

Abstract
Background and purpose:
Due to their potent bronchodilator properties, β2-adrenoceptor agonists are a mainstay of therapy in asthma. However, the effects of β2-adrenoceptor agonists on inflammation are less clear. Accordingly, we have investigated the effects of β2-adrenoceptor agonists on inflammatory mediator release.
Experimental approach:
Transcription factor activation, and both release and mRNA expression of IL-6 and IL-8 were examined by luciferase reporter assay, elisa and real-time RT–PCR in bronchial human epithelial BEAS-2B cells or primary human bronchial epithelial cells grown at an air–liquid interface.
Key results:
Pre-incubation with β2-adrenoceptor agonists (salbutamol, salmeterol, formoterol) augmented the release and mRNA expression of IL-6 and IL-8 induced by IL-1β and IL-1β plus histamine, whereas NF-κB-dependent transcription was significantly repressed, and AP-1-dependent transcription was unaffected. These effects were mimicked by other cAMP-elevating agents (PGE2, forskolin). Enhancement of cytokine release by β2-adrenoceptor agonists also occurred in primary bronchial epithelial cells. Addition of dexamethasone with salmeterol repressed IL-6 and IL-8 release to levels that were similar to the repression achieved in the absence of salmeterol. IL-6 release was enhanced when salmeterol was added before, concurrently or after IL-1β plus histamine stimulation, whereas IL-8 release was only enhanced by salmeterol addition prior to stimulation.
Conclusions and implications:
Enhancement of IL-6 and IL-8 release may contribute to the deleterious effects of β2-adrenoceptor agonists in asthma. As increased inflammatory mediator expression is prevented by the addition of glucocorticoid to the β2-adrenoceptor, our data provide further mechanistic support for the use of combination therapies in asthma management.
Keywords: β2-adrenoceptor agonist, inflammation, IL-6, IL-8, glucocorticoid, BEAS-2B epithelial cells, NHBE, asthma
Introduction
Asthma is a chronic inflammatory disease of the airways for which the most common treatments include inhaled glucocorticoids and β2-adrenoceptor agonists, administered either separately or in combination (Sears and Lotvall, 2005). β2-Adrenoceptor agonists represent the standard bronchodilator therapy both in emergency room admissions and in the daily management of asthma (Sears and Lotvall, 2005). However, controversy over the use of β2-adrenoceptor agonists as monotherapy arose in the 1960s when increased mortality of asthma sufferers was attributed to overuse of the non-selective β-adrenoceptor agonist, isoprenaline (Stolley and Schinnar, 1978). In the 1980s, the β2-adrenoceptor agonist, fenoterol, was associated with increased mortality in New Zealand (Crane et al., 1989). Although there were indications that this effect was due to cardiopathic side effects, via non-specific activation of the β1-adrenoceptor (Crane et al., 1989), other studies suggested that this mortality was due to increased asthma severity (Sears and Taylor, 1994). More recently, the salmeterol multicenter asthma research trial demonstrated that patients, particularly African Americans, receiving long-acting β2-adrenoceptor agonist (LABA) monotherapy have a significantly higher risk of fatal, or potentially fatal, asthma episodes (Nelson et al., 2006). Accordingly, current therapeutic guidelines recommend the use of LABAs only in combination with glucocorticoids, and, although regular long-term daily use of short-acting β2-adrenoceptor agonists is not recommended, they remain the mainstay of acute asthma treatment (Bateman et al., 2008).
The β2-adrenoceptor is a member of the seven transmembrane G-protein-coupled receptor superfamily, and couples typically with the heterotrimeric G-protein, Gs (Giembycz and Newton, 2006). Binding of ligand results in activation of adenylyl cyclase by Gs. This catalyses the conversion of ATP into cAMP, which in turn binds to the regulatory subunits of protein kinase A (PKA) (Giembycz and Newton, 2006). These disassociate from the catalytic subunits enabling the phosphorylation of numerous effector proteins, some of which lead to airway smooth muscle relaxation (Giembycz and Newton, 2006). However, agonists of the β2-adrenoceptor are now recognized to activate multiple cAMP-dependent and -independent pathways, and may actually increase the underlying inflammation in asthma (see Giembycz and Newton, 2006 and references therein).
Asthmatic inflammation involves the increased production of numerous mediators, including cytokines, such as interleukin (IL)-6, and chemokines such as IL-8, whose expression is partly controlled at the level of transcription by factors such as activator protein (AP)-1, cAMP response element (CRE) binding protein (CREB), CCAAT/enhancer binding protein and nuclear factor κB (NF-κB) (see Roebuck, 1999; Ammit et al., 2002; Holden et al., 2007).
The pulmonary epithelium is a source of many inflammatory cytokines and chemokines involved in asthmatic inflammation (Janssen-Heininger et al., 2009). Importantly, the epithelium acts as an interface between inhaled air and the respiratory system, and is the first point of contact for not only inflammatory insults, such as airborne allergens, pathogens and pollutants, but also inhaled therapies such as β2-adrenoceptor agonists and glucocorticoids (Davies and Holgate, 2002). Thus, the airway epithelium is critical to both the pathogenesis and treatment of asthma, and may hold the key to the detrimental effects observed in patients taking β2-adrenoceptor agonists as monotherapies.
In the current study, we have used human bronchial airway epithelial cells to demonstrate that the induction of IL-6 and IL-8 expression, and NF-κB-dependent transcription, by IL-1β is increased by histamine. Addition of the β2-adrenoceptor agonists salbutamol, salmeterol or formoterol significantly increased IL-6 and IL-8 release, yet had a repressive effect on NF-κB-dependent transcription. However, further addition of dexamethasone with the β2-adrenoceptor agonist abolished the enhanced release of both IL-6 and IL-8.
Methods
Cell culture
BEAS-2B cells were grown to confluence in 24-well plates using DMEM/F12 medium (Invitrogen, Burlington, Ontario, Canada) supplemented with 10% fetal calf serum as previously described (Catley et al., 2004). Cells were cultured overnight in serum-free medium before changing to fresh serum-free medium containing drugs and stimuli. Normal human bronchial epithelial (NHBE) cells (EpiAirway) (Mattek, Ashland, MA, USA) were cultured at an air–liquid interface on Millipore Millicell CM single-well tissue culture plate inserts (Millipore, Billerica, MA, USA) according to the supplier's instructions.
Adenovirus infection
BEAS-2B cells were either infected with an empty Ad5 expression vector (Ad5.CMV.Null) or a vector expressing PKIα, a highly selective PKA inhibitory peptide (Ad5.CMV.PKIα). Cells were infected with Ad5.CMV.PKIα and Ad5.CMV.Null adenoviruses at a multiplicity of infection of 30 as previously described (Meja et al., 2004).
NF-κB, CRE and AP-1 transcriptional reporters and luciferase assay
BEAS-2B cells stably harbouring the previously described, and validated, NF-κB-dependent luciferase reporter, pGL3.neo.TATA.3κBu (3κBu-luc) (Holden et al., 2007), or the previously characterized CRE reporter, pADneo2-C6-BGL, which contains six tandem CRE motifs upstream of a minimal β-globin site (Meja et al., 2004), were grown in 24-well plates to confluence before being incubated in serum-free medium overnight and subsequently treated for 6 h. The AP-1 luciferase reporter plasmid, pGL3.neo.TATA.3AP-1, was created by inserting two tandem repeats of the sequence, sense strand 5′-TCGATGTGAGTCAGTGAGTCACTGAGTCACGTCGA-3′ (the AP-1 consensus sites are in bold, and the Xho1 compatible sticky end sites are underlined), into the Xho1 site upstream of the minimal promoter of pGL3.neo,TATA (Catley et al., 2004 for details of vector). Insertion of the AP-1 binding sites was validated by sequencing (data not shown). BEAS-2B cells grown to ∼60% confluence in 24-well plates were transiently transfected with pGL3.neo.TATA.3AP-1 using 0.25 µg of plasmid and 0.25 µL of Lipofectamine 2000 (Invitrogen) per well. These were incubated for 30 min before being added to 500 µL of serum-free medium and transfered onto the cells. After 24 h, the cells were changed to fresh serum-free medium for a further 12 h prior to experiments. Cells were harvested 6 h after treatments in reporter lysis buffer (Promega, Madison, WI, USA). Luciferase activity was measured using luciferase assay kits (Biotium, Hayward, CA, USA).
Cytokine release measurement
Release of IL-6 and IL-8 from BEAS-2B cells was measured by using commercial elisa kits (R&D Systems, Hornby, ON, Canada) according to the manufacturer's instructions. Analysis of cytokine release from NHBE basolateral samples was performed using the Procarta cytokine assay (Affymetrix, Santa Clara, CA, USA). Samples were diluted in medium containing 0.5% BSA, and analysed for IL-6 and IL-8 expression according to the manufacturer's instructions (Affymetrix). MiraiBio Masterplex QT v4.0 software (MasterPlex version 1.0.1.18, Hitachi Software Engineering Co., Ltd., San Francisco, CA, USA) was used to extrapolate mean fluorescence intensity to IL-6 and IL-8 concentrations.
Real-time TaqMan PCR analysis
Total RNA was isolated using the RNeasy mini kit (Qiagen, Mississauga, ON, Canada), and 0.5 µg was used for reverse transcription (RT) reactions to prepare cDNA as previously described (Chivers et al., 2006). After cDNA synthesis, TaqMan PCR was performed using 2.5 µL of cDNA in a reaction volume of 20 µL according to the manufacturer's specification (Applied Biosystems Inc., Foster City, CA, USA) using a pre-made master mix (Applied Biosystems) and an ABI 7900HT instrument (Applied Biosystems). Analysis of GAPDH was carried out using the validated off-the-shelf assay 432631E (Applied Biosystems). IL-8 was amplified using the primers 5′-CTGGCCGTGGCTCTCTTG-3′ (forward) and 5′-TTAGCACTCCTTGGCAAAACTG-3′ (reverse) with the 5-carboxyfluorescein/5-carboxytetramethylrhodamine-linked probe 5′-CCTTCCTGATTTCTGCAGCTCTGTGTGAA-3′. IL-6 was amplified using the primers 5′-TGGCTGAAAAAGATGGATGCT-3′ (forward) and 5′-AACTCCAAAAGACCAGTGATGATTT-3′ (reverse) with the 5-carboxyfluorescein/minor groove binding protein-linked probe 5′-CAATGAGGAGACTTG-3′. All primers were designed to amplify only cDNA (from which introns had been excised), using Primer Express version 2 software (Applied Biosystems). Samples were analysed in duplicate, and relative cDNA concentrations were obtained from a cDNA standard curve of serial dilutions of an IL-1β plus histamine-stimulated cDNA sample.
Statistics
Data are presented as means ± SEM of ‘n’ independent observations. Comparison between groups of experimental data was performed using either a one-way anova with a Dunnett's post-test, or Student's t-test as appropriate. Significance was taken where P < 0.05 (*/#), P < 0.01 (**/##) and P < 0.001 (***/###).
Materials
IL-1β (R&D Systems), histamine and poly I : C (Sigma, Oakville, ON, Canada) were dissolved in sterile phosphate-buffered saline. Salbutamol, salmeterol, formoterol, forskolin and ICI 118551 (Sigma) were dissolved in dimethylsulphoxide (DMSO), and prostaglandin (PG) E2 (Sigma) was dissolved in ethanol. Final concentrations of DMSO or ethanol added to cells were <0.1%, and this had no effect on any of the responses (data not shown). Drug and molecular target nomenclature follows Alexander et al. (2009).
Results
Effect of β2-adrenoceptor agonists and other cAMP-elevating agents on IL-6 and IL-8 expression
In supernatants from untreated BEAS-2B cells, both IL-6 and IL-8 release were below the level of detection for the respective elisa kits (<7.8 pg·mL−1). Although there did appear to be an increase in IL-6 and IL-8 release when short-acting (salbutamol) or long-acting (salmeterol and formoterol), β2-adrenoceptor agonists were incubated with otherwise untreated cells. However, these increases were not significant (see Supporting Information Figure S1). Upon stimulation with IL-1β (1 ng·mL−1), IL-6 and IL-8 release were increased to 29 ± 9 and 64 ± 24 pg·mL−1 respectively. Addition of a maximally effective concentration of histamine (100 µM) with IL-1β increased IL-6 and IL-8 release to 164 ± 16 and 141 ± 14 pg·mL−1, respectively (Holden et al., 2007). The addition of either short-acting, or LABAs, increased IL-1β-stimulated IL-6 by 13.7- to 17.8-fold, and IL-8 release by 2.5- to 2.9-fold (Figure 1A). When the β2-adrenoceptor agonists were added to histamine plus IL-1β-stimulated cells, IL-6 release increased by 5.2- to 5.5-fold, and IL-8 release increased by 2.1- to 2.2-fold (Figure 1A). Likewise, the adenylyl cyclase activator, forskolin, or PGE2, an alternative activator of Gs, increased IL-1β-stimulated IL-6 release by 11.2- to 15.7-fold (Figure 1B), and IL-8 release by 3.3- to 3.5-fold (Figure 1B). When cells were stimulated with IL-1β plus histamine, the prior addition of either forskolin or PGE2 increased IL-6 release by 2.5- to 4.1-fold (Figure 1B), and IL-8 release by 2.4- to 2.8-fold (Figure 1B). In each case, possible autocrine roles for prostanoids formed via the COX pathway were excluded as neither indomethacin (10 µM) nor diclofenac (10 µM) affected the release of IL-6 or IL-8 (see Supporting Information Figure S2).
Figure 1.
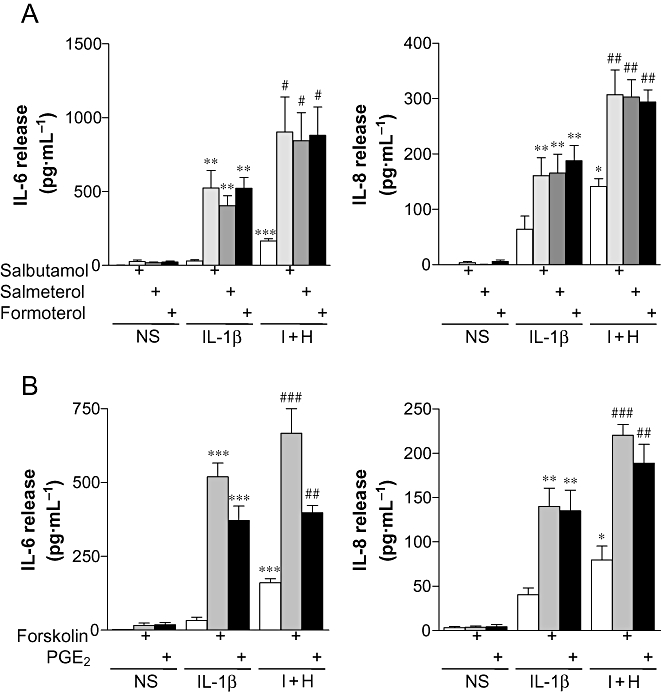
Effect of β2-adrenoceptor agonists and cAMP-elevating agents on the release of IL-6 and IL-8. BEAS-2B cells were either left untreated or were incubated with: (A) salbutamol (1 µM), salmeterol (0.1 µM) or formoterol (0.01 µM), or (B) forskolin (10 µM) or PGE2 (1 µM) for 1 h prior to either being left not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1), or IL-1β (1 ng·mL−1) and histamine (100 µM) (I + H). Cells were harvested after 6 h, and the supernatants were analysed by elisa for IL-6 and IL-8 release. Data (n= 4–6), expressed as pg·mL−1, are plotted as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus IL-1β, ##P < 0.01, ###P < 0.001 versus I + H.
In BEAS-2B cells stimulated with IL-1β, expression of IL-6 and IL-8 mRNA was increased 7.7 ± 4.7- and 74 ± 30-fold, respectively (Figure 2). The addition of histamine plus IL-1β increased IL-6 and IL-8 mRNA to 79 ± 31- and 247 ± 92-fold, respectively (Figure 2). Prior addition of either short- or LABAs to IL-1β-stimulated cells increased IL-6 mRNA to 183- to 204-fold (Figure 2), while IL-8 mRNA was increased to 522- to 619-fold (Figure 2). When either short or LABAs were added to histamine plus IL-1β-stimulated cells, IL-6 mRNA increased to 364- to 479-fold (Figure 2), and IL-8 mRNA increased to 885- to 992-fold (Figure 2).
Figure 2.
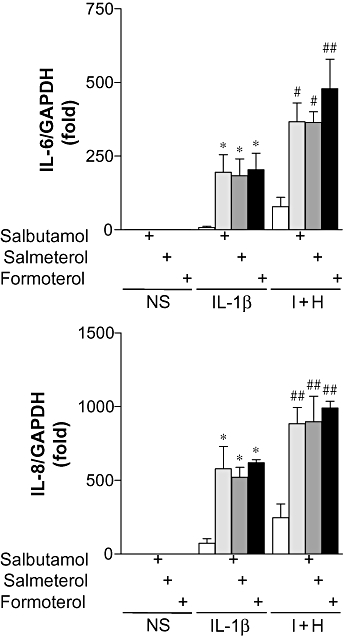
Effect of β2-adrenoceptor agonists on IL-6 and IL-8 mRNA expression. BEAS-2B cells were either left untreated or were incubated with salbutamol (1 µM), salmeterol (0.1 µM) or formoterol (0.01 µM) for 1 h prior to either being left not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1), or IL-1β (1 ng·mL−1) and histamine (100 µM) (I + H). Cells were harvested after 6 h, and TaqMan real-time RT–PCR was carried out for IL-6, IL-8 and GAPDH. Relative cDNA concentrations (n= 3), normalized to GAPDH, are plotted as mean folds ± SEM. *P < 0.05 versus IL-1β, #P < 0.05, ##P < 0.01 versus I + H.
To examine the relevance of these observations in a more physiological system, we took advantage of a model in which primary NHBE cells are grown to confluence at an air–liquid interface. In this model, the cells adopt a pseudostratified, highly differentiated phenotype that closely resembles the epithelium of the respiratory tract (Hayden et al., 2007). The cells become ciliated, secrete mucus and interact with adjacent cells to form tight junctions (Hayden et al., 2007). Moreover, these cells can contribute to innate responses, characterized by the increased secretion of chemokines and cytokines to Toll-like receptor agonists such as poly I : C (Sha et al., 2004). Therefore, poly I : C was selected as a stimulus to examine the effect of salmeterol in this system. In basolateral supernatants from untreated NHBE cells, IL-6 and Il-8 release was 61 ± 4 and 564 ± 87 pg·mL−1 respectively. Following stimulation with poly I : C (100 µg mL), IL-6 and IL-8 release was significantly increased to 2460 ± 638 and 42 100 ± 3100 pg·mL−1. When salmeterol (10 µM) was added prior to stimulation with poly I : C (100 µg mL), IL-6 release was increased by 2.9-fold to 7250 ± 483 pg·mL−1 (P < 0.01) (Supporting Information Table S1), and IL-8 release was increased by 1.5-fold to 62 500 ± 10 700 pg·mL−1 (P < 0.05) (Supporting Information Table S1). In addition, analysis of tumour necrosis factor (TNF)-α, monocyte chemotactic protein-1, IL-1β, epithelial-derived neutrophil-activating peptide 78 and granulocyte colony-stimulating factor (G-CSF) revealed robust release in response to poly I : C, and in each case this was significantly enhanced by prior treatment with salmeterol (Supporting Information Table S1).
Effect of β2-adrenoceptor agonists and other cAMP-elevating agents on NF-κB-dependent transcription
As the genes encoding IL-6 and IL-8 in BEAS-2B cells are highly dependent on NF-κB (Holden et al., 2007), we investigated the effects of β2-adrenoceptor agonists, forskolin and PGE2 on NF-κB-dependent transcription. In contrast to the effects on IL-6 and IL-8 expression, a 1 h pre-incubation with salbutamol, salmeterol or formoterol resulted in decreases of 33 ± 11, 35 ± 13 and 39 ± 16%, respectively, in IL-1β-stimulated NF-κB-dependent transcription (Figure 3). Likewise, a 1 h pre-incubation with forskolin or PGE2 resulted in 19 ± 9 or 22 ± 7% repression of NF-κB-dependent transcription, respectively (Figure 3). Similar to the previously described TNF-α plus histamine stimuli (Holden et al., 2007), treatment of BEAS-2B cells with IL-1β plus histamine resulted in a 2.1 ± 0.1-fold increase in NF-κB-dependent transcription as compared to cells treated with IL-1β alone (Figure 3). Pretreatment of cells with salbutamol, salmeterol or formoterol before stimulation with IL-1β plus histamine decreased NF-κB-dependent transcription by 41 ± 7, 47 ± 11 and 52 ± 11%, respectively (Figure 3). Pre-incubation of cells with forskolin or PGE2 decreased NF-κB-dependent transcription by 39 ± 8 and 39 ± 8%, respectively, when compared to cells treated with IL-1β plus histamine alone (Figure 3). Thus, the enhancement of IL-6 and IL-8 release by β2-adrenoceptor agonists is unlikely to be due to increases in NF-κB-dependent transcription.
Figure 3.
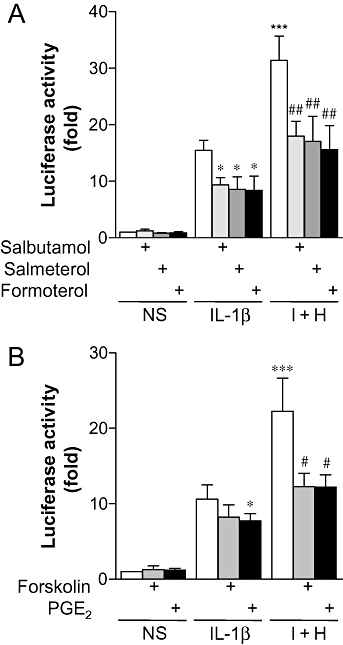
Effect of β2-adrenoceptor agonists and cAMP-elevating agents on NF-κB-dependent transcription. BEAS-2B 3κBu luciferase reporter cells were either left untreated or were incubated with: (A) salbutamol (1 µM), salmeterol (0.1 µM) or formoterol (0.01 µM), or (B) forskolin (10 µM) or PGE2 (1 µM) for 1 h prior to either being left not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1), or IL-1β (1 ng·mL−1) and histamine (100 µM) (I + H). Cells were harvested after 6 h for luciferase activity determination. Data (n= 5–6), expressed as fold activation, are plotted as means ± SEM. *P < 0.05, ***P < 0.001 versus IL-1β, ##P < 0.01 versus I + H.
Effect of LABA on AP-1- and CRE-dependent transcription
To investigate other transcription factors that may be involved in β2-adrenoceptor-mediated enhancement of IL-6 and IL-8 release, BEAS-2B cells were transiently transfected with the AP-1-dependent luciferase reporter plasmid, pGL3.neo.TATA.3AP-1. However, pre-incubation for 1 h with salmeterol had no significant effect on either histamine-, IL-1β- or IL-1β plus histamine-stimulated AP-1-dependent transcription (Figure 4A).
Figure 4.
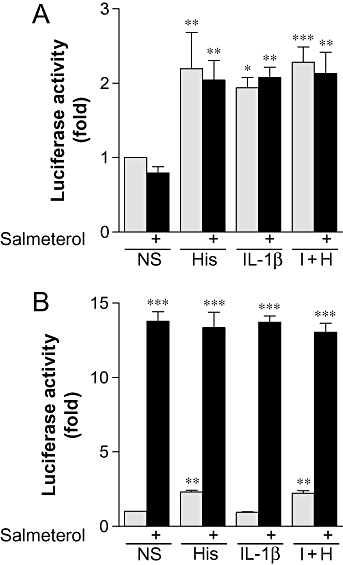
Effect of salmeterol on AP-1- and CRE-dependent transcription. BEAS-2B cells (A) transiently transfected with pGL3.neo.TATA.3AP-1 or (B) stably transfected with the CRE luciferase reporter were either left untreated or were incubated with salmeterol (0.1 µM) for 1 h prior to either being left not stimulated (NS) or stimulated with histamine (100 µM) (His), IL-1β (1 ng·mL−1) or IL-1β (1 ng·mL−1) plus histamine (100 µM) (I + H). Cells were harvested after 6 h for luciferase activity determination. Data (n= 3–5), expressed as fold activation, are plotted as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus NS.
We have previously described BEAS-2B cells as harbouring a CRE reporter that responds to salmeterol (Kaur et al., 2008). In the current study, we investigated the effects of histamine and IL-1β alone, and in combination, on basal and salmeterol-stimulated CRE-dependent transcription (Figure 4B). As previously reported (Holden et al., 2007), histamine (100 µM) stimulated CRE-dependent transcription 2.3 ± 0.1-fold, whereas IL-1β had no effect (Figure 4B). In combination, IL-1β plus histamine produced a similar response to histamine alone, with CRE-dependent transcription being stimulated 2.2 ± 0.2-fold (Figure 4B). Stimulation with salmeterol alone increased CRE-dependent transcription 13.8 ± 0.6-fold (Figure 4B). This was not significantly changed in the presence of IL-1β or histamine alone, or in combination (Figure 4B). Thus the β2-adrenoceptor agonist-dependent enhancement of IL-6 and IL-8 release is unlikely to be due to the simple enhancement of either AP-1 or CRE-dependent transcription.
Concentration dependence of LABA-enhanced IL-6 and IL-8 release
BEAS-2B cells were pre-incubated for 1 h with salmeterol (10 fM to 1 µM) prior to stimulation with IL-1β and histamine. IL-6 release was significantly enhanced by 1 pM salmeterol, and this effect reached a maximum at 100 pM with an EC50 of 280 fM (Figure 5). IL-8 release demonstrated a similar pattern and was significantly enhanced by 0.1 pM salmeterol. This response reached a maximum around 1 pM with an EC50 of 125 fM (Figure 5). Analysis of IL-1β-stimulated cells demonstrated a similar enhancement of IL-6 release by salmeterol (data not shown) with an EC50 of 7 pM. Additionally, the effects of formoterol (0.1 fM–10 nM) and forskolin (0.1 pM–10 µM) were also examined on IL-1β plus histamine-stimulated IL-6 release, yielding EC50 values of 15 pM and 704 nM, respectively (data not shown).
Figure 5.
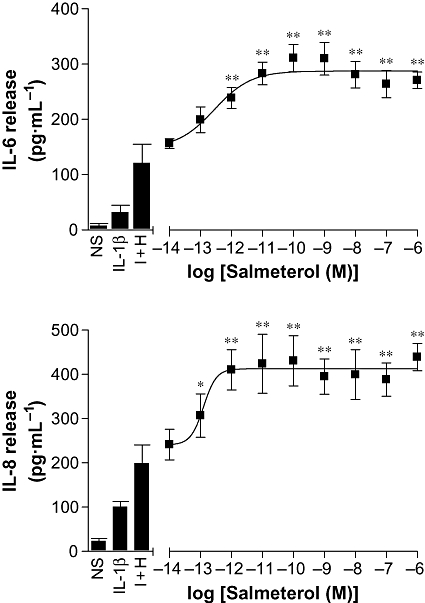
Effect of salmeterol concentration on IL-1β plus histamine-induced IL-6 and IL-8 release. BEAS-2B cells were either left untreated and either left not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1), or IL-1β (1 ng·mL−1) plus histamine (100 µM), or were incubated with various concentrations of salmeterol for 1 h prior to being stimulated with IL-1β (1 ng·mL−1) and histamine (100 µM) (I + H). Cells were harvested after 6 h, and the supernatants were analysed by elisa for IL-6 and IL-8 release. Data (n= 6), expressed as pg·mL−1, are plotted as means ± SEM. *P < 0.05, **P < 0.01 versus I + H.
Role of the β2-adrenoceptor and PKA in salmeterol-enhanced IL-6 and IL-8 release
Pretreatment of BEAS-2B cells with the selective β2-adrenoceptor antagonist, ICI 118551, revealed no significant effect on either IL-1β- or IL-1β plus histamine-stimulated IL-6 and IL-8 release (Figure 6A). Pretreatment with salmeterol significantly increased both IL-6 and IL-8 release as compared to cells stimulated with IL-1β or IL-1β plus histamine. This effect was prevented by pre-incubation with ICI 118551 and confirmed dependence on the β2-adrenoceptor (Figure 6A).
Figure 6.
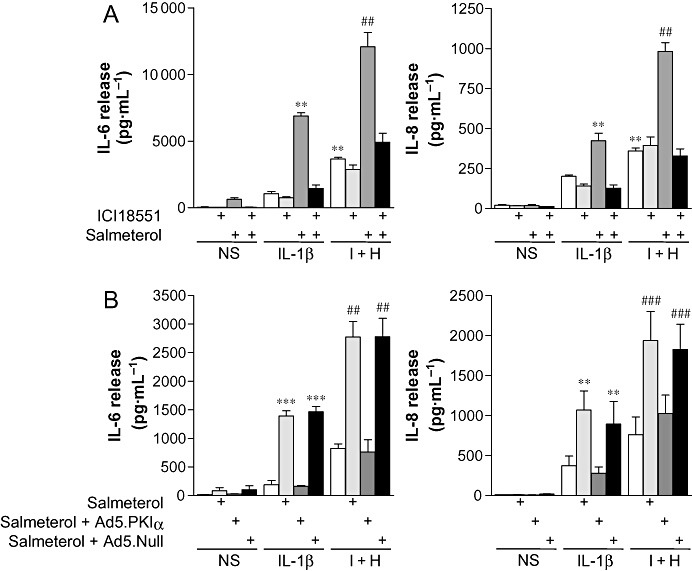
Effect of ICI 118551 and Ad5.CMV.PKIα on salmeterol-enhanced IL-6 and IL-8 release. (A) BEAS-2B cells were either left untreated or were incubated with ICI 118551 (2 µM) for 30 min. Cells were then either left untreated or were incubated with salmeterol (0.1 µM) for a further 1 h prior to either being not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1), or IL-1β (1 ng·mL−1) plus histamine (100 µM) (I + H). (B) BEAS-2B cells were either left uninfected or were infected with an MOI of 100 of Ad5.CMV.PKIα or Ad5.CMV.Null for 24 h. After incubation in serum-free media overnight, cells were then left untreated or were incubated with salmeterol (0.1 µM) for 1 h prior to either being not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1), or IL-1β (1 ng·mL−1) plus histamine (100 µM) (I + H). Cells were harvested after 6 h, and the supernatants were analysed by elisa for IL-6 and IL-8 release. Data (n= 4–5), expressed as pg·mL−1, are plotted as means ± SEM. **P < 0.01, ***P < 0.001 versus IL-1β, ##P < 0.01, ###P < 0.001 versus I + H.
In BEAS-2B cells infected with Ad5.CMV.Null virus, pre-incubation with salmeterol significantly increased IL-6 and IL-8 release when compared to cells stimulated with IL-1β alone (Figure 6B). This effect was indistinguishable from that produced by naïve cells treated in the same manner. However, when cells were infected with Ad5.CMV.PKIa, which expresses PKIα, a highly selective inhibitor of PKA, salmeterol no longer enhanced the IL-1β-stimulated release of IL-6 and IL-8 (Figure 6B). Identical effects were also found in cells stimulated with IL-1β plus histamine, with salmeterol significantly enhancing release from both naïve and Ad5.CMV.null-infected cells (Figure 6B), yet having no significant effect on Ad5.CMV.PKIα-infected cells (Figure 6B). Thus, β2-adrenoceptor agonist-dependent increases in IL-6 and IL-8 release are contingent on activation of PKA.
Effect of dexamethasone on salmeterol- and histamine-enhanced IL-1β-induced IL-6 and IL-8 release
BEAS-2B cells were stimulated with the indicated concentrations of IL-1β, which alone caused concentration-dependent increases in IL-6 and IL-8 release (Figure 7, left panels). The addition of histamine further increased IL-6 and IL-8 release at all concentrations of IL-1β studied (Figure 7, right panels). Pre-incubation of BEAS-2B cells with salmeterol prior to IL-1β treatment increased IL-6 release only at a concentration of 1 ng·mL−1 IL-1β (Figure 7, upper left panel). Similarly, salmeterol increased IL-1β-stimulated IL-8 release at concentrations of 0.3 and 1 ng·mL−1 of IL-1β (Figure 7, lower left panel). However, pre-incubation of cells with salmeterol prior to histamine stimulation in the presence of various IL-1β concentrations increased IL-6 and IL-8 release at all concentrations of IL-1β (Figure 7, right panels).
Figure 7.
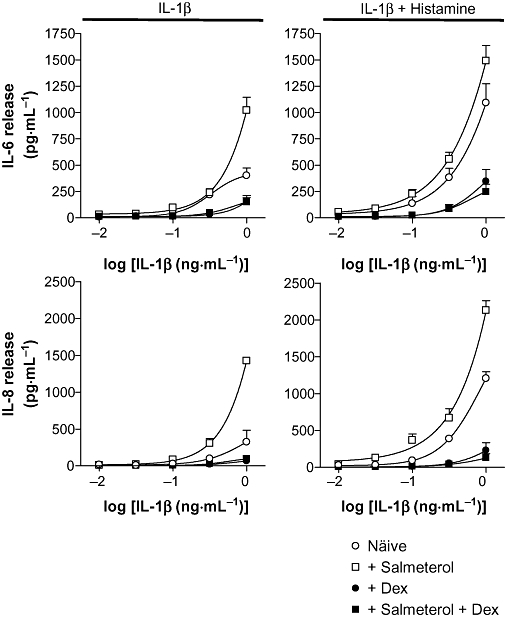
Effect of dexamethasone on salmeterol and histamine-enhanced IL-1β-induced IL-6 and IL-8 release. BEAS-2B cells were either left untreated (○) or were incubated with: salmeterol (0.1 µM) (□), dexamethasone (1 µM) (•) or a combination of salmeterol (0.1 µM) plus dexamethasone (1 µM) ( ) for 1 h, prior to stimulation with various concentrations of IL-1β (0.01–1 ng·mL−1) in the absence and presence of histamine (100 µM). Cells were harvested after 6 h, and the supernatants were analysed by elisa for IL-6 and IL-8 release. Data (n= 4), expressed as pg·mL−1, are plotted as means ± SEM.
) for 1 h, prior to stimulation with various concentrations of IL-1β (0.01–1 ng·mL−1) in the absence and presence of histamine (100 µM). Cells were harvested after 6 h, and the supernatants were analysed by elisa for IL-6 and IL-8 release. Data (n= 4), expressed as pg·mL−1, are plotted as means ± SEM.
As LABAs are typically prescribed in combination with a glucocorticoid, the effect of adding dexamethasone was examined. In each case, pre-incubation with a maximally effective concentration of dexamethasone (1 µM) prior to stimulation, both in the absence and presence of salmeterol, reduced the release of IL-6 and IL-8 to basal levels with only the highest concentration of IL-1β causing detectable release of IL-6 and IL-8 (Figure 7). In each case (IL-1β alone, or IL-1β plus histamine), the presence of dexamethasone diminished the response to salmeterol such that IL-1β- and IL-1β plus histamine-treated cells gave rise to responses that were indistinguishable from the corresponding treatment in the presence of salmeterol.
The effect of time on the salmeterol-enhanced and dexamethasone-dependent inhibition of IL-6 and IL-8 release
In the experiments above, β2-adrenoceptor agonists were added 1 h prior to cell stimulation. To investigate the effect of time of addition, BEAS-2B cells were incubated with salmeterol (0.1 µM) at various times in relation to the IL-1β plus histamine stimulus. IL-6 release was significantly increased by the addition of salmeterol at all the times examined compared to cells treated with IL-1β plus histamine alone. This effect appeared maximal when salmeterol was added simultaneously with IL-1β plus histamine (Figure 8). Even when added 4 h post-stimulation, salmeterol still significantly increased IL-6 release compared to cells treated with IL-1β plus histamine alone (Figure 8). Conversely, IL-8 release was maximally, and significantly, enhanced when salmeterol was added 4 h prior to the IL-1β plus histamine stimulation compared to that in the presence of IL-1β plus histamine alone (Figure 8). With shorter pre-incubation times, this effect was progressively lost, until the simultaneous incubation of salmeterol with the IL-1β plus histamine was indistinguishable from cells treated with IL-1β plus histamine alone (Figure 8). This loss of enhancement remained at all time-points thereafter.
Figure 8.
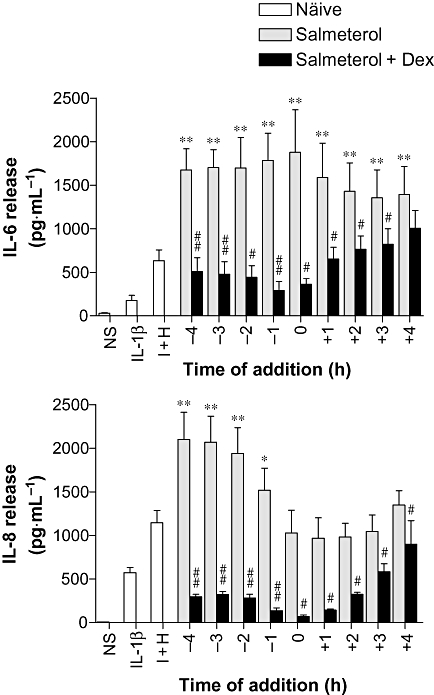
Effect of time of salmeterol ± dexamethasone addition on IL-6 and IL-8 release. BEAS-2B cells were either left untreated (naïve) or were incubated with salmeterol (0.1 µM) in the absence or presence of dexamethasone (1 µM) added at various times (-4 to +4 h) in relation to IL-1β (1 ng·mL−1) plus histamine (100 µM) (I + H) stimulus (added at time 0). Cells were also left not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1) alone. Cells were harvested 6 h post-stimulation, and the supernatants were analysed by elisa for IL-6 and IL-8 release. Data (n= 6), expressed as pg·mL−1, are plotted as means ± SEM. *P < 0.05, **P < 0.01 versus I + H, #P < 0.05, ##P < 0.01 versus corresponding salmeterol-treated sample.
To investigate whether the addition of glucocorticoid was effective at inhibiting the salmeterol-enhanced release of IL-6 and IL-8 at each of these times, BEAS-2B cells were incubated with dexamethasone (1 µM) added simultaneously with the salmeterol. For IL-8, simultaneous addition of dexamethasone and salmeterol significantly inhibited release compared to cells treated with salmeterol alone (Figure 8). Similarly, IL-6 release was also significantly inhibited by the addition of dexamethasone, except when added 4 h post-stimulation. This inhibitory effect was maximal around the time of IL-1β plus histamine treatment (t= 0), after which the addition of dexamethasone became less effective. These patterns of salmeterol-induced enhancement of cytokine release and repression by the addition of a glucocorticoid were maintained at 24 h post-stimulation (Supporting Information Figure S3). By 48 h, the relative enhancement by salmeterol was reduced, but repression in the presence of dexamethasone was maintained (Supporting Information Figure S3).
Discussion
A number of studies have indicated that β2-adrenoceptor agonists increase the release of inflammatory mediators, including IL-4, TNF-α, IL-6 and IL-8, in the context of inflammatory stimuli such as rhinovirus, IL-1β and organic dust (Edwards et al., 2007; Strandberg et al., 2007). However, inflammation is a complex process, which involves numerous mediators that may interact in a combinatorial manner. Thus, histamine can synergistically enhance TNF-α-stimulated release of both IL-6 and IL-8 (Holden et al., 2007). Consequently, in the current study, we have examined the effects of β2-adrenoceptor agonists on IL-6 and IL-8 release from human bronchial epithelial BEAS-2B cells, stimulated with IL-1β and histamine. As previously described (Edwards et al., 2007), we showed that pretreatment of BEAS-2B cells with salmeterol enhanced the release of IL-6 induced by IL-1β. Furthermore this effect extended to IL-8 release, which was also enhanced by salmeterol (Edwards et al., 2007). However, in the context of IL-1β plus histamine stimulation, the release of both IL-6 and IL-8 was substantially further increased following pre-incubation with β2-adrenoceptor agonists. Thus, the effects of β2-adrenoceptor agonists on IL-6 and IL-8 expression may have been considerably underestimated in previous biological model systems, by not taking into account the interactions of several inflammatory mediators. Paradoxically, there are indications of both pro- and anti-inflammatory effects for β2-adrenoceptor agonists. Thus, in airway smooth muscle, salbutamol and salmeterol inhibited eotaxin expression by preventing histone H4 acetylation and NF-κB activation (Nie et al., 2005). Similarly, ‘anti-inflammatory’ or repressive effects of β2-adrenoceptor agonists were attributed to the inhibition of the IκBα/NF-κB pathway in monocytic cells and myofibroblasts (Farmer and Pugin, 2000; Baouz et al., 2005). Conversely, salbutamol may induce IκBα phosphorylation and the activation of NF-κB-dependent transcription in airway smooth muscle cells (Agrawal et al., 2004). In the current study, β2-adrenoceptor agonists and other cAMP-elevating agents significantly decreased NF-κB-dependent transcription in BEAS-2B cells. Thus, despite both IL-6 and IL-8 being highly NF-κB dependent in BEAS-2B cells (Holden et al., 2007), the enhancement of IL-6 and IL-8 release cannot be readily explained by increased NF-κB-dependent transcription. In addition to NF-κB, the promoters of both IL-6 and IL-8 contain several other putative transcription factor binding sites. Thus, the IL-8 gene promoter contains sites for AP-1, C/EBP, Oct – 1 and NFAT-1 (Roebuck, 1999), whereas the IL-6 promoter contains IRF-1, AP-1, CREB, C/EBP and Sp1 binding sites (see Ammit et al., 2002; Holden et al., 2007 and references therein). However, salmeterol had no discernible effects on either basal or stimulated AP-1-dependent transcription, and, although histamine alone enhanced CRE-dependent transcription approximately twofold over basal levels, this is an effect we have previously demonstrated to occur through activation of the Gsα-linked H2 receptor (Holden et al., 2007). There was no apparent effect on salmeterol-stimulated CRE luciferase activity by either histamine or IL-1β. Thus, the increased release of IL-6 and IL-8 also cannot be readily explained by increased AP-1- or CRE-dependent transcription occurring via a single, simple, cis-acting site. Despite this, the enhancement of TNF-α-induced IL-6 release by β2-adrenoceptor agonists in airway smooth muscle cells was found to be dependent on the activation of a CRE element in the IL-6 promoter (Ammit et al., 2002). Thus, it is possible that enhancement of IL-6 transcription by cAMP-elevating agents may involve cross-talk between factors binding at a CRE site and other key sites. However, such a scheme is unlikely to account for the observed increases in IL-8 expression because no CRE has, to date, been described for this chemokine (Roebuck, 1999).
An alternative, and possibly unifying, explanation may rest in the fact that both IL-6 and IL-8 promoters contain C/EBP elements. Thus, the increased release of IL-6 and IL-8 could be due to the β2-adrenoceptor agonist enhancing C/EBP-dependent transcription. Indeed, there are several reports suggesting that the cAMP/PKA pathway plays a major role in the regulation of both C/EBP expression and function. For example, both C/EBP-β and C/EBP-δ mRNA are induced in a cAMP-dependent fashion in various cell types (Cardinaux and Magistretti, 1996; Cantwell et al., 1998; Vogel et al., 2004). In addition, the C/EBP-β protein contains a number of PKA-specific phospho-acceptor sites that can modulate the DNA binding affinity of this factor (Trautwein et al., 1994). However, the possibility of multiple transcription factors acting together in a synergistic fashion, either between themselves or with other proteins such as the CREB binding protein (CBP) must also be considered. Further studies are needed to explore this.
The marked sensitivity of BEAS-2B cells to the β2-adrenoceptor agonists studied is a novel and surprising finding. This was shown by EC50 values for enhancement of IL-6 and IL-8 that were considerably lower than previously reported for the activation of CRE-dependent transcription in these cells (Kaur et al., 2008). Furthermore, because such concentrations may be readily achievable in the lung following inhaled therapy (Johnson and Rennard, 2001; Todorova et al., 2006), it is likely that such effects could occur in individuals taking β2-adrenoceptor agonist monotherapy. This potency of salmeterol is, to our knowledge, unprecedented, and this phenomenon warrants further investigation.
The ablation of salmeterol-dependent enhancement of IL-6 and IL-8 by PKIα over-expression demonstrates that this pathway is PKA dependent. A number of previous reports have suggested a role for PKA either by using cAMP analogues, PDE inhibitors and forskolin (Edwards et al., 2007), or by use of the pharmacological PKA inhibitor H-89 (Tan et al., 2007). While the use of forskolin, PDE inhibitors and cAMP analogues clearly demonstrates that the enhancement of inflammatory mediators may be cAMP dependent, they do not per se denote PKA dependence as numerous studies now document the existence of cAMP-dependent, but PKA-independent pathways (see Giembycz and Newton, 2006). Furthermore, the use of the H-89 to investigate the role of PKA is compromised by a number of off-target effects that include the inhibition of protein kinases such as mitogen- and stress-activated protein kinase 1, p70 ribosomal protein S6 kinase 1 and Rho-dependent protein kinase II at potencies greater than, or similar to, that for PKA (Davies et al., 2000), as well as direct antagonism of the β2-adrenoceptor (Penn et al., 1999).
The cAMP dependence of the enhancement of IL-6 and IL-8 also suggests that other cAMP-elevating agents such as PDE4 inhibitors may have similar effects. In this respect, the PDE4 inhibitor rolipram was shown to increase the expression of IL-6 in BEAS-2B cells (Edwards et al., 2007). Additionally, other cAMP-elevating agents also increase inflammatory mediator release. Thus PGE2, acting via the EP2 and EP4 receptors, increases the expression of G-CSF in a PKA-dependent manner (Clarke et al., 2005). Consequently, when new cAMP-elevating therapies are evaluated, their combination with a glucocorticoid should be carefully considered in order to counteract any possible adverse pro-inflammatory effects.
Although current asthma guidelines recommend the use of LABAs only in the presence of a glucocorticoid, short-acting β2-adrenoceptor agonists are prescribed as an ‘as-needed’ medication (Bateman et al., 2008). However, more recently, there have also been moves to replace this monotherapy with a β2-adrenoceptor agonist and glucocorticoid combination inhaler (Papi et al., 2009). In the current study, we demonstrate that the simultaneous addition of dexamethasone with a β2-adrenoceptor agonist reverses the β2-adenoceptor agonist-dependent enhancement of IL-6 and IL-8 that is observed in the presence of IL-1β and IL-1β plus histamine. Thus, our data strongly support the use of combination inhalers as rescue therapy because this may prevent any β2-adrenoceptor agonist-dependent increases in the underlying inflammation.
Another intriguing aspect of this study is the different temporal patterns of IL-6 and IL-8 enhancement by β2-adrenoceptor agonists. While IL-6 release was significantly enhanced by all pre- and post-treatment times, and addition even 4 h following the IL-1β plus histamine stimulus, IL-8 release was only enhanced by β2-adrenoceptor agonist pretreatments. Thus, it is possible that the enhancement of IL-6 can occur at any stage in the gene expression process up to and most likely including cytokine translation. Conversely, the enhancement of IL-8 is totally lost once gene transcription has commenced, and this raises the prospect of a purely transcriptional mechanism. The further elucidation of these novel findings will undoubtedly shed light on different facets of the regulation of these two important inflammatory genes.
In summary, we have shown β2-adrenoceptor agonist-dependent enhancement of IL-1β-stimulated, and IL-1β plus histamine-stimulated IL-6 and IL-8 expression at concentrations that are readily achieved in clinical practice (Johnson and Rennard, 2001; Todorova et al., 2006). These effects were mimicked by forskolin and PGE2, and prevented by PKIα, thereby implicating a key role for the cAMP–PKA cascade. Despite the fact that IL-6 and IL-8 are highly NF-κB-dependent genes, β2-adrenoceptor agonists, as well as forskolin and PGE2, all inhibited NF-κB-dependent transcription. Finally, whereas β2-adrenoceptor agonists enhanced the release of IL-6 and IL-8, we also showed that addition of a glucocorticoid effectively prevented this enhancement. Because NHBE cells grown at an air–liquid interface also revealed enhanced cytokine release in the presence of LABA, these findings strongly support the clinical use of glucocorticoid and β2-adrenoceptor agonist combination inhalers to prevent adverse inflammatory effects of β2-adrenoceptor agonists.
Acknowledgments
This work was supported by an establishment grant from the Alberta Heritage Foundation for Medical Research (AHFMR) and operating funds from the Canadian Institutes of Health Research (CIHR). N.S.H. is an Izaac Walton Killam postdoctoral fellow. R.N. is an AHFMR scholar and CIHR new investigator. M.A.G. is an AHFMR senior scholar.
Glossary
Abbreviations:
- AP-1
activator protein-1
- CBP
CREB binding protein
- CRE
cyclic AMP response element
- CREB
CRE binding protein
- DMSO
dimethylsulphoxide
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IL
interleukin
- MOI
multiplicity of infection
- NF-κB
nuclear factor kappa B
- NHBE
normal human bronchial epithelial
- PKA
protein kinase A
Conflicts of interest
R.N. and M.A.G. have received research funding from GlaxoSmithKline, AstraZeneca and Nycomed Pharmaceuticals. J.V. and M.S. are employees of Gilead Sciences Inc. The authors are unaware of any other conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of β2-adrenoceptor agonists and cAMP-elevating agents on the release of IL-6 and IL-8. BEAS-2B cells were either left untreated or were incubated with: (A) salbutamol (1 µM), salmeterol (0.1 µM) or formoterol (0.01 µM), or (B) forskolin (10 µM) or PGE2 (1 µM) for 1 h prior to being left non-stimulated (NS). Cells were harvested after 6 h, and the supernatants were analysed by ELISA for IL-6 and IL-8 release. Data (n = 4–6), expressed as pg·mL−1, are plotted as means ± SEM.
Figure S2 Effect of COX inhibition on the enhancement of cytokine release. BEAS-2B CRE luciferase reporter cells were either left untreated or were pre-incubated with either indomethacin (10 µM) or diclofenac (10 µM) for 30 min prior to either being left untreated, or treatment with salmeterol (0.1 µM) (S). After a further hour, cells were either not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1) or IL-1β (1 ng·mL−1) plus histamine (100 µM) (I + H). Cells were harvested after 6 h, and IL-6 in the supernatants was analysed by ELISA. Data (n = 4), expressed as pg·mL−1, are plotted as means ± SEM. Cell lysates were analysed for luciferase activity determination. Data (n = 4), expressed as fold activation, are plotted as means ± SEM.
Figure S3 The effect of time of addition on the salmeterol-enhanced and dexamethasone-dependent inhibition of IL-6 release at 24 and 48 h. BEAS-2B cells were either left untreated (naïve) or were incubated with salmeterol (0.1 µM) in the absence or presence of dexamethasone (1 µM) added together at various times (−4 to +4 h) in relation to the IL-1β (1 ng·mL−1) plus histamine (100 µM) (I + H) stimulus (added at time 0). Cells were also not stimulated (NS) or stimulated with IL-1β (1 ng·mL−1) alone. Cells were harvested at either (A) 24 h or (B) 48 h post-stimulation, and the supernatants were analysed for IL-6 release by ELISA. Data (n = 4), expressed as pg·mL−1, are plotted as means ± SEM. *P < 0.05, **P < 0.01 versus I + H, #P < 0.05, ##P < 0.01 versus relevant salmeterol-treated sample.
Table S1 NHBE cells, grown to confluence at an air–liquid interface, were either not stimulated (untreated) or were either pre-incubated with salmeterol (10 µM) or vehicle (0.1% DMSO) for 4 h prior to stimulation with poly I:C (100 mg·mL−1) as indicated. Basolateral supernatants were harvested at 24 h post-stimulation, and IL-6, IL-8, TNFα, MCP-1, IL-1β, ENA78 and G-CSF release was measured by the Procarta cytokine assay kit. Data (n = 3–6) in pg·mL−1 are expressed as means ± SEM. Significance between untreated and poly I:C + vehicle is indicated as P < 0.05 (*), P < 0.01 (**). Significance between poly I:C + vehicle and poly I:C + salmeterol is indicated as P < 0.05 (#), P < 0.01 (##).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Agrawal DK, Ariyarathna K, Kelbe PW. (S)-Albuterol activates pro-constrictory and pro-inflammatory pathways in human bronchial smooth muscle cells. J Allergy Clin Immunol. 2004;113:503–510. doi: 10.1016/j.jaci.2003.12.039. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. 4th edn. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammit AJ, Lazaar AL, Irani C, O'Neill GM, Gordon ND, Amrani Y, et al. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smooth muscle cells: modulation by glucocorticoids and beta-agonists. Am J Respir Cell Mol Biol. 2002;26:465–474. doi: 10.1165/ajrcmb.26.4.4681. [DOI] [PubMed] [Google Scholar]
- Baouz S, Giron-Michel J, Azzarone B, Giuliani M, Cagnoni F, Olsson S, et al. Lung myofibroblasts as targets of salmeterol and fluticasone propionate: inhibition of alpha-SMA and NF-kappaB. Int Immunol. 2005;17:1473–1481. doi: 10.1093/intimm/dxh325. [DOI] [PubMed] [Google Scholar]
- Bateman ED, Hurd SS, Barnes PJ, Bousquet J, Drazen JM, FitzGerald M, et al. Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J. 2008;31:143–178. doi: 10.1183/09031936.00138707. [DOI] [PubMed] [Google Scholar]
- Cantwell CA, Sterneck E, Johnson PF. Interleukin-6-specific activation of the C/EBPdelta gene in hepatocytes is mediated by Stat3 and Sp1. Mol Cell Biol. 1998;18:2108–2117. doi: 10.1128/mcb.18.4.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinaux JR, Magistretti PJ. Vasoactive intestinal peptide, pituitary adenylate cyclase-activating peptide, and noradrenaline induce the transcription factors CCAAT/enhancer binding protein (C/EBP)-beta and C/EBP delta in mouse cortical astrocytes: involvement in cAMP-regulated glycogen metabolism. J Neurosci. 1996;16:919–929. doi: 10.1523/JNEUROSCI.16-03-00919.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catley MC, Cambridge LM, Nasuhara Y, Ito K, Chivers JE, Beaton A, et al. Inhibitors of protein kinase C (PKC) prevent activated transcription: role of events downstream of NF-kappaB DNA binding. J Biol Chem. 2004;279:18457–18466. doi: 10.1074/jbc.M400765200. [DOI] [PubMed] [Google Scholar]
- Chivers JE, Gong W, King EM, Seybold J, Mak JC, Donnelly LE, et al. Analysis of the dissociated steroid RU24858 does not exclude a role for inducible genes in the anti-inflammatory actions of glucocorticoids. Mol Pharmacol. 2006;70:2084–2095. doi: 10.1124/mol.106.025841. [DOI] [PubMed] [Google Scholar]
- Clarke DL, Belvisi MG, Smith SJ, Hardaker E, Yacoub MH, Meja KK, et al. Prostanoid receptor expression by human airway smooth muscle cells and regulation of the secretion of granulocyte colony-stimulating factor. Am J Physiol Lung Cell Mol Physiol. 2005;288:L238–L250. doi: 10.1152/ajplung.00313.2004. [DOI] [PubMed] [Google Scholar]
- Crane J, Pearce N, Flatt A, Burgess C, Jackson R, Kwong T, et al. Prescribed fenoterol and death from asthma in New Zealand, 1981–1983: case-control study. Lancet. 1989;1:917–922. doi: 10.1016/s0140-6736(89)92505-1. [DOI] [PubMed] [Google Scholar]
- Davies DE, Holgate ST. Asthma: the importance of epithelial mesenchymal communication in pathogenesis. Inflammation and the airway epithelium in asthma. Int J Biochem Cell Biol. 2002;34:1520–1526. doi: 10.1016/s1357-2725(02)00048-1. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MR, Haas J, Panettieri RA, Jr, Johnson M, Johnston SL. Corticosteroids and beta2 agonists differentially regulate rhinovirus-induced interleukin-6 via distinct cis-acting elements. J Biol Chem. 2007;282:15366–15375. doi: 10.1074/jbc.M701325200. [DOI] [PubMed] [Google Scholar]
- Farmer P, Pugin J. Beta-adrenergic agonists exert their ‘anti-inflammatory’ effects in monocytic cells through the IkappaB/NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol. 2000;279:L675–L682. doi: 10.1152/ajplung.2000.279.4.L675. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Newton R. Beyond the dogma: novel beta2-adrenoceptor signalling in the airways. Eur Respir J. 2006;27:1286–1306. doi: 10.1183/09031936.06.00112605. [DOI] [PubMed] [Google Scholar]
- Hayden P, Kubilus J, Kandárová H, Klausner M, Jackson G, Bolmarcich J. An in vitro model of human airway epithelium (EpiAirway) for in vitro metabolism and toxicity screening. Toxicol Lett. 2007;172:S79. [Google Scholar]
- Holden NS, Gong W, King EM, Kaur M, Giembycz MA, Newton R. Potentiation of NF-kappaB-dependent transcription and inflammatory mediator release by histamine in human airway epithelial cells. Br J Pharmacol. 2007;152:891–902. doi: 10.1038/sj.bjp.0707457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen-Heininger YM, Poynter ME, Aesif SW, Pantano C, Ather JL, Reynaert NL, et al. Nuclear factor kappaB, airway epithelium, and asthma: avenues for redox control. Proc Am Thorac Soc. 2009;6:249–255. doi: 10.1513/pats.200806-054RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M, Rennard S. Alternative mechanisms for long-acting beta(2)-adrenergic agonists in COPD. Chest. 2001;120:258–270. doi: 10.1378/chest.120.1.258. [DOI] [PubMed] [Google Scholar]
- Kaur M, Chivers JE, Giembycz MA, Newton R. Long-acting beta2-adrenoceptor agonists synergistically enhance glucocorticoid-dependent transcription in human airway epithelial and smooth muscle cells. Mol Pharmacol. 2008;73:203–214. doi: 10.1124/mol.107.040121. [DOI] [PubMed] [Google Scholar]
- Meja KK, Catley MC, Cambridge LM, Barnes PJ, Lum H, Newton R, et al. Adenovirus-mediated delivery and expression of a cAMP-dependent protein kinase inhibitor gene to BEAS-2B epithelial cells abolishes the anti-inflammatory effects of rolipram, salbutamol, and prostaglandin E2: a comparison with H-89. J Pharmacol Exp Ther. 2004;309:833–844. doi: 10.1124/jpet.103.060020. [DOI] [PubMed] [Google Scholar]
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM. The salmeterol multicenter asthma research trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest. 2006;129:15–26. doi: 10.1378/chest.129.1.15. [DOI] [PubMed] [Google Scholar]
- Nie M, Knox AJ, Pang L. Beta2-adrenoceptor agonists, like glucocorticoids, repress eotaxin gene transcription by selective inhibition of histone H4 acetylation. J Immunol. 2005;175:478–486. doi: 10.4049/jimmunol.175.1.478. [DOI] [PubMed] [Google Scholar]
- Papi A, Caramori G, Adcock IM, Barnes PJ. Rescue treatment in asthma. More than as-needed bronchodilation. Chest. 2009;135:1628–1633. doi: 10.1378/chest.08-2536. [DOI] [PubMed] [Google Scholar]
- Penn RB, Parent JL, Pronin AN, Panettieri RA, Jr, Benovic JL. Pharmacological inhibition of protein kinases in intact cells: antagonism of beta adrenergic receptor ligand binding by H-89 reveals limitations of usefulness. J Pharmacol Exp Ther. 1999;288:428–437. [PubMed] [Google Scholar]
- Roebuck KA. Regulation of interleukin-8 gene expression. J Interferon Cytokine Res. 1999;19:429–438. doi: 10.1089/107999099313866. [DOI] [PubMed] [Google Scholar]
- Sears MR, Lotvall J. Past, present and future – beta2-adrenoceptor agonists in asthma management. Respir Med. 2005;99:152–170. doi: 10.1016/j.rmed.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Sears MR, Taylor DR. The beta 2-agonist controversy. Observations, explanations and relationship to asthma epidemiology. Drug Saf. 1994;11:259–283. doi: 10.2165/00002018-199411040-00005. [DOI] [PubMed] [Google Scholar]
- Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–364. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- Stolley PD, Schinnar R. Association between asthma mortality and isoproterenol aerosols: a review. Prev Med. 1978;7:519–538. doi: 10.1016/0091-7435(78)90265-7. [DOI] [PubMed] [Google Scholar]
- Strandberg K, Palmberg L, Larsson K. Effect of formoterol and salmeterol on IL-6 and IL-8 release in airway epithelial cells. Respir Med. 2007;101:1132–1139. doi: 10.1016/j.rmed.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. Beta2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal. 2007;19:251–260. doi: 10.1016/j.cellsig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Todorova L, Gurcan E, Miller-Larsson A, Westergren-Thorsson G. Lung fibroblast proteoglycan production induced by serum is inhibited by budesonide and formoterol. Am J Respir Cell Mol Biol. 2006;34:92–100. doi: 10.1165/rcmb.2005-0048OC. [DOI] [PubMed] [Google Scholar]
- Trautwein C, van der Geer P, Karin M, Hunter T, Chojkier M. Protein kinase A and C site-specific phosphorylations of LAP (NF-IL6) modulate its binding affinity to DNA recognition elements. J Clin Invest. 1994;93:2554–2561. doi: 10.1172/JCI117266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Sciullo E, Park S, Liedtke C, Trautwein C, Matsumura F. Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J Biol Chem. 2004;279:8886–8894. doi: 10.1074/jbc.M310190200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.