

Abstract
Background and purpose:
Smoking cessation trials with three high-affinity partial agonists of α4β2 neuronal nicotinic acetylcholine receptors (nAChRs) have demonstrated differences in their clinical efficacy. This work examines the origin of the differences by taking into account brain exposure and pharmacological effects at human α4β2 nAChRs.
Experimental approach:
Rat plasma and brain pharmacokinetics were characterized and used to predict human steady-state plasma and brain concentrations following recommended doses of each of the three compounds. The pharmacological characterization included in vitro affinities at different nAChR subtypes, functional efficacies and potencies at the human α4β2 nAChR, as well as in vivo effects on rat mesolimbic dopamine turn-over.
Key results:
A comparison of predicted human brain concentrations following therapeutic doses demonstrated that varenicline and nicotine, but not dianicline and cytisine, can extensively desensitize and, to a lesser extent, activate α4β2 nAChRs. The limited clinical efficacy of dianicline may be accounted for by a combination of weak functional potency at α4β2 nAChRs and moderate brain penetration, while recommended doses of cytisine, despite its high in vitro potency, are predicted to result in brain concentrations that are insufficient to affect α4β2 nAChRs.
Conclusions and implications:
The data provide a plausible explanation for the higher abstinence rate in smoking cessation trials following treatment with varenicline than with the two other α4β2 nAChR partial agonists. In addition, this retrospective analysis demonstrates the usefulness of combining in vitro and in vivo parameters with estimated therapeutic human brain concentrations for translation to clinical efficacy.
Keywords: cytisine, dianicline, dopamine, nAChR partial agonists, nicotine, rat pharmacokinetics, unbound brain concentrations, varenicline, voltage clamp
Introduction
Nicotine dependence is a chronic relapsing condition often requiring multiple quit attempts with only few smokers achieving long-term abstinence. Successful quitting is extremely difficult because of the distressing nicotine withdrawal symptoms that accompany smoking cessation, and are intensified by social and environmental sensory cues. Smoking tobacco gives almost immediate relief of these withdrawal symptoms as inhaled nicotine rapidly reaches peak concentrations in the brain to produce the pleasurable experience that reinforces smoking behaviour (Fiore et al., 2008; Benowitz, 2009).
Pharmacotherapies that provide relief of nicotine withdrawal symptoms during a quit attempt have been shown to be effective, but treatments that would also reduce the pleasurable reward-producing effects of nicotine from smoking should further improve efficacy. To be maximally effective, such a treatment should not only have high affinity for the target receptor to compete with nicotine and prevent its binding to the receptor, but also possess adequate pharmacokinetic properties that provide sufficient and long-lasting brain exposure to cover the period of repeated high nicotine exposures during a relapse to smoking.
A recent approach to achieve such dual activity is the use of partial agonists of the central high affinity α4β2-containing nicotinic acetylcholine receptors (nAChRs; nomenclature follows Alexander et al., 2009), believed to play a key role in the addictive effects of nicotine (Picciotto et al., 1998; 2008; Mansvelder and McGehee, 2002; Marubio et al., 2003; Laviolette and Van der Kooy, 2004; Hogg and Bertrand, 2005). High-affinity α4β2 nAChR partial agonists should relieve craving and withdrawal symptoms during a quit attempt by activating α4β2 nAChRs, and will reduce the reinforcing effects of nicotine upon smoking by competing with nicotine for the binding site (Cohen et al., 2003; Coe et al., 2005; Hogg and Bertrand, 2007; Rollema et al., 2007a). Compounds with reduced agonist efficacy at α4β2 nAChRs with a slower onset and much longer half-life than inhaled nicotine should exhibit a low potential for abuse liability.
Three α4β2 nAChR partial agonists varenicline (Coe et al., 2005), cytisine (Papke and Heinemann, 1994; Tutka and Zatoński, 2005) and dianicline (Cohen et al., 2003; structures in Figure 1) have been studied in clinical trials, and found to exhibit differing efficacies as smoking cessation aids. Achieved abstinence rates and odds ratios (ORs) versus placebo are higher for varenicline (Cahill et al., 2008; Nides et al., 2008) than for either cytisine (Etter, 2006; Cahill et al., 2008) or dianicline (Fagerström and Balfour, 2006). The purpose of this study was to examine whether the reported differences in abstinence rates can be attributed to key pharmacokinetic and pharmacodynamic parameters that determine clinical efficacy of the agent, that is, its unbound brain concentration and functional interaction with the target receptor at therapeutic doses.
Figure 1.
Chemical structures of nicotine and the α4β2 nAChR partial agonists tested clinically as aids to smoking cessation: (A) nicotine; (B) Cytisine (Tabex); (C) Dianicline (SSR5918113); and (D) varenicline (Chantix, Champix).
To correlate pharmacologically active concentrations to human brain exposure after recommended doses of the partial agonists, rat plasma and CNS pharmacokinetics were measured in order to predict human unbound steady-state plasma and brain exposures. Disparities between central exposures of each partial agonist prompted us to study key properties that determine their penetration into the brain.
For a quantitative in vitro analysis of the receptor interactions, we determined binding affinities at different nAChR subtypes, their functional efficacies and concentration dependence for both activation and desensitization at the human α4β2 nAChRs. Finally, dose-dependent effects on mesolimbic dopamine turn-over in rat brain were determined as a relevant measure of in vivo potencies of the partial agonists.
Our results show that differences in predicted unbound human brain concentrations and in in vitro potencies to desensitize and activate central α4β2 nAChRs, provide a plausible explanation for the lower abstinence rates with dianicline and cytisine than observed with varenicline, and can be used as predictors of clinical outcome.
Methods
Animals
All animal care and experimental procedures complied with and were approved by, as appropriate, the Institutional Animal Care and Use Committees of Pfizer Global Research and Development (Groton, CT, USA) and the University of Groningen (The Netherlands), and the animal rights rules from Geneva, Switzerland. Male Sprague-Dawley rats (n= 75, 250–320 g, Charles River Laboratories, Wilmington, MA, USA and Harlan, The Netherlands), male FVB/N (n= 12) and mdr1a/1b (–/–,–/–) (n= 12) mice (Taconic Laboratories, Germantown, NY, USA) were used. The animals were housed on a 12 h light schedule (lights on at 7:00 am) in temperature-controlled (20°C) colony rooms with free access to standard chow and water. Xenopus laevis ovaries were harvested from females (n= 5) maintained at the Medical Faculty of Geneva.
Pharmacokinetic studies
In vitro protein binding
To determine unbound concentrations and to calculate brain-to-plasma ratios (B/P), concentration-time profiles were determined in rat plasma, brain, CSF and extracellular fluid (ECF), and total concentrations were corrected with the unbound fractions. Protein binding was determined with 1 µM test compound in pooled human plasma, pooled rat plasma or 20% rat brain homogenate by equilibrium dialysis in Dulbecco's phosphate-buffered saline (pH 7.4) as described previously (Reed-Hagen et al., 1999). The unbound fraction (fu) of the test compound in undiluted brain tissue was calculated from the fu in the homogenate and the homogenate dilution factor according to Kalvass and Maurer (2002).
In vivo brain disposition studies in rats and in P-glycoprotein (P-gp)-deficient mice
Plasma and brain exposures of cytisine and dianicline were measured in wild-type (WT) mice and in mice lacking the P-gp transporter to examine whether these compounds were substrates for this transporter. Male rats received a single 1 mg kg–1 p.o. dose of partial agonist or 1 mg kg–1 s.c. nicotine. The rats were killed by CO2 inhalation at 0.5, 1.5, 3 and 6 h post-p.o. dosing, and at 0.167, 0.5, 1 and 2 h post-s.c. dosing. Whole blood was collected by cardiac puncture, and centrifuged to obtain plasma. CSF was collected via puncture of the cisterna magna, and samples were immediately frozen on dry ice upon collection. Whole brains were collected, rinsed in phosphate-buffered saline, weighed and immediately frozen on dry ice upon collection.
Male FVB/N (WT) and mdr1a/1b (–/–, –/–) mice (30 g, n= 3 per genotype/time point/compound) were given a single 3 mg kg–1 p.o. dose of cytisine or dianicline in phosphate-buffered saline, and killed in a CO2 chamber at 0.5 and 2 h post-dose. Whole blood and brains were collected as described above, and all samples were stored at –20°C until analysis.
In vivo rat brain (ECF) disposition study
Free brain concentrations were measured directly in the extracellular space by ultra slow-flow microdialysis. Unbound concentrations in rat brain ECF of each test compound after 1 mg kg–1 p.o. (nicotine s.c.) were determined by ultra-slow-flow microdialysis in rat cortex, as described by Cremers et al. (2009). A 4 mm MetaQuant probe (BrainLink, Groningen, The Netherlands) was stereotaxically implanted into the prefrontal cortex (coordinates from bregma: AP 3.4 mm and ML 0.8 mm; from dura: DV –5.8 mm) under 2% isoflurane anaesthesia, and experiments were performed 48 h later. The probe was perfused with artificial CSF (aCSF) at a flow rate of 0.15 µL min–1, while ultra-purified water was delivered through the dilution inlet of the probe to create a carrier flow of 0.80 µL min–1. After equilibrating for 1 h, the test compound was administered at t= 0 min, and nine microdialysis samples were collected for 4.5 h at 30 min intervals in tared vials in an automated fraction collector (CMA 142). For in vitro recoveries, a MetaQuant probe was placed in a stirred aCSF solution containing 10−8 M of test compound, and perfused with aCSF and water at the same flow rates as in the in vivo experiment. The exact slow-flow rate was verified by weighing each vial during the experiment, and two additional vials at the end of the experiment, 15 min after stopping the slow flow. All samples were stored at –80°C until analysis.
Rat i.v. and p.o. studies for plasma pharmacokinetic and human pharmacokinetic prediction
For the estimation of human plasma exposure of dianicline and cytisine by allometric scaling, pharmacokinetic parameters were obtained from a rat i.v. and p.o. pharmacokinetic study. Male rats received a single 1 mg kg–1 i.v. or p.o. dose of dianicline or cytisine in saline. Blood samples were collected via a jugular vein catheter at 0.083, 0.167, 0.5, 1, 2, 4 and 7 h post-dose, and centrifuged to obtain plasma samples that were stored at –20°C until analysis. Unbound human clearance for dianicline and cytisine was predicted based on observed rat pharmacokinetic data using single-species allometric scaling as described by Hosea et al. (2009).
Steady-state human brain exposures following therapeutic doses can be predicted from the rat B/P and the human average steady-state plasma concentration (Css,avg), assuming that rat and human have the same B/P ratio for each compound. The predicted human Css,avg for each compound was calculated according to the equation:
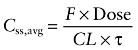 |
(1) |
where F is the oral availability, CL is the clearance and τ is the dosing interval.
Assays and pharmacokinetic analysis
All samples from the protein binding, brain disposition and pharmacokinetic studies were analysed by HPLC–mass spectrometry (detailed assay conditions for each compounds are given in the Supporting Information Appendix S1).
The i.v. and p.o. pharmacokinetic analyses were conducted using individual animal concentration data. Data from the brain penetration study were analysed using mean concentrations at each time-point. All pharmacokinetic analyses were performed using WinNonlin Enterprise version 5.2 (Pharsight Corporation, Mountainview, CA, USA).
Pharmacodynamic studies
In vitro binding affinity to nAChRs
Binding affinities of the test compounds to nAChR subtypes were determined as previously described (Rollema et al., 2007b) using [3H]-epibatidine to label α4β2, α3β4 and α6/4β4 nAChRs expressed in HEK293 cells, [125I]-α-bungarotoxin to label α7 nAChRs expressed in IMR32 cells and α1βγδ nAChRs in Torpedo electroplax membrane. Ki values (Table 1) were calculated according to Ki= IC50/(1 +[3H-ligand]/Kd), and expressed as mean ± SEM (n= 5).
Table 1.
Protein binding, unbound exposure and exposure ratios in rat plasma, brain, CSF and ECF after 1 mg kg–1 p.o. of the partial agonists, and 1 mg kg–1 s.c. of nicotine
| Nicotine | Varenicline | Cytisine | Dianicline | |
|---|---|---|---|---|
| Protein binding (fraction unbound, fu) | ||||
| fu Plasma | 0.81 | 0.55 | 1.00 | 0.95 |
| fu Brain | 1.00 | 0.67 | 1.00 | 0.44 |
| Unbound exposure (AUC0–6h, h·ng·mL−1) | ||||
| Plasmaunbound | 512 | 397 | 702 | 448 |
| Brainunbound | 1070 | 1570 | 80 | 176 |
| CSF | 639 | 495 | 190 | 150 |
| ECF | 495 | 380 | 36 | 77 |
| Brainunbound AUC/Plasmaunbound AUC | 2.10 | 3.90 | 0.11 | 0.39 |
| CSF AUC /Plasmaunbound AUC | 1.25 | 1.25 | 0.27 | 0.33 |
| ECF AUC /Plasmaunbound AUC | 0.97 | 0.96 | 0.05 | 0.17 |
fu, protein binding expressed as fraction unbound in plasma and brain; AUC0–6h, unbound exposure expressed as area under the curve over 0–6 h in h·ng·mL−1.
In vitro functional activity at α4β2 nAChRs expressed in oocytes
Experiments were carried out on human α4β2 nAChRs expressed in Xenopus laevis oocytes using complementary DNA expression. Currents evoked by ACh and nicotinic ligands were recorded using a standard two electrode voltage clamp configuration as described by Hoda et al. (2008). Ovaries were harvested from Xenopus females, and a small piece of ovary was isolated for immediate preparation, while the remaining part was placed at 4°C in a sterile Barth solution containing (in mM) NaCl, 88; KCl, 1; NaHCO3, 2.4; HEPES, 10; MgSO4.7H2O, 0.82; Ca(NO3)2.4H2O, 0.33; CaCl2.6H2O, 0.41 at pH 7.4, supplemented with 20 µg mL–1 of kanamycin, 100 unit mL–1 penicillin and 100 µg mL–1 streptomycin. All recordings were performed at 18°C, and cells were superfused with OR2 medium containing (in mM): NaCl, 82.5; KCl, 2.5; HEPES, 5; CaCl2.2H2O, 2.5; MgCl2.6H2O, 1 at pH 7.4, and 0.5 µM atropine to prevent possible activation of endogenous muscarinic receptors. Cells were held at –80 mV, and data were captured and analysed using data acquisition and analysis software running under Matlab (Mathworks Inc., Natick, MA, USA). Concentration–activation curves were fitted using either a single or dual empirical Hill equation (Buisson and Bertrand, 2001):
| (2) |
where Y is the fraction of evoked current, a is the fraction of high-affinity component, EC50H is the concentration for 50% activation of the high affinity, nHH is the apparent cooperativity for the high affinity, EC50L is the concentration for 50% activation of the low affinity, nHL is the apparent cooperativity for the low affinity and x is the agonist concentration. For the single Hill curve fitting, the second term was not used, and a is the unity.
Concentration–inhibition curves were fitted with a comparable equation:
| (3) |
where a, nHH and nHL are the same as above; Y is the fraction of remaining current, IC50H is the concentration for 50% inhibition of the high-affinity component; IC50L is the concentration for 50% inhibition of the low-affinity component; and x is the antagonist concentration.
Whenever needed, a dual Hill equation, similar to that used for the concentration–activation curves, was used. For statistical analysis, the unpaired, two-tailed Student's t-test was computed either with Excel (Microsoft) or Matlab (Mathworks Inc.).
Dopamine turn-over in rat nucleus accumbens
The dose-dependent effects of the partial agonists on dopamine turn-over in rat nucleus accumbens were determined by oral administration of test compound 2 h prior to rapid removal of the nucleus accumbens, and compared with the response to a maximally effective dose of 1 mg kg–1 s.c. nicotine (Rollema et al., 2007b). Concentrations of dopamine and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were measured in the tissue samples by HPLC and electrochemical detection (details in Supporting Information Appendix S1). Dopamine turn-over was calculated as the ratio ([DOPAC]+[HVA])/[dopamine], and expressed as percentage of controls ± SEM (n= 4–6). Statistical significance was analysed by one-way anova using Dunnett's post hoc test.
Materials
Varenicline (6,7,8,9-tetrahydro-6,10-methano-6H-pyrazino[2,3-h][3]benzazepine) was synthesized according to Coe et al. (2005), and dianicline (SSR591813; 5aS,8S,10aR)-5a,6,9,10-tetrahydro,7H,11H-8,10a-methanopyrido[2′,3′:5,6]pyrano[2,3-d]azepine) according to Galli et al. (2000). Cytisine (1R,5S)-1,2,3,4,5,6-hexahydro-1,5-methano-8H-pyrido[1,2a][1,5]diazocin-8-one) was purchased from Austin Chemical Co. (Buffalo Grove, IL, USA), and nicotine bitartrate from Sigma-Aldrich (St Louis, MO, USA). All compounds were dissolved in saline, administered as 1 mL kg–1 solutions, and doses are expressed as the active compound (base). Radioligands were purchased from PerkinElmer Life and Analytical Sciences Inc. (Boston, MA, USA); all other analytical grade chemicals were from Sigma-Aldrich.
Results
Pharmacokinetic data
Four types of pharmacokinetic studies were performed to predict human unbound brain concentrations and to account for differences observed in rat in brain penetration of the compounds.
Unbound exposure and brain penetration
Time-courses of drug concentrations measured in rat plasma, brain, CSF and ECF following p.o. administration of 1 mg kg–1 of each compound (nicotine s.c.) are shown in Figure 2, and corresponding calculated areas under the concentration time curve over 0–6 h (AUC0–6h) are shown in Table 1. Measurements of unbound fractions (fu) of each of the four compounds show that nicotine and cytisine are essentially non-protein bound in plasma and brain (fu > 0.8), that dianicline is unbound in plasma and 60% bound in brain, while varenicline is moderately protein bound (fu > 0.5) in plasma and brain (Table 1).
Figure 2.
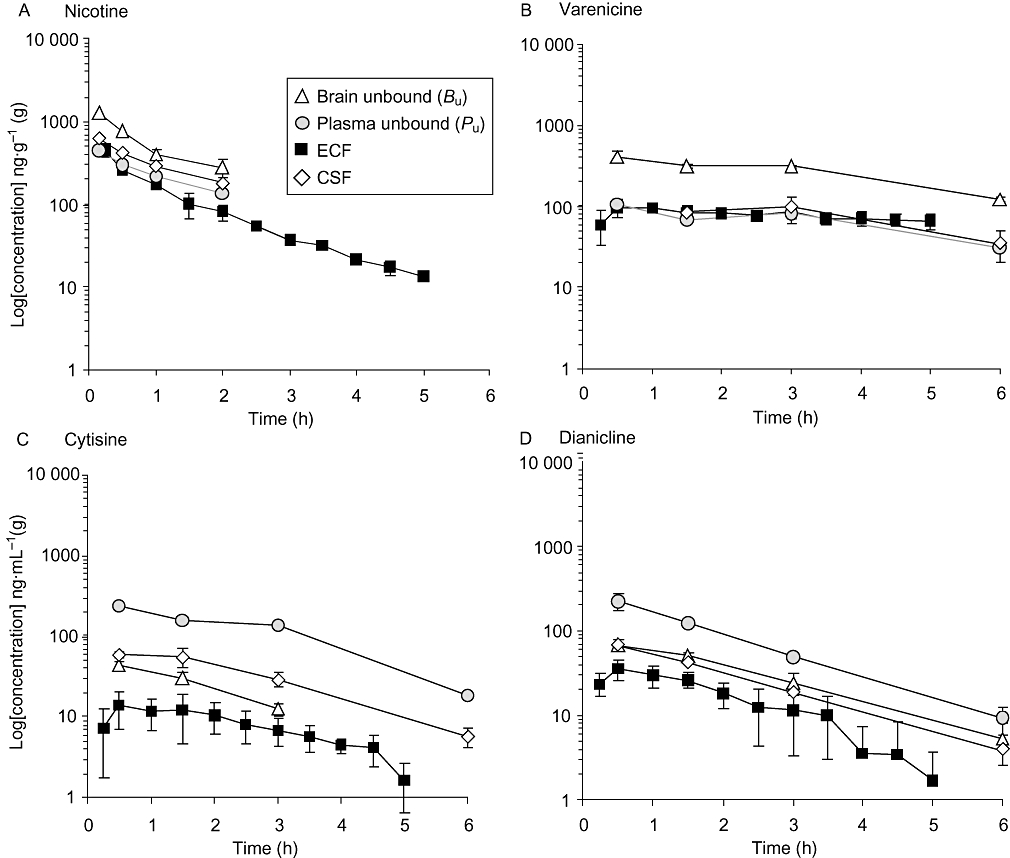
Time-courses of unbound concentrations of nicotine and partial agonists in rat plasma, brain, CSF and ECF after 1 mg kg–1 of each compound. Data are expressed as ng·mL–1 or ng·g–1 (brain) ± SD (n= 3–6). (A) Nicotine 1 mg kg–1 s.c.; (B) varenicline 1 mg kg–1 p.o.; (C) cytisine 1 mg kg–1 p.o.; and (D) dianicline 1 mg kg–1 p.o.
Oral administration to rats of 1 mg kg–1 of each compound yielded comparable unbound plasma concentrations for the four compounds, with AUC0–6h values of 512, 397, 702 and 448 h ng–1 mL–1for nicotine, varenicline, cytisine and dianicline respectively. However, CNS exposures were significantly higher for nicotine and varenicline than for dianicline and cytisine (Figure 2, Table 1). CSF exposures of nicotine and varenicline are two- to threefold lower than their corresponding unbound brain exposures, suggesting either overestimation of unbound brain exposures or active transport of both compounds into the brain or underestimation of CSF concentrations.
In vivo rat brain ECF disposition study
The direct measurement of free drug exposures in the extracellular space by ultra-slow-flow microdialysis sampling revealed that ECF exposures of the test compounds are approximately two- to fivefold lower than their unbound brain concentrations. ECF concentrations of nicotine and varenicline are thus practically identical to their unbound plasma and CSF exposures, indicating that entry of varenicline and nicotine into the brain is likely to be mediated primarily via passive diffusion. In contrast, unbound brain exposures, as well as CSF and ECF exposures of dianicline and cytisine, are several fold lower than their unbound plasma exposures, while in addition, central exposures were 4- to 19-fold lower for dianicline and cytisine than for nicotine and varenicline.
In vivo brain disposition studies in P-gp-deficient mice
Because these data could indicate either reduced brain entry or active brain efflux of cytisine and dianicline, we examined whether the two compounds are substrates for the P-gp transporter in WT and P-gp knock-out (KO) mice. The data (Supporting Information Table S2) show that CNS penetration of cytisine, with a KO-to-WT total brain-to-total plasma (KOB/P/WTB/P) ratio of 0.9, is not influenced by mdr1 P-gp, and that for dianicline, with a KOB/P/WTB/P ratio of 2.2, P-gp does not impair CNS penetration.
These data are consistent with in vitro data that predict a lack of susceptibility to human P-gp of the three compounds (Supporting Information Table S3). Other molecular properties that can affect brain penetration, such as ELogD7.4, polar surface area, membrane permeability, multidrug resistance protein (MDR1) and breast cancer resistance protein (BCRP) transporter susceptibility, do not significantly differ between the three partial agonists (Supporting Information Table S3).
Pharmacokinetics of dianicline and cytisine in rats
For the prediction of unbound brain levels of cytisine and dianicline, their human plasma levels were estimated by allometric scaling from rat pharmacokinetic parameters obtained in an i.v. and p.o. pharmacokinetic study of dianicline and cytisine in rats following a 1 mg kg–1 dose (Table 2).
Table 2.
Rat pharmacokinetic parameters (mean ± SD) for cytisine and dianicline following a 1 mg kg–1 i.v. and p.o. dose
| Route(1 mg·kg–1) | CL(mL·min–1·kg–1) | Vd(L·kg−1) | t½(h) | Cmax (ng·mL–1) | Tmax(h) | AUC0–∞ (h·ng·mL−1) | F | |
|---|---|---|---|---|---|---|---|---|
| Cytisine | i.v. | 35.0 ± 7.5 | 2.7 ± 1.2 | 1.5 ± 0.1 | 517 ± 125 | |||
| p.o. | 38.3 ± 8.3 | 4.9 ± 1.4 | 146 ± 26 | 0.63 ± 0.25 | 483 ± 94 | 0.94 | ||
| Dianicline | i.v. | 66.5 ± 14.2 | 3.8 ± 1.7 | 0.95 ± 0.1 | 233 ± 27 | |||
| p.o. | 66.1 ± 27 | 7.2 ± 1.1 | 126 ± 16 | 0.5 ± 0 | 303 ± 88 | 1.30 |
Vd, volume of distribution; t½, half-life; Cmax, maximum observed concentration; Tmax, time when maximum concentration is observed; AUC0–∞, area under the curve from t= 0 to end of dosing interval; F, oral bioavailability.
Cytisine showed a moderate plasma clearance (CL∼35 mL min–1 kg–1), whereas the CL of dianicline approximates rat hepatic blood flow (CL∼66 mL min–1 kg–1). Both compounds showed moderate volumes of distribution (Vd) with values ranging from 2.7 to 3.8 L kg–1, and short half-lives of 1.5 and 0.95 h respectively. Following oral administration, both compounds were rapidly and fully absorbed into the systemic circulation as indicated by the short time to maximum plasma concentration (Tmax) (0.63 and 0.5 h) and complete oral bioavailability.
Prediction of human steady-state brain concentrations after recommended doses
The predicted human free Css,avg of cytisine after 1.5 mg four times a day (QID) (Etter, 2006) and of dianicline after 40 mg twice a day (BID) (http://clinicaltrials.gov/ct2/results?term=dianicline) was calculated from estimated human pharmacokinetic parameters (Table 2) and measured human plasma protein binding (Table 1), to be 36 and 216 nM, respectively (Table 3). The human free Css,avg of varenicline following oral administration of the recommended 1 mg BID dose has been reported to be approximately 33 nM (Burstein et al., 2006; Faessel et al., 2006a,b;). Subsequently, an upper and lower range of predicted unbound exposure in human brain was calculated for each compound by multiplying the human free plasma Css,avg with the highest and lowest unbound B/P determined in rats with the methods described above and shown in Table 1, that is, unbound brain-to-unbound plasma ratio (Bu/Pu), CSF-to-unbound plasma ratio (CSF/Pu) and ECF-to-unbound plasma ratio (ECF/Pu). In this way, the unbound Css,avg in human brain after therapeutic doses was predicted to range from 32 to 131 nM for varenicline, from 2 to 10 nM for cytisine and from 37 to 84 nM for dianicline (Table 3).
Table 3.
Measured human pharmacokinetic data for varenicline and predicted human pharmacokinetic data for cytisine and dianicline, after recommended therapeutic doses of each compound
| Compounda | Dose(mg) | CL(mL·min–1·kg–1) | Vd(L·kg–1) | t½(h) | fu,PL | F | Css,avg(nM) | [Bu]predicted(nM) |
|---|---|---|---|---|---|---|---|---|
| Varenicline | 2 × 1 | 2.3b | nd | 31.5 | 0.80 | nd | 33 | 32–131 |
| Cytisine | 4 × 1.5 | 5.2 | 1.6 | 3.6 | 0.64 | 1 | 36 | 2–10 |
| Dianicline | 2 × 40 | 14.1 | 3.3 | 2.7 | 0.82 | 1 | 214 | 37–84 |
Varenicline data from Faessel et al. (2006b); cytisine and dianicline data were predicted by allometric scaling of rat pharmacokinetic data (Supporting Information Table S3).
Calculated from dose/AUC0–τ (area under the curve from t= 0 to end of dosing interval) for 70 kg body weight.
nd, not determined; Vd, volume of distribution; t½, half-life; fu,PL, unbound plasma fraction; F, oral bioavailability; Css,avg, average unbound concentration in human plasma at steady state; [Bu], unbound brain concentration.
Pharmacodynamic data
In vitro binding affinities of the compounds were measured at several nAChR subtypes to assess their binding potency and selectivity. Functional studies determined concentration-dependent activation and desensitization profiles at the human α4β2 nAChR to provide functional potencies and functional efficacies. These in vitro experiments were carried out with a mixed population of α4β2 nAChRs not intentionally enriched for any particular stoichiometry of α and β subunits. Effects on dopamine turn-over in the rat mesolimbic system were determined to assess the functional activities and potencies of the compounds in vivo.
Receptor binding affinities
In vitro receptor binding data (Table 4) show that the test compounds have some selectivity for α4β2 nAChRs, and that varenicline binds to the α4β2 subtype with higher receptor affinity (Ki= 0.4 nM) than the other compounds. It has 15-fold higher affinity than nicotine (Ki= 6.1 nM), a requirement for effectively competing with nicotine for the α4β2 nAChR. Although varenicline has significantly lower affinity for α7 (Ki= 125 nM) than for α4β2 nAChRs, it binds to this nAChR subtype with at least 20-fold higher affinity than the other compounds (Ki values > 2.1 µM). Cytisine is the only compound showing moderate affinity for the muscle α1-containing nAChRs (Ki= 430 nM) to which nicotine binds with low affinity (Ki= 2 µM), while varenicline and dianicline do not bind to this subtype (Ki values > 8 µM).
Table 4.
In vitro receptor binding affinities of test compounds and radioligands used for binding assays at different nAChR subtypes
| Ki (means ± SEM, nM) | ||||
|---|---|---|---|---|
| nAChR Subtype radioligand | Nicotine | Varenicline | Cytisine | Dianicline |
| α4β2 ([3H]-epibatidine) | 16.1 ± 4.9 | 0.4 ± 0.1 | 2.0 ± 0.2 | 105 ± 14 |
| α7 ([3H]-α bungarotoxin) | 2110 ± 852 | 125 ± 18 | 5890 ± 1250 | >12 500 |
| α3β4 ([3H[-epibatidine) | 520 ± 120 | 86 ± 16 | 480 ± 63 | 5130 ± 180 |
| α6/α4β4 ([3H[-epibatidine) | 270 ± 75 | 110 ± 13 | 329 ± 33 | 2320 ± 875 |
| α1β1γδ ([3H]-α bungarotoxin) | 2090 ± 620 | 8200 ± 1530 | 492 ± 11 | >12 500 |
Data represent mean Ki values ± SEM in nM.
Functional efficacies and potencies at α4β2 nAChRs
To correlate the predicted human free brain concentrations after recommended doses to drug concentrations required to desensitize and activate α4β2 nAChRs, we performed voltage clamp analyses of the functional interaction of each compound at the human α4β2 nAChR expressed in Xenopus oocytes (Table 5, Figure 3). Brief applications of nicotine or partial agonists cause activation of the receptors, whereas sustained exposures desensitize the receptor. Examples of concentration-dependent responses evoked by the partial agonists and of concentration-dependent desensitization by the compounds are illustrated in Figure 4. Consistent with data from earlier studies (Briggs and McKenna, 1998; Hogg and Bertrand, 2005; Picciotto et al., 2008), receptor desensitization was observed at lower concentrations than those needed for activation. The EC50 values for high-affinity activation of α4β2 nAChRs ranged from 0.06 to 18 µM, while the IC50 values for high-affinity desensitization were 0.05–2.8 nM (Table 5, Figure 3).
Table 5.
Agonist efficacies and functional potencies of nicotine, varenicline, cytisine and dianicline at human α4β2 nAChRs expressed in oocytes
| Compound | High affinity | Low affinity | Efficacy versus ACh | |||
|---|---|---|---|---|---|---|
| Activation | a | EC50µM | nH | EC50µM | nH | % |
| Nicotine | 0.25 | 0.06 ± 0 | 1 ± 0.0 | 4.6 ± 1 | 1.4 ± 0.03 | 100 ± 1 |
| Varenicline | 1.0 | 1.4 ± 0.1 | 1.3 ± 0.01 | 22 ± 2.5 | ||
| Cytisine | 1.0 | 2.0 ± 0.1 | 1.2 ± 0.02 | 6.5 ± 0.2 | ||
| Dianicline | 1.0 | 18.4 ± 1.2 | 1.24 ± 0.04 | 8.0 ± 0.3 | ||
| Desensitization | a | IC50 nM | nH | IC50 nM | nH | |
| Nicotine | 0.25 ± 0.01 | 2.8 ± 1.1 | 1.5 ± 0.45 | 167 ± 63 | 1.3 ± 0.05 | |
| Varenicline | 0.48 ±.0.04 | 0.07 ± 0.01 | 1.3 ± 0.01 | 50 ± 9 | 1 | |
| Cytisine | 0.53 ± 0.07 | 0.05 ± 0.01 | 1.3 ± 0.10 | 28 ± 6 | 1 | |
| Dianicline | 0.33 ± 0.05 | 2.0 ± 0.1 | 1.3 ± 0.08 | 3000 ± 860 | 1 | |
Parameters are obtained from best fits of the data using a dual empirical Hill equation (solid lines in Figure 3).
a, fraction of high-affinity component (Hill equation); %, relative agonist efficacy versus ACh or versus nicotine; EC50, concentration in µM for 50% activation; IC50, concentration in nM for 50% inhibition of ACh- or nicotine-evoked currents; nH, Hill coefficient.
Figure 3.
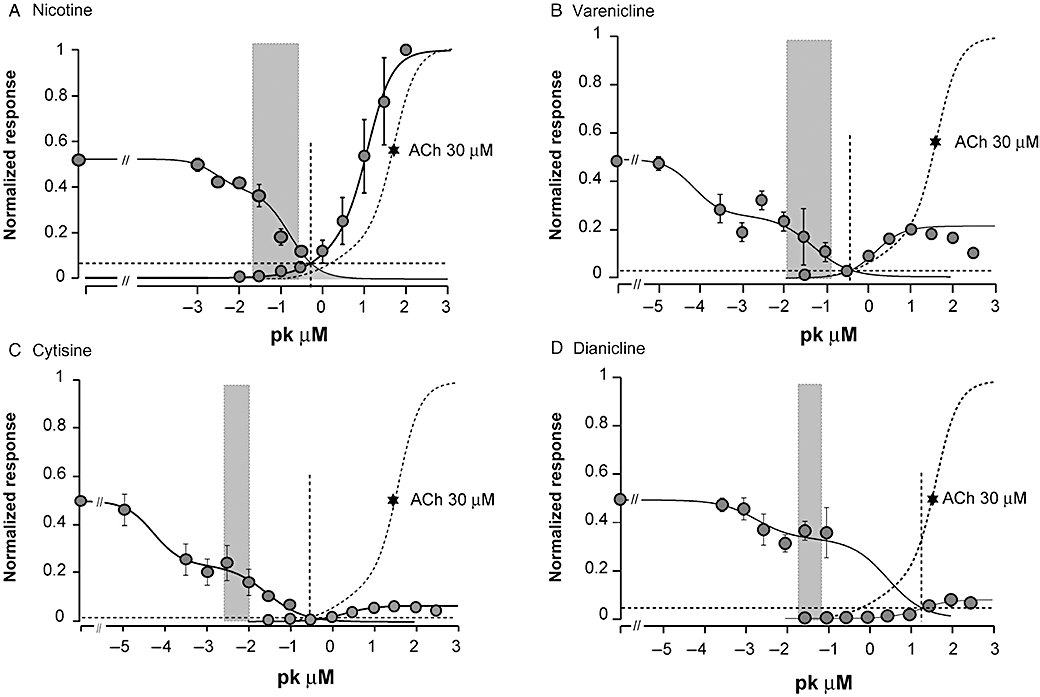
Plots of concentration activation and concentration inhibition curves obtained for nicotine, varenicline, cytisine and dianicline at the human α4β2 nAChR expressed in Xenopus oocytes measured by voltage clamp. The activation responses are normalized to the maximal response to 100 µM ACh (=1); inhibition responses are normalized to the half maximal response (=0.5) obtained with 30 µM ACh. Continuous lines through the data points are the best fits obtained for each compound (parameters in Table 5). The dotted vertical lines indicate the midpoint concentration of the area defined by the overlap of activation and concentration inhibition curves. The grey bars represent the predicted unbound brain concentration of nicotine after smoking and of each partial agonist after recommended therapeutic doses from Table 3. (A) Nicotine, (B) varenicline, (C) cytisine and (D) dianicline.
Figure 4.
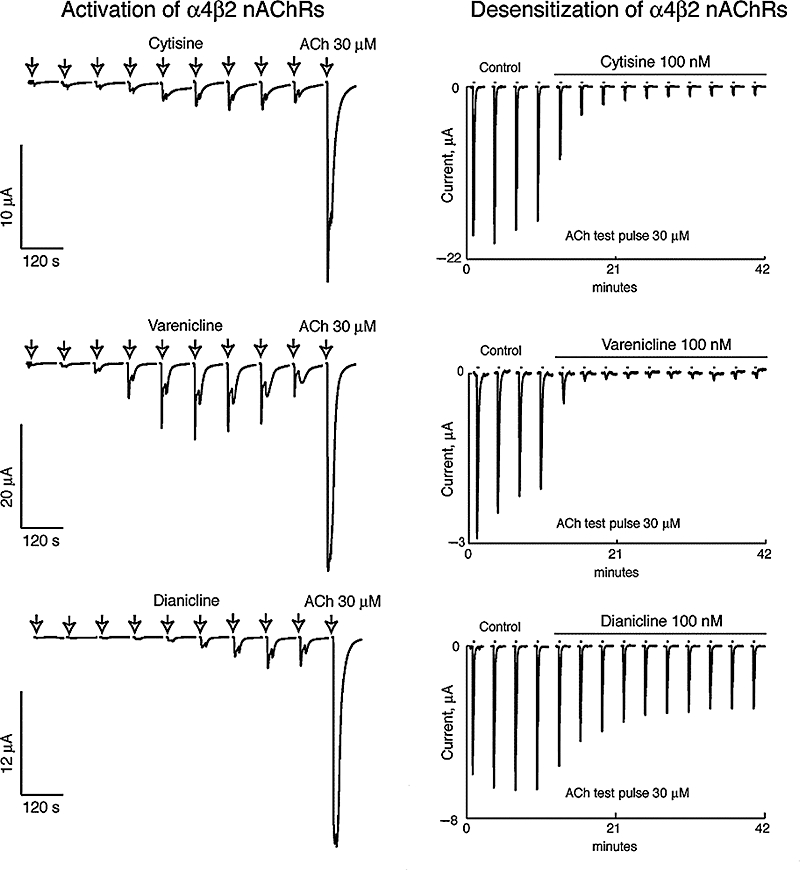
Activation and desensitization experiments at human α4β2 nAChRs expressed in Xenopus oocytes. Typical concentration-dependent responses evoked by cytisine, varenicline and dianicline are illustrated in the left panels. Cells were exposed once every 2 min for 10 s to increasing concentrations of partial agonist (0.03, 0.1, 0.3, 1, 3, 10, 30, 100 and 300 µM) followed by a pulse of ACh (30 µM) to determine the maximal response. Typical concentration-dependent desensitization data obtained for cytisine, varenicline and dianicline are illustrated in the right panels. To evaluate the desensitization effects caused by sustained agonist exposure, a protocol of repetitive challenges with a fixed ACh pulse (30 µM, 10 s, applied every 2 min) was used. To assess stability of the recordings, cells were first challenged with ACh (30 µM, 10 s) four times in the control medium and then with the same ACh test pulse in the presence of a fixed concentration of agonist (100 nM).
The concentration–activation curves show that agonist efficacies relative to the effect of 100 µM ACh are 22 ± 2.5% for varenicline, 6.5 ± 0.2% for cytisine and 8.0 ± 0.3% for dianicline. Parallel studies in HEK293 cells expressing human α4β2 nAChRs, using calcium flux measurements with a fluorimetric imaging plate reader (FLIPR; data not shown), yielded comparable results with higher functional efficacies relative to the response evoked by 100 µM nicotine of 48 ± 1.8% for varenicline, 46 ± 5% for cytisine and 34 ± 5.6% for dianicline. Table 5 shows desensitization–activation potencies and efficacies, as well as Hill parameters obtained from voltage clamp data in Figure 3, using the sum of two isotherms for the curve fitting (Buisson and Bertrand, 2001). Overall, the in vitro binding and functional data are in good agreement with binding potencies and functional efficacies relative to either nicotine or ACh that have been reported for these compounds previously (Cohen et al., 2003; Coe et al., 2005; Mihalak et al., 2006; Rollema et al., 2007b; Smith et al., 2007).
The overlap of the concentration-dependent activation and desensitization curves defines a concentration range for each compound thought to represent a pharmacologically active range where the receptor can be partially activated without complete desensitization (Hogg and Bertrand, 2005). This concentration range between onset of activation and complete desensitization is approximately 10–100 µM for dianicline, and 0.01–10 µM for the other three compounds (Figure 3). The predicted unbound human brain concentrations (Table 3) are compared with this pharmacological range for each compound (grey bars in Figure 3).
Effect on dopamine turn-over in rat nucleus accumbens
In vivo activities of the three compounds were assessed by measuring effects on dopamine turn-over, compared with the maximal effect of nicotine. Each compound, in a dose-dependent manner, increased dopamine turn-over in rat nucleus accumbens. The highest test dose of each partial agonist produced increases to less than 140% of controls, compared with the maximal nicotine effect of 180% of controls after 1 mg kg–1 s.c. nicotine (Figure 5). Higher doses of the partial agonists were either poorly tolerated and not further assessed, or caused lower turn-over increases than after the previous test dose, resulting in an inverted U-shaped dose–response curve, consistent with previous findings (Rollema et al., 2007b). Relative potencies of the three partial agonists were estimated by comparing oral doses required to produce a 20% increase in dopamine turn-over above control levels (‘ED20’). These data revealed that varenicline (ED20∼0.12 mg kg–1) is about 20-fold more potent in increasing dopamine turn-over than cytisine (ED20∼2.5 mg kg–1), and at least 100-fold more potent than dianicline (ED20∼20 mg kg–1). From the dose–response curves, we also estimated the unbound rat brain peak concentrations associated with a dose that significantly increased dopamine turn-over. These doses and unbound concentrations were for nicotine 1 mg kg–1 s.c. and ∼4 µM, for varenicline 1 mg kg–1 p.o. and ∼0.4 µM, for cytisine 10 mg kg–1 p.o. and ∼0.7 µM and for dianicline 20 mg kg–1 p.o. and ∼3.2 µM.
Figure 5.
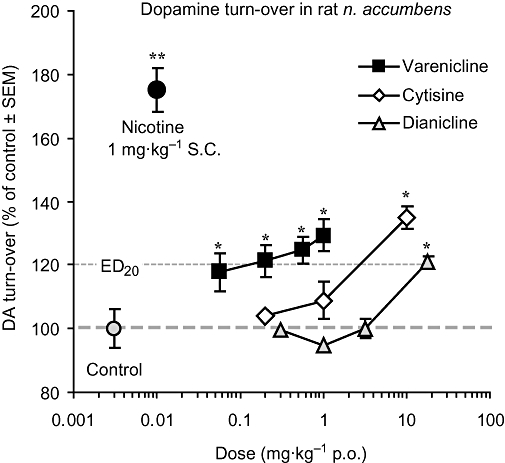
Effects of test compounds on dopamine (DA) turn-over in rat nucleus accumbens. Dose–response curves for the effects of orally administered varenicline, cytisine and dianicline, compared with the effect of 1 mg kg–1 s.c. nicotine and controls. Data are expressed as % of controls ± SEM. *P < 0.05, **P < 0.01 compound versus controls.
Discussion and conclusions
Because of their inherent activation and desensitization properties, α4β2 nAChR partial agonists are believed to relieve craving during abstinence and to reduce nicotine reinforcement during smoking lapses, resulting in improved efficacy as aids to smoking cessation compared with other treatments. This hypothesis was borne out from the observation of higher abstinence rates in smokers treated with varenicline, a partial agonist approved as a smoking cessation aid (Chantix, Champix), than with any other treatment (Cahill et al., 2008; Nides et al., 2008). Although no head-to-head comparisons have been made, the available data from smoking cessation trials with two other α4β2 nAChR partial agonists, cytisine (Tabex) and dianicline (SSR591813), show lower abstinence rates (Etter, 2006; Fagerström and Balfour, 2006; Cahill et al., 2008) than for varenicline, raising questions about optimal requirements for efficacy. As with other centrally acting drugs, the clinical outcome of treatment with partial agonists is determined both by their unbound human brain concentrations and their functional potencies at the target α4β2 nAChR, and our aim was to evaluate whether key pharmacokinetic and pharmacodynamic properties of the three partial agonists would have predicted differences in their clinical efficacies.
In vivo pharmacokinetic studies in rat demonstrated that oral administration of varenicline, cytisine or dianicline resulted in comparable unbound plasma exposures, but there are important differences between the unbound brain concentrations of the agents. Varenicline is the only compound for which human plasma levels after recommended therapeutic doses are available (Faessel et al., 2006a,b;). For the prediction of unbound brain levels of cytisine and dianicline, it was therefore necessary to estimate their human plasma levels by allometric scaling. Rat single species allometry predicted that human unbound Css,avg after 1.5 mg QID cytisine was in the same range as reported values for unbound Css,avg after 1 mg BID varenicline, while 40 mg BID dianicline was estimated to result in a proportionally higher unbound Css,avg (Table 2). However, in brain, human steady-state unbound concentrations (Bu) after recommended doses of varenicline are predicted to be much higher than those of cytisine due to the reduced brain entry of cytisine (Table 2). The poor brain penetration of cytisine has previously been attributed to its high hydrophilicity, that is, a lower logP value than nicotine and the other agonists (Reavill et al., 1990; Tutka and Zatoński, 2005; Supporting Information Table S3). It is, however, unlikely that hydrophilicity is the sole or major reason for the low brain exposure of cytisine. At physiological pH, the partial agonists have approximately the same distribution coefficient (ElogD7.4), because at pH 7.4 much less cytisine, which has an acid dissociation constant (pKa) of 7.8, is in the protonated form than varenicline and dianicline (pKa 9.3–9.8; Supporting Information Table S3). Moreover, there are very small differences in other molecular properties of the partial agonists that impact permeability and transport, and none of the compounds was found to be a substrate for P-gp or BCRP efflux transporters (Supporting Information Tables S2 and S3). We speculate that the low brain concentrations of cytisine could be due to an active, non-P-gp, non-BCRP brain efflux mechanism. Dianicline has comparable permeability and hydrophilicity as varenicline, and is not a substrate for P-gp or BCRP efflux transporters (Supporting Information Tables S2 and S3), but has limited brain penetration. Recommended doses of 40 mg BID dianicline are predicted to result in relatively low unbound human brain concentrations, comparable to those of varenicline after 1 mg BID (Table 3).
To correlate the predicted human Bu of the partial agonists and nicotine with their in vitro properties, binding and functional studies were conducted at human nAChRs. Results of these experiments confirmed that each compound binds selectively to α4β2 nAChRs, and that varenicline, cytisine and dianicline act as partial agonists (Figure 3; Tables 4 and 5). However, there are substantial quantitative differences in binding affinities and functional potencies of dianicline versus the other two partial agonists, and much higher dianicline concentrations are required for an interaction with α4β2 nAChRs. This pharmacologically relevant range between the activation and desensitization curves is 10–100 µM for dianicline, compared to 0.01–10 µM for nicotine, varenicline and cytisine (Figure 3). Plots of predicted Bu values of each compound over their desensitization and activation concentration–response curves at the human α4β2 nAChR reveal whether drug concentrations are sufficient for functional receptor interactions. These data (grey bars in Figure 3) show that unbound nicotine concentrations from tobacco smoking (50–300 nM; Rose et al., 1999) and varenicline concentrations after therapeutic doses (30–130 nM) will markedly, but not fully, desensitize the receptors. Thus, steady-state concentrations of varenicline are expected to result in low, but sustained activation of the α4β2 receptor. In contrast, the low Bu for 1.5 mg QID cytisine (2–10 nM) is predicted to cause significant desensitization, but not α4β2 nAChR activation. Finally, treatment with 40 mg BID dianicline is predicted to result in unbound brain concentrations of 40–80 nM, which will cause only minimal desensitization of α4β2 nAChRs. This is likely insufficient to significantly reduce craving when quitting, and will only marginally reduce nicotine-evoked reinforcing effects.
The translation of these data to the clinic is based on the hypothesis that differences in functional interactions between the partial agonist and the α4β2 nAChR will result in varying degrees of relief of craving and attenuation of nicotine reinforcement, and hence in clinical efficacy. Fluctuating nicotine concentrations in smokers, with a fast rising phase in seconds and a slow decline over hours, are expected to produce receptor activation followed by desensitization that likely contributes to the wide range of effects of nicotine (Picciotto et al., 2008). In contrast, drug concentrations with a slow onset and long half-life will mainly cause receptor desensitization and limited activation of α4β2 nAChRs, provided steady-state drug concentrations are near the area of overlap between the desensitization and activation profiles. The present data could therefore explain the higher abstinence rates observed with varenicline in smoking cessation trials than with cytisine and dianicline. Compared with placebo, the OR for varenicline for end-of-treatment continuous abstinence during weeks 9–12 of a 12 week treatment period is 3.7 [95% confidence interval (CI): 2.86–4.68; Nides et al., 2008]. Cytisine was reported to have an OR versus placebo of 1.9 (95% CI: 1.2–3.1), based on a meta-analysis of point prevalence data of self-reported abstinence at weeks 3–8 of an 8 week treatment (Etter, 2006). At present, only preliminary phase 2 data are available for dianicline with end-of-treatment quit rates for 40 mg BID dianicline of 16 versus 8% for placebo (Fagerström and Balfour, 2006). Because in addition, further development of dianicline was halted in 2008 (Sanofi-Aventis press release, 2008), it is reasonable to assume that the OR of dianicline is comparable to or lower than that of cytisine. The abstinence rates of varenicline are also higher than those observed with various nicotine replacement therapies, where nicotine acts as a full agonist at the nAChR. A comparative meta-analysis of abstinence rates versus placebo at 6 months post-quit for available smoking cessation medications showed an estimated OR (95% CI) of 3.1 (2.5–3.8) for varenicline, while those for individual nicotine replacement therapies were 2.3 (1.7–3.0) or lower (Fiore et al., 2008).
The notion that the capacity to interact with α4β2 nAChRs significantly contributes to differences in pharmacological activity is supported by the relative in vivo potencies to increase rat mesolimbic dopamine turn-over. After oral administration, varenicline was found to be 20- and 100-fold more potent than cytisine and dianicline, respectively, in stimulating dopamine turn-over in rat nucleus accumbens (Figure 5). Interestingly, each compound produced a significantly increased dopamine turn-over in nucleus accumbens after doses at which unbound brain concentrations fall at the centre between their α4β2 nAChR desensitization and activation curves. This is consistent with nAChR-mediated dopamine release being predominantly mediated via activation and desensitization of α4β2-containing nAChRs located on dopaminergic and GABAergic neurons (Picciotto et al., 1998; 2008; Mansvelder and McGehee, 2002; Mansvelder et al., 2002; Laviolette and Van der Kooy, 2004; Maskos et al., 2005).
These data illustrate that correlating unbound brain concentrations with in vitro functional potencies can translate to clinical efficacy of partial agonists based on studies with α4β2 nAChRs. In addition to β2-containing subtypes, the α7 nAChR has been implicated in the effects of nicotine (Mansvelder and McGehee, 2002; Mansvelder et al., 2002). As varenicline has moderate affinity for α7 nAChRs (Ki= 125 nM; Table 4), we are also investigating the interactions of varenicline with this receptor subtype. Functional data confirm that varenicline acts as a full α7 nAChR agonist (107% v ACh) (Mihalak et al., 2006), but at concentrations that are significantly higher than estimated human brain concentrations. At therapeutic doses, varenicline is predicted to minimally desensitize α7 nAChRs and not causing detectable activation of this receptor. This suggests that α7 nAChRs plays a minor role in the clinical effects of varenicline as a smoking cessation aid. Additional nAChR subtypes, including α3*, α5* and α6* nAChRs, have been proposed to contribute to mesolimbic activity and nicotine dependence (Drenan et al., 2008; Exley et al., 2008; Pons et al., 2008; Grady et al., 2009; Livingstone et al., 2009; Ramiro et al., 2009). Further work is therefore needed for a more thorough evaluation of the effects of varenicline, but the good correlation between the present pre-clinical and clinical data argues in favour of a dominant role of α4β2-containing nAChRs in mediating effects of varenicline.
Taken together, translation of pre-clinical pharmacokinetic and pharmacodynamic data to the clinic suggests that an nAChR partial agonist will be most efficacious as an aid for smoking cessation if four criteria are simultaneously met. The compound should exhibit potent binding affinity to the α4β2 nAChR, should reach sufficiently high unbound brain concentrations to allow desensitization and some activation of α4β2 nAChRs and should prevent inhaled nicotine from binding at nAChRs to block its reinforcing effect. Working together, these properties result in a significant increase in efficacy over other treatments that do not meet these criteria, either because of insufficient CNS drug exposure at the target site or because of inadequate functional potency.
In conclusion, this retrospective analysis for the first time identified predictors of clinical efficacy of three α4β2 nAChR partial agonists as smoking cessation aids, providing a basis for further studies aimed at designing more efficacious compounds for the treatment of nicotine dependence. The study also illustrates that in vitro properties of centrally acting drugs have limited predictive value for clinical outcome without taking into account pharmacokinetic data to estimate human brain exposures.
Acknowledgments
Electrophysiology studies by S.B. and D.B. were supported by funding from Pfizer Inc. and by the Swiss National Science Foundation to D.B. We greatly appreciate the contributions and helpful discussions with Marina Shalaeva, Gus Campos, Christopher Shaffer, Hélène Faessel, Bo Feng and Charles Potter (Pfizer Global Research and Development).
Glossary
Abbreviations:
- aCSF
artificial CSF
- AUC
area under the curve
- BCRP
breast cancer resistance protein
- B/P
brain-to-plasma ratio
- Bu
unbound brain concentrations
- CL
plasma clearance
- Css,avg
steady-state plasma concentration
- DOPAC
3,4-dihydroxyphenylacetic acid
- ECF
extracellular fluid
- FLIPR
fluorimetric imaging plate reader
- fu
unbound fraction
- HPLC
high-performance liquid chromatography
- HVA
homovanillic acid
- Ka
acid dissociation constant
- Ki
receptor affinity
- MDR1
multidrug resistance protein
- P-gp
P-glycoprotein
- Tmax
time to maximum plasma concentration
- Vd
volume of distribution
Conflicts of interests
All the authors, with the exception of the last four authors M.dV, T.C., S.B. and D.B., are employed by Pfizer Inc., the manufacturer of varenicline (Chantix). Editorial support for the development of this paper was provided by Penny Gorringe of UBC Scientific Solutions, and was funded by Pfizer, Inc.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1 Summary of assay conditions.
Table S2 Total plasma and total brain concentrations (mean ± SD) at 0.5 and 2 h after 3 mg kg–1 p.o. cytisine or dianicline in FVB/N (WT) and mdr1a/1b (–/–) (P-gp-deficient) mice.
Table S3 Molecular properties of nicotine and α4β2 nAChR partial agonists.
Appendix S1 Supplementary materials and methods.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL. Pharmacology of nicotine: addiction, smoking-induced disease, and therapeutics. Annu Rev Pharmacol Toxicol. 2009;49:57–71. doi: 10.1146/annurev.pharmtox.48.113006.094742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs CA, McKenna DG. Activation and inhibition of the human α7 nicotinic acetylcholine receptor by agonists. Neuropharmacology. 1998;37:1095–1102. doi: 10.1016/s0028-3908(98)00110-5. [DOI] [PubMed] [Google Scholar]
- Buisson B, Bertrand D. Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J Neurosci. 2001;21:1819–1829. doi: 10.1523/JNEUROSCI.21-06-01819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein AH, Fullerton T, Clark DJ, Faessel HM. Pharmacokinetics, safety, and tolerability after single and multiple oral doses of varenicline in elderly smokers. J Clin Pharmacol. 2006;46:1234–1240. doi: 10.1177/0091270006291837. [DOI] [PubMed] [Google Scholar]
- Cahill K, Stead LF, Lancaster T. Nicotine receptor partial agonists for smoking cessation. Cochrane Database of Systematic Reviews 2008, Issue 3. Art. No.: CD006103. DOI: 10.1002/14651858.CD006103.pub3. [DOI] [PubMed]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, et al. An α4β2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- Cohen C, Bergis OE, Galli F, Lochead AW, Jegham S, Biton B, et al. SSR591813, a novel selective and partial α4β2 nicotinic receptor agonist with potential as an aid to smoking cessation. J Pharm Exp Therap. 2003;306:407–420. doi: 10.1124/jpet.103.049262. [DOI] [PubMed] [Google Scholar]
- Cremers TIFH, DeVries MG, Huinink KD, van Loon JP, van der Hart M, Westerink BHC, et al. Quantitative microdialysis using modified ultraslow microdialysis; direct rapid and reliable determination of free brain concentrations with the MetaQuant technique. J Neurosci Methods. 2009;178(2):249–254. doi: 10.1016/j.jneumeth.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, et al. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha6* nicotinic acetylcholine receptors. Neuron. 2008;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter JF. Cytisine for smoking cessation. A literature review and meta-analysis. Arch Intern Med. 2006;166:1553–1559. doi: 10.1001/archinte.166.15.1553. [DOI] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ. α6 Containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology. 2008;33:2158–2166. doi: 10.1038/sj.npp.1301617. [DOI] [PubMed] [Google Scholar]
- Faessel HM, Smith BJ, Gibbs MA, Gobey JS, Clark DJ, Burstein AH. Single-dose pharmacokinetics of varenicline, a selective nicotinic receptor partial agonist, in healthy smokers and nonsmokers. J Clin Pharmacol. 2006a;46:991–998. doi: 10.1177/0091270006290669. [DOI] [PubMed] [Google Scholar]
- Faessel HM, Gibbs MA, Clark DJ, Rohrbacher K, Stolar M, Burstein AH. Multiple-dose pharmacokinetics of the selective nicotinic receptor partial agonist, varenicline, in healthy smokers. J Clin Pharmacol. 2006b;46:1439–1448. doi: 10.1177/0091270006292624. [DOI] [PubMed] [Google Scholar]
- Fagerström K, Balfour DJK. Neuropharmacology and potential efficacy of new treatments for tobacco dependence. Expert Opin Investig Drugs. 2006;15:107–116. doi: 10.1517/13543784.15.2.107. [DOI] [PubMed] [Google Scholar]
- Fiore MD, Jaén CS, Baker TB, Bailey WC, Benowitz NL, Curry SJ, et al. Treating Tobacco Use and Dependence: 2008 Update. Clinical Practice Guideline. Rockville, MD: U.S. Department of Health and Human Services. Public Health Service; 2008. [Google Scholar]
- Galli F, Samir J, Alistair L, Axelle S. Preparation of pyridopyranoazepines as α4β2 nicotinic receptor ligands. 2000. WO 2000053608 A1, World Intellectual Property Organization.
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, et al. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the α3β4* and α3β3β4* subtypes mediate acetylcholine release. J Neurosci. 2009;229:2272–2228. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoda J-C, Gu W, Friedli M, Phillips HA, Bertrand S, Antonarakis SE, et al. Human nocturnal frontal lobe epilepsy: pharmocogenomic profiles of pathogenic nicotinic acetylcholine receptor β-subunit mutations outside the ion channel pore. Mol Pharmacol. 2008;74:379–391. doi: 10.1124/mol.107.044545. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Bertrand D. Nicotinic acetylcholine receptors as drug targets. Curr Drug Targets CNS Neurol Disord. 2005;3:123–130. doi: 10.2174/1568007043482507. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Bertrand D. Partial agonists as therapeutic agents at neuronal nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;73:459–564. doi: 10.1016/j.bcp.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Hosea NA, Collard WT, Cole S, Maurer TS, Fang RX, Jones H, et al. Prediction of human pharmacokinetics from preclinical information: comparative accuracy of quantitative prediction approaches. J Clin Pharmacol. 2009;49:513–533. doi: 10.1177/0091270009333209. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos. 2002;23:327–338. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Van der Kooy KD. The neurobiology of nicotine addiction: bridging the gap from molecules to behaviour. Nat Rev Neurosci. 2004;5:55–65. doi: 10.1038/nrn1298. [DOI] [PubMed] [Google Scholar]
- Livingstone PD, Srinivasan J, Kew JNC, Dawson LA, Gotti C, Moretti M, et al. α7 And non-α7 nicotinic acetylcholine receptors modulate dopamine release in vitro and in vivo in the rat prefrontal cortex. Eur J Neurosci. 2009;29:539–550. doi: 10.1111/j.1460-9568.2009.06613.x. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Marubio LM, Gardier AM, Durier S, David D, Klink R, Arroyo-Jimenez MM, et al. Effects of nicotine in the dopaminergic system of mice lacking the α4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci. 2003;17:1327–1337. doi: 10.1046/j.1460-9568.2003.02564.x. [DOI] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux J-P, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at α4β2 and a full agonist at α7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70:801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- Nides M, Glover ED, Reus VI, Christen AG, Make BJ, Billing CB, et al. Varenicline versus bupropion SR or placebo for smoking cessation: a pooled analysis. Am J Health Behav. 2008;32:664–675. doi: 10.5555/ajhb.2008.32.6.664. [DOI] [PubMed] [Google Scholar]
- Papke RL, Heinemann SF. Partial agonist properties of cytisine on neuronal nicotinic receptors containing the α2 subunit. Mol Pharmacol. 1994;45:142–149. [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the β2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not ‘either/or’: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, et al. Crucial role of α4 and β6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro S, Sturm R, Boulter J, De Biasi M. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci. 2009;29:3014–3018. doi: 10.1523/JNEUROSCI.4934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reavill C, Walther B, Stolerman I, Testa B. Behavioral and pharmacokinetic studies on nicotine, cytisine and lobeline. Neuropharmacology. 1990;29:619–624. doi: 10.1016/0028-3908(90)90022-j. [DOI] [PubMed] [Google Scholar]
- Reed-Hagen AE, Tsuchiya M, Shimada K, Wentland JA, Obach RS. Pharmacokinetics of ezlopitant, a novel non-peptidic neurokinin-1 receptor antagonist in preclinical species and metabolite kinetics of the pharmacologically active metabolites. Biopharm Drug Dispos. 1999;20:429–439. doi: 10.1002/1099-081x(199912)20:9<429::aid-bdd209>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Rollema H, Coe JW, Chambers LK, Hurst RS, Stahl SM, Williams KE. Rationale, pharmacology and clinical efficacy of partial agonists of alpha4beta2 nACh receptors for smoking cessation. Trends Pharmacol Sci. 2007a;28:316–325. doi: 10.1016/j.tips.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RA, Lebel LA, et al. Pharmacological profile of the alpha4beta2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology. 2007b;52:985–994. doi: 10.1016/j.neuropharm.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman EC, Coleman RE. Arterial nicotine kinetics during cigarette smoking and intravenous nicotine administration: implications for addiction. Drug Alcohol Depend. 1999;56:99–107. doi: 10.1016/s0376-8716(99)00025-3. [DOI] [PubMed] [Google Scholar]
- Sanofi-Aventis press release, February 12, 2008, p.14. http://en.sanofi-aventis.com/press/press%20releases/2008/ppc14630.asp.
- Smith JW, Mogg A, Tafi E, Peacey E, Pullar IA, Szekeres P, et al. Ligands selective for α4β2 but not α3β4 or α7 nicotinic receptors generalise to the nicotine discriminative stimulus in the rat. Psychopharmacology. 2007;190:157–170. doi: 10.1007/s00213-006-0596-8. [DOI] [PubMed] [Google Scholar]
- Tutka P, Zatoński W. Cytisine for the treatment of nicotine addiction: from a molecule to therapeutic efficacy. Pharmacol Rep. 2005;58:777–798. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.