

Abstract
This perspective article is an opportunity to explain a new genetic finding for late-onset Alzheimer s disease (LOAD). This invited perspective is specifically writtenfor physicians and scientists who are interested in LOAD, but it may be relevant to those interested in identifying susceptibility variants for other complex diseases. The significant finding discussed here is that a variable–length, deoxythymidine homopolymer (poly-T) within intron 6 of the TOMM40 gene is associated with the age of onset of LOAD [3]. This result was obtained with a phylogenetic study of the genetic polymorphisms that reside within the linkage disequilibrium [LD] block that contains TOMM40, APOE, and APOC1 genes on each chromosome from patients with LOAD and age-matched subjects without disease. While the data will have diagnostic, prognostic and therapeutic strategy implications, this perspective is meant to place the inheritance pattern for this complex human disease into context, and to highlight the potential utility of applying phylogenetic tools to the study of the genetics of complex diseases.
Introduction and Discussion
The thesis that genetic predisposition to complex diseases results from small genetic effects from multiple loci has now been tested in a very large number of genome-wide association studies (GWAS) with varying degrees of success [4]. GWAS test up to a million relatively common SNPs, individually, for association with a phenotype. This large number of association tests increases the rate of false discoveries and each test incurs a statistical correction penalty, which limits the power to detect a significant associations. These studies have reported, with a couple of notable exceptions, that common SNPs impart only small risk of disease, and that the association signal may not be readily reproduced in independent cohorts. Certainly, to date, there are no GWAS results with the robust effect size and reproducible significance of the association of the TOMM40-APOE-APOC1 region and risk of LOAD. This risk is attributed to the 4 allele of APOE (APOE4) [1].
Even now, with several thousand patients and unaffected subjects tested in GWAS of LOAD, other association intervals show only borderline statistical significance and relatively low effect size. See, for example, Table 1 which presents the results of a recent GWAS of LOAD [2]. The associations between SNPs within the APOE LD region with disease are highly significant (P values less than 10−37 to 10−157) with odds ratios ranging from 0.6 to >2. The significance values for the associations of other loci with this disease are markedly smaller (P values less than 10−8 to 10−9) with odds ratios much closer to 1. While the large size of GWAS typically allows detection of these modest association signals, and although many of the recent LOAD studies have received attention in the public press, the utility of these modest associations is not clear.
Table 1. Example of GWAS results for LOAD.
The genes in the vertical red box are within the APOE LD region. The SNP indicated in the horizontal red box is the SNP for which there is the best statistical support as a separator of the two clades on the phylogenetic tree for the TOMM40-APOE region (indicated in Figure 1). Modified from Harold et al. 2009 [2]).
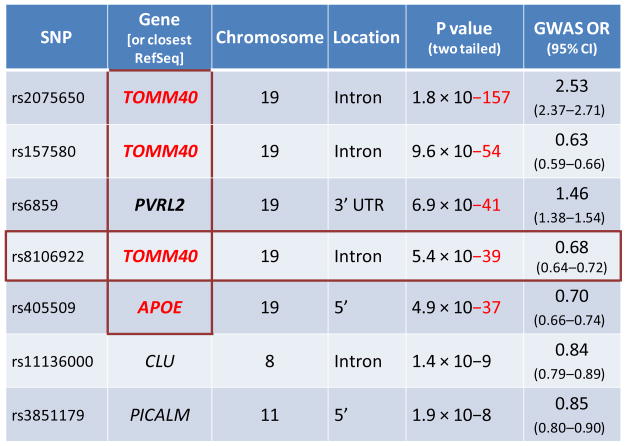 |
All nine published LOAD GWAS demonstrated a highly significant association between SNPs in the APOE LD region and LOAD [2, 5–12]. It should be emphasized, however, that the specific polymorphisms that identify the 3 and 4 alleles of APOE are not included on the platforms used in these GWAS. Nevertheless, it was assumed in eight of the studies that the highly significant SNP association signals from the “APOE” region were due to the robust disease risk signal previously described for APOE4. Only one of these GWAS studies referred to other potential candidate genes within the APOE LD region [12]
The discovery that the TOMM40 gene, encoding an essential mitochondrial protein [The Outer Mitochondrial Membrane translocase], was adjacent to and in strong LD with APOE [13] spurred a series of important biological studies. An apoE4 peptide was shown to interact with mitochondria, specifically with the outer membrane [14], and apoE or apoE fragments was shown to induce mitochondrial toxicity and reduce in mitochondrial motility [15, 16]. It has also been demonstrated that amyloid beta precursor protein (APP) interacts with mitochondrial translocase-containing complexes [17, 18] and accumulates in the outer and inner mitochondrial import channels leading to mitotoxicity [18]. In the brains of LOAD patients, the amount of APP caught in the channels varied with disease severity and patient genotype, with APOE3/4 patients having the highest amount of mitochondrial APP accumulation [18]. It is possible that apoE isoforms influence the targeting of APP to mitochondria, perhaps via differential binding between APP/apoE isomers [19–21] or differential interaction between apoE truncated peptide fragment isoforms and mitochondria. The molecular and genetic studies that implicated mitochondrial dysfunction in LOAD pathogenesis were complemented by positron emission tomography (PET) studies that demonstrated reduced glucose utilization in the brains of Alzheimer s Disease patients or in cognitively normal people who were carriers of APOE4 [22, 23]. There is also APOE4 dose-dependent decrease in glucose consumption in affected brain regions, and lack of efficacy of a PPAR agonist for the treatment of LOAD for carriers of APOE4 [23, 24].
Since coincidences do not generally occur in nature, the biological data suggest that the large, highly significant and reproducible disease susceptibility signal associated with the APOE LD region is due to contributions from both the TOMM40 and APOE genes to the disease mechanism or pathway [12, 25–27].
Phylogenetic strategies are widely used outside of human genetics for the analysis of the evolution of simpler organisms with smaller genomes and shorter generation times, for viruses and bacteria for example [28–31]. Practical uses of phylogenetics in human medicine include the study of the evolution of the H. influenza virus to generate the annual vaccines used to prevent infection and for elucidating the evolution of drug resistance in the human immunodeficiency virus [31–34]. Phylogenetic approaches for analyzing human genomic DNA sequences reveal the spatial relationships of genetic variants that occur on chromosomes with different mutation histories. In the special case of a region of high LD (or low recombination), the mutations stay connected during evolution. This pattern of co-inheritance can provide a view of the order (or timing) for each mutation in a region of interest.
A phylogenetic approach to ordering mutations that occurred over evolutionary time can show how mutations, rare and common, relate to each other and these relationships are presented in the form of a phylogenetic tree. The tree or phylogeny illustrated in Figure 1 is more appropriately a genealogy since it is constructed from DNA sequences from one species, human, and is constructed from DNA sequences from the APOE LD region from LOAD patients and controls. Each node in the tree represents the ancestral sequence of the two lineages that branch from it. At the tips of this tree are clusters of highly similar sequences from the APOE LD region of all of the sampled chromosomes.
Figure 1. Phylogenetic tree that resolved with strong bootstrap support and without prior consideration of APOE allele for a 10 Kb region in the TOMM40-APOE LD block.

A and B refer to the two clades at the first branch point; the boxed regions identify haplogroups of the indicated APOE allele and poly-T length polymorphism at rs10524523. Note that APOE3-long “523” and APOE4 haplotypes separate into clade A.
Events such as mutation and recombination introduce diversity in each chromosome and each of these events is subsequently inherited by chromosomes in that lineage. If a mutation causes susceptibility to a disease, then this variant will tend to be clustered into one lineage on a phylogenetic tree. It then follows that chromosomes of affected and non-affected individuals will be non-randomly distributed on the phylogeny [35]. If a phylogeny is developed from a genomic region that is known to be associated with disease then each cluster of related haplotypes on the tree can be tested for association with a phenotype of interest, and the variants that distinguish each cluster can be further analyzed. This clustering technique ensures that infrequent or rare mutations are not lost to the analysis. In a GWAS, where a single SNP is used to tag a complex LD region that may be more appropriately represented by groups of evolutionarily-related haplotypes of variants, the genetic locus may appear to impart only a small genetic effect if the effects of different haplotypes essentially cancel each other.
Molecular evolutionary analysis techniques, and specifically the development of phylogenetic trees, are being used with increasing frequency in a variety of different applications. Public domain computer programs are readily available to produce phylogenetic trees from DNA and protein sequence data. The availability of extensive genetic variation data makes it possible to study evolution that occurs over shorter time scales, particularly to test for selective pressures in recent human history. A very good book, entitled Phylogenetic Trees Made Easy: A How-to Manual [36], describes in practical terms the steps for producing and interpreting phylogenetic trees.
The phylogenetic tree from the APOE-TOMM40 region (Figure 1) was published in our recent paper entitled, “A TOMM40 variable length polymorphism determines the age of late-onset Alzheimer’s disease”. For the reader interested in LOAD but uninitiated to phylogenetics, it may be useful to walk through Figure 1.
Multiple mutations occurred within the TOMM40-APOE locus during the course of human evolution. Some of these mutations created branching points in the Caucasian phylogenetic tree whereas others occurred on the branches. Together or independently, may these mutations may be associated with disease phenotype. A number of mutations apparently occurred independently on chromosomes that carry APOE3 or APOE4 and, because there has been limited recombination in this region over the course of evolutionary history represented by the Caucasians that we sampled, each mutational event was preserved in the context of the APOE allele in which it occurred.
One of the polymorphisms in this region, a poly-T repeat at rs10524523 (“523”), appears to have mutated more than once in Caucasian history (See the five boxes in Figure 1). Based on the number of T residues, three categories (alleles) of repeat length are described, short [<20], long [>20], or very long [>30] (Figures 1 and 2). Each allele is represented by a range of poly-T lengths but the means of each length category are significantly different.
Figure 2. Poly-T repeats linked to APOE3 and APOE4.

These histograms show the frequency distributions of lengths of the deoxythymidine homopolymers (number of T residues) at rs10524523 in linkage with APOE3 (top panel) or APOE4 (bottom panel). The three different categories of length are significantly different from each other. This is the analysis from cohort 1 cases and controls representing 210 chromosomes.
The major findings of our phylogenetic analysis of LOAD were: 1) that APOE4 was, almost without exception, always linked to a long poly-T repeat; 2) that the ε3 allele of APOE was linked to either a short poly-T or a very long poly- T repeat (Figures 1 and 2); and 3) that linkage to a short or very long poly-T for an APOE3 carrier resulted in later or earlier onset of LOAD, respectively, regardless of risk imparted by APOE4.
In virtually every publication of age of LOAD onset for APOE3/4 patients, the average age of onset is reported to be about 75 years. Potentially, a very long poly-T repeat in linkage with APOE3 may equal, or even exceed, the disease risk associated with APOE4. If this is true, then individuals with the APOE3/3 genotype and two copies of the very long “523” polymorphism may be at risk of developing LOAD at a very early age, perhaps <70 years. Patients who develop LOAD in their 50 s, but do not carry the rare autosomal dominant mutations that cause early-onset Alzheimer s disease, may be homozygous for the APOE3-very long “523” haplotype. As an interesting note, in the second cohort that we examined, we observed two, rare, APOE4 haplotypes in clade B that were linked to short “523” alleles and we speculate that these might represent the ancestral scenario. The intriguing observation was that the age of disease onset for both of these people was 78 years, which is similar to the age of onset for individuals who possess the short-APOE3/long-APOE4 genotype.
The poly-T variant was discovered because we deep sequenced the APOE LD region. Deep sequencing was essential for accurately measuring the length of the poly-T allele in each instance and also for unequivocally determining the link between the poly-T polymorphism and the APOE allele. The discovery of the “523” poly-T variant suggests that susceptibility to LOAD behaves as an autosomal co-dominant trait with the age of onset phenotype determined by the combined effects of each allele at the rs10524523 (“523”) poly-T locus [3]. An interesting question is, then, which is the engine of the LOAD endophenotype (age of onset)? Is it the APOE allele as was previously assumed, or the “523” poly-T length?
Beyond LOAD, a more general hypothesis is that, in the evolutionary history of other complex diseases, multiple evolutionarily-linked variants within a genomic interval, that are not necessarily in high LD [nor acting as coincident genetic loci], may determine risk of disease. In other words, haplotypes of evolutionarily-related variants may associate more significantly with the disease phenotype than individual, common SNPs that represent the average effect of a genomic interval. The approach that we have adopted in studying other complex diseases is to use phylogenetic analysis to fine-map regions that are reproducibly flagged by GWAS or linkage studies but which impart only small effect or narrowly miss genome-wide significance. In the case of LOAD, the phenotype (age of disease onset) was highly associated with multiple polymorphisms that arose over the course of human evolution at a single site (rs10524523) in the APOE LD region (see Figure 1). However, phylogenetic approaches may also provide a powerful mechanism for capturing the effects of disease susceptible variants that occur at multiple loci within a larger, disease-associated region.
Experimental data: Phylogenetic trees
Phylogenetic trees can be rooted or unrooted. Rooting of the tree is accomplished by including an incontrovertibly ancestral group, for example, sequence information from an ancestral species. Unrooted trees, as exemplified in Figure 1, do not explicitly speak to the chronological ordering of the events that created the different branches, but do cluster the most related sequences into clades, or haplogroups. Because the phylogenetic tree provides a picture of the relationships between groups of sequences and the mutations that define the subgroups or clades, we are able to make the observation that a specific locus, “523,” has mutated multiple times over the course of human evolution.
The goal of our analysis was to identify clades, composed of groups of haplotypes of rare and common SNPs and structural variants, which were associated with increased and coordinately decreased risk for AD. Windows of sizes <10Kb within the TOMM40-APOE region were sequenced and analyzed using a number of phylogenetic algorithms. We searched for genomic regions where the clades of the phylogenetic tree had strong statistical support and had different patient/control ratios. A region of TOMM40 that spanned exon 6 through exon 10 (Figure 3) provided the strongest phylogenetic signal and gave the tree structure shown in Figure 1. The topology of the tree illustrated in Figure 1 was quite exceptional in that there was very high statistical support (bootstrap support was 97%, 1000 replicates) for the first branch point that divided the tree into the two major clades. It is unusual to obtain such a robust signal with intra-species data because of the relatively limited sequence diversity that typically occurs over such a short period of evolutionary history. A higher case/control ratio was associated with clade A compared to clade B and this suggested that at least some of the polymorphisms that defined the differences between the clades in this region would be disease susceptibility variants. While the phylogenetic tree structure shown in Figure 1 presents the data from one cohort of Caucasian LOAD patients and unaffected controls (105 patients or 210 chromosomes), the tree structure was robust and was confirmed for a second cohort of Caucasian cases and controls (150 subjects or 300 chromosomes).
Figure 3. Diagram of exons 6 (E6) to 10 (E10) of TOMM40.

The vertical lines represent SNPs that distinguish clades A and B; purple boxes in intron 6 indicate the locations of two poly-T tracts. The histograms indicate the frequency distributions of poly-T lengths at two loci.
By using a phylogenetic approach, an important observation was made: 98% of the APOE4/4 individuals (patients or controls) mapped to clade A while APOE3/3 and APOE3/4 individuals were present in both clades on this phylogenetic tree (Figure 1). We subsequently mapped the polymorphisms that distinguished the two clades. For example, in 97% of all cases the “G” allele of SNP rs8106922 mapped to clade A. It is interesting to note that this SNP is one of the most significant disease risk SNPs subsequently identified by Harold et al. in a GWAS (Table 1) [2]. The polymorphism, rs10524523, in intron 6 of TOMM40 was also very characteristic; this locus was polymorphic with respect to the number of T residues (poly-T) (Figure 3). Long poly-T tracts separated almost exclusively into clade A, regardless of APOE allele. Further, the length of the poly-T variant distinguished the APOE3 chromosomes that segregate to clade A (very long poly-T) or clade B (short poly-T) (Figure 2).
A third study was conducted using an independent collection of LOAD patients, all with the APOE3/4 genotype, who had reliable age of disease onset recorded in their medical records. In the two previous sets of subjects all APOE4-containing chromosomes were, almost with exception, attached to long “523” poly-T repeats. It seemed reasonable, therefore, to ask whether age of disease onset for these APOE3/4 heterozygotes was associated with the presence of a long or short “523” allele on the APOE3 chromosomes. The results were striking and statistically significant (Figure 4). We observed that when a short “523” repeat was linked to APOE3 the average age of onset was 78 years of age. When a long repeat was linked to APOE3 the average age of onset was 70 years of age, which is very close to the average age of disease onset for APOE4/4 (i.e. “523” long/long) LOAD patients.
Figure 4. Average age of LOAD diagnosis for APOE3/4 heterozygous individuals.

Long refers to APOE3-very long poly-T haplotype; Short refers to APOE3-short poly-T haplotype. All individuals carried one copy of the APOE4-long poly-T haplotype.
In virtually every publication of age of LOAD onset for APOE3/4 patients, the average age of onset is reported to be about 75 years. Potentially, a very long poly-T repeat in linkage with APOE3 may equal, or even exceed, the disease risk associated with APOE4. If this is true, then individuals with the APOE3/3 genotype and two copies of the very long “523” polymorphism may be at risk of developing LOAD at a very early age, perhaps <70 years. Patients who develop LOAD in their 50 s, but do not carry the rare autosomal dominant mutations that cause early-onset Alzheimer s disease, may be homozygous for the APOE3-very long “523” haplotype. As an interesting note, in the second cohort that we examined, we observed two, rare, haplotypes in clade B where APOE4 was linked to a short “523” allele and we speculate that these might represent the ancestral scenario. These unusual APOE4- short “523” haplotypes belonged to two patients who were homozygous for APOE4 but were heterozygous for poly-T length at the “523” locus (i.e., these individuals were short-APOE4/long-APOE4). The intriguing observation was that the age of disease onset for both of these people was 78 years, which is similar to the age of onset for individuals who possess the short-APOE3/long-APOE4 genotype.
These data are now being confirmed in other populations where, similar to our third cohort, normal subjects were followed prospectively in several medical centers and age of disease onset and LOAD phenotype were accurately recorded. The results from these studies will allow us to generate a family of age of onset curves for APOE3/4, APOE3/3 and APOE2/3 patients rather than the averaged curves now seen in the literature that are based on APOE genotype alone (Figure 5).
Figure 5. Age of onset curves for individuals with the indicated genotypes using the information available in 1993.

From [1]. Today we propose that each of the age of onset curves is an average of sub-curves. There are two curves for APOE3/4 individuals due to the presence of either a short or a long 523 polymorphisms linked to APOE3. There are likely three curves for APOE3/3 individuals due to the possible combination of alleles at 523, i.e. short/short, short/very long, and very long/very long.
Genetic parsimony
The most parsimonious explanation for the genetics of age of LOAD onset is that this endophenotype is associated with length of the poly-T tract. The discovery of APOE4 as a genetic risk factor for LOAD in 1992 [1, 37, 38] may have occurred because it is almost always associated with a long “523” poly-T in intron 6 of TOMM40 (98% of the time in our combined data). The length of the T homopolymer defines two evolutionary divergent forms of APOE3 and further stratifies the age of disease onset for carriers of the APOE3 allele. This is significant for the public since eighty percent of all Caucasians are APOE3 carriers. We suggest that, by including the TOMM40 “523” allele information with APOE genotype, there will be enhanced accuracy for prediction of age-related disease risk.
Validation studies of our data are important and this experiment must be repeated for other races and ethnicities. Unfortunately, to determine the length of the poly-T is not as simple as genotyping a SNP and an appropriate high-throughput assay does not exist on the market, as did APOE genotyping as a cardiology risk factor in 1993. This may slow down replication studies but we are developing a high throughput assay to be available as a CLIA-based, FDA-approved, test available for academic research at a very low cost.
Shiraz Pharmaceuticals, Inc. was established in 2009 to commercialize the intellectual property from this new discovery. Zinfandel Pharmaceuticals, Inc. was also founded in 2009 to plan and execute a prospective diagnostic validation study of the “523” variable length poly-T diagnostic for predicting risk of AD onset in the next 5–7 years for individuals aged 60–87 years. Planning for a combination diagnostic validation study and prevention (delay of age of onset) clinical trial in unaffected individuals is currently in progress [http://www.opalstudy.org/index.html]. As of October 2009, a Voluntary Exploratory Data Submission discussion with the FDA regarding the design of the clinical trial and the use of the “523” diagnostic was completed. Figure 6 outlines the proposed design of a combined diagnostic validation/delay of disease therapeutic clinical study. Five epidemiologic-based recruitment sites for Caucasians without cognitive impairment have been organized and are piloting subject recruitment in order to decrease the recruitment time once the trial commences. It is anticipated that the ability to predict risk will be confirmed and become integral to other LOAD delay of onset prevention and therapeutic treatment trials.
Figure 6. Proposed clinical trial design.

This study is designed to simultaneously validate the genetic marker and to test the efficacy of a therapeutic that delays the onset of LOAD.
Finally, evolutionary history differs in different ethnic groups, and it is likely that the topology of the phylogenetic tree of the TOMM40-APOE region, and the frequency of each TOMM40-APOE haplotype, will differ among ethnic groups. An understanding of the these allele frequencies is especially important for designing drug trials and interpreting pharmacogenetic results now that Phase III trials are increasing conducted world-wide pulling into one study patients of diverse genetic backgrounds. If the major genetic determinants of disease are not accounted for in these studies a drug may appear to fail when actually it is the trial design that fails. We are now planning phylogenetic studies of the TOMM40-APOE region in Asian and African populations.
Acknowledgments
The work described in this article would not have been possible without the generous contribution of DNA samples from the Netherlands Brain Bank (under the direction of Dr. Rivka Ravid), the Banner Sun Health Research Institute (under the direction of Dr. Thomas Beach), the Arizona Alzheimer s Disease Core Center (Arizona ADCC) and the Joseph and Kathleen Bryan Alzheimer s Disease Research Center (Bryan ADRC). Work at the Arizona ADCC was supported in part by grants from the National Institute on Aging (NIA) to Dr. Eric Reiman (P30 AG019610 and R01 AG031581); grants from the National Institute of Neurological Disorders and Stroke (R01 NS059873) and Science Foundation Arizona to Dr. Matthew Huentelman; the Arizona Alzheimer’s Consortium, and the State of Arizona. The work at the Joseph and Kathleen Bryan ADRC was supported in part by a NIA grant to Dr. Kathleen Welsh-Bohmer (P30 AG028377). Dr. Roses is supported in part by a grant from the NIA (1RC1 AG035635-01).
Footnotes
Conflict of interest statement: Dr. Roses is the President of three companies filed as S-Corporations in the state of North Carolina: Cabernet Pharmaceuticals, Inc is a pipeline pharmacogenetic consultation and project management company that has other pharmaceutical companies as clients; Shiraz Pharmaceuticals, Inc. is focused on the commercialization of diagnostics, including companion diagnostics, for universities, pharmaceutical companies, and biotechnology companies; Zinfandel Pharmaceuticals is the sponsor of OPAL [Opportunity to Prevent Alzheimer’s Disease] which is a combined clinical validation of a diagnostic and a pharmacogenetic-assisted delay of onset clinical trial.
These companies are independent of Duke University but any intellectual property generated by Dr. Roses or his team is intended to be treated as Deane Drug Discovery Institute property once there is an established commercial value.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 2.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable length polymorphism determines the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2009 doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khoury MJ, Bertram L, Boffetta P, Butterworth AS, Chanock SJ, Dolan SM, Fortier I, Garcia-Closas M, Gwinn M, Higgins JP, et al. Genome-wide association studies, field synopses, and the development of the knowledge base on genetic variation and human diseases. Am J Epidemiol. 2009;170:269–79. doi: 10.1093/aje/kwp119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abraham R, Moskvina V, Sims R, Hollingworth P, Morgan A, Georgieva L, Dowzell K, Cichon S, Hillmer AM, O’Donovan MC, et al. A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Med Genomics. 2008;1:44. doi: 10.1186/1755-8794-1-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beecham GW, Martin ER, Li Y-J, Slifer MA, Gilbert JR, Haines JL, Pericak-Vance MA. Genome-wide Association Study Implicates a Chromosome 12 Risk Locus for Late-Onset Alzheimer Disease. Am J Hum Genet. 2009;84:35–43. doi: 10.1016/j.ajhg.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BMM, Hooli B, DiVito J, Ionita I, et al. Genome-wide Association Analysis Reveals Putative Alzheimer’s Disease Susceptibility Loci in Addition to APOE. Am J Hum Genet. 2008;83:623–32. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrasquillo MM, Zou F, Pankratz VS, Wilcox SL, Ma L, Walker LP, Younkin SG, Younkin CS, Younkin LH, Bisceglio GD, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nat Genet. 2009;41:192–8. doi: 10.1038/ng.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, Zismann VL, Beach TG, Leung D, Bryden L, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry. 2007;68:613–8. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- 10.Grupe A, Abraham R, Li Y, Rowland C, Hollingworth P, Morgan A, Jehu L, Segurado R, Stone D, Schadt E, et al. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum Mol Genet. 2007;16:865–73. doi: 10.1093/hmg/ddm031. [DOI] [PubMed] [Google Scholar]
- 11.Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, Hosford D, Barnes MR, Briley JD, Borrie M, et al. Candidate Single-Nucleotide Polymorphisms From a Genomewide Association Study of Alzheimer Disease. Arch Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 13.Lai E, Riley J, Purvis I, Roses A. A 4-Mb High-Density Single Nucleotide Polymorphism-Based Map around Human APOE. Genomics. 1998;54:31–8. doi: 10.1006/geno.1998.5581. [DOI] [PubMed] [Google Scholar]
- 14.Chang S, Ma Tr, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102:18694–9. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brodbeck J, Balestra M, Freedman S, Weisgraber KH, Mahley RW, Huang Y. O3-03-03: Apolipoprotein E4 and its fragment impair mitochondrial dynamics in neuronal cultures. Alzheimers Dement. 2008;4:T163-T. [Google Scholar]
- 16.Brodbeck J, Miranda RD, Freedman SB, Weisgraber KH, Mahley RW, Huang Y. Apolipoprotein E4 impairs mitochondrial function and increases reactive oxygen species in neuronal cells. Alzheimers Dement. 2009;5:P307-P. [Google Scholar]
- 17.Anandatheerthavarada HK, Biswas G, Robin M-A, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK, Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–68. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma J, Yee A, Brewer HB, Das S, Potter H. Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature. 1994;372:92–4. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 20.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:8098–102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–8. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 23.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci U S A. 2005;102:8299–302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Risner ME, Saunders AM, Altman JFB, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6:246–54. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 25.Roses A, Gilbert J, Xu P, Sullivan P, Popko B, Burkhart D, Christian-Rothrock T, Saunders A, Maeda N, Schmechel D. Cis-acting human ApoE tissue expression element is associated with human pattern of intraneuronal ApoE in transgenic mice. Neurobiol Aging. 1998;19:S53–8. doi: 10.1016/s0197-4580(98)00030-x. [DOI] [PubMed] [Google Scholar]
- 26.Roses AD. Alzheimer diseases: a model of gene mutations and susceptibility polymorphisms for complex psychiatric diseases. Am J Med Genet. 1998;81:49–57. doi: 10.1002/(sici)1096-8628(19980207)81:1<49::aid-ajmg10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 27.Roses AD, Saunders AM, Huang Y, Strum J, Weisgraber KH, Mahley RW. Complex disease-associated pharmacogenetics: drug efficacy, drug safety, and confirmation of a pathogenetic hypothesis (Alzheimer’s disease) Pharmacogenomics J. 2007;7:10–28. doi: 10.1038/sj.tpj.6500397. [DOI] [PubMed] [Google Scholar]
- 28.Jordan IK, Kondrashov FA, Rogozin IB, Tatusov RL, Wolf YI, Koonin EV. Constant relative rate of protein evolution and detection of functional diversification among bacterial, archaeal and eukaryotic proteins. Genome Biol. 2001;2:research0053.1.9. doi: 10.1186/gb-2001-2-12-research0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan QH, Zeng CJ, Ni XW, Pan HJ, Fang SG. Giant panda genomic data provide insight into the birth-and-death process of mammalian major histocompatibility complex class II genes. PLoS One. 2009;4:e4147. doi: 10.1371/journal.pone.0004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watts S, Vogel J, Harriman W, Itoh T, Stauss H, Goodenow R. DNA sequence analysis of the C3H H-2Kk and H-2Dk loci. Evolutionary relationships to H-2 genes from four other mouse strains. J Immunol. 1987;139:3878–85. [PubMed] [Google Scholar]
- 31.Yu H, Zhang PC, Zhou YJ, Li GX, Pan J, Yan LP, Shi XX, Liu HL, Tong GZ. Isolation and genetic characterization of avian-like H1N1 and novel ressortant H1N2 influenza viruses from pigs in China. Biochem Biophys Res Commun. 2009;386:278–83. doi: 10.1016/j.bbrc.2009.05.056. [DOI] [PubMed] [Google Scholar]
- 32.Chen JM, Sun YX, Chen JW, Liu S, Yu JM, Shen CJ, Sun XD, Peng D. Panorama phylogenetic diversity and distribution of type A influenza viruses based on their six internal gene sequences. Virol J. 2009;6:137. doi: 10.1186/1743-422X-6-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehlmann M, Dawson ED, Townsend MB, Smagala JA, Moore CL, Smith CB, Cox NJ, Kuchta RD, Rowlen KL. Robust Sequence Selection Method Used To Develop the FluChip Diagnostic Microarray for Influenza Virus. J Clin Microbiol. 2006;44:2857–62. doi: 10.1128/JCM.00135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zaslavsky L, Bao Y, Tatusova T. Visualization of large influenza virus sequence datasets using adaptively aggregated trees with sampling-based subscale representation. Bmc Bioinformatics. 2008;9:237. doi: 10.1186/1471-2105-9-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zöllner S, Pritchard JK. Coalescent-based association mapping and fine mapping of complex trait loci. Genetics. 2005;169:1071–92. doi: 10.1534/genetics.104.031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall BG. Phylogenetic Trees Made Easy: A How-To Manual. 3. Sunderland, MA: Sinauer Associates, Inc; 2007. p. 203. [Google Scholar]
- 37.Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, et al. Association of apolipoprotein E allele e4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 38.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]