

Summary
Metabolic monitoring at the cellular level in live tissues is important for understanding cell function, disease processes and potential therapies. Multiphoton imaging of the relative amounts of NADH and FAD (the primary electron donor and acceptor, respectively, in the electron transport chain) provides a non-invasive method for monitoring cellular metabolic activity with high resolution in three dimensions in vivo. NADH and FAD are endogenous tissue fluorophores, and thus this method does not require exogenous stains or tissue excision. We describe the principles and protocols of multiphoton redox ratio imaging in vivo.
Keywords: Reduced nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide (FAD), redox ratio, metabolism, mitochondria, multiphoton microscopy
1. Introduction
The metabolic rate of a cell is an important marker for the diagnosis, staging and treatment of diseases ranging from Alzheimer's to cancer, and can serve as a general marker of cell health. Optical imaging of endogenous tissue fluorophores provides a non-invasive, fast, and inexpensive method for evaluating the metabolic rate of cells in vivo. The electron transport chain is the primary means of energy production in the cell. The electron transport chain produces energy in the form of adenosine triphosphate (ATP) by transferring electrons to molecular oxygen. There are two endogenous (occurring naturally in the cell) fluorophores in tissue related to cellular metabolism in the electron transport chain. The first fluorophore is the reduced form of nicotinamide adenine dinucleotide (NADH), which transfers electrons to molecular oxygen. NADH has fluorescence excitation and emission maxima at 350 nm and 460 nm, respectively [1]. The second fluorophore is flavin adenine dinucleotide (FAD), which is an electron acceptor. FAD has fluorescence excitation and emission maxima at 450 nm and 535 nm, respectively [1]. An approximation of the oxidation-reduction ratio of the mitochondrial matrix space can be determined from the “redox ratio”, which is the fluorescence intensity of FAD divided by the fluorescence intensity of NADH [2]. This optical redox ratio provides relative changes in the oxidation-reduction state in the cell without the use of exogenous stains or dyes, and can thus be measured in vivo in both human and animal studies. This advantage is important because it eliminates possible artifacts in metabolic measurements that can be introduced by tissue excision, processing or staining. The redox ratio is sensitive to changes in the cellular metabolic rate and vascular oxygen supply [2-5]. A decrease in the redox ratio usually indicates increased cellular metabolic activity [6].
Multiphoton microscopy is an attractive method for imaging the redox ratio in vivo, because it provides high resolution (∼400 nm) three-dimensional images deep within living tissue (∼1mm). Multiphoton excitation occurs when a fluorophore is excited simultaneously by two photons of half the absorption energy of the fluorophore (or by three photons of one-third the absorption energy of the fluorophore, etc.), and probes the same biological fluorophores as single-photon fluorescence [7]. Multiphoton excitation of NADH and FAD occurs in the near infrared (NIR) wavelength region [8], and these wavelengths of non-ionizing radiation are relatively benign [9]. The NIR wavelength range between 650 nm and 900 nm is called the “optical window” where light can penetrate deep into tissue, due to reduced tissue scattering and minimal absorption from water and hemoglobin. Thus, multiphoton excitation also allows for increased imaging depth compared to single photon excitation.
A typical multiphoton microscope includes a titanium sapphire (Ti-Sapphire) laser, a raster scan unit, a dichroic mirror, a microscope objective and a photomultiplier tube (PMT). The most common excitation sources are femtosecond titanium sapphire (Ti-Sapphire) lasers that generate 100 femtosecond pulses at a repetition rate of about 80 MHz. This allows for sufficient two-photon excitation without excessive heat or photodamage to the sample. The tuning range of Ti-Sapphire systems are 700 to 1000 nm, sufficient to excite both NADH and FAD. The raster scan unit scans the excitation beam across the x-y plane so that two-dimensional images can be created at each image depth. The focal point of the objective can be moved in the z- (depth) direction by a z-stage motor. A short pass dichroic mirror reflects the longer-wavelength IR light onto the sample, and transmits the shorter-wavelength fluorescence (usually in the visible wavelength range) to the detector. High NA (numerical aperture) microscope objectives are used to maximize the excitation efficiency. PMTs are a popular choice for detector because they are robust, low cost, and relatively sensitive. Multiphoton microscopes can be custom built or purchased from most microscope companies. Multiphoton endoscopes [10-12] are required for redox ratio measurements in organ sites that are not accessible with a microscope.
In vivo metabolic measurements require that anesthesia minimally perturbs the metabolic state of cells, or at least, that the anesthesia has the same effect on experimental and control groups. A previous study found that Isoflurane (1.5%) produced constant muscle pO2 and blood perfusion in mice, and thus Isoflurane is a good choice for metabolic imaging using the redox ratio [13]. Baudelet et al also found that the pO2 of both tumors and normal muscle tissue decreased approximately the same percentage with ketamine/xylazine anesthesia in mice [13].
2. Materials
The advantage of multiphoton redox imaging is that no sample processing is necessary. The required components include a multiphoton microscope, a live sample, and a computer for analysis.
2.1. Equipment
Multiphoton microscope can be custom built [14] or purchased from vendors including Lavision Biotech (Pittsford, NY), Zeiss (Thornwood, NY), Prairie Ultima (Middleton, WI), Olympus (Center Valley, PA) and Leica (Bannockburn, IL)
Titanium-Sapphire lasers can be purchased from vendors including PicoQuant (Berlin, Germany) and Newport (Mountain View, CA) if they do not come with your microscope
x-y translator for the microscope stage
Anesthesia machine or injectable anesthesia
2.2 Reagents and Supplies
5.8 × 10-5 g/ml Rhodamine B in distilled water
Standard coverslips
3. Methods
3.1. Image collection
Select appropriate excitation wavelength(s) and emission filters. Huang et al. found that NADH and FAD fluorescence is isolated at 750 nm and 900 nm excitation, respectively [8]. Optical filters, which only allow wavelengths in a selected range to pass, can also be used with one or more detectors to further isolate NADH and FAD fluorescence. Huang et al. found that a 410-490 bandpass filter isolated NADH emission and a 510-560 nm bandpass filter isolated FAD fluorescence at 800 nm excitation [8]. Mitochondrial uncouplers and inhibitors can be used to further verify the isolation of NADH and FAD fluorescence [8].
Collect calibration standard images at the NADH excitation wavelength (see Note 1). Daily calibration standards must be measured to account for fluctuations in the throughput and excitation efficiency of the system. A standard calibration sample is Rhodamine B, and previous in vivo multiphoton studies have used a concentration of 5.8 × 10-5 g/ml Rhodamine B in distilled water [15]. For added precision, the mean of three intensity images of the Rhodamine standard can be used to correct for system variations. The Rhodamine standard images should be collected under conditions identical to those of the in vivo NADH data from that day. Alternatively, an internal microscope reference that reports the two-photon excitation efficiency can be used to correct the measured fluorescence intensities for incident power [16].
Place the anesthetized animal on the imaging stage. Anesthesia that minimally alters the metabolic rate of the tissue should be chosen. The tissue of interest should be secured flush with a coverslip to ensure the deepest possible imaging depth. Bulk motion can be avoided by properly securing the tissue on the microscope stage. However, it is important that normal circulation be maintained so as not to alter the metabolic rate of the cells of interest.
Collect multiphoton image stack number one at the NADH excitation wavelength (see Note 2 and 3). Use the NADH emission filter, if necessary. The resolution, field of view and imaging depth can be optimized with the appropriate objective. Resolution and field of view are determined from the numerical aperture (NA) of the objective, and the imaging depth is determined from the working distance of the objective. Water immersion objectives allow for deeper imaging depths than oil immersion objectives due to index matching with the tissue. The number of slices in the stack is determined by the z-stack slice separation, which can be as small as the axial resolution of the system. Typical z-stack slice separations range from 2 to 10 microns.
Collect additional image stacks at the NADH excitation wavelength, noting the position of each image stack on an x- y- translator.
Collect multiphoton image stack number one at the FAD excitation wavelength (use the FAD emission filter, if necessary). Collect additional image stacks at the FAD excitation wavelength from the remaining x-y positions.
Collect calibration standard images at the FAD excitation wavelength under conditions identical to those of the in vivo FAD data from that day.
3.1. Image analysis
ImageJ software is sufficient for all image analysis steps, and is available free online at http://rsbweb.nih.gov/ij/.
Calculate the redox image. Choose your region of interest and divide the FAD image by its corresponding NADH image. Multiply the resulting image by a scalar value that accounts for the Rhodamine standard measured for the NADH and FAD images.
| (1) |
In equation 1, [Redox] is the redox ratio image, [FAD] is the FAD intensity image, [NADH] is the corresponding NADH intensity image, and RFAD and RNADH are the mean Rhodamine intensity values measured under identical experimental conditions as [FAD] and [NADH], respectively. Note that the ratio of RFAD and RNADH is a scalar value, and the ratio of [FAD] and [NADH] is a matrix value. The division of [FAD] and [NADH] should be done pixel-by-pixel.
An example of relative redox ratio images from the normal and pre-cancerous hamster cheek pouch in vivo are shown in Fig. 1. Three-dimensional redox ratio images can be useful for quantifying changes in the distribution of the redox ratio within cells, and with depth in tissue, as well as volume-averaged changes in the bulk tissue (see Note 4). Statistical analysis on multiple images from the study shown in Fig. 1 [15] indicate increased heterogeneity of the redox ratio within pre-cancerous cells compared to normal cells, a decrease in the redox ratio with depth within normal tissues and no change in the redox ratio with depth within pre-cancerous tissues. There was no change in the volume-averaged redox ratio with pre-cancer compared to normal tissues in this study, which indicates the importance of high resolution, depth resolved imaging of the redox ratio in vivo (see Note 5).
Figure 1.
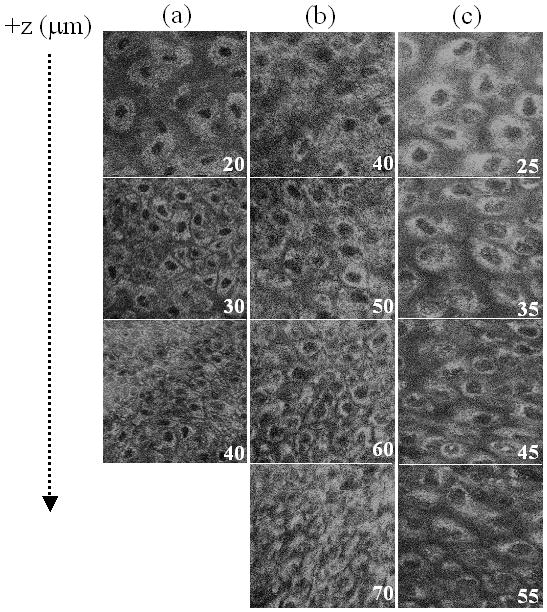
Representative in vivo three-dimensional multiphoton images of the redox ratio (fluorescence intensity of FAD/NADH) from tissues diagnosed as normal (a), low-grade precancer (b), and high-grade precancer (c) in the 7,12-dimethylbenz(a)anthracene (DMBA)-treated hamster cheek pouch model of oral cancer. The numbers in the corner of each image indicate the depth below the tissue surface in microns, and each image is 100 × 100 μm. From ref [15], © 2007, National Academy of Sciences, U.S.A.
4. Notes
If the incident power and detector gain settings are identical for all measurements, a Rhodamine calibration is not necessary. In cases where the dynamic range of the NADH and FAD fluorescence intensities across all samples is large, then the incident power and detector gain settings must change between samples, and a Rhodamine calibration is necessary.
Monitor photon count rates while collecting multiphoton images, to ensure that photobleaching does not occur. Alternatively, after all images in the multiphoton image stack have been collected, collect the first image in the stack again to be sure that the image intensity has not diminished due to photobleaching.
Previous multiphoton measurements of tissue NADH at 780nm excitation used an average power, peak power, and the focused fluence incident on the sample of approximately 15 mW, 300 GW/cm2, and 3.7 MW/cm2, respectively, with a pixel dwell time of 11.5 μs [16]. Incident powers and integration times such as these should give sufficient signal for in vivo redox measurements using multiphoton microscopy.
This in vivo optical redox ratio is a relative measurement, so careful consideration of the appropriate controls must be made. All changes in the redox ratio must be considered relative to these controls.
These methods can be used to monitor metabolic changes in cell culture and excised tissue. However, these measurements may not reflect the true in vivo metabolic state, which is largely determined by oxygen delivery from the vasculature.
Acknowledgments
This work was supported by the NIH (R01 EB000184). M.S. acknowledges individual fellowship support from the DOD (W81XWH-04-1-0330) and the NIH (F32 CA130309).
References
- 1.Ramanujam N. Fluorescence spectroscopy of neoplastic and non-neoplastic tissues. Neoplasia. 2000;2:89–117. doi: 10.1038/sj.neo.7900077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chance B, Schoener B, Oshino R, Itshak F, Nakase Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J Biol Chem. 1979;254:4764–71. [PubMed] [Google Scholar]
- 3.Drezek R, Brookner C, Pavlova I, Boiko I, Malpica A, Lotan R, Follen M, Richards-Kortum R. Autofluorescence microscopy of fresh cervical-tissue sections reveals alterations in tissue biochemistry with dysplasia. Photochem Photobiol. 2001;73:636–41. doi: 10.1562/0031-8655(2001)073<0636:AMOFCT>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 4.Gulledge CJ, Dewhirst MW. Tumor oxygenation: a matter of supply and demand. Anticancer Res. 1996;16:741–9. [PubMed] [Google Scholar]
- 5.Ramanujam N, Kortum RR, Thomsen S, Jansen AM, Follen M, Chance B. Low temperature fluorescence imaging of freeze-trapped human cervical tissues. Opt Express. 2001;8:335–343. doi: 10.1364/oe.8.000335. [DOI] [PubMed] [Google Scholar]
- 6.Chance B. Metabolic heterogeneities in rapidly metabolizing tissues. J Appl Cardiol. 1989;4:207–221. [Google Scholar]
- 7.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–6. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 8.Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J. 2002;82:2811–25. doi: 10.1016/S0006-3495(02)75621-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Squirrell JM, Wokosin DL, White JG, Bavister BD. Long-term two-photon fluorescence imaging of mammalian embryos without compromising viability. Nat Biotechnol. 1999;17:763–7. doi: 10.1038/11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bird D, Gu M. Two-photon fluorescence endoscopy with a micro-optic scanning head. Opt Lett. 2003;28:1552–1554. doi: 10.1364/ol.28.001552. [DOI] [PubMed] [Google Scholar]
- 11.Helmchen F. Miniaturization of fluorescence microscopes using fibre optics. Exp Physiol. 2002;87:737–45. doi: 10.1113/eph8702478. [DOI] [PubMed] [Google Scholar]
- 12.Jung JC, Schnitzer MJ. Multiphoton endoscopy. Opt Lett. 2003;28:902–4. doi: 10.1364/ol.28.000902. [DOI] [PubMed] [Google Scholar]
- 13.Baudelet C, Gallez B. Effect of anesthesia on the signal intensity in tumors using BOLD-MRI: comparison with flow measurements by Laser Doppler flowmetry and oxygen measurements by luminescence-based probes. Magn Reson Imaging. 2004;22:905–12. doi: 10.1016/j.mri.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Wokosin DL, Squirrell JM, Eliceiri KW, White JG. An optical workstation with concurrent independent multiphoton imaging and experimental laser microbeam capabilities. Rev Sci Instrum. 2003;74:193–201. doi: 10.1063/1.1524716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, Ramanujam N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci U S A. 2007;104:19494–9. doi: 10.1073/pnas.0708425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skala MC, Squirrell JM, Vrotsos KM, Eickhoff JC, Gendron-Fitzpatrick A, Eliceiri KW, Ramanujam N. Multiphoton microscopy of endogenous fluorescence differentiates normal, precancerous, and cancerous squamous epithelial tissues. Cancer Res. 2005;65:1180–6. doi: 10.1158/0008-5472.CAN-04-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]