

Abstract
Each year, pediatric traumatic brain injury (TBI) accounts for 435,000 emergency department visits, 37,000 hospital admissions, and approximately 2,500 deaths in the United States. TBI results in immediate injury from direct mechanical force and shear. Secondary injury results from the release of biochemical or inflammatory factors that alter the loco-regional milieu in the acute, subacute, and delayed intervals after a mechanical insult. Preliminary preclinical and clinical research is underway to evaluate the benefit from progenitor cell therapeutics, hypertonic saline infusion, and controlled hypothermia. However, all phase III clinical trials investigating pharmacologic monotherapy for TBI have shown no benefit. A recent National Institutes of Health consensus statement recommends research into multimodality treatments for TBI. This article will review the complex pathophysiology of TBI as well as the possible therapeutic mechanisms of progenitor cell transplantation, hypertonic saline infusion, and controlled hypothermia for possible utilization in multimodality clinical trials.
Keywords: Traumatic brain injury, Treatment, Therapy, Stem cells, Progenitor cells, Hypothermia, Hypertonic saline, Review
Each year, pediatric traumatic brain injury (TBI) accounts for 435,000 emergency department visits, 37,000 hospital admissions, and approximately 2,500 deaths in the United States.1 The incidence of TBI-related hospitalization is 679 of 100,000 children.2 Of those patients affected, up to 48% are impaired by physical, cognitive, and psychosocial deficits.3 Beginning aggressive rehabilitation early in the patient’s hospital course has shown improvement in functional status4,5; however, neurons show little ability to repair and no treatment modality is currently available to reverse acute brain injury. A large body of work has pursued pharmacologic neuroprotective strategies. Single agent pharmacologic neuroprotective approaches to treat TBI have not proven successful clinically. Both clinical and preclinical research is underway to show possible benefit from progenitor cell therapeutics, hypertonic saline (HTS) treatment of intracranial hypertension, and controlled hypothermia for treatment of TBI in the acute setting.
TBI Classification
The development of neuroprotective treatments for TBI requires a clear classification of injury severity and understanding of pathophysiologic mechanisms. A common method of classification separates immediate (primary) injury from delayed (secondary) injury. Primary injury results from direct mechanical force that leads to compression and shearing of neural, glial, and vascular cells.6 A central core of cellular necrosis is surrounded by a diffuse area of axonal cell injury (penumbra). Secondary injury consists of the delayed deleterious effects resulting from the release of biochemical or inflammatory factors that alter the loco-regional milieu in the acute, subacute, and delayed intervals after a mechanical insult.
A recent consensus statement from the Society of Neurotrauma classified TBI according to acute clinical monitoring, pathoanatomic injury, neuroimaging, and biomarkers. The Glasgow Coma Scale (GCS) is used to clinically classify TBI as mild, moderate, or severe. Although useful in the acute phase (0–4 hours), GCS scores are often altered by the use of sedatives. In addition, severe injury as indicated by GCS scoring fails to correlate with a specific mechanism or neuropathology. Therefore, additional modalities such as high-resolution computed tomography (CT), intracranial pressure (ICP) measurement, and magnetic resonance imaging can be used to further classify TBI in the acute setting.7
TBI can be further classified using pathoanatomic injury types. Multiple in vivo models have been developed to investigate the biomechanics of TBI allowing for the development of novel neuroprotective therapies. Animal models include direct injury, acceleration-deceleration injury, and impact injury. Table 1 outlines the advantages and disadvantages of each injury model.8 The efficacy of many neuroprotective therapeutics developed in animal models has been limited in human trials. A common thought is that the pathophysiology of injury in the lissencephalic rat brain used in most models does not translate to the biomechanics of human TBI. However, in vivo models continue to give insight into anatomic location of injury, cell types at risk, and injury progression allowing for the investigation of novel therapies.7
TABLE 1.
Advantages Versus Disadvantages of Various in Vivo TBI Models8
| Direct injury | Fluid percussion injury | Reproducible injury severity |
Neurogenic pulmonary edema, brain stem involvement |
| Controlled cortical impact |
Reproducible injury severity focused injury area |
Skull fractures hemorrhage |
|
| Penetrating impact injury |
Projectile missile | Characterization of missile wound |
Difficult to evaluate biologic outcomes |
| Nonpenetrating impact injury |
Controlled concussion models |
Reducible mild to moderate injury no skull fractures |
Some models produce frequent hemorrhages |
| Unrestrained impact acceleration models |
Produces diffuses axonal injury |
Poor reproducibility | |
| Constrained impact acceleration models |
Graded diffuse axonal injury reproducibility |
||
| Nonimpact injury | Head acceleration models |
Produces diffuse axonal injury |
Expensive technically difficult |
Neuroimaging offers additional information for TBI classification that is independent of the level of consciousness. Advantages of high-resolution CT include wide scale availability, short testing time, and ability to detect mass effect, cerebral contusion, and hemorrage reliably.7 Magnetic resonance imaging is used to detect diffuse or traumatic axonal injury that is often not appreciated on CT in the subacute time frame.9 When combined with an accurate clinical assessment, neuroimaging is a powerful tool for TBI classification and the initiation of therapeutic intervention.
Serum and cerebrospinal fluid (CSF) biomarker concentrations offer a theoretical method to classify TBI severity and add a biological correlate to the loss of consciousness and imaging potentially allowing for serial measurements and risk stratification. Recent clinical trials have shown increased levels of serum myelin basic protein, neuron specific enolase, and the biomarker S100B after TBI in a pediatric population.10,11 However, at the time of this review, no published data were available to compare the efficacy of myelin basic protein, neuron specific enolase, and S100B as biomarkers for TBI. Initial research has shown that biomarkers could aid in the classification of TBI severity, but further investigation into the role of specific markers needs to be completed.
Astute clinical assessment combined with neuroimaging, measurement of biomarkers, and further investigation into pathoanatomic injury enables clinicians to initially measure the severity of TBI and stratify patients according to risk. Classification of TBI directs patients to the most effective therapeutic groups and allows the development of further clinical trials investigating novel neuroprotective therapies.
TBI Pathophysiology: Implications for Future Targets
When compared with adults, the pediatric population is more susceptible to severe TBI because of a more pliable and immature skull, weak cervical musculature, and proportionally increased head mass.12 After the primary mechanical insult, changes in the nervous system microenvironment such as increased membrane permeability and calcium concentration, formation of edema, decreased cerebral blood flow (CBF), increase in oxidative species, and enhanced levels of inflammation could account for significant secondary injury. Figure 1 outlines the timing of cytokine formation, cerebral edema formation, scar formation, and delayed neuronal death after TBI.
Figure 1.
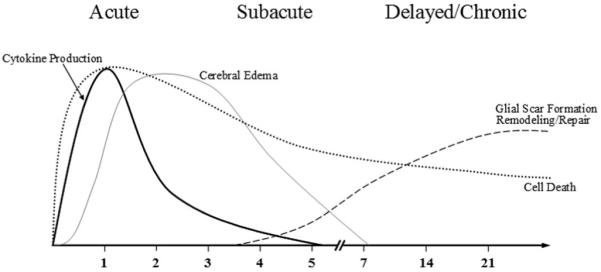
Timing in days of cytokine production, cerebral edema, scar formation, and delay cell death after TBI.
Post-Injury Neural Exitotoxicity
A large amount of preclinical research has linked neuronal cell excitotoxicity and elevated extracellular glutamate with increased cellular membrane permeability and subsequent neuronal injury. After TBI, elevated levels of excitatory amino acids, such as glutamate, have been found in both animal and human models.13,14 Increased glutamate levels lead to neuronal exitotoxicity and alteration of normal cellular metabolism. Previous work done in the Robertson laboratory using a rat TBI model showed elevated lactate or creatinine ratios consistent with increased glycolysis and decreased oxidative metabolism for as long as 7 days after injury.15 In addition, [18F]fluorodeoxyglucose-positron emission tomography of human patients after TBI has shown both regional and diffuse cerebral hyperglycolysis.16
The reliance of neuronal cells on anaerobic respiration depletes adenosine triphosphate (ATP) stores leading to active ion pump failure. The loss of electrochemical equilibrium supplied by the active Na+ or K+ pump and activation of ion channels by increased glutamate leads to massive Ca2+ influx.17 Elevation of intracellular calcium has been shown to induce calpain-mediated spectrin proteolysis causing increased levels of membrane breakdown and axonal injury.18
The complex interactions between metabolism, glutamate, and membrane permeability offer many targets for neuroprotection. Controlled hypothermia has been shown to slow metabolism and decrease ATP utilization (discussed later).19,20 Additional research has shown improved neurologic severity score as well as decreased brain edema and neuronal injury after infusion of glutamate receptor antaganonists.21 Ideally, maintenance of the normal metabolic rate and antagonism of glutamate channels could preserve the electrochemical gradient leading to retained membrane integrity and decreased levels of calpain-induced axonal injury.
Cerebral Edema
Neuronal injury after TBI is largely mediated by increased ICP. Elevated ICP results from increased brain tissue water derived from vasogenic and cytotoxic edema.22 Vasogenic edema is due to uncontrolled ion and protein flux from the intravascular to extracellular space due to disruption of the cerebral vascular endothelial cell lining.23 Work completed in the Halley lab has shown vacuole formation in cerebral endothelial cell lining leading to breakdown of the blood brain barrier causing increased transvascular protein flux and vasogenic interstitial edema.17,24
As previously discussed, TBI is associated with active ion pump failure and increased membrane permeability leading to elevation of intracellular ions. Influx of such ions leads to the development of an osmotic gradient causing concurrent flow of water into the intracellular space. The increase in intracellular water termed cytotoxic edema may be independent of endothelial cell integrity.17
Increased ICP derived from vasogenic and cytotoxic edema is indirectly related to CBF. Investigation of comatose patients after TBI with xenon-enhanced CT has shown decreased CBF.25 In addition, clinically significant cerebral vasospasm leading to decreased CBF has also been noted for up to 13 days in patients with severe TBI.26
The relationships between increased ICP, CBF, and cerebral vasospasm yield possible opportunities for neuroprotection. HTS infusion has been shown to decrease ICP while augmenting mean arterial pressures leading to an increase in CBF (discussed later).27-29 Maintenance of cerebral perfusion pressure by controlling ICP and vasospasm could significantly decrease neuronal cell injury.
Mitochondrial Injury
A common hypothesis implicates the role of mitochondrial oxidative damage in neuronal cell death. Analysis of murine hippocampal mitochondria after controlled cortical impact (CCI) injury showed increased protein oxidation and lipid peroxidation. Reactive nitrogen species played a significant role in oxidation as evidenced by elevated levels of mitochondrial 3-nitrotyrosine.30 The Hall laboratory investigated the role of peroxynitrite radicals on mitochondrial respiration. The peroxynitrite donor SIN-1 (3-morpholinosydnonimine) was administered to brain mitochondria showing a decrease in respiratory control ratio and increase in oxidation. In addition, penicillamine, a peroxynitrite scavenging agent, and tempol, a free radical scavenger, were shown to protect against oxidative damage.31 The generation of oxidative radicals associated with TBI leads to increased secondary neuronal injury making investigation into additional anti-oxidant treatments key for enhanced neuroprotection.
Neuronal Inflammation
TBI initiates an array of inflammatory cascades that contribute to secondary neuronal cell injury. In vivo analysis of rat intracerebral fluid after CCI has shown elevation of the pro-inflammatory cytokines TNFα, IL-1α, IL-1β, and IL-632 (Fig. 2). Further investigation of human CSF after TBI has shown increased levels of the cytokines IL-1β, IL-6, IL-8, IL-10, and TNFα as well as intracellular adhesion molecule 1 (ICAM-1) when compared with plasma.33-36 To test this hypothesis, the Wainwright laboratory administered the experimental therapeutic agent, Minozac (Mzc), to mice after CCI to decrease glial activation and proinflammatory cytokine production. The results demonstrated that the attenuation of cytokine production was associated with decreased astrocytic activation and improved functional neurologic outcomes.37 Investigation into the administration of N-acetylcysteine has shown attenuation of IL-1β, ICAM-1, and TNFα levels as well as reduction in blood brain barrier permeability, apoptotic index, and cerebral edema when given within 1 hour of injury.33,38 Modulation of the inflammatory response could significantly decrease neuronal injury making further research into anti-inflammatory therapeutics imperative.
Figure 2.

Elevated intracerebral cytokines identified in specific areas and at specified time points relative to the TBI. The pro-inflammatory cytokines IL-1α (A), IL-1β (B), IL-6 (C), and TNF-α (D) were significantly elevated six hours after CCI in the injury and penumbral regions when compared to sham animals (*p < 0.01 for all). IL-1α, IL-1β, and IL-6 remained elevated through 12, 12, and 24 hrs, respectively (*p < 0.01 or †p < 0.05). In the frontal area, IL-6 was significantly increased at 24 hours (33–50 fold, p < 0.01, Dunnett’s test), but not at 6 or 12 hours after. TBI Reprinted with permission.32
TREATMENT MODALITIES
Hypothermia
Clinical Significance
Controlled hypothermia after TBI could offer neuroprotection by decreasing neuronal metabolism. Cerebral hypoperfusion and subsequent ischemia evident during cardiovascular surgery or arrest provides a model for comparison with TBI. Controlled hypothermia with cardiopulmonary bypass and hypothermic circulatory arrest during pediatric cardiovascular surgery has shown a clear time dependent neuroprotective effect. In addition, clinical trials investigating the effect of controlled hypothermia on adult cardiac arrest patients showed decreased mortality (41 vs. 55%) and improved neurologic functional status.39 The observed benefit of hypothermia in cardiac patients has led to multiple studies involving TBI. A recent meta-analysis using eight clinical trials of controlled hypothermia in TBI showed a significant decrease in mortality and improvement in neurologic function.40 The role of hypothermia in the treatment of TBI remains controversial requiring further research into possible therapeutic mechanisms with particular emphasis on examining the therapeutic window.
Possible Therapeutic Mechanisms
Controlled hypothermia is believed to decrease neuronal metabolic rate after TBI. Although trials investigating the clinical effect of hypothermia for treatment of TBI have been completed, the majority of basic science analysis has been based on cardiac arrest models. In vivo research by the Griepp laboratory, measured CBF as well as venous and arterial oxygen saturations during human hypothermic circulatory arrest to calculate a temperature coefficient that was used to compare a distinct metabolic rate separated by 10°C. Their results showed a significant decrease in metabolic rate associated with hypothermia.20 An in vivo hypothermic piglet cardiac arrest model showed decreased cerebral ATP utilization and acidosis in association with attenuation of the metabolic rate.19,41 A growing body of preclinical and clinical research has given insight into the effect of hypothermia on neuronal metabolism in arrest models; however, more investigation involving TBI needs to be conducted to derive optimal strategies to enhance the therapeutic benefit.
TBI is associated with increased levels of extracellular glutamate and neuronal excitotoxicity.13,14 The observed alteration in normal membrane electrochemistry and glutamate activated ion channels increase intracellular calcium thereby stimulating calpain mediated neuronal injury.18,42 Investigation into the effect of controlled hypothermia in rat global cerebral ischemia models has shown decreased calpain activation and glutamate levels.43,44 Reduction of calpain activation could decrease the neuronal apoptotic fraction while depressed function of glutamate, activated ion channels, would stabilize the membrane electrochemical gradient leading to a therapeutic benefit of controlled hypothermia.
Preclinical research studies have shown elevated levels of oxidative radicals associated with TBI.30 Observation of rats undergoing cerebral ischemia-reperfusion injury found no increase in oxidative radicals when maintained at hypothermic temperatures compared with a dramatic, 2.5 fold increase noted in normothermic controls.45 Further investigation of cerebral ischemia-reperfusion injury in gerbils treated with controlled hypothermia has shown decreased H2O2 levels.46 Neuroprotection by controlled hypothermia seems to be partially mediated by the reduction in oxidative radicals. Further research into treatment of TBI with hypothermia and free radical scavengers such as tempol (BIOMOL International) could provide an additive effect ultimately enhancing the neuroprotective effect.
TBI stimulates multiple inflammatory cascades leading to the increase in pro-inflammatory and anti-inflammatory cytokines such as IL-1β, IL-6, IL-8, IL-10, TNFα, and ICAM-1.33-36 Significant increase in pro-inflammatory cytokines could lead to neuronal injury making modulation of the inflammatory response a target for neuroprotective therapy. Preclinical research has shown treatment with controlled hypothermia to decrease levels of ICAM-1 thereby attenuating neutrophil migration.47 However, analysis of CSF from children after TBI showed no change in cytokine production after treatment with controlled hypothermia.48 Overall, the effect of hypothermia on inflammatory modulation remains controversial and further investigation using both preclinical and clinical models needs to be completed.
Barriers to Treatment
The development of further clinical trials investigating the role of controlled hypothermia in treatment of TBI requires careful assessment of the possible untoward effects. A recent phase II clinical trial of moderate hypothermia after severe TBI has shown no statistical difference in coagulopathy, arrythmias, or infectious complications in a pediatric population.49 In addition, the timing of hypothermic treatment needs to be evaluated. Review of current literature shows average initiation of therapy within 24 hours of TBI followed by up to 48 hours of treatment; however, no trials exist comparing multiple treatment timing parameters.
A recent meta-analysis of eight clinical trials investigating adult TBI showed statistically significant reduced mortality (relative risk, 0.51) and improved neurologic outcome (relative risk, 1.91) in patients treated with moderate hypothermia for greater then 48 hours. However, a significant increase in pneumonia (relative risk, 2.37) was also noted.40 Although still controversial, prolonged hypothermia (greater than 48 hours) offers a promising treatment to attenuate neuronal injury after TBI requiring further research into therapeutic safety and usage as a multimodality treatment.
Hypertonic Saline
Clinical Significance
Previous research has shown that HTS infusion after TBI offers neuroprotection via reduction of increased ICP leading to improved CBF.29,50 HTS resuscitation also increased mean arterial pressures thereby attenuating a significant increase of poor neurologic outcomes seen with hypotension within 6 hours of TBI.51 HTS infusion decreased the complications, required interventions, and length of intensive care unit stay after TBI in a pediatric population.52 Despite the promising results noted in the acute and subacute setting, some trials have failed to show improved neurologic outcomes 6 months after TBI.53 To organize further clinical trials and optimize the therapeutic efficacy of HTS resuscitation, research into the role of HTS in control of ICP, maintenance of membrane electrochemical potential, and modulation of the inflammatory response needs to be completed.
Possible Therapeutic Mechanisms
HTS reduces ICP by increasing the plasma osmolarity (largely Na+) thereby reducing the flow of water into the intracellular and interstitial spaces. Neuronal injury associated with TBI leads to accumulation of organic osmolytes within the cell.54 Subsequent hyperosmolarity and influx of water is associated with increased brain edema and ICP.55 HTS resuscitation increases the serum osmolarity reducing the flow of water from the intravascular space into the extracellular space to attenuate the accumulation of brain edema.29 In addition, HTS infusions increase intravascular volume28 leading to increased CBF.27 The overall increase in CSF with decreased ICP leads to improved neuronal oxygenation with reduction of ischemia.
TBI is associated with elevated levels of glutamate and Ca2+ as well as failure of active ion pumps leading to disruption of the normal electrochemical gradient. HTS infusion increases plasma sodium levels restoring normal function of the Na+ or glutamate pump thereby reducing levels of extracellular glutamate.56 Normalization of intracellular Na+ leads to activation of the Na+ or Ca2+ pump decreasing levels of intracellular Ca2+.57 Work completed in the Zaaroor laboratory has shown decreased calpain activity in rats resuscitated with HTS after cortical injury.58 Normalization of the membrane electrochemical gradient ultimately leading to decreased calpain activated neuronal damage could account for the neuroprotection see with HTS resuscitation.
TBI is associated with increased systemic inflammation and leukocyte migration leading to secondary neuronal injury.33-36 In vivo, HTS infusion has shown decreased penumbral neutrophil density after CCI in a rat model.58 A recent trial of HTS infusion in TBI patients has also shown attenuation of tissue ICAM-1 and mitogen-activated protein kinase leading to decreased neutrophil migration and neuronal apoptosis. In addition, a significant decrease in multi organ dysfunction was observed.59 Preliminary research has shown benefit from HTS resuscitation through modulation of neutrophil migration; however, further investigation into the effect on the overall pro inflammatory response has yet to be completed.
Barriers to Treatment
Mannitol infusion has long been the standard of care for elevated ICPs in the setting of TBI; however, because of increasing knowledge of cerebral physiology, multiple in vivo trials have been completed to compare HTS to mannitol.60 Single, equi-osmolar bolus doses of HTS and mannitol infused after TBI in a rat model showed significant reduction in ICP for up to 500 minutes after treatment in the HTS group. Conversely, after a transient decrease in ICP, the mannitol group was found to return to or overshoot the baseline-elevated ICP.61 In addition, HTS infusion after TBI in a rat model has shown significant decrease in calpain-mediated apoptosis and neuroinflammation compared with a mannitol treatment group. Of note, the additional benefit of HTS when compared with mannitol was independent of the infused volume indicating a likely mechanism separate from the effect on brain tissue edema.58 Initial in vivo research has shown the additional benefit from HTS therapy for TBI; however, before acceptance as standard of care, prospective randomized trials with comparison to mannitol will likely be needed.
Central pontine myelinolysis is a demyelinating disease of the pons that has been associated with rapid changes in serum Na+ levels.62 It is believed that HTS resuscitation with TBI could rapidly increase serum Na+ causing central pontine myelinolysis; however, small clinical trials have shown no evidence of demyelination disorders.29
HTS resuscitation has been linked to increased rates of renal failure in burn patients63 with evidence of reversible renal insufficiency in a pediatric TBI resuscitation trial.29 Pleural effusions of unknown etiology have also been observed.64 HTS resuscitation after hypertensive intracerebral hemorrhage has shown rebound malignant cerebral edema after 24 hours of therapy.65 Although a review of the literature has shown some untoward effects from HTS resuscitation, the observed therapeutic benefit seems to outweigh the potential risks.
Cell Therapy
Clinical Significance
A large amount of research is currently underway investigating the role of cell therapeutics in the treatment of TBI. By definition, stem or progenitor cells are capable of self-renewal and are multipotent meaning the cells are able to differentiate into multiple cell lines.66 Progenitor cell populations are maintained in niches throughout the body to protect against cell depletion or over proliferation.67 Adult stem cell niches and respective populations currently under investigation include bone marrow-derived cells (mesenchymal stem cells [MSCs] and hematopoeitic stem cells), subventricular zone (neural stem cells), umbilical cord blood mononuclear fraction (MSCs and hematopoeitc stem cells), and adipose tissue. Progenitor cell capacity to self-renew, multipotency, and ready availability make them an attractive treatment modality for multiple diseases.
Preclinical studies evaluating the efficacy of stem cell therapeutics are in their infancy; however, many promising results have already been published. Intravenous and direct transplantation of MSCs, human umbilical cord blood, neural stem cells, and adipose-derived stem cells after TBI in animal models have shown cell engraftment and functional neurologic improvement.68-71 The development of clinical models requires careful investigation into therapeutic mechanisms including transdifferentiation, growth factor secretion, cell to cell contact, and modulation of the inflammatory response.
Possible Therapeutic Mechanisms
Initial preclinical research into cell therapeutics for TBI has shown clear functional neurologic improvement, however, the exact mechanisms of action have yet to be clearly delineated. Tissue specific stem cells such as MSCs have shown the ability to differentiate into loco-regional cell lines and promote tissue regeneration.72 In addition, MSCs have been shown to phenotypically adopt neural characteristics.73 Early thought indicated such cell plasticity and engraftment could account for the observed therapeutic benefit. However, the frequency of engraftment and clinical significance of transdifferentiation remains controversial.74
Additional investigation has shown progenitor cell migration to the site of injury and secretion of various trophic factors. Culture of MSCs with rat brain supernatant after CCI injury and induced middle cerebral artery stroke have shown secretion of brain-derived neurotrophic factor, nerve growth factor, epidermal growth factor, hepatocyte growth factor, vascular endothelial growth factor, insulin-like growth factor (IGF-1), and fibroblast growth factor (FGF-2).75,76 Increased vascular endothelial growth factor levels have been shown to increase penumbral angiogenesis in a rat stroke model.77 Progenitor cell secretion of growth factors could alter the local neuronal cellular milieu stimulating resident cell regeneration or repair accounting for the observed therapeutic benefit.
Cell-to-cell contact or fusion allowing transfer of proteins or genes is another theoretical mechanism accounting for the efficacy of cell therapeutics. Preliminary investigation of heat shocked epithelial airway cells treated with MSCs has shown 1% incidence of cellular fusion.78 Current research to investigate the usage of progenitor cells as vehicles to carry deficient genes that could be transferred via direct cellular contact is underway.79
As previously discussed, induction of a systemic inflammatory response plays a significant role in secondary injury with TBI. A large amount of preclinical work has shown a decrease in inflammation after treatment with progenitor cells in both in vivo and in vitro models. Culture of MSCs with purified immune cells has shown an increase in the anti-inflammatory cytokines IL-4 and IL-10 while decreasing levels of TNF-α and IFN-γ. Increased levels of IL-4 in accordance with decreaed INF-γ has been shown to cause a shift from cytotoxic TH1 cells to TH2 helper T cells. In addition, decreased TNF-α with increased IL-10 promotes a more tolerant or anti-inflammatory immune response.80 In addition, transfusion of human umbilical cord blood in rats after induced ischemic stroke prevented loss of splenic mass and a decrease in CD8+ T cells that had previously been observed. Of note, overall T cell proliferation was decreased in accordance with decreased INF-γ and increased IL-10 levels. An 85% reduction in cerebral infarct volume was noted.81 Modulation of the systemic inflammatory response offers a possible explanation for the therapeutic benefit of cell therapeutics and is an attractive avenue for neuroprotection.
Barriers to Treatment
Both emboli and tumor development have been associated with progenitor cell transplantation. MSC transplantation in a murine model has shown malignant transformation into fibrosarcomas82 and osteosarcomas located in the lung.83 In addition, pulmonary thromboemboli have been reported with intravenous transfusion of progenitor cells.84 Intracarotid transfusion of progenitor cells in a stroke model has been associated with decreased CBF.85 Overall, progenitor cell therapeutics offers exciting new therapies to enhance neuroprotection; however, further research into cell delivery, treatment dosage, and timing of therapy need completed before multi-center clinical trials.
Future considerations
The National Institutes of Health (NIH) recently convened a meeting to discuss the future investigation of TBI therapeutics.86 The conference noted the complex nature to neuronal injury and failure of all clinical trials based on monotherapy to date. Recommendations included the development of in vitro models to research multimodality treatments ultimately leading to preclinical and clinical selection of therapies that could have an additive effect by targeting multiple mechanisms of TBI’s complex pathophysiology (www.nih.gov).
TBI is a major source of morbidity and mortality in the American health system. Although previous clinical and preclinical research has shown therapeutic benefit from controlled hypothermia, HTS resuscitation, and cell therapeutics, multimodal therapy could potentially offer the most effective treatment. The beneficial effects of hypothermia, HTS resuscitation, and cell therapeutics could prove to be synergistic enhancing overall neuroprotection.
Acknowledgments
Supported by grants NIH T32 GM 08 79201; NIH P018/N01 HB 37163; M01 RR 02558; Texas Higher Education Coordinating Board; Children’s Memorial Hermann Hospital Foundation.
REFERENCES
- 1.Langlois JA, Rutland-Brown W, Thomas KE. The incidence of trau-matic brain injury among children in the United States: differences by race. J Head Trauma Rehabil. 2005;20:229–238. doi: 10.1097/00001199-200505000-00006. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy ML, Serpi T, Kufera JA, Demeter LA, Paidas C. Factors influencing admission among children with a traumatic brain injury. Acad Emerg Med. 2002;9:684–693. doi: 10.1111/j.1553-2712.2002.tb02146.x. [DOI] [PubMed] [Google Scholar]
- 3.Hawley CA, Ward AB, Magnay AR, Long J. Outcomes following childhood head injury: a population study. J Neurol Neurosurg Psychiatry. 2004;75:737–742. doi: 10.1136/jnnp.2003.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray DS, Burnham RS. Preliminary outcome analysis of a long-term rehabilitation program for severe acquired brain injury. Arch Phys Med Rehabil. 2000;81:1447–1456. doi: 10.1053/apmr.2000.16343. [DOI] [PubMed] [Google Scholar]
- 5.Cowen TD, Meythaler JM, DeVivo MJ, Ivie CS, III, Lebow J, Novack TA. Influence of early variables in traumatic brain injury on functional independence measure scores and rehabilitation length of stay and charges. Arch Phys Med Rehabil. 1995;76:797–803. doi: 10.1016/s0003-9993(95)80542-7. [DOI] [PubMed] [Google Scholar]
- 6.Moppett IK. Traumatic brain injury: assessment, resuscitation and early management. Br J Anaesth. 2007;99:18–31. doi: 10.1093/bja/aem128. [DOI] [PubMed] [Google Scholar]
- 7.Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT, Workshop Scientific Team and Advisory Panel Members Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cernak I. Animal models of head trauma. NeuroRx. 2005;2:410–422. doi: 10.1602/neurorx.2.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topal NB, Hakyemez B, Erdogan C, et al. MR imaging in the detection of diffuse axonal injury with mild traumatic brain injury. Neurol Res. 2008;30:974–978. doi: 10.1179/016164108X323799. [DOI] [PubMed] [Google Scholar]
- 10.Berger RP, Adelson PD, Pierce MC, Dulani T, Cassidy LD, Kochanek PM. Serum neuron-specific enolase, S100B, and myelin basic protein concentrations after inflicted and noninflicted traumatic brain injury in children. J Neurosurg. 2005;103:61–68. doi: 10.3171/ped.2005.103.1.0061. [DOI] [PubMed] [Google Scholar]
- 11.Berger RP, Beers SR, Richichi R, Wiesman D, Adelson PD. Serum biomarker concentrations and outcome after pediatric traumatic brain injury. J Neurotrauma. 2007;24:1793–1801. doi: 10.1089/neu.2007.0316. [DOI] [PubMed] [Google Scholar]
- 12.Noppens R, Brambrink AM. Traumatic brain injury in children–clinical implications. Exp Toxicol Pathol. 2004;56:113–125. doi: 10.1016/j.etp.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Bullock R, Zauner A, Myseros JS, Marmarou A, Woodward JJ, Young HF. Evidence for prolonged release of excitatory amino acids in severe human head trauma. Relationship to clinical events. Ann N Y Acad Sci. 1995;765:290–297. doi: 10.1111/j.1749-6632.1995.tb16586.x. discussion 298. [DOI] [PubMed] [Google Scholar]
- 15.Casey PA, McKenna MC, Fiskum G, Saraswati M, Robertson CL. Early and sustained alterations in cerebral metabolism after traumatic brain injury in immature rats. J Neurotrauma. 2008;25:603–614. doi: 10.1089/neu.2007.0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergsneider M, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. 1997;86:241–251. doi: 10.3171/jns.1997.86.2.0241. [DOI] [PubMed] [Google Scholar]
- 17.Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 18.Buki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Laptook AR, Corbett RJ, Sterett R, Garcia D, Tollefsbol G. Quantitative relationship between brain temperature and energy utilization rate measured in vivo using 31P and 1H magnetic resonance spectroscopy. Pediatr Res. 1995;38:919–925. doi: 10.1203/00006450-199512000-00015. [DOI] [PubMed] [Google Scholar]
- 20.McCullough JN, Zhang N, Reich DL, et al. Cerebral metabolic suppression during hypothermic circulatory arrest in humans. Ann Thorac Surg. 1999;67:1895–1899. doi: 10.1016/s0003-4975(99)00441-5. discussion 1919–1821. [DOI] [PubMed] [Google Scholar]
- 21.Fei Z, Zhang X, Bai HM, Jiang XF, Wang XL. Metabotropic glutamate receptor antagonists and agonists: potential neuroprotectors in diffuse brain injury. J Clin Neurosci. 2006;13:1023–1027. doi: 10.1016/j.jocn.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 22.Marmarou A, Fatouros PP, Barzo P, et al. Contribution of edema and cerebral blood volume to traumatic brain swelling in head-injured patients. J Neurosurg. 2000;93:183–193. doi: 10.3171/jns.2000.93.2.0183. [DOI] [PubMed] [Google Scholar]
- 23.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 24.Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- 25.Bouma GJ, Muizelaar JP, Stringer WA, Choi SC, Fatouros P, Young HF. Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J Neurosurg. 1992;77:360–368. doi: 10.3171/jns.1992.77.3.0360. [DOI] [PubMed] [Google Scholar]
- 26.Oertel M, Boscardin WJ, Obrist WD, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg. 2005;103:812–824. doi: 10.3171/jns.2005.103.5.0812. [DOI] [PubMed] [Google Scholar]
- 27.Walsh JC, Zhuang J, Shackford SR. A comparison of hypertonic to isotonic fluid in the resuscitation of brain injury and hemorrhagic shock. J Surg Res. 1991;50:284–292. doi: 10.1016/0022-4804(91)90192-o. [DOI] [PubMed] [Google Scholar]
- 28.Moss GS, Gould SA. Plasma expanders. An update. Am J Surg. 1988;155:425–434. doi: 10.1016/s0002-9610(88)80106-5. [DOI] [PubMed] [Google Scholar]
- 29.Khanna S, Davis D, Peterson B, et al. Use of hypertonic saline in the treatment of severe refractory posttraumatic intracranial hypertension in pediatric traumatic brain injury. Crit Care Med. 2000;28:1144–1151. doi: 10.1097/00003246-200004000-00038. [DOI] [PubMed] [Google Scholar]
- 30.Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- 31.Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: protective effects of peroxynitrite scavengers. J Neurosci Res. 2007;85:2216–2223. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- 32.Harting MT, Jimenez F, Adams SD, Mercer DW, Cox CS., Jr. Acute, regional inflammatory response after traumatic brain injury: implications for cellular therapy. Surgery. 2008;144:803–813. doi: 10.1016/j.surg.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008;2008:716458. doi: 10.1155/2008/716458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bell MJ, Kochanek PM, Doughty LA, et al. Interleukin-6 and interleukin-10 in cerebrospinal fluid after severe traumatic brain injury in children. J Neurotrauma. 1997;14:451–457. doi: 10.1089/neu.1997.14.451. [DOI] [PubMed] [Google Scholar]
- 35.Whalen MJ, Carlos TM, Kochanek PM, et al. Interleukin-8 is increased in cerebrospinal fluid of children with severe head injury. Crit Care Med. 2000;28:929–934. doi: 10.1097/00003246-200004000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Singhal A, Baker AJ, Hare GM, Reinders FX, Schlichter LC, Moulton RJ. Association between cerebrospinal fluid interleukin-6 concentrations and outcome after severe human traumatic brain injury. J Neurotrauma. 2002;19:929–937. doi: 10.1089/089771502320317087. [DOI] [PubMed] [Google Scholar]
- 37.Lloyd E, Somera-Molina K, Van Eldik LJ, Watterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiong Y, Peterson PL, Lee CP. Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J Neurotrauma. 1999;16:1067–1082. doi: 10.1089/neu.1999.16.1067. [DOI] [PubMed] [Google Scholar]
- 39.Hypothermia after Cardiac Arrest Study Group Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- 40.Peterson K, Carson S, Carney N. Hypothermia treatment for traumatic brain injury: a systematic review and meta-analysis. J Neurotrauma. 2008;25:62–71. doi: 10.1089/neu.2007.0424. [DOI] [PubMed] [Google Scholar]
- 41.Laptook AR, Corbett RJ, Burns D, Sterett R. Neonatal ischemic neuroprotection by modest hypothermia is associated with attenuated brain acidosis. Stroke. 1995;26:1240–1246. doi: 10.1161/01.str.26.7.1240. [DOI] [PubMed] [Google Scholar]
- 42.Froehler MT, Geocadin RG. Hypothermia for neuroprotection after cardiac arrest: mechanisms, clinical trials and patient care. J Neurol Sci. 2007;261:118–126. doi: 10.1016/j.jns.2007.04.042. [DOI] [PubMed] [Google Scholar]
- 43.Busto R, Globus MY, Dietrich WD, Martinez E, Valdés I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20:904–910. doi: 10.1161/01.str.20.7.904. [DOI] [PubMed] [Google Scholar]
- 44.Harada K, Maekawa T, Tsuruta R, et al. Hypothermia inhibits translocation of CaM kinase II and PKC-alpha, beta, gamma isoforms and fodrin proteolysis in rat brain synaptosome during ischemia-reperfusion. J Neurosci Res. 2002;67:664–669. doi: 10.1002/jnr.10159. [DOI] [PubMed] [Google Scholar]
- 45.Globus MY, Busto R, Lin B, Schnippering H, Ginsberg MD. Detection of free radical activity during transient global ischemia and recirculation: effects of intraischemic brain temperature modulation. J Neurochem. 1995;65:1250–1256. doi: 10.1046/j.1471-4159.1995.65031250.x. [DOI] [PubMed] [Google Scholar]
- 46.Lei B, Adachi N, Arai T. The effect of hypothermia on H2O2 production during ischemia and reperfusion: a microdialysis study in the gerbil hippocampus. Neurosci Lett. 1997;222:91–94. doi: 10.1016/s0304-3940(97)13349-3. [DOI] [PubMed] [Google Scholar]
- 47.Inamasu J, Suga S, Sato S, et al. Intra-ischemic hypothermia attenuates intercellular adhesion molecule-1 (ICAM-1) and migration of neutrophil. Neurol Res. 2001;23:105–111. doi: 10.1179/016164101101198217. [DOI] [PubMed] [Google Scholar]
- 48.Buttram SD, Wisniewski SR, Jackson EK, et al. Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. J Neurotrauma. 2007;24:1707–1717. doi: 10.1089/neu.2007.0349. [DOI] [PubMed] [Google Scholar]
- 49.Adelson PD, Ragheb J, Kanev P, et al. Phase II clinical trial of moderate hypothermia after severe traumatic brain injury in children. Neurosurgery. 2005;56:740–754. doi: 10.1227/01.neu.0000156471.50726.26. discussion 740–754. [DOI] [PubMed] [Google Scholar]
- 50.Pinto FC, Capone-Neto A, Prist R, E Silva MR, Poli-de-Figueiredo LF. Volume replacement with lactated Ringer’s or 3% hypertonic saline solution during combined experimental hemorrhagic shock and traumatic brain injury. J Trauma. 2006;60:758–763. doi: 10.1097/01.ta.0000214581.89316.73. discussion 763–764. [DOI] [PubMed] [Google Scholar]
- 51.Samant UB IV, Mack CD, Koepsell T, Rivara FP, Vavilala MS. Time of hypotension and discharge outcome in children with severe traumatic brain injury. J Neurotrauma. 2008;25:495–502. doi: 10.1089/neu.2007.0491. [DOI] [PubMed] [Google Scholar]
- 52.Simma B, Burger R, Falk M, Sacher P, Fanconi S. A prospective, randomized, and controlled study of fluid management in children with severe head injury: lactated Ringer’s solution versus hypertonic saline. Crit Care Med. 1998;26:1265–1270. doi: 10.1097/00003246-199807000-00032. [DOI] [PubMed] [Google Scholar]
- 53.Cooper DJ, Myles PS, McDermott FT, et al. HTS Study Investigators Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: a randomized controlled trial. JAMA. 2004;291:1350–1357. doi: 10.1001/jama.291.11.1350. [DOI] [PubMed] [Google Scholar]
- 54.Nonaka M, Yoshimine T, Kohmura E, Wakayama A, Yamashita T, Hayakawa T. Changes in brain organic osmolytes in experimental cerebral ischemia. J Neurol Sci. 1998;157:25–30. doi: 10.1016/s0022-510x(98)00062-8. [DOI] [PubMed] [Google Scholar]
- 55.Battistella FD, Wisner DH. Combined hemorrhagic shock and head injury: effects of hypertonic saline (7.5%) resuscitation. J Trauma. 1991;31:182–188. [PubMed] [Google Scholar]
- 56.Tyagi R, Donaldson K, Loftus CM, Jallo J. Hypertonic saline: a clinical review. Neurosurg Rev. 2007;30:277–289. doi: 10.1007/s10143-007-0091-7. discussion 289–290. [DOI] [PubMed] [Google Scholar]
- 57.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 58.Soustiel JF, Vlodavsky E, Zaaroor M. Relative effects of mannitol and hypertonic saline on calpain activity, apoptosis and polymorphonuclear infiltration in traumatic focal brain injury. Brain Res. 2006;1101:136–144. doi: 10.1016/j.brainres.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 59.Bing M, Zheng-lu H, Hui C, Xian D, Jian H. Variations of p38 MAPK and sICAM-1 with therapeutic effect of different resuscitation fluids on severe traumatic patients. Chin J Traumatol. 2007;10:263–268. [PubMed] [Google Scholar]
- 60.Infanti JL. Challenging the gold standard: should mannitol remain our first-line defense against intracranial hypertension? J Neurosci Nurs. 2008;40:362–368. [PubMed] [Google Scholar]
- 61.Mirski AM, Denchev ID, Schnitzer SM, Hanley FD. Comparison between hypertonic saline and mannitol in the reduction of elevated intracranial pressure in a rodent model of acute cerebral injury. J Neurosurg Anesthesiol. 2000;12:334–344. doi: 10.1097/00008506-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 62.Lampl C, Yazdi K. Central pontine myelinolysis. Eur Neurol. 2002;47:3–10. doi: 10.1159/000047939. [DOI] [PubMed] [Google Scholar]
- 63.Huang PP, Stucky FS, Dimick AR, Treat RC, Bessey PQ, Rue LW. Hypertonic sodium resuscitation is associated with renal failure and death. Ann Surg. 1995;221:543–554. doi: 10.1097/00000658-199505000-00012. discussion 554–s547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maeda H, Tsuruya K, Yotsueda H, et al. A case report of syndrome of inappropriate secretion of antidiuretic hormone with marked edema due to administration of hypertonic saline. Ther Apher Dial. 2007;11:309–313. doi: 10.1111/j.1744-9987.2007.00469.x. [DOI] [PubMed] [Google Scholar]
- 65.Qureshi AI, Suarez JI, Bhardwaj A. Malignant cerebral edema in patients with hypertensive intracerebral hemorrhage associated with hypertonic saline infusion: a rebound phenomenon? J Neurosurg Anesthesiol. 1998;10:188–192. doi: 10.1097/00008506-199807000-00010. [DOI] [PubMed] [Google Scholar]
- 66.Weiner LP. Definitions and criteria for stem cells. Methods Mol Biol. 2008;438:3–8. doi: 10.1007/978-1-59745-133-8_1. [DOI] [PubMed] [Google Scholar]
- 67.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 68.Gao J, Prough DS, McAdoo DJ, et al. Transplantation of primed human fetal neural stem cells improves cognitive function in rats after traumatic brain injury. Exp Neurol. 2006;201:281–292. doi: 10.1016/j.expneurol.2006.04.039. [DOI] [PubMed] [Google Scholar]
- 69.Kim JM, Lee ST, Chu K, et al. Systemic transplantation of human adipose stem cells attenuated cerebral inflammation and degeneration in a hemorrhagic stroke model. Brain Res. 2007;1183:43–50. doi: 10.1016/j.brainres.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 70.Mahmood A, Lu D, Wang L, Li Y, Lu M, Chopp M. Treatment of traumatic brain injury in female rats with intravenous administration of bone marrow stromal cells. Neurosurgery. 2001;49:1196–1203. discussion 1203–1194. [PubMed] [Google Scholar]
- 71.Lu D, Sanberg PR, Mahmood A, et al. Intravenous administration of human umbilical cord blood reduces neurological deficit in the rat after traumatic brain injury. Cell Transplant. 2002;11:275–281. [PubMed] [Google Scholar]
- 72.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276:71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 73.Mahmood A, Lu D, Qu C, Goussev A, Chopp M. Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. J Neurosurg. 2006;104:272–277. doi: 10.3171/jns.2006.104.2.272. [DOI] [PubMed] [Google Scholar]
- 74.Castro RF, Jackson KA, Goodell MA, Robertson CS, Liu H, Shine HD. Failure of bone marrow cells to transdifferentiate into neural cells in vivo. Science. 2002;297:1299. doi: 10.1126/science.297.5585.1299. [DOI] [PubMed] [Google Scholar]
- 75.Qu R, Li Y, Gao Q, et al. Neurotrophic and growth factor gene expression profiling of mouse bone marrow stromal cells induced by ischemic brain extracts. Neuropathology. 2007;27:355–363. doi: 10.1111/j.1440-1789.2007.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen X, Katakowski M, Li Y, et al. Human bone marrow stromal cell cultures conditioned by traumatic brain tissue extracts: growth factor production. J Neurosci Res. 2002;69:687–691. doi: 10.1002/jnr.10334. [DOI] [PubMed] [Google Scholar]
- 77.Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98:1076–1084. doi: 10.1002/jcb.20886. [DOI] [PubMed] [Google Scholar]
- 78.Spees JL, Olson SD, Ylostalo J, et al. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc Natl Acad Sci USA. 2003;100:2397–2402. doi: 10.1073/pnas.0437997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wallace GQ, Lapidos KA, Kenik JS, McNally EM. Long-term survival of transplanted stem cells in immunocompetent mice with muscular dystrophy. Am J Pathol. 2008;173:792–802. doi: 10.2353/ajpath.2008.080259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 81.Vendrame M, Gemma C, Pennypacker KR, et al. Cord blood rescues stroke-induced changes in splenocyte phenotype and function. Exp Neurol. 2006;199:191–200. doi: 10.1016/j.expneurol.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 82.Miura M, Miura Y, Padilla-Nash HM, et al. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells. 2006;24:1095–1103. doi: 10.1634/stemcells.2005-0403. [DOI] [PubMed] [Google Scholar]
- 83.Aguilar S, Nye E, Chan J, et al. Murine but not human mesenchymal stem cells generate osteosarcoma-like lesions in the lung. Stem Cells. 2007;25:1586–1594. doi: 10.1634/stemcells.2006-0762. [DOI] [PubMed] [Google Scholar]
- 84.Baker KS, DeFor TE, Burns LJ, Ramsay NK, Neglia JP, Robison LL. New malignancies after blood or marrow stem-cell transplantation in children and adults: incidence and risk factors. J Clin Oncol. 2003;21:1352–1358. doi: 10.1200/JCO.2003.05.108. [DOI] [PubMed] [Google Scholar]
- 85.Walczak P, Zhang J, Gilad AA, et al. Dual-modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. Stroke. 2008;39:1569–1574. doi: 10.1161/STROKEAHA.107.502047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Health NIo [Accessed September 26, 2008];Combination therapies for traumatic brain injury workshop. 2008 http://www.ninds.nih.gov/news_and_events/proceedings/Combination_Therapies_for_Traumatic_Brain_Injury_Workshop.htm.