

Abstract
Background and purpose:
In the present paper we studied the effect of shikonin on ear oedema induced by 12-O-tetradecanoylphorbol-13-acetate (TPA), and determined the mechanisms through which shikonin might exert its topical anti-inflammatory action.
Experimental approach:
Acute ear oedema was induced in mice by topical application of TPA. The in vitro assays used macrophages RAW 264.7 cells stimulated with lipopolysaccharide. Cyclooxygenase-2, inducible nitric oxide synthase, protein kinase Cα, extracellular signal-regulated protein kinase (ERK), phosphorylated ERK (pERK), c-Jun N-terminal kinase (JNK), pJNK, p38, p-p38, p65, p-p65, inhibitor protein of nuclear factor-κB (NF-κB) (IκBα) and pIκBα were measured by Western blotting, activation and binding of NF-κB to DNA was detected by reporter gene and electrophoretic mobility shift assay, respectively, and NF-κB p65 localization was detected by immunocytochemistry.
Key results:
Shikonin reduced the oedema (inhibitory dose 50 = 1.0 mg per ear), the expression of cyclooxygenase-2 (70%) and of inducible nitric oxide synthase (100%) in vivo. It significantly decreased TPA-induced translocation of protein kinase Cα, the phosphorylation and activation of ERK, the nuclear translocation of NF-κB and the TPA-induced NF-κB-DNA-binding activity in mouse skin. Moreover, in RAW 264.7 cells, shikonin significantly inhibited the binding of NF-κB to DNA in a dose-dependent manner and the nuclear translocation of p65.
Conclusions and implications:
Shikonin exerted its topical anti-inflammatory action by interfering with the degradation of IκBα, thus inhibiting the activation of NF-κB.
Keywords: shikonin, TPA, cyclooxygenase-2, ear mouse, RAW 264.7 macrophages, NF-κB, p65, inducible nitric oxide synthase, MAPK
Introduction
The inflammatory process is a non-specific response against external insults that takes place in vascular tissues and involves a cascade of physiological and immunological reactions in response to harmful stimuli, such as pathogens, damaged cells or irritants (Staniforth et al., 2004). The overexpression and activity of pro-inflammatory enzymes such as inducible nitric oxide synthase and cyclooxygenase-2 is associated with autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, Alzheimer's disease and septic shock (Park et al., 2007; Kim et al., 2008). A growing body of evidence indicates that the principal signalling pathway involved in the initiation and amplification of the inflammatory response is the activation of the nuclear factor-κB (NF-κB) (Handel and Girgis, 2001; Surh et al., 2001; Lappas et al., 2002), a ubiquitous and well-characterised protein responsible for the regulation of the transcription of a large number of genes, particularly those involved in immune, inflammatory and anti-apoptotic responses (Ghosh et al., 1998). In most cell types, dimeric NF-κB is trapped in the cytoplasm of unstimulated cells, existing mainly as a heterodimer made up of subunits of the Rel family p50 and p65. The inhibitor proteins of NF-κB (IκB) retain NF-κB in the cytoplasm by masking its nuclear localization sequence, which is contained in the Rel homology domain (Baeuerle, 1998; Lee et al., 2006). A great number of cellular stimuli, including lipopolysaccharide, tumour necrosis factor-α (TNF-α), reactive oxygen species and ultraviolet light, are able to activate the IκB kinase (IKK) complex, thus triggering site-specific phosphorylation of IκBα at two critical serine residues (Ser32, Ser36) (Didonato et al., 1996; Park et al., 2007). These are then ubiquitinated and subsequently cleaved by the 26S proteosome. The resulting free NF-κB is translocated to the nucleus, where it binds specifically to κB enhancer elements of DNA and induces the transcription of pro-inflammatory mediators (Baeuerle and Baltimore, 1996; Bell et al., 2003). Inappropriate regulation of NF-κB is directly involved in a wide range of human diseases, including a variety of cancers, neurodegenerative diseases, ataxiatelangiectasia, arthritis, asthma, inflammatory bowel disease and numerous other inflammatory conditions (Bremner and Heinrich, 2002; Celec, 2004; Uwe, 2008). The development of strategies for modulating and/or inhibiting NF-κB activity has thus generated a great deal of interest.
The protein kinase C family of enzymes comprises critical components of cellular signal transduction cascades. Various signals that stimulate members of G-protein coupled receptors, tyrosine kinase receptors, or non-receptor tyrosine kinases can, in turn, induce diacylglycerol production, which is the endogenous activator of protein kinase C (Newton, 1995). Because different external agents such as 12-O-tetradecanoylphorbol 13-acetate (TPA) and other phorbol esters, which are not readily metabolised, can mimic the role of diacylglycerol in human cells, the treatment of tissues or cells with these esters can bring about a lasting activation of protein kinase C (Castagna et al., 1982). The isoforms of protein kinase C have been classified into three subfamilies of enzymes on the basis of structural similarities and cofactor requirements. Among these, the protein kinase Cα is present in mouse skin, having a high functional relevance as it is implicated in keratinocyte differentiation, epidermal tumour promotion and cutaneous inflammation (Szaefer et al., 2007). Earlier reports suggest that topical application of TPA results in increased expression of cyclooxygenase-2 that catalyses the rate-limiting step in prostaglandin biosynthesis and inflammation in mouse skin (Afaq et al., 2004). TPA-induced cyclooxygenase-2 expression, in turn, is regulated by transcription factors such as NF-κB and upstream kinases including inhibitory-κB kinase (IκB) and mitogen-activated protein kinases (MAPKs) (Lee et al., 2006). Both protein kinase C and MAPKs are critical components of cellular signal transduction cascades. Accumulating evidence indicates that these kinases are implicated in the up-regulation of cyclooxygenase-2 expression by TPA, through a mechanism dependent on NF-κB activation. Dhawan and Richmond (2002) revealed a close association between the extracellular signal-regulated protein kinase (ERK) activity and phosphorylation and degradation of IκB, which leads to the traslocation of p65 to the nucleus and the activation of the transcription of pro-inflammatory mediators.
The red naphthoquinone pigment shikonin (Figure 1) is a major component of the root of Lithospermum erythrorhyzon Sieb. et Zucc. (Boraginaceae), which is used in traditional Chinese medicine for treating macular eruptions, measles, sore throat, carbuncles and burns (Papageorgiou et al., 1999). This compound has been reported to possess different medicinal properties such as antibacterial, wound healing, anti-inflammatory, antithrombotic and antitumour effects (Wu et al., 2004). Several studies suggest that the anti-inflammatory effects of shikonin and its derivatives can be explained by invoking different mechanisms of action, including the inhibition of leukotriene B4 biosynthesis (Wang et al., 1994), suppression of mast cell degranulation (Wang et al., 1995), inhibition of neutrophil respiratory burst (Kawakami et al., 1996), impaired phosphatidylinositol signalling (Wang and Kuo, 1997), blocking of chemokine binding to the chemokine receptor 1 (Baggiolini, 1998), and inhibition of TPA-induced cyclooxygenase-2 expression (Subbaramaiah et al., 2001). Recently, Cheng et al. (2008) demonstrated that shikonin inhibits inducible nitric oxide synthase expression by down-regulating NF-κB activation in RAW 264.7 cells stimulated with lipopolysaccharide.
Figure 1.
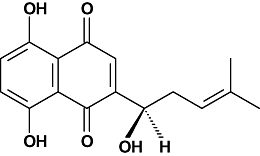
Chemical structure of shikonin.
The aim of this work is to study the effect in vivo of shikonin in TPA-induced acute inflammation, and to determine, with the aid of both functional and molecular procedures, some of the mechanisms through which shikonin exerts its topical anti-inflammatory action on TPA-induced skin inflammation in mice to provide insight into its possible mechanisms of action. This is the first time that the effect in vivo of shikonin on NF-κB activation is reported.
Methods
Animals
All animal care and experimental protocols were approved by the Institutional Ethics Committee of the University of Valencia, Spain. Female Swiss mice weighing 25–30 g (Harlan Interfauna Iberica, Barcelona, Spain) were used for in vivo experiments. All animals were fed a standard diet ad libitum and housed under a 12-h light/dark cycle at 22°C and 60% humidity.
TPA-induced acute ear oedema
Skin inflammation was induced in the right ears of the mice through topical application of TPA (2.5 µg per ear; Sigma-Aldrich, St. Louis, MO, USA) dissolved in 20 µL of acetone (Panreac, Barcelona, Spain). Control animals received the same volume of acetone applied to the right ear. Shikonin (0.3, 0.6, 1.0 and 2.0 mg per ear) was dissolved in 20 µL of acetone and applied topically in conjunction with TPA. Indomethacin (Sigma-Aldrich) was used as a reference drug (0.5 mg per ear). Control animals were given the same volume of vehicle. Animals were killed by cervical dislocation 1, 2 or 4 h after TPA treatment. Right and left ear punches 7 mm in diameter were taken from each mouse. Oedema was expressed as the difference between the weight of the right ear punch and that of the left. The increase in oedema was directly proportional to the degree of inflammation. Tissues were frozen and stored at −80°C until use.
Preparation of cytosolic and nuclear fractions from ear punches
Protein extraction from the skin was performed as described previously by Lai et al. (2007). In brief, tissues were homogenised for 1 min with a polytron tissue homogeniser (Kinematica AG, Lucerne, Switzerland) in 1.5 mL of ice cold buffer A [10 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulphonic acid) (HEPES) pH 7.8, 10 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol, 0.1 mM EDTA and 0.1 mM phenylmethylsulphonyl fluoride]. The homogenates were chilled on ice with gentle shaking for 15 min. The membrane fraction was precipitated by means of centrifugation at 106×g for 5 min. The supernatant containing the cytosolic fraction was stored at −80°C until use. The pellet was resuspended with vortex in 50 µL of buffer A with 10% Nonidet P-40 (Sigma-Aldrich). After centrifugation at 20 800×g for 2 min, the supernatant was discarded and the pellet was resuspended in 200 µL of buffer B (50 mM HEPES pH 7.8, 50 mM KCl, 300 mM NaCl, 1 mM dithiothreitol, 0.1 mM EDTA, 0.1 mM phenylmethylsulphonyl fluoride, and 10% glycerol) and kept on ice for 30 min. After additional centrifugation at 20 800×g for 5 min, the supernatant containing the nuclear fraction was removed and stored at −80°C until use. Cell lysates (30 µg protein) were boiled in sodium dodecyl sulfate sample buffer for 5 min before undergoing electrophoresis (see Western blot assay). The nuclear extracts were made into aliquots and equal amounts of protein (5 µg) were added to the reaction mixture to facilitate the electrophoretic mobility shift assay (EMSA), in accordance with the manufacturer's instructions.
Preparation of cytosolic and membrane fractions to determine protein kinase Cα levels
To measure the expression of protein kinase Cα in mouse skin, cytosolic and membrane fractions were prepared as described by Medeiros et al. (2007). Briefly, tissues were homogenised in ice cold buffer A (10 mM HEPES pH 7.4, 1.5 mM MgCl2, 10 mM KCl, 1 mM phenylmethylsulphonyl fluoride, 1 µg mL−1 leupeptin, 1 µg mL−1 pepstatin A, 1 µg mL−1 aprotinin, 1 mM sodium orthovanadate, 10 mM β-glycerophosphate, 50 mM sodium fluoride and 0.5 mM dithiothreitol). The homogenates were chilled on ice vigorously shaken for 15 min. The membrane fraction was precipitated by centrifugation at 14 000×g for 60 min. The supernatant containing the cytosolic fraction was stored at −80°C until use. The membrane pellet was resuspended in 50 µL of buffer A containing 1% Triton X-100 (Sigma-Aldrich) and incubated under continuous shaking at 4°C for 30 min. The membrane fraction was finally precipitated by centrifugation at 14 000×g and the supernatant as stored at −80°C until use.
Cell cultures
For all experiments in vitro, we used the murine macrophage cell line RAW 264.7 (ECACC, Salisbury, UK). The cells were maintained in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum, penicillin (100 U mL−1) and streptomycin sulfate (100 µg mL−1) in a humidified 5% CO2 atmosphere.
Cell viability assay
The effect of shikonin on cell viability was evaluated with the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay. Thus, murine RAW 264.7 macrophages were exposed to shikonin at a range of concentrations (100–0.1 µM) in a 96-well microplate at various assay times for the different experiments, after which 100 µL per well of a 0.5 mg mL−1 solution of MTT (Sigma-Aldrich) was added.
The resulting solution was incubated at 37°C until blue deposits were visible and then this coloured metabolite was dissolved in dimethyl sulfoxide. Absorbance was measured at 490 nm with a Labsystems Multiskan EX plate reader (Helsinki, Finland). The results were expressed in absolute absorbance readings; a decrease in absorbance indicated a reduction in cell viability.
Preparation of total protein extract from cells
RAW 264.7 macrophages at a density of 1 × 106 cells per well were placed in a 6-well cell culture plate along with 2 mL of culture medium and then incubated for 24 h. The cells were pre-treated with shikonin for 1 h and subsequently stimulated with lipopolysaccharide (1 µg mL−1) for the specified periods. Cell lysates were obtained with lysis buffer [1% Triton X-100, 1% deoxycholic acid, 20 mM NaCl, and 25 mM Tris, pH 7.4 (all from Sigma-Aldrich), and a complete mini EDTA-free protease inhibitor cocktail]. After centrifugation (10 000×g, Eppendorf centrifuge 5810R), proteins were determined in the supernatant by the Bradford method with bovine serum albumin as the standard (Slomiany and Slomiany, 2002).
Transient transfection and NF-κB-dependent reporter gene expression assay
To examine the effect of shikonin on the transcriptional activity of NF-κB, RAW 264.7 cells (5 × 105 cells per well) were placed in 24-well plates and transiently transfected with pNF-κB-Luc expression plasmid (0.8 µg) and the control plasmid TK-Renilla (0.2 µg), both kindly donated by Dr. Lidija Klampfer (Albert Einstein Cancer Center, New York, USA). Transfections were performed with Lipofectamine™ 2000, following the instructions of the manufacturer (Invitrogen, Carlsbad, CA, USA). Twenty-four hours after transfection, the cells were treated with shikonin (1 µM) for 24 h. The luciferase assay was performed with the Dual-Luciferase Assay System, following the instructions of the manufacturer (Promega, Madison, WI, USA). Luciferase activity was normalised to TK-Renilla activity to control for transfection efficiency.
Western Blot analysis for cyclooxygenase-2, inducible nitric oxide synthase, protein kinase Cα, p65, phosphorylated-p65 (p-p65), IκBα, p-IκBα, ERK, phosphorylated ERK (pERK), c-Jun N-terminal kinase (JNK), p-JNK, p38 and p-p38
After extraction, the presence of proteins in the supernatants was determined by the Bradford method with bovine serum albumin as the standard (Slomiany and Slomiany, 2002). Equal amounts of protein (30 µg) were then loaded onto 10% sodium dodecyl sulfate polyacrylamide electrophoresis gel and were transferred onto polyvinylidene difluoride membranes at 125 mA for 90 min. The membranes were then blocked in phosphate-buffered saline-Tween 20 containing 3% w/v defatted milk. For ERK, the membranes were incubated with anti-ERK1 antibody (SC-93) (1/500 dilution); for pERK1/2, the membranes were incubated with anti-pERK1/2 (SC-101760) (1/500); for JNK2, the membranes were incubated with anti-JNK2 (SC-46013) (1/500); for pJNK, the membranes were incubated with anti-pJNK (SC-12882) (1/500); for p38, the membranes were incubated with anti-p38 (SC-7972) (1/500); for p-p38, the membranes were incubated with anti-p-p38 (SC-101759) (1/500); for inducible nitric oxide synthase, the membranes were incubated with anti-inducible nitric oxide synthase polyclonal antibody (1/1000) while for cyclooxygenase-2, they were incubated with anti-cyclooxygenase-2 polyclonal antibody (1/1000) (both antibodies were obtained from Cayman, Ann Arbor, MI, USA). For p65, the membranes were incubated with anti-p65 polyclonal antibody (1/500) (SC-7151); for p-p65, the membranes were incubated with anti-p-p65 polyclonal antibody (1/500) (SC-33020-R); for IκB, the membranes were incubated with anti-IκB polyclonal antibody (1/500) (SC-1643); for p-IκB, they were incubated with anti-p-IκB polyclonal antibody (1/500) (SC-8404), for protein kinase Cα, they were incubated with anti-protein kinase Cα polyclonal antibody (1/500) (SC-8393); and for poly (ADP-ribose) polymerase (PARP), the membranes were incubated with anti-PARP polyclonal antibody (1/400) (SC-1562). These antibodies were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Finally, for β-actin, the membranes were incubated with anti-β-actin polyclonal antibody (1/12000) (Sigma-Aldrich). The blots were washed and incubated with peroxidase-conjugate anti-rabbit, anti-mouse or anti-goat immunoglobulin G (1/20000 dilution; Cayman). The immunoreactive bands were visualised with the aid of an enhanced chemiluminiscence system (GE Healthcare, Fairfield, CT, USA).
EMSA
RAW 264.7 macrophages at 1 × 106 cells mL−1 were co-incubated in a petri dish (5 mL) with shikonin (0.1 and 1 µM) at 37°C. After 1 h, 1 µg mL−1 of lipopolysaccharide was added and incubated at 37°C for an additional 1 h. The cells were washed with cold phosphate-buffered saline and resuspended in 1.5 mL of ice cold buffer A (10 mM HEPES pH 7.6, 0.1 mM EDTA pH 8.0, 15 mM KCl, and 2 mM MgCl2).
The cellular lysate was incubated for 10 min on ice and then centrifuged at 2150×g for 5 min at 4°C. The pellet was resuspended in 200 µL of ice cold buffer B (buffer A + 0.2% Nonidet-P40), incubated for 10 min on ice, and centrifuged at 2150×g for 5 min at 4°C. Finally, the pellet was incubated in 50 µL of ice cold buffer C (255 mM HEPES pH 7.6, 0.1 mM EDTA pH 8.0, 50 mM KCl, 10% glycerol, 1 µM dithiothreitol, 5 ppm phenylmethylsulphonyl fluoride, 0.01 mg L−1 aprotinin, and complete mini EDTA-free protease inhibitor cocktail from Roche, Mannheim, Germany) for 5 min on ice. Five microlitre of NaCl 5 M was added, after which the cell lysate was incubated for 30 min under continuous shaking. After centrifugation (20 800×g, 15 min, 4°C), the presence of proteins in the supernatants was determined by means of the Bradford method. Equal amounts of protein (5 µg) were then added to the reaction mixture in accordance with the instructions of the manufacturer (Roche). The specificity of the binding reaction was also examined through competition with the unlabeled consensus oligonucleotide. Electrophoresis was performed in a non-denaturant polyacrylamide gel (6%) in Tris–borate–EDTA buffer at pH 8.0. The immunoreactive bands were visualised with the aid of an enhanced chemiluminiscence system (GE Healthcare).
Immunocytochemistry for NF-κB p65 localization
The effect of shikonin on the nuclear translocation of p65 was examined by means of immunocytochemistry techniques. Cells were grown on chamber slides at a concentration of 1 × 105 cells mL−1 and were either left untreated or were treated with shikonin (0.5 and 1 µM). The cells were then fixed in a solution of methanol : acetic acid (95:5) for 20 min at −20°C. After being washed in phosphate-buffered saline, the cells were permeabilised with 0.3% Triton X-100 at 4°C overnight and then blocked with 5% bovine serum albumin for 15 min. The slides were then incubated with rabbit polyclonal anti-p65 in a 1:500 dilution. After 2 h of incubation in a humectation chamber at 37°C, the slides were washed with phosphate-buffered saline and incubated at 37°C for 40 min with a secondary goat anti-rabbit antibody conjugated to Alexa Fluor® 488 (Invitrogen). The samples were examined under a fluorescent microscope and images were taken with a camera (Nikon Microscope Eclipse E800, Badhoevedorp, the Netherlands).
Software
Images for all Western blot and EMSA experiments were acquired with the image analysis system LAS-3000 mini (Fujifilm, Tokyo, Japan). Digital images were processed and band density measurements were made with the aid of a Multi Gauge V3.0 software package (Fujifilm).
Data analysis
The results are presented as the mean ± standard error of the mean statistically significant differences between the groups were assessed by means of a one-way analysis of variance (anova) and Dunnett's test.
Materials
Shikonin was purchased from TCI Europe (Zwijndrecht, Antwerp, Belgium). A 10 mM stock solution in dimethyl sulphoxide was prepared and stored at −20°C. All chemical and biochemical reagents were purchased from Fluka Chemika–Biochemika (Buchs, Switzerland) and Baker (Deventer, Holland).
Results
Effect of shikonin on TPA-induced ear oedema
Application of a single dose of TPA induces an acute inflammatory reaction in mouse ears which reaches its peak at 6 h. In earlier studies (Payáet al., 1993), we found that the oedema observed 4 h after TPA was similar to that observed after 6 h; but the maximum effect of a standard anti-inflammatory drug applied simultaneously with TPA was observed at 4 h after TPA. In the present experiments, simultaneous treatment with shikonin was found to significantly reduce the oedema (Figure 2A) in a dose-dependent fashion. The inhibitory dose 50 calculated by means of linear regression in this experimental model was 1.0 mg per ear (dose range: 0.25–2.0 mg per ear, r2= 0.9645, P= 0.0179, significant).
Figure 2.
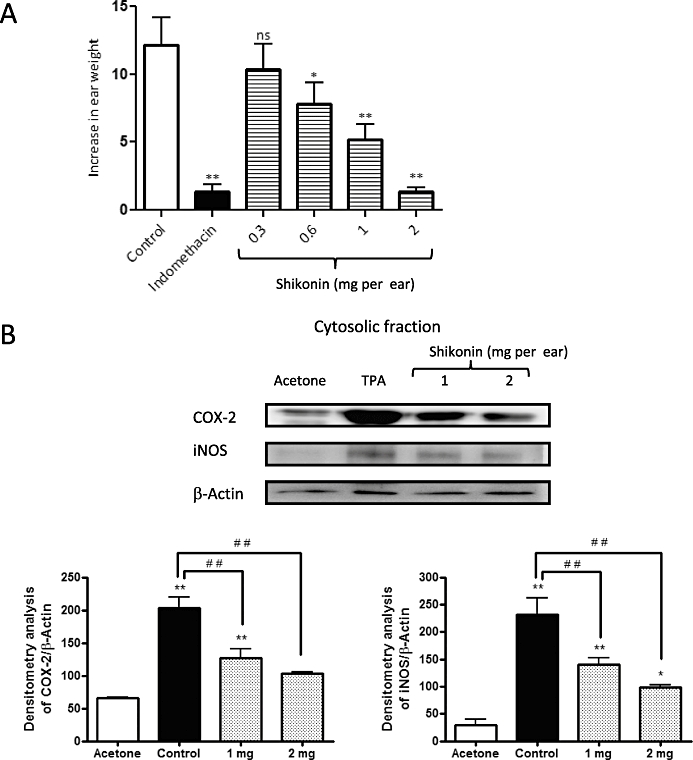
Inhibitory effects of shikonin (Sh) on 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced mouse ear oedema. Mice topically treated with shikonin (0.3, 0.6, 1 and 2 mg per ear) were killed 1, 2 or 4 h after TPA application. (A) Oedema was measured after 4 h of TPA application; values are expressed as the increase in ear weight (ΔW) ± standard error of the mean, n= 6; *P < 0.05, **P < 0.01. ns, not significantly different; Dunnett's test. (B) Effect of shikonin treatment on levels of cyclooxigenase-2 and inducible nitric oxide synthase in ear tissue. The upper panels show an example of Western blot following probing with the corresponding antibody. The lower histograms represent the data derived from the Western blots following densitometry analysis. *P < 0.05, **P < 0.01, significantly different from the acetone group; #P < 0.05, ##P < 0.01, significantly different between the control group and the groups treated with shikonin; Dunnett's test. n= 3.
Shikonin decreases expression of cyclooxygenase-2 and inducible nitric oxide synthase protein in TPA-induced ear oedema.
To gain further insight into the anti-inflammatory mechanisms involved in the inhibitory effects of shikonin on TPA-induced inflammation in mouse ears, we evaluated the possible influence of this compound on cyclooxygenase-2 and inducible nitric oxide synthase expression, comparing the effects with those in animals treated with acetone only. Topical application of TPA is known to induce expression of these enzymes in mouse skin in vivo (Chun et al., 2003; Lai et al., 2007). In agreement with these findings, Western blot analysis revealed that topical application of TPA in the ears of mice resulted in a significant up-regulation of cyclooxygenase-2 and inducible nitric oxide synthase in tissue extracts (Figure 2B). Topical application of shikonin (1 and 2 mg) caused a dose-dependent reduction of both enzymes in the mouse ear extracts (Figure 2B). The maximal inhibition obtained at 2 mg per ear was 70% and 100% for cyclooxygenase-2 and inducible nitric oxide synthase, respectively (P < 0.01, n= 3).
Inhibitory effect of shikonin on TPA-induced activation of NF-κB in mouse skin
As inducible nitric oxide synthase and cycloxygenase-2 are frequently regulated through activation of the NF-κB signalling pathway and, as NF-κB activation and nuclear translocation are preceded by the phosphorylation and proteolytic degradation of IκBα, we decided to examine the inhibitory activity of shikonin against the activation and nuclear translocation of p65, the functionally active subunit of NF-κB in mouse skin. As depicted in Figure 3A, NF-κB activity was detectable in nuclear proteins obtained from acetone-treated mouse ears. However, DNA binding activity was found to increase significantly (P < 0.01, n= 3) in nuclear extracts 4 h after TPA treatment. After topical application of shikonin to the mouse ears, we found that TPA-induced NF-κB nuclear translocation was inhibited in a dose-dependent manner, as determined with Western blot analysis (Figure 3A). The effect of shikonin treatment on TPA-induced NF-κB-DNA-binding was evaluated with an EMSA (Figure 3B), in which shikonin was found to suppress TPA-induced NF-κB-DNA-binding activity in mouse skin (n= 4).
Figure 3.
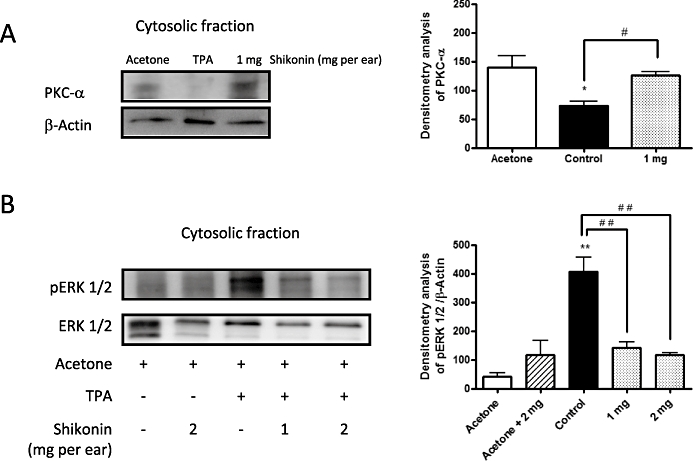
(A) Effect of shikonin on protein kinase Cα (PKC-α) enzymes in extracts of ear tissue after 12-O-tetradecanoylphorbol-13-acetate (TPA). (B) Effect of shikonin treatment on levels of phosphorylated and non-phosphorylated extracellular signal-regulated protein kinase (ERK 1/2) after 1 h of TPA application. The upper panels in Figures 3A,B show an example of Western blot following probing with the corresponding antibody. The lower histograms represent the data derived from the Western blots following densitometry analysis. *P < 0.05, **P < 0.01, significantly different from the acetone group; #P < 0.05, ##P < 0.01, significantly different between the control group and the groups treated with shikonin; Dunnett's test. n= 3–5.
Shikonin decreases TPA-induced protein kinase Cα translocation in TPA-induced ear oedema
Protein kinase C isoforms are major regulators of cutaneous homeostasis that mediate inflammation in response to TPA (Mueller, 2006). The results, shown in Figure 4A, indicate that topical treatment of mouse ears with TPA for 4 h was able to activate the protein kinase Cα isoform, causing it to translocate from the cytosol to the membrane. Topical application of shikonin (1 mg per ear) significantly decreased the TPA-induced translocation of protein kinase Cα (Figure 4A). At a dose of 1 mg per ear, the calculated percentage of inhibition caused by shikonin was 96% (P < 0.01, n= 3).
Figure 4.
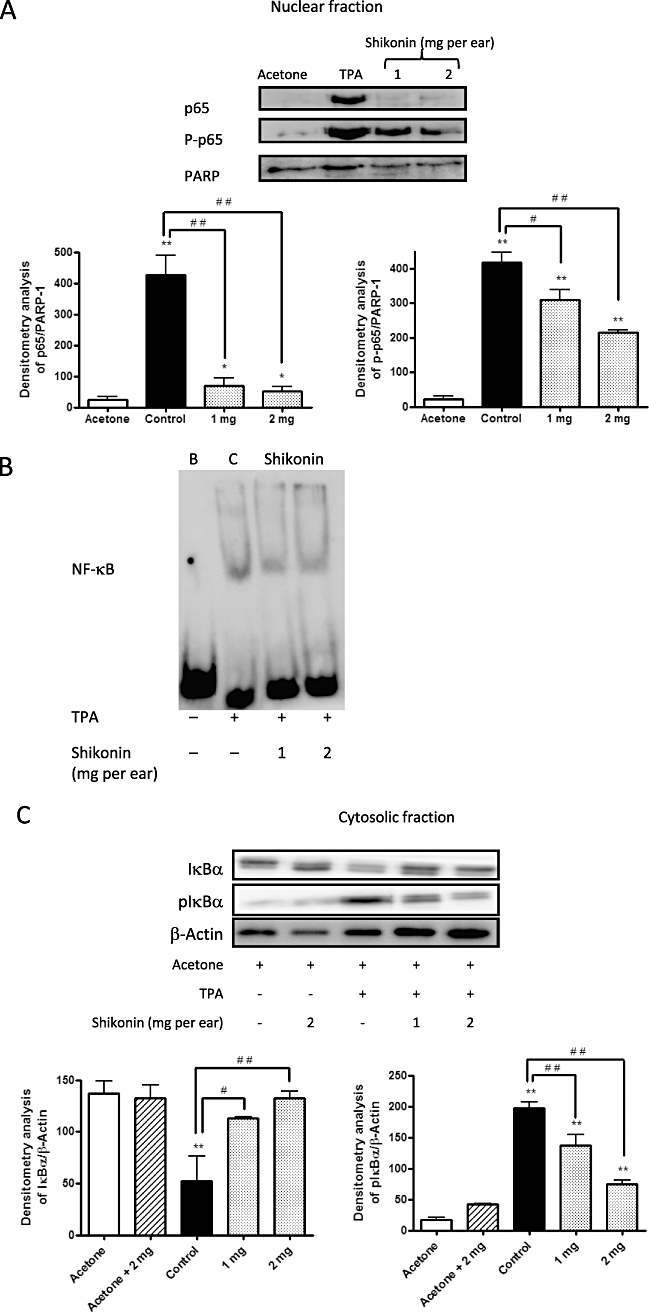
( A) Nuclear cell extract prepared from the ears after 4 h of 12-O-tetradecanoylphorbol-13-acetate (TPA) application was analysed for p65 and P-p65. (B) Effect of shikonin on nuclear factor-κB (NF-κB) binding activity. B, blank (non-treated); C, control (TPA treated); Shikonin (TPA and shikonin treated). (C) Two hours after TPA application, the effect of shikonin treatment on levels of IκBα and pIκBα was assayed with the aid of Western blot techniques. The upper panels in Figures 4A,C show an example of Western blot following probing with the corresponding antibody. The lower histograms represent the data derived from the Western blots following densitometry analysis. *P < 0.05, **P < 0.01, significantly different from the acetone group; #P < 0.05, ##P < 0.01, significantly different between the control group and the groups treated with shikonin; Dunnett's test. n= 3–4.
Effect of shikonin on MAPK phosphorylation in TPA-induced ear oedema
MAPKs are involved in the activation of NF-κB in mouse skin (Chun et al., 2003). To assess the effect of shikonin on the phosphorylation of these MAPKs, Western blot analysis of cytosolic extracts from 1 h TPA-treated mice was carried out. As shown in Figure 4B, shikonin clearly suppressed ERK 1/2 activation (n= 5), without affecting JNK or p38 phosphorylation (data not shown).
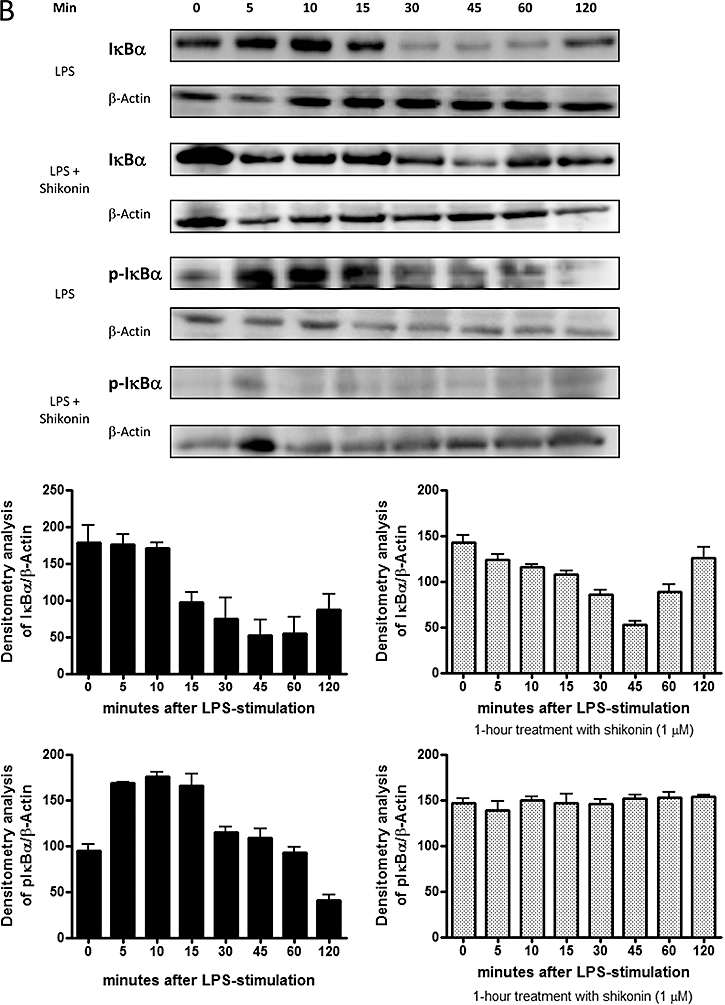
Shikonin interferes with the degradation of IκBα and the DNA binding activity of NF-κB in RAW 264.7 cells
Several studies disclose a relation between ERK activation and the degradation due to phosphorylation of IκBα (Briant et al., 1998; Foehr et al., 2000; Dhawan and Richmond, 2002) which subsequently leads to the activation and translocation of p65 to the nucleus, initiating the transcription of pro-inflammatory mediators. When examining the effect of the treatment with shikonin on the cytosolic levels of IκBα and pIκBα, we observed that shikonin-treated animals showed a dose-dependent decrease in the levels of pIκBα, compared with those which only received treatment with TPA (Figure 3C) (n= 4).
To confirm these results, we carried out an experiment in RAW 264.7 macrophages stimulated with lipopolysaccharide to determine if this inhibition also occurred in vitro and the exact moment at which it was occurring. As seen in Figure 5B, lipopolysaccharide induces IκBα degradation within 30 min in control cells. Levels of IκBα start to recover after 2 h. However, in cells treated with shikonin at 1 µM, this degradation was prevented. Because IκBα phosphorylation precedes its degradation (Perkins, 2007), we also studied the effect of shikonin on the phosphorylation of IκBα. We observed that phosphorylation and, consequently, subsequent degradation of IκBα started 5 min after stimulation with lipopolysaccharide. When cells were pre-treated with shikonin, however, almost no phosphorylated IκBα was produced. These results confirm that shikonin exerts it inhibitory effect in a step previous to IκBα degradation (n= 4).
Figure 5.
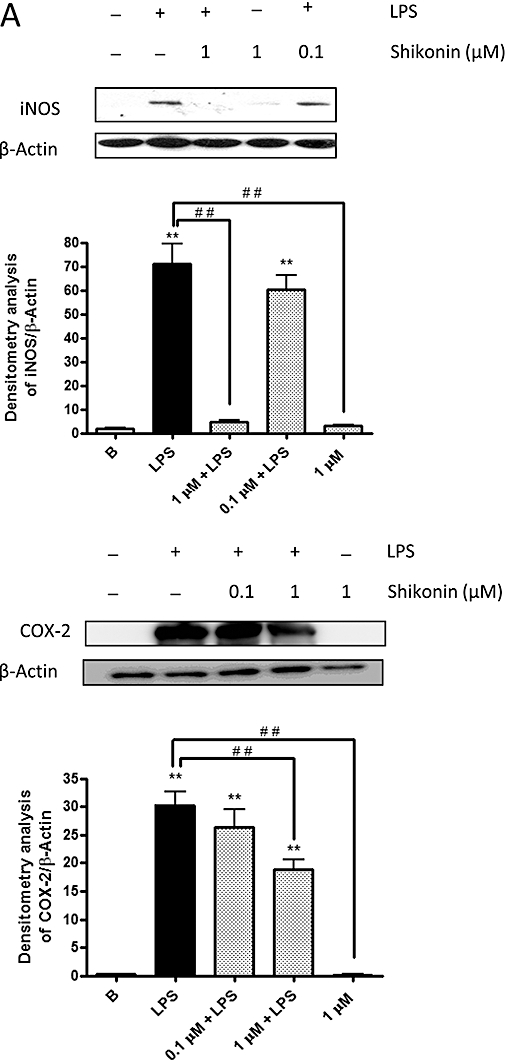
(A) Effect of shikonin treatment on levels of inducible nitric oxide synthase and cyclooxygenase-2 in RAW 264.7 macrophages. The data given are the mean values ± standard error of the mean taken from six independent experiments for each enzyme. *P < 0.05, **P < 0.01, significantly different from the blank group; #P < 0.05, ##P < 0.01, significant difference between the control group and the groups treated with shikonin; Dunnett's test. (B) Shikonin inhibits lipopolysaccharide (LPS)-dependent IκBα degradation. The data given are the means ± standard error of the mean of values taken from four independent experiments. A representative experiment is shown in the upper panel. The lower histograms represent the data derived from the Western blots following densitometry analysis. In A and B, levels were normalised against β-actin.
Effect of shikonin on the expression of inducible nitric-oxide synthase and cyclooxygenase-2 proteins in RAW 264.7 cells
To confirm the results obtained in vivo, the expression of inducible nitric oxide and cyclooxygenase-2 was analysed in cytosolic extracts from RAW 264.7 cells by the Western blot techniques. As shown in Figure 5A, inducible nitric-oxide synthase and cyclooxygenase-2 were undetectable in unstimulated cells. However, upon 24 h treatment with lipopolysaccharide, the expressions of both proteins showed a marked increase. A clear inhibition of the expression of inducible nitric-oxide synthase and of cyclooxygenase-2 was observed in the presence of 1 µM shikonin.
Shikonin inhibits p65 translocation into the nucleus and p65 phosphorylation in RAW 264.7 cells
The effect of shikonin on lipopolysaccharide-induced nuclear translocation of p65 was determined by means of Western blot techniques. As shown by Cheng et al. (2008), while lipopolysaccharide induced nuclear translocation of p65 in RAW 264.7 cells, pre-treating the cells with shikonin (1 µM) inhibited this translocation (data not shown). To confirm this inhibitory effect of shikonin, an immunofluorescence assay was carried out. In non-stimulated cells, p65 was localised in the cytoplasm (Figure 6A, blank); whereas, in cells treated with lipopolysaccharide, p65 suffered nuclear translocation from the cytoplasm to the nucleus (Figure 6A, control). However, when RAW 264.7 cells were pre-treated with shikonin, this lipopolysaccharide-induced nuclear translocation did not occur (Figure 6A, shikonin) (n= 4).
Figure 6.
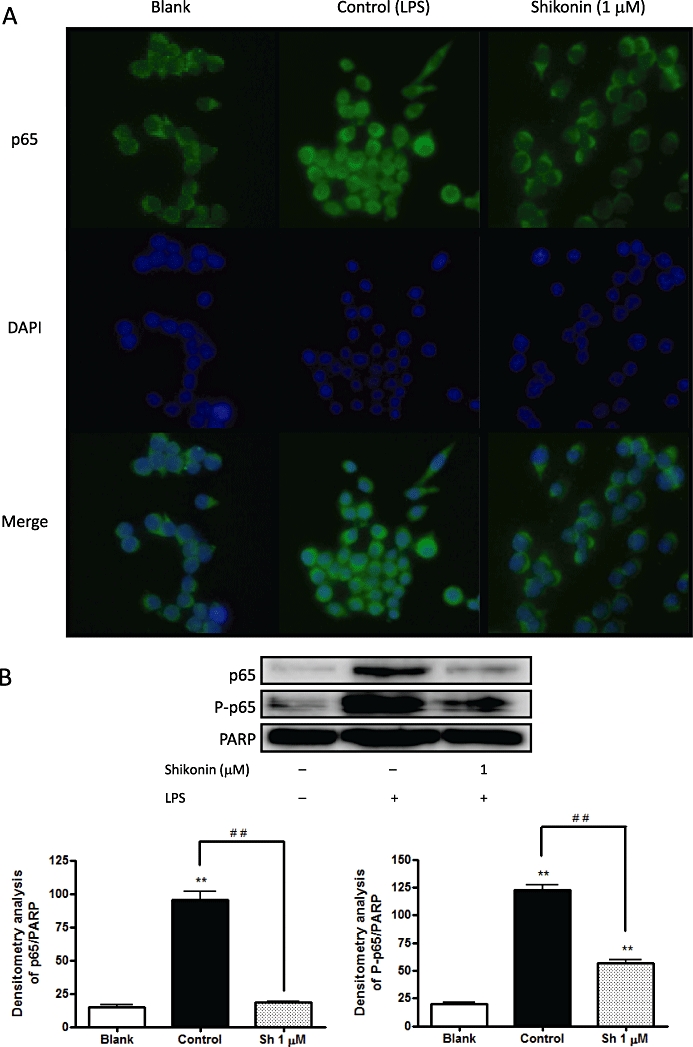
(A) Shikonin suppresses p65 translocation to the nucleus in RAW 264.7 macrophages, as shown by an immunocytochemical analysis of p65 localization. p65 was indicated by the presence of fluorescent green, DNA by fluorescent blue. In non-stimulated cells (blank) and shikonin-treated RAW 264.7 cells, p65 was localised in the cytoplasm, whereas after treatment with lipopolysaccharide, p65 suffered nuclear translocation (control). (B) Shikonin inhibits lipopolysaccharide-induced nuclear translocation of p65 in RAW 264.7 macrophages. The upper panels show a representative Western blot following probing with the corresponding antibody. The bars represent the data derived from the Western blots following densitometry analysis. Levels were normalised against poly (ADP-ribose) polymerase-antibody. *P < 0.05, **P < 0.01; significantly different from the blank group; #P < 0.05, ##P < 0.01; significantly different between the control group and the groups treated with shikonin; Dunnett's test. n= 5.
When the catalytic subunit of protein kinase A is associated with complex NF-κB : IκBα, its catalytic activity is inhibited. Stimulation of NF-κB activity through IκBα degradation can induce the activation of protein kinase Ac, which leads to the phosphorylation of p65, thereby increasing the transactivating activity of the DNA-bound NF-κB (Zhong et al., 1997; Ghosh et al., 1998). When we studied the effect of shikonin on the phosphorylation of p65 using Western blot techniques, we found that lipopolysaccharide induced protein phosphorylation whereas shikonin strongly suppressed it (Figure 6B) (n= 5).
Effect of shikonin on lipopolysaccharide-mediated NF-κB transcriptional activity in RAW 264.7 cells
To examine the effect of shikonin on lipopolysaccharide-induced NF-κB activation, we performed a NF-κB reporter gene assay in RAW 264.7 cells (Figure 7). First, the cells were transiently transfected with NF-κB-Luc plasmids. While subsequent treatment of the transfected cells with lipopolysaccharide for 18 h increased the luciferase activity over the basal level (Figure 7), pre-treating the cells with shikonin (1–1.5 µM) was found to inhibit this activity in a dose-dependent manner. Thus, at 1 µM, shikonin suppressed the gene expression by 28%; whereas, at 1.5 µM, gene expression was reduced by 46% (n= 4).
Figure 7.
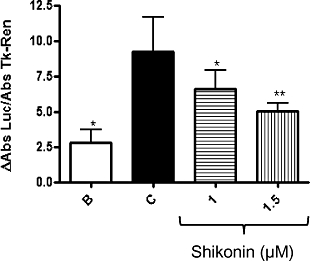
Shikonin suppresses lipopolysaccharide (LPS)-induced NF-κB activity, as shown in the luciferase assay in RAW 264.7 macrophages. Luciferase activity was normalised to TK-Renilla activity. The data were obtained from four independent experiments and expressed as the mean (ΔAbs) ± standard error of the mean differences in absorbance of each group with the control group were determined by means of a one-way analysis of variance followed by Dunnett's test, n= 4; *P < 0.05, **P < 0.01.
Discussion and conclusions
Although several research groups have investigated the biological and pharmacological effects of shikonin and its derivatives, the precise mode of action of these compounds remains undetermined. Among the various biological activities of shikonin, its pharmacological properties have interested a number of researchers, who have proposed several possible mechanisms of action for the compound (Chen et al., 2002). In this context, and in relation with the compound's anti-inflammatory activity, shikonin has been demonstrated to inhibit the transcriptional activity of human cyclooxygenase-2 and TNF-α promoters (Subbaramaiah et al., 2001; Staniforth et al., 2004; Chiu and Yang, 2007). Cheng et al. (2008) demonstrated that shikonin inhibits nitric oxide synthase induction in lipopolysaccharide-stimulated RAW 264.7 cells via down-regulation of mitogen-activated protein kinase/NF-κB signalling. However, little information is available in the literature about the anti-inflammatory effects of shikonin in vivo (Papageorgiou et al., 1999). In our study, we have investigated the anti-inflammatory effect of shikonin against TPA-induced acute ear oedema in mice and analysed its effect on the protein expression of several pro-inflammatory enzymes such as cyclooxygenase-2, inducible nitric oxide synthase, protein kinase C and MAPKs, as well as its effect on the activation of NF-κB.
The protein kinase C family of compounds comprises critical components of cellular signal transduction cascades. These are directly involved in different diseases, such as cancer and inflammation, and they have thus become one of the most important targets for drug development (Surh et al., 2001). After application of TPA, protein kinase C translocates from the cytoplasm to the cell membrane, where it interacts with diacylglycerol and phosphatidylserine (Szaefer et al., 2007). Once stimulated, protein kinase C induces a pleiotropic tissue response encompassing a strong inflammatory reaction due to the stimulation of a wide variety of inflammatory factors (Mueller, 2006). In particular, protein kinase Cα has been implicated in a wide variety of cellular functions, including proliferation, apoptosis, differentiation and inflammation (Nakashima, 2002).
In our experimental work with mouse skin, we have demonstrated that shikonin at 1 mg per ear decreases the TPA-induced translocation of protein kinase Cα from cytosol to the membrane. This effect may prevent the transmembrane signal transduction and thus lead to the inhibition of TPA-induced acute inflammation in mice.
Topical application of TPA induces acute ear oedema, producing a maximal expression of cyclooxygenase-2, 4 h after application (Garg et al., 2008). For its part, the maximal protein expression of nitric oxide synthase occurs 2 h after TPA application (Lai et al., 2007). We observed that 4 h after TPA application, the protein levels were still higher than those in control animals treated with acetone only. For this reason, we used the same time point to assay both proteins. Our data show that, for the first time, the topical application of shikonin can inhibit the expression of the cyclooxygenase-2 in vivo in a dose-dependent fashion. Moreover, topical application of shikonin together with TPA inhibits nitric oxide synthase expression in a manner similar to that of cyclooxygenase-2. These results have been confirmed in lipopolysaccharide-stimulated RAW 264.7 macrophages, where shikonin inhibits the expression of both enzymes.
The expression of nitric oxide synthase and cyclooxygenase-2 is regulated by NF-κB in both cultured cell lines and skin inflammation after TPA-application (Surh et al., 2001; Chun et al., 2003). The activation of NF-κB is a multistep process which includes IKK complex activation, which facilitates the movement of the NF-κB heterodimer from the cytoplasm to the nucleus, where it activates the transcription of genes encoding pro-inflammatory proteins such as nitric oxide synthase and cyclooxygenase-2. The degree of activation by NF-κB is likely to result from a combination of p65 nuclear translocation and post-translational modifications of p65, such as phosphorylation of serine residues (Jones et al., 2007). In this paper, we have demonstrated that topical application of shikonin in vivo inhibited NF-κB activation in mouse ears stimulated with TPA. Although maximal activation of NF-κB occurs 1 h after TPA administration, we have observed that even 4 h after TPA application, the levels of NF-κB activation are still considerable as are those of p65 translocated to the nucleus. The analysis of the protein extracts from ears treated with shikonin showed a significant reduction in both the nuclear translocation of p65 and the levels of phosphorylated p65. We corroborated these results in vitro, demonstrating that the suppression of NF-κB activation in RAW 264.7 macrophages is correlated with the inhibition of lipopolysaccharide-induced IκBα degradation, p65 phosphorylation, p65 translocation, and NF-κB-dependent reporter gene expression. Sandur et al. (2006) studied in vitro a structurally-related compound, plumbagin, finding it to be a potent inhibitor of the NF-κB activation pathway that leads to the suppression of NF-κB regulated gene products involved in cell proliferation and anti-apoptosis through inhibition of the binding of the p65 subunit to DNA and inhibition of IKK activity.
MAP kinase signalling pathways have been extensively studied in cultured cell lines, although much less is known about specificity and the extent to which they are activated during the inflammation in mouse skin. Topical application of TPA on the ears of CD1 mice induced a rapid and sustained activation of ERK but not of p38 MAPK (Jaffee et al., 2000; Liu et al., 2001). We have found that treatment of ears of female mice with TPA significantly enhanced phosphorylation of ERK ½; whereas, topically applied shikonin was able to inhibit it. Chun et al. (2003) support the idea that ERK may play an important role in the signalling pathway of TPA-induced cyclooxygenase-2 expression and NF-κB activation in mouse skin. Dhawan and Richmond (2002) demonstrated that in HS294T cells, ERK regulation of NF-κB activation involves increased IκB phosphorylation with concomitant elevation in the NF-κB DNA binding activity. Most inhibitors of NF-κB activation such as curcumin (Chun et al., 2003) or a-amyrin (Medeiros et al., 2007) exert their anti-inflammatory effects through suppression of phosphorylation and degradation of IκBα. We have observed that topical application of shikonin suppressed induced IκBα phosphorylation in a dose-dependent manner both in vivo in TPA-treated mice, as well as in vitro in RAW 264.7 cells stimulated with lipopolysaccharide. It seems that the ERK signalling pathway regulates NF-κB activation through inhibition of IκBα phosphorylation (Chun et al., 2003). These results may provide the molecular mechanism responsible for the inhibitory effect of shikonin on NF-κB activation in TPA-treated mouse skin.
Our findings thus provide additional, useful mechanistic explanations for the anti-inflammatory effect of shikonin and highlight its pharmaceutical importance.
Acknowledgments
This work was supported by the Spanish Government (SAF2006-06726) and Generalitat Valenciana (GVPRE/2008/387). I. Andújar is grateful for a fellowship from the Generalitat Valenciana, grant BFPI/2008/040.
Glossary
Abbreviations:
- DMEM
Dulbecco's modified Eagle's medium
- EMSA
electrophoretic mobility shift assay
- ERK
extracellular signal-regulated protein kinase
- IκB
inhibitor protein of NF-κB
- IKK
IκB kinase
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide
- NF-κB
nuclear factor-κB
- PARP
poly (ADP-ribose) polymerase
- TNF-α
tumour necrosis factor-α
- TPA
12-O-tetradecanoylphorbol-13-acetate
Conflict of interest
The authors state no conflict of interest.
References
- Afaq F, Saleem M, Aziz MH, Mukhtar H. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced tumor promotion markers in CD-1 mouse skin by oleandrin. Toxicol Appl Pharmacol. 2004;195(3):361–369. doi: 10.1016/j.taap.2003.09.027. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA. IkB-NF-κB structures: at the interface of inflammation control. Cell. 1998;95:729–731. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- Bell S, Degitz K, Quirling M, Jilg N, Page S, Brand K. Involvement of NF-κB signalling in skin physiology and disease. Cell Signal. 2003;15:1–7. doi: 10.1016/s0898-6568(02)00080-3. [DOI] [PubMed] [Google Scholar]
- Bremner P, Heinrich M. Natural products as targeted modulators of the nuclear factor-κB pathway. J Pharm Pharmacol. 2002;54:453–472. doi: 10.1211/0022357021778637. [DOI] [PubMed] [Google Scholar]
- Briant L, Robert-Hebmann V, Sivan V, Brunet A, Pouyssegur J, Devaux C. Involvement of extracellular signal-regulated kinase module in HIV-mediated CD4 signals controlling activation of nuclear factor-kappa B and AP-1 transcription factors. J Immunol. 1998;160:1875–1885. [PubMed] [Google Scholar]
- Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257(13):7847–7851. [PubMed] [Google Scholar]
- Celec P. Nuclear factor kB – molecular biomedicine: the next generation. Biomed Pharmacother. 2004;58:365–371. doi: 10.1016/j.biopha.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Chen X, Yang L, Oppenheim JJ, Howard MZ. Cellular pharmacology studies of shikonin derivatives. Phytother Res. 2002;16:199–209. doi: 10.1002/ptr.1100. [DOI] [PubMed] [Google Scholar]
- Cheng YW, Chang CY, Lin KL, Hu CM, Lin CH, Kang JJ. Shikonin derivatives inhibited LPS-induced NOS in RAW 264.7 cells via downregulation of MAPK/NF-κB signaling. J Ethnopharmacol. 2008;120:264–271. doi: 10.1016/j.jep.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Chiu SC, Yang NS. Inhibition of tumor necrosis factor-a through selective blockade of Pre-mRNA splicing by shikonin. Mol Pharmacol. 2007;71:1640–1645. doi: 10.1124/mol.106.032821. [DOI] [PubMed] [Google Scholar]
- Chun KS, Keum YS, Han SS, Song YS, Kim SH, Surh YJ. Curcumin inhibits phorbol ester-induced expression of cyclooxygenase-2 in mouse skin through suppression of extracellular signal-regulated kinase activity and NF-κB activation. Carcinogenesis. 2003;24:1515–1524. doi: 10.1093/carcin/bgg107. [DOI] [PubMed] [Google Scholar]
- Dhawan P, Richmond A. A novel NF-κB-inducing kinase-MAPK signalling pathway up-regulates NF-κB activity in melanoma cells. J Biol Chem. 2002;277:7920–7928. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, et al. Mapping of the inducible IkB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehr ED, Bohuslav J, Chen LF, DeNoronha C, Geleziunas R, Lin X, et al. The NF-κB-inducing kinase induces PC12 cell differentiation and prevents apoptosis. J Biol Chem. 2000;275:34021–34024. doi: 10.1074/jbc.C000507200. [DOI] [PubMed] [Google Scholar]
- Garg R, Ramchandani AG, Maru GB. Curcumin decreases 12-O-tetradecanoylphorbol-13-acetate-induced protein kinase C translocation to modulate downstream targets in mouse skin. Carcinogenesis. 2008;29:1249–1257. doi: 10.1093/carcin/bgn114. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Handel ML, Girgis L. Transcription factors. Best Pract Res Clin Rheumatol. 2001;15:657–675. doi: 10.1053/berh.2001.0186. [DOI] [PubMed] [Google Scholar]
- Jaffee BD, Manos EJ, Collins RJ, Czerniak PM, Favata MF, Magolda RL, et al. Inhibition of MAP kinase kinase (MEK) results in an anti-inflammatory response in vivo. Biochem Biophys Res Commun. 2000;268:647–651. doi: 10.1006/bbrc.2000.2184. [DOI] [PubMed] [Google Scholar]
- Jones E, Adcock IM, Ahmed BY, Punchard NA. Modulation of LPS stimulated NF-κB mediated Nitric Oxide production by PKCe and JAK2 in RAW macrophages. J Inflamm. 2007;4:23. doi: 10.1186/1476-9255-4-23. DOI 10.1186/1476-9255-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami N, Koyama Y, Tanaka J, Ohara A, Hayakawa T, Fujimoto S. Inhibitory effect of acetylshikonin on the activation of NADPH oxidase in polymorphonuclear leukocytes in both whole cell and cell-free systems. Biol Pharm Bull. 1996;19:1266–1270. doi: 10.1248/bpb.19.1266. [DOI] [PubMed] [Google Scholar]
- Kim JY, Park SJ, Yun KJ, Cho YW, Park HJ, Lee KT. Isoliquiritigenin isolated from the roots of Glycyrrhiza uralensis inhibits LPS-induced iNOS and COX-2 expression via the attenuation of NF-κB in RAW 264.7 macrophages. Eur J Pharmacol. 2008;584:175–184. doi: 10.1016/j.ejphar.2008.01.032. [DOI] [PubMed] [Google Scholar]
- Lai CS, Li S, Chai CY, Lo CY, Ho CT, Wang YJ, et al. Inhibitory effect of citrus 5-hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone on 12-O-tetradecanoylphorbol 13-acetate-induced skin inflammation and tumor promotion in mice. Carcinogenesis. 2007;28:2581–2588. doi: 10.1093/carcin/bgm231. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Georgiou HM, Rice GE. Regulation of proinflammatory cytokines in human gestational tissues by peroxisome proliferator-activated receptor-gamma: effect of 15-deoxy-Δ12,14-PGJ2 and troglitazone. J Clin Endocrinol Metab. 2002;87:4667–4672. doi: 10.1210/jc.2002-020613. [DOI] [PubMed] [Google Scholar]
- Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Liu Y, Duysen E, Yaktine AL, Au A, Wang W, Birt DF. Dietary energy restriction inhibits ERK but not JNK or p38 activity in the epidermis of SENCAR mice. Carcinogenesis. 2001;4:607–612. doi: 10.1093/carcin/22.4.607. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Otuki MF, Avellar MCW, Calixto JB. Mechanisms underlying the inhibitory actions of the pentacyclic triterpene α-amyrin in the mouse skin inflammation induced by phorbol ester 12-O-tetradecanoylphorbol-13-acetate. Eur J Pharmacol. 2007;559:227–235. doi: 10.1016/j.ejphar.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Mueller MM. Inflammation in epithelial skin tumours: old stories and new ideas. Eur J Cancer. 2006;42:735–744. doi: 10.1016/j.ejca.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Nakashima S. Protein kinase Cα (PKCα): regulation and biological function. J Biochem. 2002;132:669–675. doi: 10.1093/oxfordjournals.jbchem.a003272. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- Papageorgiou VP, Assimopoulou AN, Couladouros EA, Hepworth D, Nicolaou KC. The chemistry and biology of alkannin, shikonin, and related naphthazarin natural products. Angew Chem Int Ed. 1999;38:270–300. doi: 10.1002/(SICI)1521-3773(19990201)38:3<270::AID-ANIE270>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Park HJ, Kim IT, Won JH, Jeong SH, Park EY, Nam JH, et al. Anti-inflammatory activities of ent-16aH,17-hydroxy-kauran-19-oic acid isolated from the roots of Siegesbeckia pubescens are due to the inhibition of iNOS and COX-2 expression in RAW 264.7 macrophages via NF-κB inactivation. Eur J Pharmacol. 2007;558:185–193. doi: 10.1016/j.ejphar.2006.11.036. [DOI] [PubMed] [Google Scholar]
- Payá M, Ferrándiz ML, Sanz MJ, Bustos G, Blasco R, Ríos JL, et al. Study of the antioedema activity of some seaweed and sponge extracts from the Mediterranean coast in mice. Phytother Res. 1993;7:159–162. [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Sandur SK, Ichikawa H, Sethi G, Ahn KS, Aggarwal BB. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-κB activation and NF-κB-regulated gene products through modulation of p65 and IkBa kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem. 2006;281:17023–17033. doi: 10.1074/jbc.M601595200. [DOI] [PubMed] [Google Scholar]
- Slomiany BL, Slomiany A. Activation of peroxisome proliferator-activated receptor gamma suppresses inducible cyclooxygenase and nitric oxide synthase during oral mucosal ulcer healing. J Physiol Pharmacol. 2002;53:159–169. [PubMed] [Google Scholar]
- Staniforth V, Wang SY, Shyur LF, Yang NS. Shikonins, phytocompounds from Lithospermum erythrorhizon, inhibit the transcriptional activation of human tumor necrosis factor alpha promoter in vivo. J Biol Chem. 2004;279:5877–5885. doi: 10.1074/jbc.M309185200. [DOI] [PubMed] [Google Scholar]
- Subbaramaiah K, Bulic P, Lin Y, Dannenberg AJ, Pasco DS. Development and use of a gene promoter-based screen to identify novel inhibitors of cyclooxygenase-2 transcription. J Biomol Screen. 2001;6:101–110. doi: 10.1177/108705710100600206. [DOI] [PubMed] [Google Scholar]
- Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat Res. 2001;480/481:243–268. doi: 10.1016/s0027-5107(01)00183-x. [DOI] [PubMed] [Google Scholar]
- Szaefer H, Kaczmarek J, Rybczynska M, Baer-Dubowska W. The effect of plant phenols on the expression and activity of phorbol ester-induced PKC in mouse epidermis. Toxicology. 2007;230:1–10. doi: 10.1016/j.tox.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Uwe S. Anti-inflammatory interventions of NF-κB signaling: potential applications and risks. Biochem Pharmacol. 2008;75:1567–1579. doi: 10.1016/j.bcp.2007.10.027. [DOI] [PubMed] [Google Scholar]
- Wang JP, Kuo SC. Impairment of phosphatidylinositol signaling in acetylshikonin-treated neutrophils. Biochem Pharmacol. 1997;53:1173–1177. doi: 10.1016/s0006-2952(97)00098-1. [DOI] [PubMed] [Google Scholar]
- Wang JP, Raung SL, Chang LC, Kuo SC. Inhibition of hind-paw oedema and cutaneous vascular plasma extravasation in mice by acetylshikonin. Eur J Pharmacol. 1995;272:87–95. doi: 10.1016/0014-2999(94)00627-j. [DOI] [PubMed] [Google Scholar]
- Wang WJ, Bai JY, Liu DP, Xue LM, Zhu XY. The antiinflammatory activity of shikonin and its inhibitory effect on leukotriene B4 biosynthesis. Yao Xue Xue Bao. 1994;29:161–165. [PubMed] [Google Scholar]
- Wu Z, Wu L, Li L, Tashiro S, Onodera S, Ikejima T. p53-mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375-S2 cells. J Pharmacol Sci. 2004;94:166–176. doi: 10.1254/jphs.94.166. [DOI] [PubMed] [Google Scholar]
- Zhong H, Suyang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-κB is regulated by the IkB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]