

Abstract
Background and purpose:
Benznidazole (Bz) is the therapy currently available for clinical treatment of Chagas' disease. However, many strains of Trypanosoma cruzi parasites are naturally resistant. Nitric oxide (NO) produced by activated macrophages is crucial to the intracellular killing of parasites. Here, we investigate the in vitro and in vivo activities against T. cruzi, of the NO donor, trans-[RuCl([15]aneN4)NO]2+.
Experimental approach:
Trans-[RuCl([15]aneN4)NO]2+was incubated with a partially drug-resistant T. cruzi Y strain and the anti-proliferative (epimastigote form) and trypanocidal activities (trypomastigote and amastigote) evaluated. Mice were treated during the acute phase of Chagas' disease. The anti-T. cruzi activity was evaluated by parasitaemia, survival rate, cardiac parasitism, myocarditis and the curative rate.
Key results:
Trans-[RuCl([15]aneN4)NO]2+ was 10- and 100-fold more active than Bz against amastigotes and trypomastigotes respectively. Further, trans-[RuCl([15]aneN4)NO]2+ (0.1 mM) induced 100% of trypanocidal activity (trypomastigotes forms) in vitro. Trans-[RuCl([15]aneN4)NO]2+ induced permanent suppression of parasitaemia and 100% survival in a murine model of acute Chagas' disease. When the drugs were given alone, parasitological cures were confirmed in only 30 and 40% of the animals treated with the NO donor (3.33 µmol·kg−1·day−1) and Bz (385 µmol·kg−1·day−1), respectively, but when given together, 80% of the animals were parasitologically cured. The cured animals showed an absence of myocarditis and a normalisation of cytokine production in the sera. In addition, no in vitro toxicity was observed at the tested doses.
Conclusions and implications:
These findings indicate that trans-[RuCl([15]aneN4)NO]2+is a promising lead compound for the treatment of human Chagas' disease.
This article is commented on by Machado et al., pp. 258–259 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00662.x and to view a related paper in this issue by Silva et al. visit http://dx.doi.org/10.1111/j.1476-5381.2010.00524.x
Keywords: NO donors, trans-[RuCl([15]aneN4)NO]2+, benznidazole, Trypanosoma cruzi, Chagas' disease
Introduction
Trypanosoma cruzi is an intracellular protozoan that is able to cause human disease and constitutes a major cause of heart disease in Latin America. In Central and South America, Chagas' disease affects 13 million people, with 75 million at risk for infection (WHO, 2005). Moreover, transmission through transfusions and transplants represents a public health concern in non-endemic countries such as Australia, Canada, Spain and the United States (Schmunis, 2007). The current anti-parasitic chemotherapy with benznidazole (Bz) (Rochagan and Rodanil; Roche, Rio de Janeiro, Brazil) or nifurtimox (Lampit; Bayer, Wuppertal, Germany) is relatively effective during the acute and sub-chronic stages of Chagas' disease (Russomando et al., 1998; Sosa Estani et al., 1998; Cancado, 2002; Altclas et al., 2005). The adequacy of anti-parasitic treatment for patients in the indeterminate a nd chronic stages of the disease has not been confirmed. Some non-randomised studies have suggested that trypanocidal treatment with Bz slowed the progression of the clinical condition and cardiac function, as indicated by the prevention of worsening ECG, of the patients (Viotti et al., 1994; 2006; Fabbro De Suasnabar et al., 2000; Lauria-Pires et al., 2000), while others yielded inconclusive results (Lauria-Pires et al., 2000). Unfortunately, Bz treatment is unsatisfactory during the chronic phase of the disease. Therefore, the development of new drugs is necessary.
Parasite elimination depends largely on the production of pro-inflammatory cytokines, such as interferon-γ (IFN-γ), tumour necrosis factor-α (TNF-α) and interleukin (IL)-12, as they act in concert to activate macrophages that kill intracellular parasites through the production of nitric oxide (NO) and its derived nitrogen and oxygen radicals (Aliberti et al., 1999; 2001; Machado et al., 2000). Studies using experimental models of acute infection have demonstrated that the anti-parasitic activity of Bz involves the participation of these cytokines (Michailowsky et al., 1998; Molina et al., 2000) as well as covalent modifications of macromolecules by intermediates of nitro-reducers (reductive stress) (Docampo, 1990). Conversely, nifurtimox acts by reducing the nitro group to form unstable nitro anion radicals, which, in turn, react to produce highly toxic reduced oxygen metabolites, such as superoxide anion and hydrogen peroxide (Docampo, 1990).
Both Bz and nifurtimox have significant side effects, including anorexia, vomiting, peripheral polyneuropathy and allergic dermopathy (Rassi et al., 1999). Moreover, several strains of T. cruzi do not respond to these treatments, even during the acute phase of the disease (Filardi and Brener, 1987; Galvao et al., 1993; Urbina, 1999). The rate of cure observed in patients with these drugs is 50–70% during the acute phase and 0–20% during the chronic phase (Guedes et al., 2006). This situation is severely aggravated by the absence of a gold standard for diagnosis, which throws doubt on all the parameters used for evaluating the outcome of trypanocidal therapies. Thus, there is an imperative requirement for the development of new therapeutic agents for Chagas' disease.
Ruthenium NO donor compounds have recently emerged as an interesting and important alternative treatment of experimental infections with T. cruzi (Silva et al., 2007). These compounds showed low toxicities in vitro and in vivo and stability in aqueous media in the presence of oxygen and NO, which are released by reducing agents present in the host environment (Bogdan, 2001; Silva et al., 2007). Our group recently reported the trypanocidal activity, in vitro and in vivo, of a series of ruthenium nitrosyls, including trans-[RuII(NO+)(NH3)4L]X3, L = imidazole (imidazole coordinated by nitrogen (imN) or imidazole coordinated by carbon, pyridine, L-histidine, sulphite (SO32−), pyrazine, nicotinamide, 4-picoline, triethylphosphite ([P(OEt)3]), isonicotinamide (isn), isonicotinic acid, X = BF4-, Cl- or PF6-, and [RuII(NO+)(Hedta)] against the Y strain of T. cruzi. Such compounds were efficient in reducing both parasitaemia and cardiac inflammation, and also led to the total survival of infected mice that were treated (Silva et al., 2007). The therapeutic schedule, however, was used only for 15 days of treatment and did not focus on the evaluation of parasitological cure. Here, we used a new and more potent NO donor, trans-[RuCl([15]aneN4)NO]2+complex ([15]aneN4= 1,4,8,12-tetraazacyclopentadecane, a macrocyclic quadridentate amine ligand), over a therapeutic schedule of 20 days of treatment in mice infected with the Y strain of T. cruzi. The parasitological cure of mice treated with trans-[RuCl([15]aneN4)NO]2+ was evaluated and compared with that after treatment with Bz, as well as after a combination of both drugs.
Methods
Parasite and experimental infection
The Y strain of T. cruzi was used in all experiments (Silva and Nussenzweig, 1953). Mice were infected by i.p. injection with 1 × 103 blood trypomastigote stages of T. cruzi. Culture trypomastigotes were obtained from a fibroblast cell line (LLC-MK2). Epimastigote forms were obtained during the exponential phase of growth in liver infusion tryptose (LIT) medium.
Nitric oxide measurement in bone marrow-derived macrophages
Bone marrow-derived macrophages (BMDM) were generated from bone marrow stem cells that were cultured as previously described (Celada et al., 1984). These cells were then loaded with the selective NO fluorescent dye 4,5-diaminofluorescein diacetate (DAF-2 DA) (10 µM) for 30 min at room temperature (Rodrigues et al., 2008). The membrane permeable DAF-2 DA readily enters the cells and is subsequently hydrolysed by cytosolic esterases to release free DAF-2, which does not leave the cell. At physiological pH, DAF-2 is relatively non-fluorescent, but it forms the fluorescent product DAF-2 triazole (DAF-2T) in the presence of NO and oxygen. This approach allows the direct visualisation and semi-quantitative analysis of the basal NO availability at the cellular level. The excess dye was removed by washing in a bath solution. The cytosolic NO concentration was assessed by a Leica TCS SP5 spectral scanning confocal microscope (Leica Microsystems, Wetzlar, Germany). DAF-2T fluorescence was excited with the 488 ηm line of an argon ion laser, and the emitted fluorescence was measured at 515 ηm. Time course software was used to capture images of the cells at 2 s intervals using the Live Data Mode acquisition. The intensities of the intracellular maximum or minimum fluorescence were measured through the application of the LSCM computer software. From these data, the basal fluorescence intensity value (basal) was registered at time zero of the experiment. The final FI value (15-ane) was registered at 300 s after the addition of both trans-[RuCl([15]aneN4)NO]2+) (1 mM) and the reducing agent phenylephrine (0.1 µM) to the medium.
Culture of mouse leukaemic monocyte macrophage cell line 264.7 macrophages and luciferase assays
The murine mouse leukaemic monocyte macrophage cell line (RAW) 264.7 Luc macrophage, bearing the luciferase vector inserted in the NF-κB promoter (pNF-κB-Luc), was routinely cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin, at 37°C in a humidified atmosphere of 5% CO2. For luciferase reporter assays, RAW 264.7 macrophages (5 × 105 cells·mL−1) were grown in 24-well plates to 60–70% confluence. After culturing for 24 h, cells were stimulated with lipopolysaccharide (1 µg·mL−1) following the addition of trans-[RuCl([15]aneN4)NO]2+(1.0, 0.5 and 0.1 mM), ascorbic acid (AA; 1.0, 0.5 and 0.1 mM) and Bz (1.0 mM) to the growth medium. Cells were treated at different drug concentrations and kept overnight under incubation conditions. Cells were harvested and extracted in lysis buffer (TNT), and the luciferase activities in the cell lysates were determined using the Dual Luciferase Reporter assay system (Promega). Data were then expressed as a ratio of light units.
Surface molecule expression and cytotoxicity of treated macrophage
The expression of co-stimulatory molecules (CD40, CD80 and CD86) and MHC-II was assessed in BMDM after being incubated for 24 h with trans-[RuCl([15]aneN4)NO]2+. Surface staining of untreated- and drug-treated macrophages was performed, in each case, by cell incubation with specific phycoerythrin- or fluorescein isothiocyanate-labelled monoclonal antibodies: anti-CD40, anti-CD86, anti-CD86 and anti-MHC-II (BD-Pharmingen, San Diego, CA, USA). For cytotoxicity assays, triplicates of BMDM were incubated with trans-[RuCl([15]aneN4)NO]2+ (0.1, 0.5 and 1.0 mM) and/or AA (0.1, 0.5 and 1.0 mM) diluted in phosphate buffered saline (PBS) at 37°C in a humidified atmosphere of 5% CO2 for 24 h. Bz (1.0 mM) was used as the reference trypanocidal drug, and Tween 20 (5.0, 0.5, and 0.005%) was used as a positive control for the death of cells. Cells were harvested and incubated with propidium iodide at 50 µg·mL−1. Data were acquired after 15 min, using a FACSorter apparatus (Becton-Dickinson Immunocytometry System Inc., San Jose, CA, USA). Multivariate data analysis was performed in the FlowJo software (Ashland, OR, USA).
In vivo lethal dose of trans-[RuCl([15]aneN4)NO]2+
For the determination of the half-lethal dose (LD50) in vivo, three Swiss mice (30 g) per group were injected by i.p. injection with trans-[RuCl([15]aneN4)NO]2+ or AA plus trans-[RuCl([15]aneN4)NO]2+ diluted in PBS at doses of 83, 250, 750, 2000 and 6000 µmol·kg−1, respectively, with animal survival observed over the course of 48 h, as previously described (Silva et al., 2007).
Trypanocidal activity of trans-[RuCl([15]aneN4)NO]2+ in vitro
The in vitro anti-proliferative and trypanocidal activities of trans-[RuCl([15]aneN4)NO]2+ were initially evaluated against the epimastigote and trypomastigote forms, respectively, of the T. cruzi Y strains, as previously described (Silva et al., 2007). Trypomastigote and epimastigote cultures were re-suspended at a concentration of 6.5 106 parasites·mL−1 in Roswell Park Memorial Institute (RPMI) medium with 10% FBS and LIT, respectively. Triplicate cultures were treated with trans-[RuCl([15]aneN4)NO]2+ (0.1, 0.5 and 1.0 mM) and/or AA (0.1, 0.5 and 1.0 mM) diluted in PBS at 37°C in a humidified atmosphere of 5% CO2. Bz (1.0 mM) (Roche) was used as the reference trypanocidal drug (positive control). Parasite viability was subsequently tested by determining the number of motile forms, according to the method established by Brener (1962b). The concentration of compound corresponding to 50% anti-proliferative or trypanocidal activity after 24 h of incubation was expressed as the IC50epi (inhibitory concentration for the epimastigote form) or IC50try (inhibitory concentration for the trypomastigote form), respectively. The same protocol was used to determine the trypanocidal activity of the aqueous complex, trans-[RuCl([15]aneN4)H2O]+.
The intracellular action of the drugs was also determined on the amastigote forms. Subconfluent monolayers of Vero cells (American Type Culture Collection CLL-81) were plated at 1.25 × 104 cells in RPMI (GIBCO, Grand Island, NY, USA) supplemented with 5% FBS (Nutricell, Campinas, São Paulo, Brazil) in each well of a eight-well chamber slide (chamber slide, Nunc Inc., Naperville, IL, USA) and incubated at 37°C in a humidified atmosphere of 5% CO2 for 24 h prior to infection. Cell subconfluent monolayers were then infected with tissue culture trypomastigote forms (12.5 × 104 parasites per well) for 24 h. After infection, the chamber slides were washed twice with cold PBS in order to remove parasites adhering to the outside of cells, and the cells were then re-incubated in culture medium with drug tested. These experiments were performed in triplicate. The media was removed after 24 h incubation with drug and the chamber was stained with Giemsa, as previously described (Guedes et al., 2007). The percent of infected cells was determined by randomly examining the slides and counting a minimum of 500 cells under the microscope using high magnification (400×).
Treatment of mice with NO donor and Bz
All animal care and experimental protocols were approved by the Ethics Committee on Animal Research of the University of São Paulo. The mice that were used in these experiments belonged to the animal stock of the Department of Biochemistry and Immunology, Medicine School of Ribeirão Preto at the University of São Paulo in São Paulo, Brazil. Animals were housed in temperature-controlled rooms (22–25°C) and received water and food ad libitum in the animal facilities. Female Swiss mice 6–8 weeks old were infected with 1.0 × 103 blood trypomastigotes per animal. A total of eight experimental groups, each consisting of 10 Swiss mice that weighed 25–30 g, were used. Treatment started soon after the detection of parasitaemia, which occurred on day 4 post inoculation (p.i.). Bz, in a suspension in 4% gum arabica, was administered orally at 385 µmol·kg−1 of body weight per day (equivalent to 100 mg·kg−1) for 20 consecutive days. Trans-[RuCl([15]aneN4)NO]2+ (3.33 µmol·kg−1·day−1) and AA (3.33 µmol·kg−1·day−1) diluted in PBS were given by i.p. injection for 20 consecutive days. The groups received the following treatments: Group 1 = PBS, Group 2 = AA, Group 3 = Bz, Group 4 =trans-[RuCl([15]aneN4)NO]2+, Group 5 =trans-[RuCl([15]aneN4)NO]2+plus Bz, Group 6 =trans-[RuCl([15]aneN4)NO]2+ plus AA, Group 7 =trans-[RuCl([15]aneN4)NO]2+ plus Bz plus AA and Group 8 = not infected and not treated. Ten animals from each group were killed at the end of the treatment (25 days p.i) for quantification of the levels of heart inflammation and cytokine production. Another 10 animals were used to evaluate parasitaemia, mortality and parasitological cure. The evaluation of parasitaemia was performed as previously described (Brener, 1962a).
Cytokine quantification by ELISA
The cytokine levels in serum samples obtained on day 25 p.i. were determined using ELISA kits for IL-10, IFN-γ and TNF-α (all R&D Duoset, R&D, Minneapolis, MN, USA), according to the manufacturers' instructions. The reaction was detected by peroxidase-conjugated streptavidin followed by a substrate mixture containing hydrogen peroxide and ABTS (Sigma Aldrich, St. Louis, MO, USA) as a chromogen.
Nitrite measurement in the sera of treated animals
Nitrite production in the sera of non-infected and infected mice was measured by accumulation of nitrite, which is a NO metabolite, in sera collected on day 25 p.i (the last day of treatment), as described previously (Green et al., 1981). Briefly, 0.05 mL of Griess reagent [0.1% naphthyl ethylenediamine (Sigma Aldrich) and 1% sulfanilamide in 2.5% phosphoric acid (Sigma Aldrich)] was added to 0.05 mL of sera, and absorbance was read at 540 nm using an automated plate reader. Nitrite concentration was calculated from a NaNO2 standard curve.
Evaluation of parasitological cure
Parasitological cure was assessed by a battery of three independent tests, performed 30 days after the end of treatment, that is, 55 days p.i, including parasitological assays, such as haemoculture and real-time PCR, and serological assays, such as ELISA. The parasitological cure was determined based upon a negative result with both the parasitological and serological methods.
The haemoculture assay was performed as follows. Animals were bled from the orbital venous sinus, and 400 µL of blood were collected and divided into two tubes containing 3 mL of LIT medium. Tubes were incubated at 28°C for 90 days and examined monthly for the presence of parasites (Caldas et al., 2008).
Real-time PCR was performed using the Platinum® SYBR® Green qPCR SuperMix UDG with ROX system (Invitrogen, Carlsbad, CA, USA). The reaction mix included 2 µg of DNA plus 25 pmol of primers S35 (5′AAATAATGTACGGG (T/G)GAGATGCATGA3′) and S36 (5′GGGTTCGATTGGGGTTGGTGT3′) (OPERON Technologies, INC). Samples were amplified for 40 cycles in a 7000 Sequence Detection Systems (Applied Biosystems, Foster City, CA, USA), which generated a product of 330 bp.
ELISA and flow cytometry were performed with sera obtained on the first month after treatment. ELISA plates were coated with T. cruzi antigen that was prepared from alkaline extraction of the T. cruzi Y strain during the exponential phase of growth in LIT medium. Anti-mouse IgG-peroxidase conjugated antibody (Sigma Aldrich) was used. The mean absorbance for 10 negative control samples plus two standard deviations were used as the cut-off to discriminate between positive and negative results.
Quantification of myocardial inflammation
Myocarditis was assessed as previously described (Guedes et al., 2007). Briefly, total nuclei numbers were determined in 50 microscopic fields, using at least four representative, non-consecutive, haematoxylin and eosin-stained sections (thickness of 5 µm) from each mouse. Sections were examined with a Zeiss Integrationsplatte II eyepiece (Zeiss Co., Oberkochen, Germany) reticule used in conjunction with an Olympus BHS microscope (Olympus, Miami, FL, USA) at a final magnification of 400×.
Statistical analysis
Data are expressed as the mean ± SEM. Student's t-test was used to analyse the statistical significance of the differences observed between the infected and control assays. In the time course studies, two-way anova was used, followed by Tukey–Kramer post hoc analysis. The Kaplan–Meier method was used to compare survival curves of the studied groups. Differences were considered statistically significant when P < 0.05. All analyses were performed using the PRISM 3.0 software (GraphPad, San Diego, CA, USA)
Materials
Trans-[RuCl([15]aneN4)NO]2+ was synthesised as described by Bonaventura et al. (2004). The chemical structure of trans-[RuCl([15]aneN4)NO]2+ is shown in Figure 1. Bz (N-benzyl-2-nitro-1-imidazolacetamide) was from Roche, AA and phenylephrine from Sigma
Figure 1.
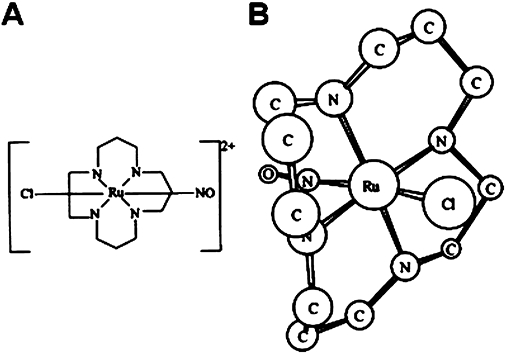
(A) Chemical structure of trans-[RuCl([15]aneN4)NO]2+; (B) 3Dδ calculated, structure of trans-[RuCl([15]aneN4)NO]2+ (hydrogen atoms are omitted).
Results
Trans-[RuCl([15]aneN4)NO]2+has low toxicity and high LD50
Bz, which is the drug available for the clinical treatment of Chagas' disease, is used at high doses and for long-term therapy, with significant side effects. The toxicity of the new NO donor trans-[RuCl([15]aneN4)NO]2+was determined and compared with that of Bz using BMDM. AA, trans-[RuCl([15]aneN4)NO]2+ and Bz, all at doses of 1.0, 0.5 and 0.1 mM, did not produce in vitro cytotoxicity (Figure 2). The LD50 of trans-[RuCl([15]aneN4)NO]2+for mice was approximately 2000 µmol·kg−1, with the lethal dose ranging from 2000 to 6000 µmol·kg−1.
Figure 2.
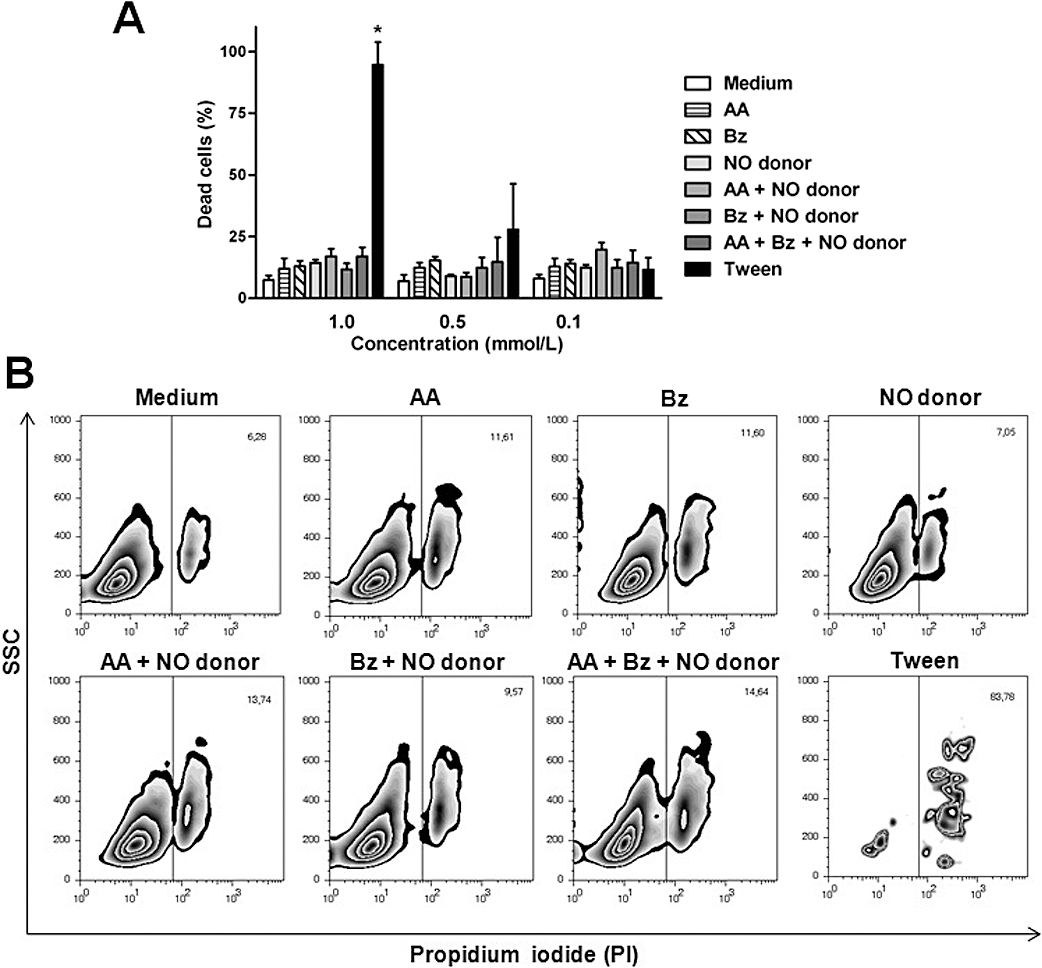
Trans-[RuCl([15]aneN4)NO]2+ showed no in vitro toxicity in cultures of bone marrow macrophages. The in vitro toxicity assay of benznidazole (Bz; 1.0 mM) or the NO donor trans-[RuCl([15]aneN4)NO]2+ (NO donor, 0.1, 0.5 and 1.0 mM) alone, in combination with ascorbic acid (AA; 0.1, 0.5 and 1.0 mM), or in combination with AA (0.1, 0.5 and 1.0 mM) plus Bz (1.0 mM) in bone marrow-derived macrophages (BMDM) after a 24-h incubation. Tween 20 (5.0, 0.5 and 0.005%) was used as positive control for cell death. (A) Percent macrophage death at different concentrations and combinations of the compounds that were tested. (B) Representative FACS analysis plots of BMDM after propidium iodide staining. Note the lack of damage on the BMDM target cells by trans-[RuCl([15]aneN4)NO]2+ Data represent means ± SEM and are representative of three independent experiments. SSC, side-scattered light; PBS, phosphate buffered saline. *Significant difference (P < 0.05).
Next, we tested whether the NO donor enhances the intracellular NO concentration, which affects the intracellular forms of T. cruzi. Macrophages that were cultured in the presence of trans-[RuCl([15]aneN4)NO]2+and 0.1 µM phenylephrine (reducing agent) exhibited enhanced intracellular concentrations of NO (Figure 3). To verify if this effect was due to the direct activation of macrophages by trans-[RuCl([15]aneN4)NO]2+or by its biologically reduced by-products, BMDM were cultured in the presence of the NO donor, and the expression levels of the co-stimulatory molecules CD40, CD80, CD86 and MHC-II were analysed. The addition of drugs (NO donor, AA, and Bz) did not enhance the expression of the co-stimulatory molecules in macrophages (Figure S1A). This absence of direct activation of macrophages by the NO donor was confirmed by using murine RAW macrophage-like cells that stably expressed a NF-κB luciferase reporter. After being cultured with the drugs, these reporter cells did not show enhanced luciferase expression (Figure S1B). These results showed that the enhancement of NO concentration in macrophages did not result from a generalised activation of macrophages by the drugs, but rather as a result of the release of NO by trans-[RuCl([15]aneN4)NO]2+.
Figure 3.
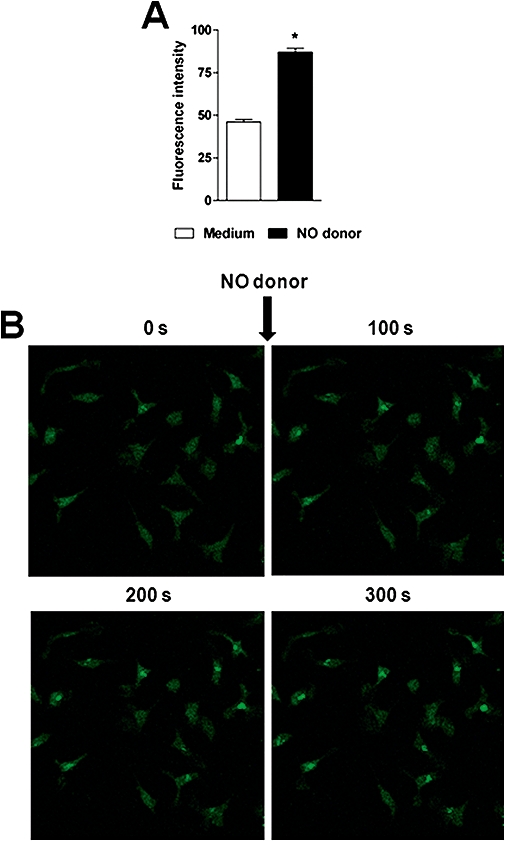
Trans-[RuCl([15]aneN4)NO]2+ enhances cytosolic NO concentration in macrophage cells from mice. Cultures of bone marrow-derived macrophages (BMDM), treated or untreated with trans-[RuCl([15]aneN4)NO]2+ (1.0 mM, for 100, 200 and 300 s) were pre-incubated in DAF-2DA (10 µM) for 30 min at room temperature and examined under fluorescent light. (A) Significant enhancement of the DAF-2DA fluorescent green staining 300 s after NO donor addition along with 0.1 µM phenylephrine (reducing agent), compared with the control culture (0.1 µM phenylephrine alone). Bars represent the mean ± SEM of five different samples. *denotes statistical difference in the fluorescence intensity, P < 0.001. (B) Serial confocal images from BMDM in culture medium (Control) and NO donor-treated macrophages. Microphotographs representative of fluorescence in macrophage cells were recorded before (medium alone) and 100, 200 and 300 s after addition of the combination of 1.0 mM of trans-[RuCl([15]aneN4)NO]2+ and 0.1 µM phenylephrine.
Trans-[RuCl([15]aneN4)NO]2+has potent in vitro trypanocidal activity
After establishing the safety of trans-[RuCl([15]aneN4)NO]2+ through its low in vitro toxicity and wide therapeutic window, we evaluated its ability to inhibit all life cycle stages of T. cruzi. An assessment of the in vitro anti-proliferative activity against epimastigotes revealed that Bz showed a 100% anti-proliferative effect at 1.0 mM. Treatment with the NO donor alone showed an approximately 10–20% anti-proliferative activity against the epimastigote form of T. cruzi. As the NO-releasing activity of the NO donor depends upon the presence of reducing agents in the medium, treatment with a combination of the NO donor and AA showed 100% activity against the epimastigote form, similar to that of 1.0 mM Bz (Figure 4A). Interestingly, trans-[RuCl([15]aneN4)NO]2+was more efficient at concentrations that were 100-fold lower than those of Bz against trypomastigotes, which are the T. cruzi forms present in the blood of vertebrate hosts (Figure 4B). At a concentration of 1.0 mM, Bz killed 53% of trypomastigote forms after 24 h of incubation, whereas the trans-[RuCl([15]aneN4)NO]2+plus AA at 0.1, 0.5 and 1.0 mM killed 100% of trypomastigote forms after 8 h (Figure 4B). A Bz concentration of 10 mM was required to kill 100% of parasites (data not shown). As observed against epimastigote forms, the action of the NO donor against trypomastigote forms also depended on the presence of reducing agents.
Figure 4.
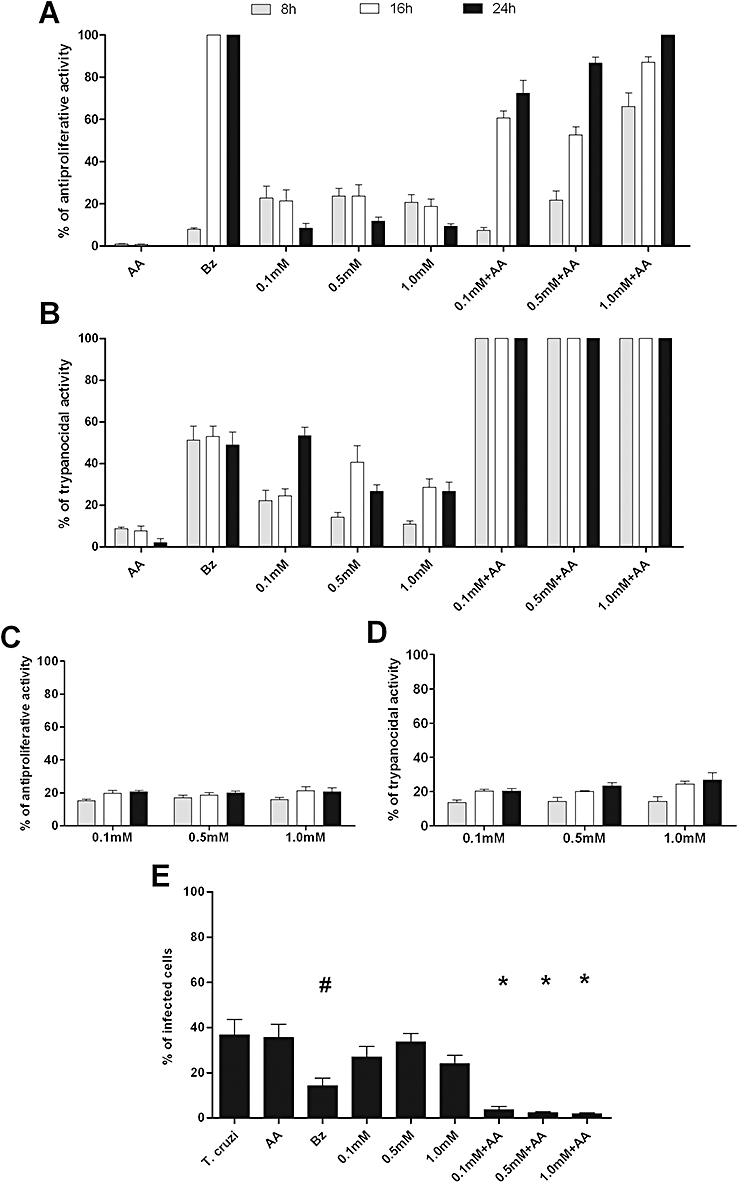
Trans-[RuCl([15]aneN4)NO]2+ shows potent trypanocidal activity in vitro. The antiproliferative and trypanocidal activities (shown as %) of the NO donor trans-[RuCl([15]aneN4)NO]2+ (0.1, 0.5 and 1.0 mM) on epimastigote (A) and trypomastigote (B) forms of the Trypanosoma cruzi Y strain in culture media. Parasites were treated with benznidazole (Bz, 1.0 mM) or NO donor alone, in combination with ascorbic acid (AA, 0.1, 0.5 and 1.0 mM), or in combination with AA plus Bz. The antiproliferative (C) and trypanocidal (D) activities of the aqueous compound trans-[RuCl([15]aneN4)H2O]+ (0.1, 0.5 and 1.0 mM) were administered alone. Antiproliferative and trypanocidal activities of trans-[RuCl([15]aneN4)NO]2+ and of the aqueous compound trans-[RuCl([15]aneN4)H2O]+ on epimastigote and trypomastigote forms of T. cruzi were determined by counting viable parasites at 8, 12,and 24 h post-treatment. The same concentrations described above for trans-[RuCl([15]aneN4)NO]2+ were also used against amastigote forms (E). Cultures were stained by Giemsa dye. Each point is the mean ± SEM and is representative of three independent experiments with similar results (n= 10). Statistically significant differences from control (P < 0.05) are denoted by *, #.
After NO release from trans-[RuCl([15]aneN4)NO]2+, the NO in the complex ion is replaced by H2O to form the complex ion trans-[RuCl([15]aneN4)H2O+]. In order to determine if this ion, devoid of NO, also has trypanocidal activity, the aqueous complex was incubated with the trypomastigote and epimastigote forms of T. cruzi. Throughout the range of concentrations that were used (0.1, 0.5 and 1.0 mM), approximately 20% of the trypomastigotes were killed, and 30% of the epimastigotes displayed a reduction in proliferation (Figure 4C,D). The NO donor is thus likely to show a synergism between NO release and the nucleus of the metallic compound.
Furthermore, in cultured Vero cells, trans-[RuCl([15]aneN4)NO]2+, in the presence of a reducing agent, was more efficient than Bz against the intracellular amastigote form (Figure 4E). Vero cells were infected with T. cruzi and were treated with drugs after parasite internalisation. The NO donor did not show a significant effect against amastigote forms of the parasite when it was added alone to the culture. After treatment with 1.0 mM of Bz or trans-[RuCl([15]aneN4)NO]2+plus AA, the proportion of infected cells was 14 ± 4% and 2 ± 0.5%, respectively, whereas, in untreated cultures, there were 38 ± 9% infected cells. Taken together, these results indicate that the in vitro trypanocidal effect of trans-[RuCl([15]aneN4)NO]2+ in the presence of a reducing agent was greater than that of Bz.
The NO donor suppressed the parasitaemia, mortality and myocarditis
The control untreated animals and those treated with AA alone showed similar parasitaemias, with the level of parasitaemia peaking on the day 9 p.i. The parasitaemia was suppressed between the second and fourth days of treatment in all animals that were treated with either the NO donor (3.33 µmol·kg−1·day−1) alone or Bz (385 µmol·kg−1·day−1) alone. A combined treatment with both drugs suppressed the parasitaemia on the first day of treatment. All control (untreated) animals showed significantly higher parasitaemia levels and patent periods (P < 0.001) than animals treated with NO donor and/or Bz (Figure 5A). In contrast to the in vitro effects, similar results were observed in mice treated with the NO donor either alone or in combination with AA. This was due to the presence of reducing agents in the blood and host tissues. A survival rate of 100% of the T. cruzi- infected and trans-[RuCl([15]aneN4)NO]2+plus Bz-treated animals was observed for 6 months after the treatment, for all the therapeutic schemes used. In contrast, untreated controls and animals treated with AA alone showed a survival rate of 0 and 10% respectively (Figure 5B).
Figure 5.
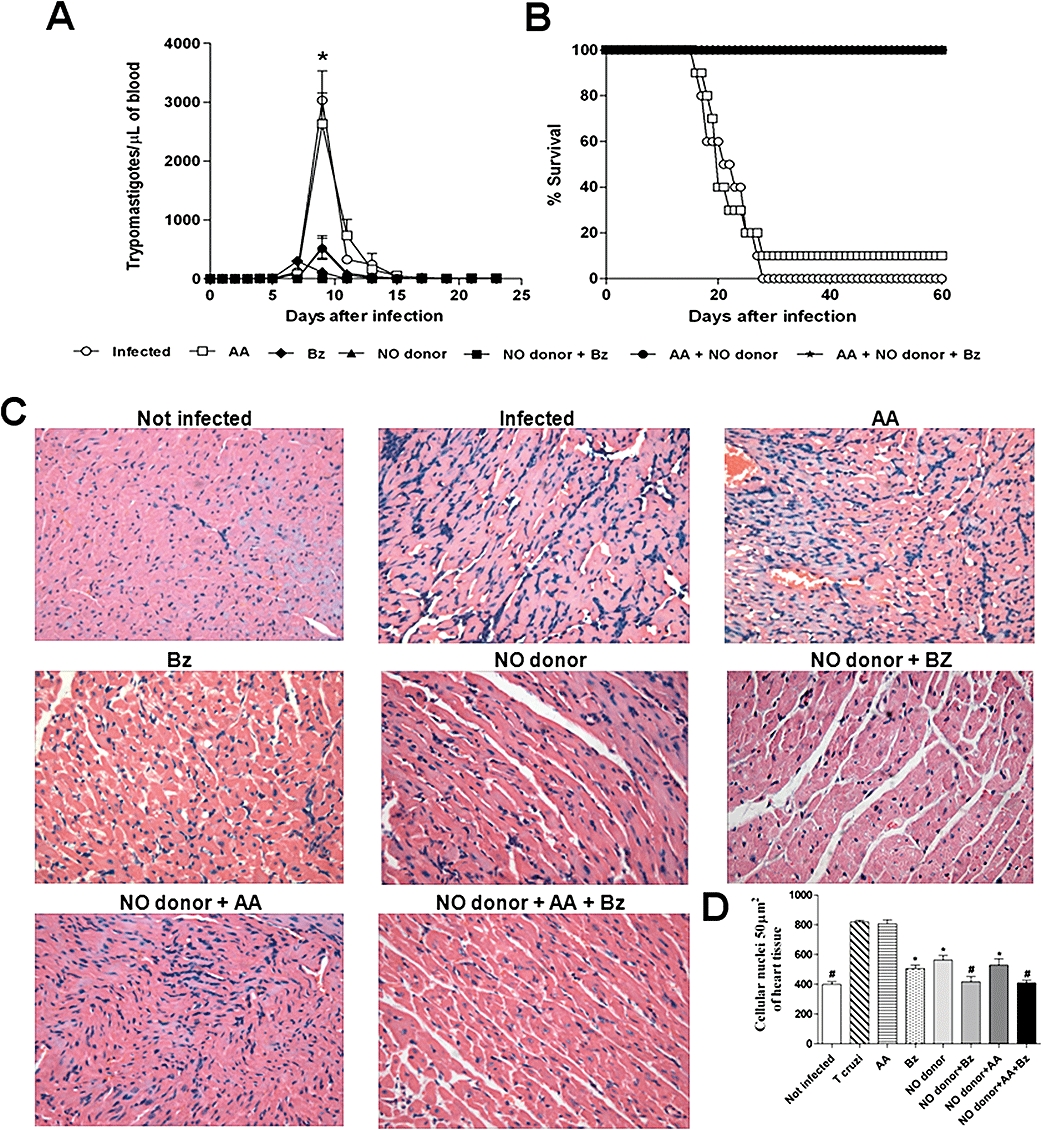
Treatment with trans-[RuCl([15]aneN4)NO]2+ suppresses parasitaemia, mortality and myocarditis of mice infected with T. cruzi. Swiss mice were infected with 1 × 103 blood trypomastigotes of T. cruzi Y strain, followed by different treatment schedules: ascorbic acid (AA; 3.33 µmol·kg−1·day−1) alone, benznidazole (Bz; 385 µmol·kg−1·day−1) alone, NO donor trans-[RuCl([15]aneN4)NO]2+ (3.33 µmol·kg−1·day−1) alone, NO donor (3.33 µmol·kg−1·day−1) plus benznidazole (385 µmol·kg−1·day−1), NO donor (3.33 µmol·kg−1·day−1) in combination with ascorbic AA (3.33 µmol·kg−1·day−1) and the NO donor (3.33 µmol·kg−1·day−1) in combination with AA (3.33 µmol·kg−1·day−1) and Bz (385 µmol·kg−1·day−1). Treatment was started on day 5 p.i and was given daily on 20 consecutive days. The control group received PBS. Parasitaemia (A) and survival (B) were evaluated daily over a period of 60 days post-infection. Representative sections of cardiac tissue (C) 25 days after infection (the last day of treatment). Original magnification of microphotographs, ×400. Quantification of cellular nuclei per 50 µm2 of heart tissue (D) of non-treated and treated animals. Note the synergic effect of the NO donor with Bz in eliminating myocarditis in T. cruzi-infected mice (Figure 5C,D), regardless of the presence or absence of AA. Each point is the mean ± SEM and is representative of three independent experiments with similar results (n= 10). Statistically significant differences from control (P < 0.05) are denoted by *, #. For experimental details, refer to the legend in Table 1.
Cardiac inflammation was evaluated at the end of the treatment (25 days p.i.). When administered separately, Bz and the NO donor were not able to achieve full clearance of the cardiac inflammation (Figure 5C,D), although these treatments showed significantly lower levels of myocardial inflammation (P < 0.05) than control animals treated with AA or untreated animals. Interestingly, when administered together, Bz and trans-[RuCl([15]aneN4)NO]2+ eliminated myocarditis in mice (Figure 5C,D), whether or not AA was also added. AA was used as reducing agent to assist NO release mainly in vitro.
Two months after each treatment, the animals were separated into cured or not cured groups, according to the results of the parasitological and serological tests. Both the cured and the not cured animals that had been treated with trans-[RuCl([15]aneN4)NO]2+ and/or Bz showed no cardiac inflammation or significantly decreased cardiac inflammation, respectively, when compared with those animals that were treated with AA (P < 0.05) (data not shown). The treated group could not be compared with the infected and untreated animals, because all the untreated mice died, as did those treated with AA, with only a single survivor in the latter group at 1 month after treatment. The reduced myocarditis observed at the end of the treatment correlated with the poor production of NO and cytokines (IL-10, IFN-γ and TNF-α) detected in the sera of all treated animals (Figure S2A,B). As observed in the cardiac inflammatory process 25 days p.i, nitrite and inflammatory cytokine production levels were high in the infected and untreated animals and in the mice treated with AA when compared with the animals treated with trans-[RuCl([15]aneN4)NO]2+ and/or Bz (Figure S2A,B). The parasitological cure was correlated with the absence of myocarditis and normalisation of serum cytokine and NO production in animals that were treated with a combination of trans-[RuCl([15]aneN4)NO]2+ and Bz (Figure 5 and Figure S2A,B).
Parasitological cure was verified by three independent criteria, including haemoculture, real-time PCR and the presence of specific IgG anti-T. cruzi antibodies detected by ELISA. In mice treated with AA (group 1), Bz (group 2), trans-[RuCl([15]aneN4)NO]2+ (group 3), trans-[RuCl([15]aneN4)NO]2+ plus Bz (group 4), AA plus trans-[RuCl([15]aneN4)NO]2+ (group 5) and AA plus trans-[RuCl([15]aneN4)NO]2+plus Bz (group 6), the parasitological and serological tests were negative for 0, 40, 30, 80, 20 and 80% of the animals respectively (Table 1). Serum levels of T. cruzi-specific immunoglobulin G antibodies and parasite DNA levels in the animal groups treated with the NO donor plus Bz were absent or remarkably low, at levels that were very similar to those in the uninfected control group (Figure S3). Similar to the untreated control group, deaths occurred among mice treated with AA during the acute phase of the infection, with only one mouse surviving through to the cure rate evaluation period.
Table 1.
The NO donor trans-[RuCl([15]aneN4)NO]2+ produced parasitological cure of experimental Chagas' disease
|
Positive results/testb |
|||||
|---|---|---|---|---|---|
| Therapeutic schedulea | No of positive ELISA/total | No of positive haemoculture/total | No of positive real-time PCR/total | No of positive tests post-treatment/total (%) | Cure rate (%) |
| Ascorbic acidc (AA) | 1 | 1 | 1 | 1/1 (100) | 0 |
| Benznidazole (Bz) | 6 | 5 | 6 | 6/10 (60) | 40 |
| NO donor | 7 | 6 | 7 | 7/10 (70) | 30 |
| NO donor + Bz | 2 | 1 | 2 | 2/10 (20) | 80 |
| NO donor + AA | 8 | 4 | 8 | 8/10 (80) | 20 |
| NO donor + AA + Bz | 2 | 2 | 2 | 2/10 (20) | 80 |
The effects of the NO donor, trans-[RuCl([15]aneN4)NO]2+, given alone and/or: AA, Bz as well as the NO donor plus ascorbic acid plus benznidazole on mice infected with T. cruzi Y strain on measures of cure are shown.
Swiss female mice, 28–30 g, were infected with 1 × 103 blood trypomastigotes of T. cruzi Y strain. Treatment started after detection of patent parasitaemia, on day 5 p.i. given daily over a period of 20 days. The NO donor trans-[RuCl([15]aneN4)NO]2+, and AA were injected i.p. at 3.33 µmol·kg−1·day−1. Bz was administered orally at 385 µmol·kg−1. The infected control group received PBS. Data are expressed as the mean ± SEM and are representative of three independent experiments with similar results, n= 10. PBS, phosphate buffered saline.
Results shown are the number of positive ELISA, haemoculture and real time PCR tests. Ten mice were used for each treatment group and for the not infected and T. cruzi-infected controls.
Only one animal remained alive until the period of the cure rate evaluation in the control group. No survivals were observed in the PBS, untreated control group.
Overall, these results indicate that treated but not cured animals still had reductions in cardiac parasitism and in cardiac damage, demonstrating the importance of treatment, even when the animals were not totally cured.
Discussion and conclusions
In this study, treatment with the new NO donor trans-[RuCl([15]aneN4)NO]2+reduced parasitic load, cardiac inflammation, and allowed 100% survival in mice during the acute phase of an experimental infection with a highly virulent strain of T. cruzi. In addition, this drug was able to induce a parasitological cure in 40% of the treated mice. Moreover, treatment in combination with Bz enhanced the rate of parasitological cure up to 80%.
Currently available antiparasitic agents for the etiological treatment of Chagas' disease include Bz and nifurtimox. The outcomes obtained with both drugs are diverse, and their cure rates depend on the stage of the disease as well as on the sensitivity of the parasite strain, which varies geographically (Filardi and Brener, 1987; Sosa Estani and Segura, 1999; Guedes et al., 2006). Despite being beneficial during the acute stage of infection, the outcomes of these antiparasitic therapies during chronic infection have not been systematically evaluated. Most Chagas' disease cases are detected during the chronic phase of the disease, when cardiovascular and gastrointestinal damage may be irreversible and when the only available therapeutic option may be surgery. Thus, the development of new therapeutic approaches is highly important for the treatment of Chagas' disease. At the moment, a multicentre, randomised double blind clinical trial is being conducted to test whether the administration of Bz to patients with non-acute forms of the disease will prevent chronic cardiovascular damage (Marin-Neto et al., 2008).
NO plays multiple important roles in the immune response, including antiviral, antimicrobial, immunostimulatory, immunosuppressive, cytotoxic and cytoprotective effects. The use of iNOS−/− mice demonstrated that most of the above-described effects were mediated by iNOS-derived NO (Bogdan et al., 2000a,b; Nathan and Shiloh, 2000). NO, which is produced by activated macrophages, is crucial to the intracellular killing of parasites, including T. cruzi (Martins et al., 2001). Many protozoa are able to inhibit NO production, which thus allows them to evade the immune system. T. cruzi induces apoptosis in lymphocytes, and regulation of iNOS mediated by cell–cell contact has been seen in apoptotic lymphocytes (Freire-de-Lima et al., 2000). Uptake of apoptotic cells drives the growth of T. cruzi within macrophages. The uptake of apoptotic, but not necrotic, lymphocytes by macrophages through fibronectin receptors and CD36 leads to the down-regulated expression of iNOS and, thus, shifts L-arginine metabolism towards the arginase pathway. In turn, there is increased production of ornithine and putrescine, which are known growth factors for T. cruzi. These effects result from the induction of endogenous TGF-β and immune system evasion (Freire-de-Lima et al., 2000). As NO produced within phagocytes is crucial to parasite killing, therapeutic approaches that use NO-releasing compounds could improve the protective immune response and are thus promising agents for the treatment of this intracellular pathogen.
In fact, the treatment of T. cruzi-infected, immunosuppressed mice with Bz generates lower survival and cure rates than the same treatment in immunocompetent animals, which indicates that the protective effects of Bz, particularly the processes of macrophage activation, NO production and parasite destruction, are dependent on the integrity of the immune system (Silva et al., 1995; Molina et al., 2000). Recently, our group demonstrated that NO donors administered at low concentrations are capable of killing T. cruzi (Silva et al., 2007). The rate of parasitological cure for these compounds has not been determined. Moreover, the NO donor used in the present study showed better effects against T. cruzi than those previously tested by our group, which is likely due to the longer period of NO release (Bonaventura et al., 2004) and synergistic activity with the aqueous ion formed from trans-[RuCl([15]aneN4)NO]2+.
We demonstrated that trans-[RuCl([15]aneN4)NO]2+did not show in vitro toxicity to bone marrow macrophages. In addition, the cytosolic NO concentration within macrophages in culture was remarkably enhanced after the addition of trans-[RuCl([15]aneN4)NO]2+along with the reducing agent phenylephrine to the culture medium. The therapeutic dose used in vivo was well below its LD50(3.33 µmol·kg−1 vs. 2000–6000 µmol·kg−1). Even when administered at lower concentrations, trans-[RuCl([15]aneN4)NO]2+ was more efficient than Bz in killing the epimastigote, trypomastigote and amastigote forms of T. cruzi. Although trans-[RuCl([15]aneN4)NO]2+was very effective in vitro, it only generated a 30% parasitological cure rate in vivo when administered alone. The cure rate of a drug correlates with its pharmacokinetic characteristics, such as bioavailability, frequency, pathway of administration and other factors. This particular NO donor drug increases the time over which NO is released, which may have an effect on its cure rate. Evaluation of these factors in the murine model will constitute the next steps in our research. As the NO-releasing activity of trans-[RuCl([15]aneN4)NO]2+ lasts for 2 h (Bonaventura et al., 2004), a schedule of giving trans-[RuCl([15]aneN4)NO]2+ two or three times a day would probably improve its pharmacodynamic properties. Based on the findings reported in this paper, future studies can be conducted to determine if and how the NO release period can be extended.
We observed 100% of survival in mice treated with 3.33 µmol·kg−1·day−1 of trans-[RuCl([15]aneN4)NO]2+, either alone or in combination with 385 µmol·kg−1·day−1 of Bz, while all the infected and untreated animals died. The NO donors tested against T. cruzi, however, are not bioequivalent, since the survival rates of mice treated with Ru(NO)imN or Ru(NO)isn at the same molar dose were 40 and 60% respectively (Silva et al., 2007). These results demonstrate that the trans-[RuCl([15]aneN4)NO]2+and other NO donors that had been previously tested are more potent and safer than Bz. Our results are promising, given that the NO donor trans-[RuCl([15]aneN4)NO]2+displays characteristics that comply with the essential criteria for antiparasitic drugs specified by the WHO and other organisations running control programmes in affected countries.
The cardiac inflammatory process was decreased at the end of the treatment with trans-[RuCl([15]aneN4)NO]2+, with Bz or with their combination (26 d.p.i.). A less intense reduction of myocarditis is observed with other NO donors and Bz (Andrade et al., 1991; Segura et al., 1994; Silva et al., 2007). However, the combination of NO donor and Bz resulted in the absence of myocarditis by the end of the treatment. Moreover, animals that were treated but not considered to be cured showed a reduced level of cardiac inflammation when compared with the control (animals treated with AA alone). These animals also showed decreased parasitism. As the presence of parasites is believed to play an important role in causing cardiac lesions, specific drug-combination therapies might reduce the myocardial damage and morbidity during the acute and chronic phases of the disease in human beings (Coura et al., 1997). Data from the literature also indicate the reversibility of cardiac fibrosis in mice chronically infected with T. cruzi and undergoing specific chemotherapy (Andrade et al., 1991).
New drugs for treating Chagas' disease should be able to improve the quality of life for those infected, through a reduction of the chronic morbidity, the subsequent expenditure by government health systems and early retirement in developing countries. In conclusion, our results indicate that trans-[RuCl([15]aneN4)NO]2+ has both in vitro and in vivo trypanocidal activity and is capable of inducing a radical parasitological cure in a mouse model of acute infection. Furthermore, this compound can be used at high doses and represents a potentially useful candidate for the treatment of Chagas' disease in human beings.
Acknowledgments
We thank Cristiane Maria Milanezi for her excellent technical assistance. This project received support from Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP 2007/53940-0), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 410467/2006-5) and a Research fellowship from Coordenação de Aperfeiçoamento de Pesssoal de Nível Superior (CAPES).
Glossary
Abbreviations:
- 4-pic
4-picoline
- AA
ascorbic acid
- ABTS
(2,2′-azino-di-(3-ethyl benzthiazoline-6-sulphonic acid)
- BMDM
bone marrow-derived macrophages
- BT
bloodstream trypomastigote forms of Trypanosoma cruzi
- Bz
benznidazole; DAF-2 DA, 4,5-diaminofluorescein diacetate
- DAF-2T
DAF-2 triazole
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- FI
fluorescence intensity
- FITC
fluorescein isothiocyanate
- IFN-γ
interferon-γ
- IL
interleukin
- imC
imidazole coordinated by carbon
- imN
imidazole coordinated by nitrogen
- ina
isonicotinic acid
- isn
isonicotinamide
- L-hist
L-histidine
- LIT
liver infusion tryptose
- LPS
lipopolysaccharide
- NF-κβ
nuclear factor-κβ
- NHE
normal hydrogen electrode
- nic
nicotinamide
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- PE
phycoerythrin
- py
pyridine
- pz
pyrazine
- SNP
sodium nitroprusside
- T. cruzi
Trypanosoma cruzi
- TGF-β
transforming growth factor β
- TNF-α
tumour necrosis factor-α
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Trans-[RuCl([15]aneN4)NO]2+ did not enhance the expression of co-stimulatory molecules and did not activate macrophages. In vitro effect of different drug schedules on the expression of CD40, CD80 and CD86 co-stimulatory and MHC-II molecules in blood bone marrow-derived macrophages.1 Drug treatments corresponding to each set of bars standing for a co-stimulatory or MHC-II molecule in the histogram: medium – no drug, 1 µg/mL LPS, 1.0 mM trans-[RuCl([15]aneN4)NO]2+ plus 1.0 mM ascorbic acid, 0.5 mM ascorbic acid, 0.5 mM trans-[RuCl([15]aneN4)NO]2+, 1.0 mM trans-[RuCl([15]aneN4)NO]2+ plus 1.0 mM benznidazole, 0.5 mM trans-[RuCl([15]aneN4)NO]2+ plus 0.5 mM ascorbic acid plus 0.5 mM benznidazole. LPS was used as a positive control for macrophage activation. Macrophage cultures were maintained for 24 h under incubation conditions following the expression analysis of the co-stimulatory and MHC-II molecules by flow cytometry (A). The murine macrophage-like cell line RAW 264.7 Luc, bearing the luciferase vector inserted in the NF-kB promoter, was treated with the same therapeutic schedule as above under incubation conditions for 24 h, prior to the assessment of luciferase activity (B). Data (means ± standard error of the mean) are representative of three independent experiments with similar results (n = 10). Statistically significant differences from control (unpaired t-test, P < 0.05) are denoted by *.
Figure S2 Treatment with trans-[RuCl([15]aneN4)NO]2+ reduced cytokine and NO production at the end of the therapeutic schedule. Swiss mice were inoculated with 1 × 103 blood trypomastigotes of T. cruzi Y strain and treated with trans-[RuCl([15]aneN4)NO]2+ (3.33 mmol/kg/day), and/or ascorbic acid (3.33 mmol/kg/day), and/or benznidazole (385 µmol/kg/day) for 20 consecutive days. The control group received only PBS. For details on the experimental protocol, refer to the legend in Figure 5. Nitrite (A) and IL-10, IFN-g and TNF-α (B) production were determined in the blood sera on day 25 post-infection (the last day of treatment) by ELISA and the Griess method, respectively. Data are representative of three independent experiments with similar results, n = 10. Data represent means ± SEM. *, + and # P < 0.05, different symbols indicate significant difference.
Figure S3 The treatment with trans-[RuCl([15]aneN4)NO]2+ induces a parasitological cure in T. cruzi-infected mice. Swiss mice were inoculated with 1 × 103 blood trypomastigotes of T. cruzi Y strain and treated with trans-[RuCl([15]aneN4)NO]2+ (3.33 mmol/kg/day), and/or ascorbic acid (3.33 mmol/kg/day), and/or benznidazole (385 µmol/kg/day) for 20 consecutive days. The control group received only PBS. For details on the experimental protocol, refer to the legend in Figure 5. The quantification of T. cruzi-specific immunoglobulin G antibodies in the blood sera (A) and real-time PCR analysis for the presence of T. cruzi DNA in the myocardium (B) were performed 55 days after infection (1 month after the end of treatment). Note that the serum levels of T. cruzi-specific immunoglobulin G antibodies as well as the levels of parasite DNA in the animal groups treated with the NO donor plus Bz are remarkably low and are very similar to what is seen for the uninfected control group. Data represent means ± SEM and are representative of three independent experiments with similar results (unpaired t-test, P < 0.05) (n = 10). PBS, phosphate buffered saline.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aliberti JC, Machado FS, Souto JT, Campanelli AP, Teixeira MM, Gazzinelli RT, et al. beta-Chemokines enhance parasite uptake and promote nitric oxide-dependent microbiostatic activity in murine inflammatory macrophages infected with Trypanosoma cruzi. Infect Immun. 1999;67(9):4819–4826. doi: 10.1128/iai.67.9.4819-4826.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliberti JC, Souto JT, Marino AP, Lannes-Vieira J, Teixeira MM, Farber J, et al. Modulation of chemokine production and inflammatory responses in interferon-gamma- and tumor necrosis factor-R1-deficient mice during Trypanosoma cruzi infection. Am J Pathol. 2001;158(4):1433–1440. doi: 10.1016/s0002-9440(10)64094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altclas J, Sinagra A, Dictar M, Luna C, Veron MT, De Rissio AM, et al. Chagas disease in bone marrow transplantation: an approach to preemptive therapy. Bone Marrow Transplant. 2005;36(2):123–129. doi: 10.1038/sj.bmt.1705006. [DOI] [PubMed] [Google Scholar]
- Andrade SG, Stocker-Guerret S, Pimentel AS, Grimaud JA. Reversibility of cardiac fibrosis in mice chronically infected with Trypanosoma cruzi, under specific chemotherapy. Mem Inst Oswaldo Cruz. 1991;86(2):187–200. doi: 10.1590/s0074-02761991000200008. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2(10):907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- Bogdan C, Rollinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000a;12(1):64–76. doi: 10.1016/s0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- Bogdan C, Rollinghoff M, Diefenbach A. The role of nitric oxide in innate immunity. Immunol Rev. 2000b;173:17–26. doi: 10.1034/j.1600-065x.2000.917307.x. [DOI] [PubMed] [Google Scholar]
- Bonaventura D, de SOF, Togniolo V, Tedesco AC, da Silva RS, Bendhack LM. A macrocyclic nitrosyl ruthenium complex is a NO donor that induces rat aorta relaxation. Nitric Oxide. 2004;10(2):83–91. doi: 10.1016/j.niox.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Brener Z. Observations on immunity to superinfections in mice experimentally inoculated with Trypanosoma cruzi and subjected to treatment. Rev Inst Med Trop Sao Paulo. 1962a;4:119–123. [PubMed] [Google Scholar]
- Brener Z. Therapeutic activity and criterion of cure on mice experimentally infected with Trypanosoma cruzi. Rev Inst Med Trop Sao Paulo. 1962b;4:389–396. [PubMed] [Google Scholar]
- Caldas IS, Talvani A, Caldas S, Carneiro CM, de Lana M, da Matta Guedes PM, et al. Benznidazole therapy during acute phase of Chagas disease reduces parasite load but does not prevent chronic cardiac lesions. Parasitol Res. 2008;103(2):413–421. doi: 10.1007/s00436-008-0992-6. [DOI] [PubMed] [Google Scholar]
- Cancado JR. Long term evaluation of etiological treatment of chagas disease with benznidazole. Rev Inst Med Trop Sao Paulo. 2002;44(1):29–37. [PubMed] [Google Scholar]
- Celada A, Gray PW, Rinderknecht E, Schreiber RD. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J Exp Med. 1984;160(1):55–74. doi: 10.1084/jem.160.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coura JR, de Abreu LL, Willcox HP, Petana W. Comparative controlled study on the use of benznidazole, nifurtimox and placebo, in the chronic form of Chagas' disease, in a field area with interrupted transmission. I. Preliminary evaluation. Rev Soc Bras Med Trop. 1997;30(2):139–144. doi: 10.1590/s0037-86821997000200009. [DOI] [PubMed] [Google Scholar]
- Docampo R. Sensitivity of parasites to free radical damage by antiparasitic drugs. Chem Biol Interact. 1990;73(1):1–27. doi: 10.1016/0009-2797(90)90106-w. [DOI] [PubMed] [Google Scholar]
- Fabbro De Suasnabar D, Arias E, Streiger M, Piacenza M, Ingaramo M, Del Barco M, et al. Evolutive behavior towards cardiomyopathy of treated (nifurtimox or benznidazole) and untreated chronic chagasic patients. Rev Inst Med Trop Sao Paulo. 2000;42(2):99–109. doi: 10.1590/s0036-46652000000200007. [DOI] [PubMed] [Google Scholar]
- Filardi LS, Brener Z. Susceptibility and natural resistance of Trypanosoma cruzi strains to drugs used clinically in Chagas disease. Trans R Soc Trop Med Hyg. 1987;81(5):755–759. doi: 10.1016/0035-9203(87)90020-4. [DOI] [PubMed] [Google Scholar]
- Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 2000;403(6766):199–203. doi: 10.1038/35003208. [DOI] [PubMed] [Google Scholar]
- Galvao LM, Nunes RM, Cancado JR, Brener Z, Krettli AU. Lytic antibody titre as a means of assessing cure after treatment of Chagas disease: a 10 years follow-up study. Trans R Soc Trop Med Hyg. 1993;87(2):220–223. doi: 10.1016/0035-9203(93)90501-g. [DOI] [PubMed] [Google Scholar]
- Green LC, Tannenbaum SR, Goldman P. Nitrate synthesis in the germfree and conventional rat. Science. 1981;212(4490):56–58. doi: 10.1126/science.6451927. [DOI] [PubMed] [Google Scholar]
- Guedes PM, Veloso VM, Caliari MV, Carneiro CM, Souza SM, de Lana M, et al. Trypanosoma cruzi high infectivity in vitro is related to cardiac lesions during long-term infection in Beagle dogs. Mem Inst Oswaldo Cruz. 2007;102(2):141–147. doi: 10.1590/s0074-02762007005000003. [DOI] [PubMed] [Google Scholar]
- Guedes PMM, Fietto JLR, Lana M, Bahia MT. Advances in Chagas disease chemotherapy anti-infective agents in medicinal. Chemistry. 2006;5(2):11. [Google Scholar]
- Lauria-Pires L, Braga MS, Vexenat AC, Nitz N, Simoes-Barbosa A, Tinoco DL, et al. Progressive chronic Chagas heart disease ten years after treatment with anti-Trypanosoma cruzi nitroderivatives. Am J Trop Med Hyg. 2000;63(3–4):111–118. doi: 10.4269/ajtmh.2000.63.111. [DOI] [PubMed] [Google Scholar]
- Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation. 2000;102(24):3003–3008. doi: 10.1161/01.cir.102.24.3003. [DOI] [PubMed] [Google Scholar]
- Marin-Neto JA, Rassi A, Jr, Morillo CA, Avezum A, Connolly SJ, Sosa-Estani S, et al. Rationale and design of a randomized placebo-controlled trial assessing the effects of etiologic treatment in Chagas' cardiomyopathy: the BENznidazole Evaluation For Interrupting Trypanosomiasis (BENEFIT) Am Heart J. 2008;156(1):37–43. doi: 10.1016/j.ahj.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Martins GA, Petkova SB, Machado FS, Kitsis RN, Weiss LM, Wittner M, et al. Fas-FasL interaction modulates nitric oxide production in Trypanosoma cruzi-infected mice. Immunology. 2001;103(1):122–129. doi: 10.1046/j.1365-2567.2001.01216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailowsky V, Murta SM, Carvalho-Oliveira L, Pereira ME, Ferreira LR, Brener Z, et al. Interleukin-12 enhances in vivo parasiticidal effect of benznidazole during acute experimental infection with a naturally drug-resistant strain of Trypanosoma cruzi. Antimicrob Agents Chemother. 1998;42(10):2549–2556. doi: 10.1128/aac.42.10.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina J, Martins-Filho O, Brener Z, Romanha AJ, Loebenberg D, Urbina JA. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob Agents Chemother. 2000;44(1):150–155. doi: 10.1128/aac.44.1.150-155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA. 2000;97(16):8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassi A, Amato Neto V, de Siqueira AF, Ferriolli Filho F, Amato VS, Rassi Junior A. Protective effect of benznidazole against parasite reactivation in patients chronically infected with Trypanosoma cruzi and treated with corticoids for associated diseases. Rev Soc Bras Med Trop. 1999;32(5):475–482. doi: 10.1590/s0037-86821999000500002. [DOI] [PubMed] [Google Scholar]
- Rodrigues GJ, Lunardi CN, Lima RG, Santos CX, Laurindo FR, da Silva RS, et al. Vitamin C improves the effect of a new nitric oxide donor on the vascular smooth muscle from renal hypertensive rats. Nitric Oxide. 2008;18(3):176–183. doi: 10.1016/j.niox.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Russomando G, de Tomassone MM, de Guillen I, Acosta N, Vera N, Almiron M, et al. Treatment of congenital Chagas' disease diagnosed and followed up by the polymerase chain reaction. Am J Trop Med Hyg. 1998;59(3):487–491. doi: 10.4269/ajtmh.1998.59.487. [DOI] [PubMed] [Google Scholar]
- Schmunis GA. Epidemiology of Chagas disease in non-endemic countries: the role of international migration. Mem Inst Oswaldo Cruz. 2007;102(Suppl. 1):75–85. doi: 10.1590/s0074-02762007005000093. [DOI] [PubMed] [Google Scholar]
- Segura MJ, Genovese OM, Segura E, Sanz OP, Sica RE. Central motor conduction in human chronic Chagas' disease. Arq Neuropsiquiatr. 1994;52(1):29–31. doi: 10.1590/s0004-282x1994000100005. [DOI] [PubMed] [Google Scholar]
- Silva JJ, Osakabe AL, Pavanelli WR, Silva JS, Franco DW. In vitro and in vivo antiproliferative and trypanocidal activities of ruthenium NO donors. Br J Pharmacol. 2007;152(1):112–121. doi: 10.1038/sj.bjp.0707363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JS, Vespa GN, Cardoso MA, Aliberti JC, Cunha FQ. Tumor necrosis factor alpha mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected gamma interferon-activated macrophages. Infect Immun. 1995;63(12):4862–4867. doi: 10.1128/iai.63.12.4862-4867.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LHP, Nussenzweig V. Sobre uma cepa de Trypanosoma cruzi altamente virulenta para o camundongo branco. Folia Clin Biol. 1953;20:191–203. [Google Scholar]
- Sosa Estani S, Segura EL. Treatment of Trypanosoma cruzi infection in the undetermined phase. Experience and current guidelines of treatment in Argentina. Mem Inst Oswaldo Cruz. 1999;94(Suppl. 1):363–365. doi: 10.1590/s0074-02761999000700070. [DOI] [PubMed] [Google Scholar]
- Sosa Estani S, Segura EL, Ruiz AM, Velazquez E, Porcel BM, Yampotis C. Efficacy of chemotherapy with benznidazole in children in the indeterminate phase of Chagas' disease. Am J Trop Med Hyg. 1998;59(4):526–529. doi: 10.4269/ajtmh.1998.59.526. [DOI] [PubMed] [Google Scholar]
- Urbina JA. Parasitological cure of Chagas disease: is it possible? Is it relevant? Mem Inst Oswaldo Cruz. 1999;94(Suppl. 1):349–355. doi: 10.1590/s0074-02761999000700068. [DOI] [PubMed] [Google Scholar]
- Viotti R, Vigliano C, Armenti H, Segura E. Treatment of chronic Chagas' disease with benznidazole: clinical and serologic evolution of patients with long-term follow-up. Am Heart J. 1994;127(1):151–162. doi: 10.1016/0002-8703(94)90521-5. [DOI] [PubMed] [Google Scholar]
- Viotti R, Vigliano C, Lococo B, Bertocchi G, Petti M, Alvarez MG, et al. Long-term cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: a nonrandomized trial. Ann Intern Med. 2006;144(10):724–734. doi: 10.7326/0003-4819-144-10-200605160-00006. [DOI] [PubMed] [Google Scholar]
- WHO. Seventeenth Programme Report of the UNICEF/UNDP/World (Progress 2003–2004) Geneva: World Health Organization; 2005. pp. 31–33. Special programme for research and training in tropical disease. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.