) treated with piracetam for 14 days. Soluble Aβ load was markedly diminished in the brain of those animals after treatment with piracetam. Aβ production by HEK cells stably transfected with mutant human APP was elevated by oxidative stress and this was reduced by piracetam. Impairment of neuritogenesis is an important consequence of Aβ-induced mitochondrial dysfunction and Aβ-induced reduction of neurite growth in PC12 cells was substantially improved by piracetam.
Conclusion and implications:
Our findings strongly support the concept of improving mitochondrial function as an approach to ameliorate the detrimental effects of Aβ on brain function.
This article is commented on by Moncada, pp. 217–219 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00706.x and to view related papers by Pravdic et al. and Puerta et al. visit http://dx.doi.org/10.1111/j.1476-5381.2010.00698.x and http://dx.doi.org/10.1111/j.1476-5381.2010.00663.x
Keywords: piracetam, mitochondrial function, ATP production, neurite growth, β-amyloid, Alzheimer's disease
Introduction
Piracetam, the prototype of the so-called ‘nootropic’ drugs (Giurgea, 1982), is used in many countries to treat cognitive impairment in aging or brain injury, as well as in dementia states. Although its clinical usefulness is still a matter of dispute, a large meta-analysis of all available (published and not published) clinical studies provided compelling evidence for the global efficacy of piracetam in a diverse group of older subjects with cognitive impairment (Waegemans et al., 2002).
As in the studies with humans, piracetam has also been shown to improve cognitive function in animals, but its mode of action is not yet finally known (Giurgea, 1982; Müller et al., 1999). Findings that the efficacy of piracetam in animals or humans is usually associated with conditions of disturbed energy supply like aging (Valzelli et al., 1980; Müller et al., 1997), or various hypoxic conditions (Saletu et al., 1995; Uebelhack et al., 2003; He et al., 2008; Holinski et al., 2008) have led to the proposal that piracetam's mechanism of action is associated with biochemical deficits typical of the senescent brain. This proposal was later supported by observations that piracetam specifically enhances membrane fluidity in aged brain material, showing no effect in membranes from young brains (Müller et al., 1997). Since piracetam's effects at the membrane level were observed at concentrations, comparable to those used in pharmacological experiments to improve cognition (Müller et al., 1997) or in patients treated with piracetam (Müller et al., 1997; Saletu et al., 1995), we proposed that by restoring age-related membrane alterations, piracetam improves brain function and finally cognition (Scheuer et al., 1999). At the subcellular level, piracetam's effects on membrane fluidity could be demonstrated for synaptosomal plasma membranes and also for membranes from brain mitochondria (Müller et al., 1999). Exactly how piracetam stabilizes membranes is not known. Since it does not interact with any specific target structure known, the most likely explanation seems to be its binding to the polar head groups of membrane phospholipids (Müller et al., 1997; Müller et al., 1999).
Evidence that piracetam's beneficial effects on the fluidity of aged membranes might lead to enhanced mitochondrial function originated from observations that piracetam improved glucose uptake and utilization as well as ATP production (Domanska-Janik and Zaleska, 1977; Benzi et al., 1985; Heiss et al., 1988; Naftalin et al., 2004). Even if these effects led to the term ‘metabolic enhancer’ sometimes used to characterize piracetam and related nootropics, the mechanism of this effect and its possible relationship to mitochondrial function remain obscure.
As already mentioned, piracetam's beneficial effects are usually associated with impaired brain function under conditions such as aging, hypoxia, glucose deprivation, injuries or free radical damage (Giurgea, 1982; Müller et al., 1999). It is quite remarkable that all these conditions, even if other deficits are also present, are associated with the vicious cycle involving energy (ATP) deficit, oxidative stress and mitochondrial dysfunction (Sastre et al., 2003; Mattson and Magnus, 2006; Fukui and Moraes, 2008) which also lead to impaired measures of synaptic plasticity like spine density and neurite outgrowth and finally to cell death. These data are in agreement with our previous observations of significant mitochondrial protection by piracetam against experimentally induced oxidative as well as nitro-oxidative stress in vitro and after ex vivo treatment, where again aged animals with well-characterized mitochondrial dysfunction benefited most (Keil et al., 2006). Compatible with the proposal of mitochondrial membranes as a primary target for piracetam, the beneficial effect of this compound was similar after experimental impairment of each of the five respiratory chain complexes (Keil et al., 2006).
Oxidative stress and mitochondrial dysfunction have been repeatedly demonstrated as a major causative factor for neurodegeneration in Alzheimer's disease (AD) (Eckert et al., 2003; Reddy and Beal, 2008) where aging, as the most important risk factor, and disease-specific histopathological lesions [β-amyloid (Aβ), hyperphosphorylated tau protein] seem to lead synergistically to mitochondrial dysfunction even in the early stages of the disease (Pereira et al., 1999; Leuner et al., 2007; Eckert et al., 2008; Hauptmann et al., 2009). Accordingly, we investigated the possible beneficial effects of piracetam on mitochondrial function impaired by Aβ, measuring mitochondrial membrane potential (MMP), ATP production and neuritogenesis.
Methods
Preparation of Aβ peptides
The preparation of aged fibrillar Aβ1-42 was performed as previously reported (Keil et al., 2004) and that of oligomeric Aβ, according to Stine et al. (2003) and Peters et al. (2009). Aβ1-42 (1 mg) was dissolved in Tris-buffered saline (pH 7.4) at a concentration of 1 mM and stored at −20°C. The stock solution was diluted in Tris-buffered saline to the desired concentrations and incubated at 37°C for 24 h to have aged fibrillar preparations of Aβ1-42. To prepare oligomeric Aβ, 1 mg of Aβ1-42 was dissolved in 222 µL 1,1,1,3,3,3-hexafluoro-isopropanol. Solutions were evaporated using a speed vacuum for 45 min. The dried film was re-suspended in 2 µL dimethylsulphoxide and diluted in 98 µL Dulbecco's modified Eagle's medium (DMEM) medium to achieve a working solution of 100 µM. The solution was vortexed for 30 s and incubated at 4°C for 24 h. Aβ1-42 peptides were characterized using native gel electrophoresis, followed by silver staining and electron microscopy as described in Peters et al. (2009). Aβ25-35 was dissolved in Tris-buffered saline (TBS) pH 7.4 at a concentration of 1 mM and stored at −20°C. The stock solution was diluted in TBS to the desired concentrations and incubated at 37°C for 24 h to obtain aged, aggregated preparations of Aβ25-35.
Cell culture
PC12 cells were stably transfected with DNA constructs harboring human mutant APP (the Swedish mutation, K670M/N671L) gene (APPsw PC12 cells), the wildtype human gene (APPwt PC12 cells) or the corresponding vector (vctPC12 cells), as described by Keil et al. (2004). They were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum and 5% heat-inactivated horse serum, 50 units·mL−1 penicillin, 50 µg·mL−1 streptomycin, and 400 µg·mL−1 G418 at 37°C in a humidified incubator containing 5% CO2. HEK293 cells stably expressing APPwt were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum, 50 units·mL−1 penicillin, 50 mg·mL−1 streptomycin, and 400 µg·mL−1 G418 at 37°C in a humidified incubator containing 5% CO2.
Animals
All animal care and experimental procedures were in concordance with the German law on animal care and handling of transgenic animals. All animals were housed in plastic cages with water and food ad libitum and were maintained on a 12 h light/dark cycle. Young (2–3 months) and old (22–24 months) female NMRI mice used in this study were from Harlan-Winkelmann GmbH, Borchen (Germany). Animals belonging to the aged group were obtained at an age of 12 months and maintained at the Biocenter's animal care facility until use. Female and male heterozygous C57BL/6J mice (from Charles River) bearing the human Swedish mutation (KM670/671NL) and London mutation (V717I) in the 751 amino acid form of human amyloid precursor protein (tgAPP) under the control of a murine Thy1.2 promoter (Blanchard et al., 2003) were bred in our animal facilities and were used in these experiments at an age of 3 months (Hauptmann et al., 2009). At weaning, the animals were genotyped from tail biopsies by means of an appropriate digest and polymerase chain reaction (data not shown).
Preparation of dissociated brain cells
Mice were killed by decapitation and brains were quickly dissected on ice. Dissociated brain cells were prepared according to Stoll et al. (1992). Briefly, after removing the cerebellum, tissue was minced into 2 mL of medium I (NaCl 138, KCl 5.4, Na2HPO4 0.17, K2HPO4 0.22, glucose 5.5 and sucrose 58.4 all in mM, pH 7.35) with a scalpel and further dissociated by trituration through a nylon mesh (pore diameter 1 mm) using a Pasteur pipette. The resulting suspension was filtered by gravity through a fresh nylon mesh with a pore diameter of 0.102 mm. The dissociated cell aggregates were washed twice with medium II (composition, in mM: NaCl 110, KCl 5.3, CaCl2× H2O 1.8, MgCl2× 6 H2O 1, glucose 25, sucrose 70 and HEPES 20; pH 7.4) by centrifugation (400 × g for 3 min at 4°C). 100 µL of the suspension were used for protein determination. After centrifugation, cells were resuspended in 6 mL DMEM, maintained for incubation of 37°C in a humidified atmosphere of 5% CO2 : 95% air. Dissociated brain cells were distributed into a 48 well plate (250 µL per well) for measurement of mitochondrial membrane potential. For measurement of ATP levels, 100 µL per well was used in a white 96 well plate.
Data are expressed as fluorescence unit per mg protein. The protein content was determined by the method of Lowry et al. using BSA as standard (Lowry et al., 1951).
Piracetam treatment
PC12 cells were treated for 24 h with 10 nM Aβ1-42. 30 min after the onset of Aβ1-42 exposure, piracetam, aniracetam or oxiracetam were added. Dissociated brain cells were treated for 4 h with 50 nM Aβ1-42 or 25 µM Aβ25-35, piracetam was added 1 h after the onset of exposure.
Treatment of animals
The treated animals received 0.1, 0.25 or 0.5 g·kg−1 piracetam in 0.9 % NaCl solution p.o. once daily for 2 weeks. Control animals were treated with 0.9 % NaCl alone. The animals were killed 24 h after the last treatment. The dissociated brain cells of untreated and treated mice were incubated for 4 h with 50 nM Aβ1-42 or 25 µM Aβ25-35.
Measurement of mitochondrial membrane potential (MMP)
PC12 cells were plated the day before at a density of 2 × 105 cells per well in a 24 well plate. The MMP of PC12 cells was measured using the fluorescence dye Rhodamine 123 (R123). The experimental conditions used have been shown previously to detect even small changes (Keil et al., 2004, 2006; Hauptmann et al., 2009). Transmembrane distribution of the dye depends on the MMP. The dye was added to the cell culture medium in a concentration of 0.4 µM for 15 min (Keil et al., 2004, 2006). The cells were washed twice with Hanks' balanced salt solution (HBSS) and the fluorescence was determined with a fluorescence reader (Victor® multilabel counter, Perkin-Elmer, Waltham, MA, USA) at 490/535 nm. The MMP of dissociated neurons was also measured using R123 in a concentration of 0.4 µM for 15 min and washed twice with HBSS (Hauptmann et al., 2009).
Determination of ATP levels with a bioluminescence assay (ViaLight™ HT)
Brain cells of mice were plated in a white 96 well plate. The assay is based upon the bioluminescent measurement of ATP (Crouch et al., 1993) and the bioluminescent method utilizes the enzyme luciferase that catalyses the formation of light from ATP and luciferin. The emitted light is linearly related to the ATP concentration and is measured using a luminometer (Victor® multilabel counter, Perkin-Elmer).
Detection of Aβ levels in transgenice mice
For the detection of secreted soluble Aβ1-40 in cell supernatants, a specific sandwich enzyme-linked immunosorbent assay employing monoclonal antibodies was used. The elisa was performed according to the instructions given in the Abeta-elisa by Invitrogen. This standard sandwich elisa utilizes a monoclonal mouse anti-human Abeta1-16 capture antibody, a cleavage-site-specific rabbit anti-human Aβ1-40 C-terminal detection antibody and anti-rabbit IgG peroxidase-conjugated secondary antibody. Colour development is started by addition of tetramethylbenzidine yielding a yellow chromophore with absorbance at 450 nm. Absorbance was measured in Victor® plate reader using 450 nm filter with 7 nm bandpass. Sample concentrations were determined from the Aβ1-40 standard curve, which was obtained from plotting the absorption of the standard dilutions versus the standard concentrations and calculation of the best-fit polynomial equation (Prism Graph Pad®, GraphPad Software, La Jolla, CA, USA).
Detection of Aβ levels in HEKwt 293 cells
In order to assess the prophylactic potency of piracetam in reducing Aβ production after nitrosative stress, HEKwt cells were pre-incubated with 1 mM piracetam for 24 h, then the culture medium was changed and the incubation with piracetam was continued for another 24 h in the presence of 0.5 mM sodium nitroprusside (SNP). The Aβ secretion in the presence of piracetam was compared with that from HEKwt cells treated with SNP alone.
Neurite outgrowth assay in PC12 cells
PC12 cells were plated at a density of 104 cells per plate (85 mm, polylysin coated) in 15% serum containing medium overnight. The next day, medium was changed to a medium containing 2% serum and nerve growth factor (NGF; 50 ng·mL−1). Cells were stressed every day with oligomeric Aβ 1 µM or SNP 0.05 mM in the presence or absence of piracetam 1 mM. PC12 cells stably expressing human mutant APP gene (APPsw PC12) or the APPwt gene (APPwt PC12), inserted downstream of a cytomegalovirus promoter, were treated as described above. For 6 days, cells were treated with different NGF concentrations (1–50 ng·mL−1) in the absence or presence of piracetam 1 mM. The neurite length was examined 6 days after different treatment regimes. After 6 days PC12 cells were fixed with paraformaldehyde solution (4%) and stained with Mayer's haematoxylin and eosin solution. Thirty cells from each sample (n= 1) were arbitrarily investigated and neurite length was detected by using Nikon NIS Elements AR 2.1 software, Nikon Instruments, Inc., Melville, NY, USA.
Statistical analysis
Data are shown as mean ± SEM For statistical comparison, Students t-test or two-way anova followed by Bonferroni's post hoc test was used. P values less than 0.05 were considered statistically significant.
Materials
R123 was purchased from Invitrogen, Karlsruhe (Germany), and the ViaLight HT kit from Cambrex, Vervies (Brussels). Hydrogen peroxide and SNP were obtained from Sigma, Munich (Germany). The Aβelisa kit was purchased from Invitrogen, Karlsruhe (Germany). BSA was obtained from BIO-RAD, Munich (Germany). Aβ1-42 and Aβ25-35 were purchased from Bachem, Bubendorf (Switzerland).
Drug and molecular target nomenclature conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2009).
Results
Aβ-induced alterations of MMP are improved by treatment in vitro with piracetam and two structurally related nootropics
When PC12 cells were treated with fibrillar Aβ1-42 10 nM (concentration for maximal effect according to Keil et al., 2004) for 24 h, a reduction of MMP was observed as described previously (Keil et al., 2004). The addition of piracetam 30 min after Aβ1-42 substantially protected MMP (Figure 1A) at concentrations as low as 0.1 mM (P < 0.05, Figure 1B). Comparable protective effects of piracetam were observed for dissociated brain cells of NMRI mice following incubation with fibrillar Aβ1-42 (50 nM) (Figure 1B). For these experiments, we used a slightly higher Aβ1-42 concentration, which showed maximal MMP reduction for these cells in preliminary experiments. In both cell types, piracetam alone has no effect on MMP (data not shown). The two structurally related metabolic enhancers, oxiracetam and aniracetam, also protected PC12 cells against fibrillar Aβ1-42 induced mitochondrial dysfunction (Figure 1C and D). In line with their nootropic potency, protection by oxiracetam was seen at slightly higher concentrations (0.5 mM, P < 0.05) (Fordyce et al., 1995; Hlinak and Krejci, 2002) while aniracetam was active at considerably lower concentrations (0.01 mM, P < 0.05) (Smith and Wehner, 2002).
Figure 1.

Piracetam, aniracetam, and oxiracetam ameliorate Aβ impaired mitochondrial function. (A) PC12 cells were treated for 24 h with fibrillar Aβ1-42, 30 min after insult piracetam (0.1 mM, 0.5 mM or 1 mM) was added and the MMP was measured using the fluorescence dye R123. Data are expressed as means ± SEM (n= 4–5). +P < 0.05 Aβ1-42 induced reduction of MMP versus untreated control; **P < 0.01, *P < 0.05 Aβ1-42 induced reduction of MMP versus piracetam treatment; student's unpaired t-test. (B) Dissociated brain cells isolated from NMRI mice were stressed for 4 h with fibrillar Aβ1-42. After 1 h, piracetam (0.1, 1, 10 mM) was added; again the mitochondrial membrane potential was detected. Data are expressed as means ± SEM (n= 4–5). +++P < 0.001 Aβ1-42 versus control; **P < 0.01 piracetam treatment versus Aβ1-42; student's unpaired t-test (C and D) PC12 cells were treated for 24 h with Aβ1-42, 30 min after insult oxiracetam (C) (0.5 mM or 1 mM), or aniracetam (D) (0.001 mM or 0.01 mM) were added and the MMP was investigated using R123. Data are expressed as means ± SEM (n= 4–5). +P < 0.05 Aβ1-42 versus control; **P < 0.01 oxiracetam or aniracetam treatment versus Aβ1-42, student's unpaired t-test (E) APPwt, APPsw and vctPC12 cells were stressed with SNP 0.5 mM in the presence and absence of piracetam (0.5 mM, 1 mM) and the MMP (E) and ATP levels (F) were measured. Data are presented as the reduction of MMP and ATP induced by SNP and the respective improvement by piracetam. PC12 cells were incubated 24 h with SNP, piracetam was added 30 min after insult. Data are expressed as means ± SEM (n= 4). *P < 0.05 SNP induced reduction of mitochondrial membrane potential in vct cells, APPwt, or APPsw cells versus vct cells, APPwt or APPsw cells treated with piracetam; *P < 0.05, **P < 0.01, ***P < 0.001 SNP induced reduction of ATP levels in vct cells, APPwt or APPsw cells versus ATP levels of vct cells, APPwt or APPsw cells treated with piracetam, student's unpaired t-test. APP, Amyloid precursor protein; APPsw, Swedish APP mutation; APPwt, Wildtype human APP; MMP, Mitochondrial membrane potential; R123, Rhodamine 123; SNP, Sodium nitroprusside.
Protection against mitochondrial damage induced by low level expression of human Aβ in PC12 cells
Many recent findings suggest mitochondrial dysfunction and reduction of MMP as very early events in the neuropathology of AD, long before the extracellular accumulation of Aβ plaques (Chan et al., 2002; Selkoe, 2002; Blanchard et al., 2003; Hauptmann et al., 2009). As a model for low-level Aβ exposure and the accompanying mitochondrial damage, we previously generated PC12 cells stably expressing either APPwt or APPsw (Leutz et al., 2002; Marques et al., 2003; Keil et al., 2004). This latter transfection results in a three- to six-fold increase in production of Aβ, compared with wildtype APP. Expression of APPsw renders these cells more vulnerable to the induction of mitochondrial dysfunction and cell death after exposure to oxidative stress (Marques et al., 2003; Keil et al., 2004).
In the present study, we investigated the efficacy of piracetam to protect mitochondria in these PC12 cells, additionally challenged by SNP (0.5 mM) (Keil et al., 2004). The decrease in MMP and ATP levels is shown in Figure 1E and F. The response of the PC12 cells to the nitrosative stress depended on the Aβ load. As expected, piracetam was also able to protect against changes in MMP and reduction of ATP levels induced by SNP in all three cell lines, at a concentration of 0.5 mM, even if the mitochondrial damage after NO exposure in APPsw PC12 cells is more pronounced than in vct cells and APPwt PC12 cells. Under the conditions chosen, piracetam alone had no effect on MMP in all three cell types (data not shown).
Piracetam treatment protects against Aβ peptides ex vivo
We have previously shown that when NMRI mice of different ages were treated with piracetam, protection against oxidative injuries was more pronounced in the brain of aged than of young animals (Keil et al., 2006). Accordingly, we similarly treated 3 months and 22 months old NMRI mice for 14 days with piracetam (0.5 g·kg−1·day−1 orally). Dissociated brain cells of control young and aged animals showed a rather similar reduction of MMP following ex vivo treatment with fibrillar Aβ1-42 (Figure 2A and B). Protection by piracetam relative to control cells also treated with Aβ1-42 was not significant in young animals. By contrast, in brain cells from aged mice, treated similarly with piracetam, there was significantly greater protection against Aβ1-42 induced mitochondrial damage (P < 0.01). The protective effect of piracetam in aged mice was dose-dependent and also protected brain cells against the neurotoxic Aβ sequence 25-35 (Aβ25-35) (Canevari et al., 1999; Casley et al., 2002; Villard et al., 2009) (Figure 2C and D). The first significant effects were seen at oral piracetam doses between 0.1 and 0.25 g·kg−1·day−1 (P < 0.05).
Figure 2.
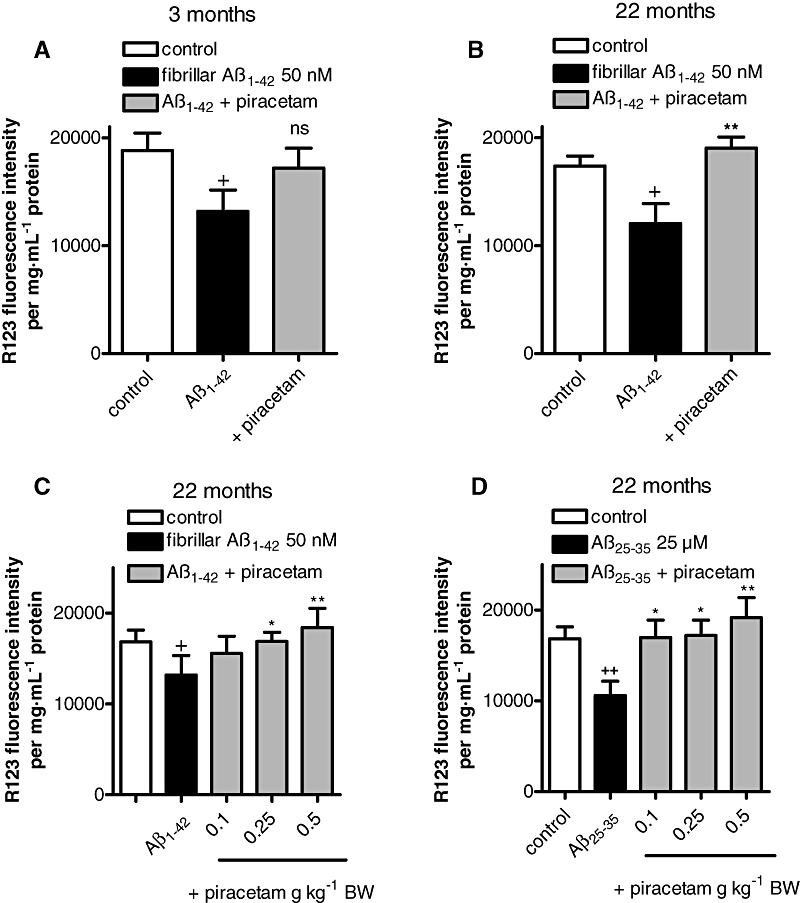
Piracetam treatment improves MMP after Aβ1-42 induced stress in vitro and ex vivo. (A and B) Young NMRI mice (A, 3 months) and aged mice (B, 22 months) were treated with 0.5 g piracetam kg−1 in 0.9% NaCl solution p.o. once daily for 2 weeks. Dissociated brain cells were prepared and stressed with fibrillar Aβ1-42 (50 nM) for 4 h in the presence or absence of piracetam (1 mM). MMP was measured using R123. Data are expressed as means ± SEM (n= 6–8).+P < 0.05, ++P < 0.01 control against Aβ1-42 treated dissociated brain cells; ns; *P < 0.01 membrane potential of dissociated brain cells stressed with Aβ1-42 against dissociated brain cells treated with Aβ1-42 in the presence of piracetam; student's unpaired t-test. (C) Aged mice (22 months) received 0.1, 0.25, 0.5 g piracetam kg−1 in 0.9% NaCl solution p.o. once daily for 2 weeks. Control animals were treated with 0.9% NaCl solution alone. Afterwards, dissociated brain cells were incubated ex vivo for 4 h with 50 nM fibrillar Aβ1-42 and MMP was measured. Piracetam protects against Aβ1-42 and Aβ25-35 induced mitochondrial damage ex vivo. Membrane potential of dissociated brain cells was measured after 4 h incubation with 50 nM Aβ1-42 (C) and 25 µM Aβ25-35 (D). Data are expressed as means ± SEM (n= 6–8) +P < 0.05, ++P < 0.01 untreated control versus Aβ1-42 induced reduction of MMP; *P < 0.05, **P < 0.01 Aβ1-42 or Aβ25-35 reduced MMP versus piracetam treated groups; student's unpaired t-test. MMP, Mitochondrial membrane potential; R123, Rhodamine 123.
Piracetam ameliorates mitochondrial dysfunction in transgenic animals overexpressing human Aβ
Isolated brain cells of mice overexpressing mutated human amyloid precursor protein (tgAPP) showed significant reductions of MMP (P < 0.05) and ATP synthesis (P < 0.05) relative to non-transgenic littermate controls (Figure 3A and B), confirming previous observations (Hauptmann et al., 2009). Piracetam treatment (0.5 g·kg−1·day−1 orally) as used in for NMRI mice (see above) substantially improved MMP and ATP production (both P < 0.05) (Figure 3A and B).
Figure 3.

Piracetam improves mitochondrial function in tgAPP mice and reduces Aβ1-40 levels in tgAPP mice and APPwt HEK290 cells. Treated animals received 0.5 g piracetam kg−1 in 0.9% NaCl solution p.o. once daily for 2 weeks. Control animals were treated with 0.9% NaCl solution alone. (A) The MMP was significantly reduced in tgAPP mice. Piracetam treatment normalizes the MMP to non-tgAPP levels. Data are expressed as means ± SEM (n= 7–8). +P < 0.05 control non-tgAPP versus control tgAPP, *P < 0.01 piracetam treated tgAPP versus tgAPP control; student's unpaired t-test. (B) ATP levels were also impaired in tgAPP mice. In contrast, piracetam treatment increases ATP levels not only in tgAPP animals but also in control animals. Data are expressed as means ± SEM (n= 7–8). *P < 0.05 control non-tgAPP versus piracetam treated non-tgAPP; +P < 0.05 control non-tgAPP versus control tgAPP, *P < 0.01 piracetam treated tgAPP versus tgAPP control; student's unpaired t-test. (C) Piracetam reduced Aβ1-40 levels in tg-APP mice. Normalized Aβ levels were quantified with elisa in Tris-buffered brain homogenates from non-tg littermate and tgAPP mice (3 months old). Data are expressed as means ± SEM (n= 7–8). +++P < 0.05 control non-tgAPP versus control tgAPP, **P < 0.01 piracetam treated tgAPP versus control tgAPP; student's unpaired t-test. (D) Piracetam reduced Aβ levels in APPwt HEK293 cells. Cells were incubated for 24 h with piracetam (1 mM) and Aβ1-40 levels were investigated using the Aβ1-40 elisa. Data are expressed as means ± SEM (n= 3–4). **P < 0.01 APPwt HEK293 control cells versus cells treated with piracetam 1 mM; student's unpaired t-test. (E) Piracetam improved MMP under basal conditions in APPwt HEK293 cells. Cells were incubated for 24 h with piracetam (1 mM) and MMP was measured using R123. Data are expressed as means ± SEM (n= 6). ***P < 0.001 APPwt HEK293 control cells versus cells treated with piracetam; student's unpaired t-test. (F) Piracetam reduced nitrosative stress-induced elevation of Aβ in APPwt HEK293 cells. Cells were pre-incubated for 24 h with piracetam (1 mM) and stressed for additional 24 h with SNP (0.5 mM). Aβ levels were again detected using the Aβ1-40 elisa. Data are expressed as means ± SEM (n= 3–4). +++P < 0.001 APPwt HEK293 control cells versus cells treated with SNP, *P < 0.01 APPwt HEK293 cells stressed with SNP 0.5 mM versus stressed cells pre-incubated with piracetam; student's unpaired t-test. (G) Piracetam ameliorated nitrosative stress-induced reduction of MMP. Cells were pre-incubated for 24 h with piracetam (1 mM) and stressed for additional 24 h with SNP (0.5 mM). Data are expressed as means ± SEM (n= 6). **P < 0.01 APPwt HEK293 cells stressed with SNP 0.5 mM versus stressed cells pre-incubated with piracetam, student's unpaired t-test. Aβ, β-Amyloid; APPwt, Wildtype human amyloid precursor protein; MMP, Mitochondrial membrane potential; tgAPP, Transgenic for the human amyloid precursor protein.
As reported earlier (Hauptmann et al., 2009), these mice express substantial level of soluble Aβ in the brain (Figure 3C) while littermates do not. Quite interestingly, piracetam treatment led to an about 25% reduction of soluble Aβ (Figure 3C, P < 0.01). In order to investigate if this effect of piracetam on Aβ levels might also be associated with improved mitochondrial function, we used APPwt HEK293 cells stably overexpressing human APP showing moderately enhanced Aβ levels (Keil et al., 2004). Piracetam lowered Aβ levels under basal conditions (Figure 3D, P < 0.01). In agreement with other findings (Guglielmotto et al., 2009), mitochondrial dysfunction induced with SNP elevated Aβ1-40 levels substantially (Figure 3F, P < 0.001). Again, treatment with piracetam lowered Aβ significantly by 15–20% (Figure 3F, P < 0.05). In addition, piracetam improved mitochondrial function under the same conditions in APPwt HEK293 when Aβ generation was decreased (Figure 3E and G).
Piracetam ameliorates Aβ induced impairment of neurite outgrowth
In agreement with the pronounced loss of neurites and synapses in AD brain as one of the functionally most relevant histopathological lesions (Dekosky and Scheff, 1990; Terry et al., 1991; Selkoe, 2002), Aβ peptides have been repeatedly demonstrated to reduce neuritic outgrowth in different neuronal cell lines in vitro including PC12 cells (Figueroa et al., 2002; Hirata et al., 2005; Kuboyama et al., 2005; Hu et al., 2007; Lacor et al., 2007; Evans et al., 2008; Petratos et al., 2008). Oligomeric Aβ seems to be more active than fibrillar Aβ (Lacor et al., 2007; Evans et al., 2008). In agreement with these observations, the addition of oligomeric Aβ1-42 to NGF treated PC12 cells reduced neurite length significantly (Figure 4A and B). More of 60% of the maximum effect (at 1 µM Aβ1-42) was seen at 100 nM oligomeric Aβ1-42 (P < 0.01). However, as in our other experiments where piracetam protection was investigated for maximum Aβ damage, we also used oligomeric Aβ1-42 at its maximally effective concentration, 1 µM (P < 0.01). When the same experiment was carried out in the presence of piracetam (1 mM), the negative effect of oligomeric Aβ1-42 was completely inhibited (Figure 4A and B, P < 0.001). In agreement with the assumption that enhanced oxidative stress might explain the Aβ induced reduction of neurite outgrowth (Guglielmotto et al., 2009) treating PC12 cells with SNP (0.05 mM) reduced neurite outgrowth even more strongly (Figure 4C, P < 0.001). Again, piracetam ameliorated this negative effect significantly, but not completely (Figure 4C, P < 0.05) under conditions of optimal NGF stimulation. A reduction of neurite outgrowth depending on Aβ load was also observed in our PC12 cells transgenic for human APP, where we observed a reduction of neurite length (Figure 5A), which could be substantially ameliorated by piracetam (Figure 5B). The enhancing effect of piracetam was observed over the whole NGF concentration range (1–50 ng·mL−1) (Figure 5C and D), still leading to increased neurite length under maximum NGF stimulation.
Figure 4.

Piracetam protects against Aβ and SNP induced impairment of neurite outgrowth. PC12 cells were treated over 6 days with NGF 50 ng·mL−1 in the presence or absence of oligomeric Aβ 1 µM or SNP 0.05 mM. In addition, piracetam 1 mM was added and the effects on neurite outgrowth were investigated. (A) Representative images from cells treated with NGF 50 ng·mL−1, NGF + oligomeric Aβ, and NGF + oligomeric Aβ and piracetam. (B) Neurite length of PC12 cells treated with NGF, NGF + oligomeric Aβ, and NGF + oligomeric Aβ. Data are expressed as means ± SEM (n= 6–7) ++P < 0.01 NGF versus oligomeric Aβ; ++P < 0.01 NGF + oligomeric Aβ versus NGF + oligomeric Aβ+ piracetam, student's unpaired t-test. (C) Neurite length of PC12 cells treated with NGF, NGF + SNP 0.05 mM, NGF + SNP 0.05 mM, NGF + SNP 0.05 mM + piracetam. Data are expressed as means ± SEM (n= 6–7) +++P < 0.001 NGF versus NGF + SNP; *P < 0.05 NGF + SNP 0.5 mM versus NGF + SNP 0.05 mM + piracetam, student's unpaired t-test. Aβ, β-Amyloid; NGF, Nerve growth factor; SNP, Sodium nitroprusside.
Figure 5.

Piracetam improves the effect of the neurotrophin NGF in APPwt and APPsw PC12 cells on neurite outgrowth. APPwt, APPsw and vct PC12 cells were treated over 6 days with NGF 50 ng·mL−1 in the presence or absence of piracetam 1 mM and neurite outgrowth was measured. (A) The neurite length in APPsw PC12 cells was significantly reduced compared to vct PC12 cells (transfected with the corresponding vector). (B) In the presence of piracetam the neurite length after treatment with NGF is improved in all three cell types. The effect is normalized to the respective control (without piracetam, 100%). (C) In the presence of different NGF concentrations (1–50 ng·mL−1) piracetam improved the neurotrophic effect of NGF in APPwt (C) and APPsw cells (D). Data are expressed as means ± SEM (n= 6–7) **P < 0.01, ***P < 0.001 versus the respective control, two-way anova with Bonferroni's post test. APPsw, Swedish amyloid precursor protein mutation; APPwt, Wildtype human amyloid precursor protein; NGF, Nerve growth factor.
Discussion
Our findings and those of others indicate that Aβ neurotoxicity is strongly associated with oxidative stress leading to mitochondrial damage and impaired synaptic function (Blanchard et al., 2003; Eckert et al., 2008; Hauptmann et al., 2009; Reddy and Beal, 2008; Shankar et al., 2008). We have previously shown that the cell models also used in the present study (PC12 and HEK cells as well as acutely dissociated mouse brain cells) are appropriate to investigate the detrimental effect of Aβ peptides applied extracellularly, on several measures of mitochondrial function including MMP, ATP synthesis, as indicators of oxidative stress, as well as measures of apoptosis (Marques et al., 2003; Keil et al., 2004; Hauptmann et al., 2009).
In agreement with our previous findings where piracetam improved mitochondrial dysfunction following oxidative stress generated by H2O2 or SNP (Keil et al., 2006), piracetam also ameliorated mitochondrial dysfunction following Aβ1-42 and Aβ25-35 exposure in PC12 cells and in acutely dissociated brain cells prepared from young and aged mice. The concentration range needed (0.1–1.0 mM) is comparable to the plasma concentrations found in patients after therapeutic doses of piracetam (200–2000 µM) (Saletu et al., 1995). In previous studies, we showed that piracetam had no effect under basal conditions, without additional insult (Keil et al., 2005). The effect of piracetam on enhanced metabolic activity was shared by two analogues (oxiracetam, aniracetam), again at concentrations close to those needed to obtain enhanced cognition (oxiracetam only somewhat, but aniracetam considerably more potent than piracetam). A comparable protection against Aβ-induced mitochondrial dysfunction was seen in dissociated brain cells of young and aged mice treated with piracetam for 2 weeks. The effect was considerably more pronounced in the aged animals. Again, the active dose needed (0.1–0.25 g·kg−1) is close to the recommended human dose (0.07 g·kg−1 or 4.8 g per day). However, the protective effect of piracetam against SNP-induced oxidative stress seemed to be less than that against Aβ-mediated toxicity.
Aβ induced mitochondrial dysfunction, which precedes the development of Aβ plaques, has been previously described by our laboratory, in the brains of mice transgenic for human APP bearing the Swedish double mutation (Hauptmann et al., 2009). At the age of 3 months, dissociated brain cells from these animal show pronounced measures of mitochondrial dysfunction (Hauptmann et al., 2009) including reduced MMP and decreased ATP synthesis, as confirmed in the present data. Consistent with its beneficial effects on mitochondrial dysfunction induced by brain aging, piracetam, when given subchronically under similar conditions, also considerably improved MMP and ATP production in tgAPP mice.
Our observation of significantly reduced brain levels of soluble Aβ (P < 0.01) in the piracetam treated tgAPP mice was unexpected. However, as increasing evidence indicates enhanced Aβ production following oxidative stress (Jin et al., 2008; Guglielmotto et al., 2009), the reduced Aβ brain levels in piracetam-treated tgAPP mice again might be a consequence of improved mitochondrial function. This concept was confirmed by experiments showing protection by piracetam against the raising of Aβ levels by SNP, in APPwt HEK cells. In addition, piracetam improved MMP under similar conditions. Even if piracetam reduced Aβ levels only in both experiments by 25%, its ability to lower Aβ is further supported by a study on plasma Aβ levels in a large population of geriatric patients where only patients treated with piracetam or with another metabolic enhancer showed reduced plasma Aβ levels (Blasko et al., 2005). Although plasma Aβ mainly seems to originate from peripheral tissues (blood cells), these findings may further confirm the concept that enhancing mitochondrial function by piracetam can reduce Aβ production.
As mitochondria are highly localized at the synaptic level, impairment of mitochondrial function is importantly associated with synaptic deficits including reduced synapse formation and impaired neuritogenesis (Schon and Manfredi, 2003; Mattson, 2007). Accordingly, reduction of synaptic plasticity by low molecular weight (oligomeric) Aβ species has been considered as a key pathomechanism of AD, beginning quite early in the course of the disease (Mattson, 2007; Walsh and Selkoe, 2007; Reddy and Beal, 2008; Shankar et al., 2008). Neurite outgrowth as an important part of synaptic plasticity has been repeatedly reported to be impaired by β-amyloid peptides in vitro and in vivo (Figueroa et al., 2002; Hirata et al., 2005; Hu et al., 2007; Petratos et al., 2008). In our initial experiments, we could confirm that Aβ1-42 reduced neurite outgrowth using PC12 cells in vitro under conditions of optimal trophic support. The effect was already present at nM concentrations and was much more pronounced for oligomeric than for fibrillar Aβ1-42, confirming other findings (Evans et al., 2008). Piracetam significantly improved neurite outgrowth under these conditions (P < 0.01). Piracetam was similarly effective when reduced neuritogenesis was induced by intracellular production of Aβ (PC12wt, PC12sw cells). Piracetam's effectiveness was seen at low up to maximum NGF concentrations, where it still significantly enhanced NGF activity, suggesting that piracetam is not merely acting by shifting the NGF dose-response curve to the left. In addition, some direct neurotrophic effects of piracetam are suggested by our findings of increased neurite length in vctPC12 cells, without Aβ impairment, and in PC12 cells after inducing oxidative stress. But these effects are rather small. As all those conditions are associated with mitochondrial dysfunction and metabolic impairment, we propose that improved mitochondrial function underlies the neurotrophic effects of piracetam.
The proposition that piracetam compensates for reduced neurotrophic support associated with cellular (mitochondrial) dysfunction may explain why piracetam seems to enhance the restitution and reorganization of neuronal circuits after periods of brain damage in experimental animals (Coq and Xerri, 1999; Xerri and Zennou-Azogui, 2003). Moreover, piracetam not only reduces neuronal loss but also enhances synaptic reorganization following chronic alcohol intake (Brandao et al., 1995, 1996). Similarly, neurotrophic properties have been associated with the restitution of EEG parameters in post-stroke patients following piracetam treatment (Szelies et al., 2001).
While the concept of Aβ-induced mitochondrial dysfunction as a major, functionally relevant, pathomechanism in AD has received substantial support over the last decade, improving mitochondrial function as a target for new drug development has not been similarly supported, as most interest has been directed to compounds leading to reduced Aβ load. However, as several compounds out of those disease-modifying drug classes have recently failed to show clinical effectiveness in AD trials (Gura, 2008), a report about substantial therapeutic effects of dimebon in a 1-year clinical trial (Doody et al., 2008) generated considerable interest. Although originally used as an antihistaminic drug, dimebon was later characterized as a mitochondrial stabilizer (Bachurin et al., 2003; Bernales et al., 2008) with properties similar to those reported for piracetam here. The concept of using mitochondrial protection as treatment strategy for dementia has recently been further supported by preliminary data of substantial clinical improvement in AD patients treated with methylene blue (Gura, 2008). Interestingly, this drug not only has been shown to enhance cognitive functions in several animal studies associated with elevated oxygen consumption, but also seems to enhance mitochondrial function by activating complex I and IV activities at the cellular level (Riha et al., 2005; Callaway et al., 2002, 2004; Atamna et al., 2008).
In conclusion, in view of the increasing interest in mitochondrial protection as treatment strategy in dementia, our findings of substantial protection by piracetam of brain mitochondria against Aβ-induced dysfunction deserve further attention.
Acknowledgments
This study was supported by an unrestricted research grant from UCB, Brussels (Belgium).
Glossary
Abbreviations:
- Aβ
β-Amyloid
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- BSA
Bovine serum albumin
- DMEM
Dulbecco's modified Eagle's medium
- HBSS
Hanks' balanced salt solution
- HEPES
(4-2-hydroxyethyl)-1-piperazineethanesufonic acid
- MMP
Mitochondrial membrane potential
- R123
Rhodamine 123
- SNP
Sodium nitroprusside
- TBS
Tris-buffered saline
- tgAPP
transgenic for the human amyloid precursor protein
Conflicts of interest
K.L. received honorarium for scientific lectures from UCB. W.E.M. is a paid consultant of several pharmaceutical companies, including UCB (Belgium), but receives no rewards from sales of any products.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008;22:703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- Bachurin SA, Shevtsova EP, Kireeva EG, Oxebkrug GF, Sablin SO. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann N Y Acad Sci. 2003;993:334–344. doi: 10.1111/j.1749-6632.2003.tb07541.x. [DOI] [PubMed] [Google Scholar]
- Benzi G, Pastoris O, Villa RR, Giuffrida AM. Influence of aging and exogenous substances on cerebral energy metabolism in posthypoglycemic recovery. Biochem Pharmacol. 1985;34:1477–1483. doi: 10.1016/0006-2952(85)90687-2. [DOI] [PubMed] [Google Scholar]
- Bernales S, Wagner S, Protter AA, Hung DT. Dimebon induces neurite outgrowth and mitochondrial statilizion. Washington, DC: Society for Neuroscience; 2008. Program No. 543.29/S12. Neuroscience Meeting Planner. Online. [Google Scholar]
- Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, et al. Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol. 2003;184:247–263. doi: 10.1016/s0014-4886(03)00252-8. [DOI] [PubMed] [Google Scholar]
- Blasko I, Kemmler G, Krampla W, Jungwirth S, Wichart I, Jellinger K, et al. Plasma amyloid ß protein 42 in non-demented persons aged 75 years: effects of concomitant medication and medial temporal lobe atrophy. Neurobiol Aging. 2005;26:1135–1143. doi: 10.1016/j.neurobiolaging.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Brandao F, Paula-Barbosa JM, Cadete-Leite A. Piracetam impedes hippocampal neuronal loss during withdrawal after chronic alcohol intake. Alcohol. 1995;12:279–288. doi: 10.1016/0741-8329(94)00107-o. [DOI] [PubMed] [Google Scholar]
- Brandao F, Cadete-Leite A, Andrade JP, Madeira MD, Paula-Barbose MM. Piracetam promotes mossy fiber synaptic reorganization in rats withdrawn from alcohol. Alcohol. 1996;13:239–249. doi: 10.1016/0741-8329(95)02050-0. [DOI] [PubMed] [Google Scholar]
- Callaway NL, Riha PD, Wrubel KM, Mccollum D, Gonzalez-Lima F. Methylene blue restores spatial memory retention impaired by an inhibitor of cytochrome oxidase in rats. Neurosci Lett. 2002;332:83–86. doi: 10.1016/s0304-3940(02)00827-3. [DOI] [PubMed] [Google Scholar]
- Callaway NL, Rihe PD, Bruchey AK, Munshi Z, Gonzalez-Lima F. Methylene blue improves brain oxidative metabolism and memory retentions in rats. Pharmacol Biochem Behav. 2004;77:175–181. doi: 10.1016/j.pbb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Canevari L, Clark JB, Bates TE. Beta-amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999;457:131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L. Beta-amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. 2002;10:258–267. doi: 10.1006/nbdi.2002.0516. [DOI] [PubMed] [Google Scholar]
- Chan SL, Furukawa K, Mattson MP. Presenilins and APP in neuritic and synaptic plasticity. Neuromol Med. 2002;2:167–196. doi: 10.1385/NMM:2:2:167. [DOI] [PubMed] [Google Scholar]
- Coq O, Xerri C. Acute reorganization of the forepaw representation in the rat. SI cortex after focal cortical injury: neuroprotective effects of piracetam treatment. Eur J Neurosci. 1999;11:2597–2608. doi: 10.1046/j.1460-9568.1999.00673.x. [DOI] [PubMed] [Google Scholar]
- Crouch P, Kozlowski R, Slater KJ, Fletcher J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods. 1993;160:81–88. doi: 10.1016/0022-1759(93)90011-u. [DOI] [PubMed] [Google Scholar]
- Dekosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Domanska-Janik K, Zaleska M. The action of piracetam on 14C-glucose metabolism in normal and posthypoxic rat cerebral cortex slices. Pol J Pharmacol Pharm. 1977;29:111–116. [PubMed] [Google Scholar]
- Doody RA, Gavrilova SI, Sana M, Thomas RG, Aisen PS, Bachurin SO, et al. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer's disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372:207–215. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]
- Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schüssel K, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol. 2003;66:1627–1634. doi: 10.1016/s0006-2952(03)00534-3. [DOI] [PubMed] [Google Scholar]
- Eckert A, Hauptmann S, Scherping E, Meinhardt J, Rhein V, Dröse S, et al. Oligomeric and fibrillar species of ß-amyloid (Aß42) both impair mitochondrial function in P301L tau transgenic mice. J Mol Med. 2008;86:1255–1267. doi: 10.1007/s00109-008-0391-6. [DOI] [PubMed] [Google Scholar]
- Evans NA, Facci L, Owen DE, Soden PE, Burgidge SA, Prinjha RK, et al. Aß1-42 reduces synapse number and inhibits neurite outgrowth in primary cortical and hippocampal neurons: a quantitive analysis. J Neurosci Methods. 2008;175:96–103. doi: 10.1016/j.jneumeth.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Figueroa DJ, Morris JA, Ma L, Kandpal G, Chen E, Li YM, et al. Presenilin-dependent gamma-secretase activity modulates neurite outgrowth. Neurobiol Dis. 2002;9:49–60. doi: 10.1006/nbdi.2001.0447. [DOI] [PubMed] [Google Scholar]
- Fordyce DE, Clark VJ, Paylor R, Wehner JM. Enhancement of hippocampally-mediated learning and protein kinase C activity by oxiracetam in learning-impaired DBA/2 mice. Brain Res. 1995;672:170–176. doi: 10.1016/0006-8993(94)01389-y. [DOI] [PubMed] [Google Scholar]
- Fukui H, Moraes CT. The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci. 2008;31:252–256. doi: 10.1016/j.tins.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgea CE. The nootropic concept and its prospective implications. Drug Dev Res. 1982;2:441–446. [Google Scholar]
- Guglielmotto M, Aragno M, Auteooi R, Giliberto L, Novo E, Colombatto S, et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of ocidative stress and HIF1α. J Neurochem. 2009;198:1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x. [DOI] [PubMed] [Google Scholar]
- Gura T. Hope in Alzheimer's fight emerges from unexpected places. Nature Med. 2008;14:894. doi: 10.1038/nm0908-894. [DOI] [PubMed] [Google Scholar]
- Hauptmann S, Scherping I, Dröse S, Brandt U, Schulz KL, Jendrach J, et al. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30(10):1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- He Z, Liao Y, Zheng M, Zeng FD, Guo LJ. Piracetam improves cognitive deficits caused by chronic cerebral hypoperfusion in rats. Cell Mol Neurobiol. 2008;28:613–627. doi: 10.1007/s10571-007-9165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss WD, Hebold I, Klinkhammer PL, Ziffling P, Szelies B, Pawlik G, et al. Effect of piracetam on cerebral glucose metabolism in Alzneimer's disease as measured by positron emission tomography. J Cereb Blood Flow Metab. 1988;8:613–617. doi: 10.1038/jcbfm.1988.104. [DOI] [PubMed] [Google Scholar]
- Hirata K, Yamaguchi H, Takaura Y, Takagi A, Fukushime T, Iwakami N, et al. A novel neurotropic agent, T-817MA [1-{3-[2-(1-benzothiophen-5-yl) Ethoxy] propyl}-2-azetidinol maleate], attenuates amyloid-ß-induced neurotoxicity and promotes neurite outgrowth in rat cultured central nervous system neurons. J Pharmacol Exp Ther. 2005;314:252–259. doi: 10.1124/jpet.105.083543. [DOI] [PubMed] [Google Scholar]
- Hlinak Z, Krejci E. Oxiracetam prevented the scopolamine but not the diazepam induced memory deficits in mice. Behav Res. 2002;133:395–399. doi: 10.1016/s0166-4328(02)00028-1. [DOI] [PubMed] [Google Scholar]
- Holinski S, Claus B, Alaaraj N, Dohmen PM, Kirilova K, Neumann K, et al. Cerebroprotective effect of piracetam in patients undergoing coronary bypass surgery. Med Sci Monit. 2008;14:1–5. [PubMed] [Google Scholar]
- Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li DJ. High content screen microscopy analysis of Aß1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of α7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007;1151:227–235. doi: 10.1016/j.brainres.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Jin SM, Cho HJ, Jung ES, Shim MY, Mook-Jung I. DNA damage-inducing agents elicit gamma-secretase activation mediated by oxidative stress. Cell Death Differ. 2008;15:1375–1384. doi: 10.1038/cdd.2008.49. [DOI] [PubMed] [Google Scholar]
- Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, et al. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004;279:50310–50320. doi: 10.1074/jbc.M405600200. [DOI] [PubMed] [Google Scholar]
- Keil U, Scherping I, Hauptmann S, Schuessel K, Eckert A, Müller WE. Piracetam improves mitochondrial dysfunction following oxidative stress. Br J Pharmacol. 2006;147:199–208. doi: 10.1038/sj.bjp.0706459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboyama T, Tohda C, Komatsu K. Neuritic regeneration and synaptic reconstruction induced by withanolide A. Br J Pharmacol. 2005;144:961–971. doi: 10.1038/sj.bjp.0706122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Sanzclemente A, Velasco PT, Wood M, et al. Aß oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27(4):796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner K, Hauptmann S, Abdel-Kader R, Scherping I, Keil U, Strosznajder JB, et al. Mitochondrial dysfunction: the first domino in brain aging and Alzheimer's disease? Antioxid Redox Signal. 2007;9:1659–1675. doi: 10.1089/ars.2007.1763. [DOI] [PubMed] [Google Scholar]
- Leutz S, Steiner B, Marques CA, Haass C, Müller WE, Eckert A. Reduction of tropic support enhances apoptosis in PC12 cells expressing Alzheimer's App mutation and sensitizes cells to staurosporine-induced cell death. J Mol Neurosci. 2002;18:189–201. doi: 10.1385/JMN:18:3:189. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Marques A, Keil U, Bonert A, Steiner B, Haass C, Müller WE, et al. Neurotoxic mechanisms caused by the Alzheimer's disease-linked Swedisch amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. J Biol Chem. 2003;278:28294–28302. doi: 10.1074/jbc.M212265200. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Mitochondrial regulation of neuronal plasticity. Neurochem Res. 2007;32:707–715. doi: 10.1007/s11064-006-9170-3. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nature. 2006;7:278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller WE, Koch S, Scheuer D, Rostock A, Bartsch R. Effects of piracetam on membrane fluidity in the aged mouse, rat, and human brain. Biochem Pharmacol. 1997;53:135–140. doi: 10.1016/s0006-2952(96)00463-7. [DOI] [PubMed] [Google Scholar]
- Müller WE, Eckert GP, Eckert A. Piracetam: novelty in a unique mode of action. Pharmacopsychiatry. 1999;32(Suppl. 1):2–9. doi: 10.1055/s-2007-979230. [DOI] [PubMed] [Google Scholar]
- Naftalin RJ, Cunningham P, Afzal-Ahmed I. Piracetam and TRH analogues antagonise inhibition by barbiturates, diazepam, melatonin and galanin of human erythrocyte D-glucose transport. Br J Pharmacol. 2004;142:594–608. doi: 10.1038/sj.bjp.0705798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira C, Santos MS, Oliveira C. Involvement of oxidative stress on the impairment of energy metabolism induced by A beta peptides on PC12 cells: protection by antioxidants. Neurobiol Dis. 1999;6:209–219. doi: 10.1006/nbdi.1999.0241. [DOI] [PubMed] [Google Scholar]
- Peters I, Igbavboa U, Schütt T, Haidari S, Hartig U, Böttner S, et al. The interaction of beta-amyloid protein with cellular membranes stimulates its own production. Biochim Biophys Acta. 2009;1788:964–972. doi: 10.1016/j.bbamem.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petratos A, Li XQ, George AJ, Hou X, Kerr LM, Unabia SE, et al. The ß-amyloid protein of Alzheimer's disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain. 2008;131:90–108. doi: 10.1093/brain/awm260. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Beal IMF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riha PD, Bruchex AK, Echevarria DJ, Gonzalez-Lima F. Memory facilitation by methylene blue: dose-dependent effect on behaviour and brain oxygen consumption. Eur J Pharmacol. 2005;511:151–158. doi: 10.1016/j.ejphar.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Saletu G, Hitzenberger G, Grunberger J, Anderer P, Zyhlarz G, Linzmayer L, et al. Double-blind, placebo-controlled, pharmacokinetic and-dynamic studies with 2 new formulations of piracetam (infusion and sirup) under hypoxia in man. Int J Clin Pharmacol Ther. 1995;33:249–262. [PubMed] [Google Scholar]
- Sastre J, Pallardo FV, Vina J. The role of mitochondrial oxidative stress in aging. Free Radic Biol Med. 2003;35:1–8. doi: 10.1016/s0891-5849(03)00184-9. [DOI] [PubMed] [Google Scholar]
- Scheuer K, Rostock A, Bartsch R, Müller WE. Piracetam improves cognitive performance by restoring neurochemical deficits of the aged rat brain. Pharmacopsychiatry. 1999;32(Suppl. 1):10–16. doi: 10.1055/s-2007-979231. [DOI] [PubMed] [Google Scholar]
- Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest. 2003;111:303–312. doi: 10.1172/JCI17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;278:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Ki S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-ß protein dimers isolated directly from Alzheimers's brains impairs synaptic plasticity and memory. Nature Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Am, Wehner JM. Aniracetam improves contextual fear conditioning and increases hippocampal γ-PKC activation. Hippocampus. 2002;12:76–85. doi: 10.1002/hipo.10008. [DOI] [PubMed] [Google Scholar]
- Stine WB, Dahlgren KN, Krafft GA, Ladu MJ. In vitro characterization conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- Stoll L, Schubert T, Müller WE. Age-related deficits of central muscarinic cholinergic receptor function in the mouse: partial restoration by chronic piracetam treatment. Neurobiol Aging. 1992;1:39–44. doi: 10.1016/0197-4580(92)90006-j. [DOI] [PubMed] [Google Scholar]
- Szelies G, Mielke R, Kessler J, Heiss WD. Restitution of alpha-topography by piracetam in post-stroke aphasie. Int J Clin Pharmacol Ther. 2001;39:152–157. doi: 10.5414/cpp39152. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, Deteresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neuro. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Uebelhack R, Vohs D, Zytowski M, Schewe HJ, Koch C, Konertz W. Effects of piracetam on cognitive performance in patients undergoing bypass surgery. Pharmacopsychiatry. 2003;36:89–93. doi: 10.1055/s-2003-39981. [DOI] [PubMed] [Google Scholar]
- Valzelli L, Bernasconi S, Sala A. Piracetam activity may differ according to the age of the recipient mouse. Int Pharmacopsychiatry. 1980;15:150–156. doi: 10.1159/000468431. [DOI] [PubMed] [Google Scholar]
- Villard V, Espallergues J, Keller E, Alkam T, Nitta A, Yamada K, et al. Anitamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEXI-41 against amyloid Aß(25–35)-induced toxicity in mice. Neuropsychopharmacology. 2009;34:1552–1566. doi: 10.1038/npp.2008.212. [DOI] [PubMed] [Google Scholar]
- Waegemans T, Wilsher CR, Danniau A, Ferris SH, Kurz A, Winblad B. Clinical efficacy of piracetam in cognitive impairment: a meta-analysis. Dement Geriatr Cogn Disord. 2002;13:217–224. doi: 10.1159/000057700. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Aß oligomers – a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Xerri C, Zennou-Azogui Y. Influence of the postlesion environment and chronic piracetam treatment on the organization of the somatotopic map in the rat primary somatosensory cortex after focal cortical injury. Neuroscience. 2003;118:161–177. doi: 10.1016/s0306-4522(02)00911-9. [DOI] [PubMed] [Google Scholar]