

Summary
Aberrant fusions between heterologous chromosomes are among the most prevalent cytogenetic abnormalities found in cancer cells. Oncogenic chromosomal translocations provide cells with a proliferative or survival advantage. They may either initiate transformation or be acquired secondarily as a result of genomic instability. Here we highlight recent advances toward understanding the origin of chromosomal translocations in incipient lymphoid cancers and how tumor-suppressive pathways normally limit the frequency of these aberrant recombination events. Deciphering the mechanisms that mediate chromosomal fusions will open new avenues for developing therapeutic strategies aimed at eliminating lesions that lead to the initiation, maintenance, and progression of cancer.
Introduction
Chromosomal translocations are the most common class of mutations found in hematological malignancies (Kuppers, 2005). In addition, recurrent chromosomal fusions have been causally implicated in sarcomas and other solid tumors (Kumar-Sinha et al., 2008). There are a few common mechanisms by which translocations provide a proliferative or survival advantage to an incipient cancer cell. First, when cis-regulatory transcriptional elements from one gene are apposed to a proto-oncogene, this causes aberrant expression of the growth-promoting oncogene. For example, Burkitt's lymphoma carries a reciprocal translocation that results in fusion of the coding region of the c-myc proto-oncogene with the immunoglobulin heavy chain (IgH) (Jankovic et al., 2007), which places c-myc under the control of the 3′ regulatory elements of IgH (Gostissa et al., 2009). c-myc is thereby deregulated and promotes cellular transformation through its effects on the cell cycle, differentiation, and apoptosis. A second mechanism by which translocations may promote transformation involves the fusion of two genes to produce a chimeric protein with oncogenic activity. A prototypical example is the Philadelphia chromosome found in a subtype of acute lymphoblastic leukemia (Ph+ ALL) and chronic myeloid leukemia (CML), in which the BCR-ABL fusion gene encodes a protein with deregulated kinase activity. BCR-ABL expression results in cytokine-independent growth, resistance to apoptosis, and genetic instability (Kuppers, 2005). In addition to protein encoding genes, chromosomal translocations can also involve microRNA genes (Calin et al., 2004). Structural and functional alterations in these small noncoding RNAs have been detected in various cancers and may play a causal role in tumorigenesis (Calin and Croce, 2007; Robbiani et al., 2009).
Translocation requires: 1) formation of paired double strand DNA breaks (DSBs) on separate chromosomes 2) proximity of broken ends (at least transiently) and 3) joining of the heterologous DNA ends, as opposed to fusion in cis (Figure 1). Although many different cancers carry recurrent chromosome translocations (see http://www.sanger.ac.uk/genetics/CGP/Census/translocation.shtml), this review will focus on the etiology of translocations in lymphocytes as these are the most well-characterized to date. We expect that most incipient cancer cells will share the basic mechanisms involved in the development of and protection against chromosomal translocations.
Figure 1. Misrepair of DNA breaks cause chromosomal translocations[rk4].
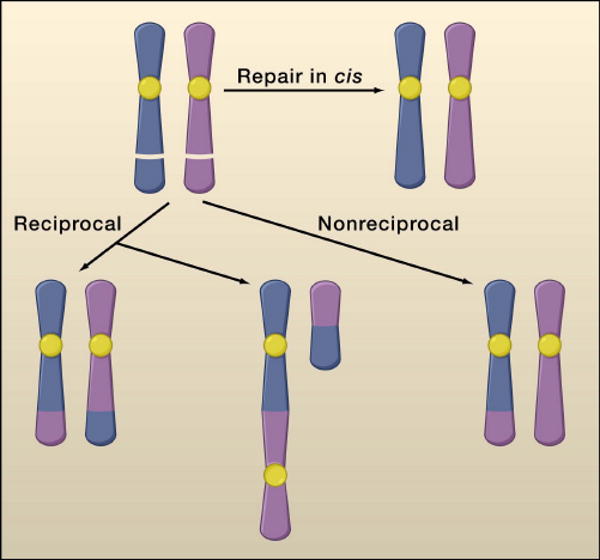
Chromosomal translocations require formation of paired double strand DNA breaks (DSBs) on different chromosomes. DSBs can be repaired in cis, or can result in chromosomal translocation by rearrangement between non homologous chromosomes. Depending on the topology of the rearrangement, the translocation can be reciprocal (balanced or unbalanced) or non-reciprocal. The majority of translocations associated with cancer in human lymphoid tumors involve balanced chromosomal translocations, whereas epithelial cancers usually carry complex nonreciprocal translocations.
Chromosomal translocations in context
Approximately 95% of all lymphomas are of B cell origin (Kuppers, 2005). These cancers are heterogeneous, involving all B cell developmental stages: from early B cells in acute lymphoblastic leukemia (ALL) to mature B cells in Burkitt's lymphoma and plasma cells in multiple myeloma. Despite their disparate origins, many of these cancers carry balanced chromosomal translocations that involve immunoglobulin (Ig) genes and oncogenic partner genes (Figure 1); in rarer cases, translocations can be non-reciprocal or join two non-Ig genes (Kuppers, 2005).
Why are B cells particularly susceptible to transformation by chromosome translocation? This issue has been the subject of much debate, beginning immediately after these abnormal cytological features were discovered. A great deal of the discussion has focused on antigen receptor gene diversification during V(D)J recombination, somatic hypermutation (SHM) and class switch recombination (CSR), as all three require programmed DNA damage (Figure 2). The notion that antibody gene diversification reactions initiate translocations was strongly bolstered when the first lymphoid cancer associated translocation was characterized as a fusion between c-myc and the switch region of the IgH locus suggesting that c-myc/IgH translocations arise as a byproduct of aberrant Ig class switching (Jankovic et al., 2007). In the ensuing years, many additional translocations have been documented in lymphoid cancers, and in most, though not all cases, at least one of the partner chromosomes was an Ig variable or switch region. Translocations involving two non-Ig genes are interesting exceptions to the rule; however, this group of translocations may also be products of “off target” genome destabilization by the Ig V(D)J recombinase, recombinase activating gene 1/2 (RAG1/2) and/or activation induced cytidine deaminase (AID) (Robbiani et al., 2009; Tsai et al., 2008) (see below).
Figure 2. Antigen diversification reactions in lymphocytes.
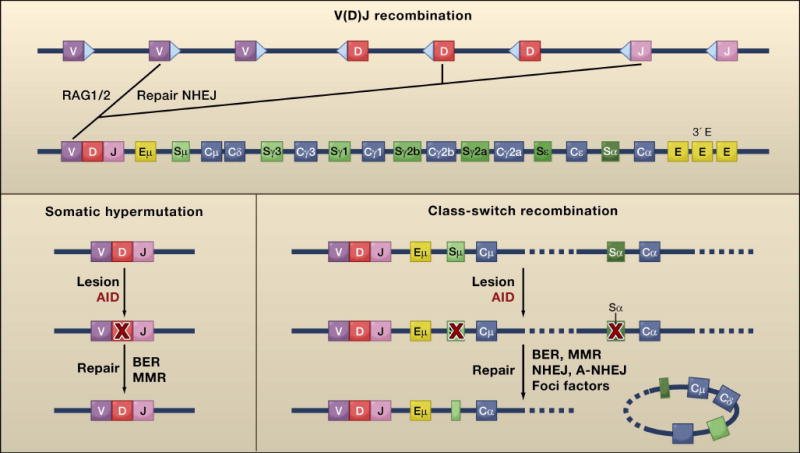
Lymphocyte antigen receptor diversity is established in developing lymphocytes by V(D)J recombination. Recombinase-activating genes 1 and 2 (RAG1 and RAG2), are transesterases that introduce double strand breaks (DSBs) at recombination signal sequences (shown in triangles) that flank V, D, and J gene segments. These DSBs are repaired by the NHEJ pathway. Mature B cells undergo two additional diversification reactions called somatic hypermutation and class switch recombination. These two processes are initiated by AID, a single strand DNA deaminase that mutates cytidine residues to uracyl. Cytdine deamination at V regions leads to somatic mutation whereas the same alteration in switch (S) regions causes class switching. AID generated mismatches in DNA are processed by base-exision repair (BER), mismatch repair (MMR), and error prone polymerases to generate mutations during somatic hypermutation. These lesions can also be converted to a DSB, an obligate intermediate during class switch recombination. These DSBs are detected and processed by foci forming factors, nonhomologous end joining (NHEJ) and alternative end joining [rk5](A-NHEJ). Accurate repair of DSBs by foci forming factors and NHEJ is necessary to prevent chromosomal translocation. Most lymphoid cancers carry chromosomal translocations that involve RAG1/2 or AID target genes. Eμ, intronic enhancer; 3′E, 3′ enhancer.
Although less is known about translocations in other cell types, they appear to be frequent events in solid tumors, especially in sarcomas and prostate cancer (Lin et al., 2009; Mitelman et al., 2007; Tomlins et al., 2007). Recently published work suggests that in prostate cells inter-chromosomal interactions and translocations are induced by transcription factors and possibly by aberrant expression of AID (Lin et al., 2009). Androgen receptors are thought to juxtapose translocating loci and recruit enzymatic activities such as AID or ORF2 endonuclease that produce DSBs (Lin et al., 2009).
DSBs Associated with Antigen Receptor Diversification
V(D)J recombination
V(D)J recombination is a site-specific DNA remodeling reaction required for the assembly of antigen receptor genes in developing B and T lymphocytes (Figure 2). This reaction is mediated by the RAG1/2 recombinase, which nicks DNA at recombination signal sequences and catalyzes a trans-esterification reaction leading to the formation of paired hairpin (coding end) and blunt (signal end) DNA ends. These breaks can trigger the DNA damage response (Chen et al., 2000) and are resolved by the non-homologous end joining (NHEJ) DNA repair machinery, whose components include the Ku70/Ku80 heterodimer, DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, XLF/Cernunnos, XRCC4, and DNA Ligase IV (Lig4)(de Villartay, 2009; Sleckman, 2005)(Figure 3)[RK1]. To date, Artemis and DNA-PKcs have been implicated in hairpin opening and are not required for the resolution of signal ends, whereas the other NHEJ components are essential for joining both coding and signal ends.
Figure 3. Repair of DNA double strand breaks.
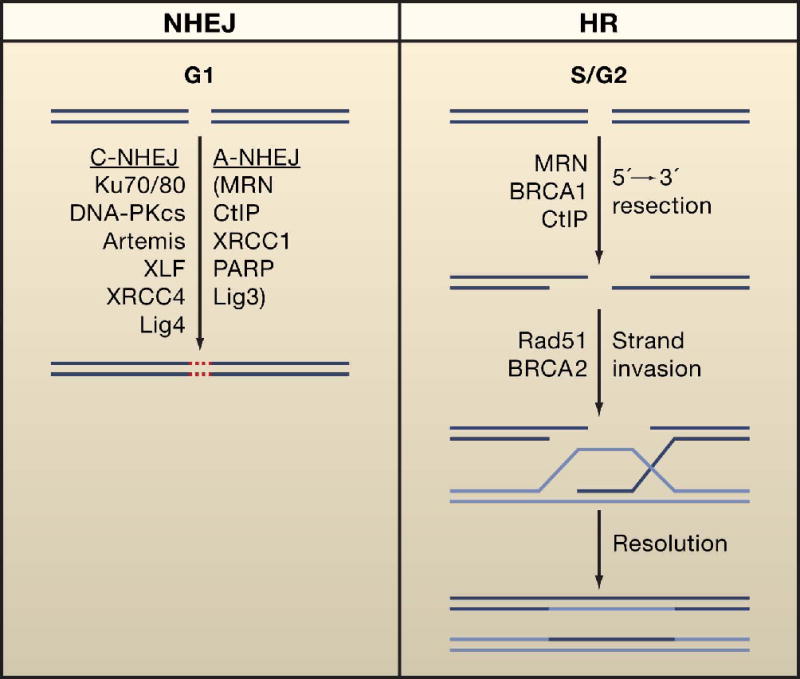
Two major mechanisms that repair double strand breaks (DSBs) include homologous recombination (HR) and non-homologous end joining (NHEJ). HR repairs DSBs by using an intact copy of the broken chromosome as template and is restricted in S and G2 phases of the cell cycle. HR is initiated by 5′-3′ resection of DSBs to form single stranded DNA. A complex of BRCA1, MRN, and CtIP is required for DSB resection. After ssDNA generation, a RAD51 nucleoprotein filament facilitated by BRCA2 is formed that initiates strand invasion of the intact sister chromatid. After DNA synthesis, the ends are eventually rejoined to yield the intact products. During NHEJ, DNA ends are recognized by the Ku70/80 heterodimer and DNA-PKcs. They are then processed by a complex consisting of Artemeis, XLF, XRCC4 and Lig4. Alternative NHEJ (A-NHEJ) pathways may also function as a backup to classical NHEJ. NHEJ and HR components are caretakers that maintain genomic stability by suppressing chromosomal translocations.
RAG1/2 can also function as a transposase in vitro (Jones and Gellert, 2004; Schatz and Spanopoulou, 2005). The transposase activity has been proposed as a means by which this enzyme can produce translocations; however, extensive sequence analysis indicates that this event is unlikely to occur frequently. Instead, the V(D)J associated translocations appear to be the result of aberrant joining of unresolved coding ends as indicated by their proximity to translocation junctions in t(11;14) bcl-1/IgH translocations in mantle cell lymphoma, t(14;18) bcl-2/IgH translocations in follicular lymphoma and in primary mouse T cells (Curry et al., 2007; Kuppers, 2005).
In addition to creating breaks in genes that contain recombination signal sequences, RAG1/2 is thought to be able to produce breaks in oncogenes, such as bcl-2, that contain non-B-DNA or methyl-CpGs (Raghavan et al., 2004; Tsai et al., 2008). Although there is as yet no direct evidence that RAG1/2 can cleave these genes in B-lymphocytes, cleavage activity on non-B-DNA lacking a recombination signal sequence (RSS) has been detected in transfected cell lines using purified substrates in vitro (Raghavan et al., 2004). In addition, the RAG1/2 recombinase is also capable of acting as a nuclease and nicking mismatches and processing nicks, gaps, and flaps into DSBs (Santagata et al., 1999). Thus, RAG1/2 creates DSBs in Ig genes and has the ability to produce lesions in other genes as well.
RAG induced translocations and cancer in mice
Direct evidence that RAG1/2 activity mediates chromosome translocations in vivo comes from analyses of mice deficient in NHEJ proteins (Jankovic et al., 2007). NHEJ deficient mice are unable to complete V(D)J recombination and therefore developing lymphocytes in these mice carry unresolved DNA breaks. However, developing NHEJ deficient lymphocytes do not usually harbor translocations, and mutant mice do not rapidly develop cancer. This is thought to be due to the fact that unresolved V(D)J breaks normally trigger p53 dependent apoptosis (Guidos et al., 1996). Consistent with this notion, mice deficient in both NHEJ and p53 invariably develop progenitor B cell lymphoma at an early age; in the majority of cases these tumors harbor clonal non-reciprocal translocations between IgH and c-myc, and both genes are commonly amplified. Importantly, these DNA rearrangements require RAG1/2-induced DNA cleavage (Difilippantonio et al., 2002; Zhu et al., 2002). The majority of breakpoints in the IgH locus are near the JH cluster, which further supports the role of failed V(D)J recombination in translocation. The breakpoints on Chr15 are localized 70-700 kb from the 3′ end of c-myc (Difilippantonio et al., 2002; Zhu et al., 2002). This is distinct from what occurs in human Burkitt's lymphoma, where translocations between IgH and c-myc are usually reciprocal (meaning that there is equal exchange between both broken chromosomes) and IgH sequences are usually joined to a sequence cluster near exon 1 of c-myc. Thus, although the RAG1/2 endonuclease catalyzes IgH breaks, the mechanism for c-myc breakage in NHEJ-deficient tumors remains unclear, as are the DNA repair pathways that catalyze these complex oncogenic fusion and amplification events in the absence of NHEJ. In addition to showing that aberrant end-joining pathways can lead to translocations, these studies also make it clear that p53 activation protects against RAG-mediated translocations.
Somatic Hypermutation and Class Switching
Somatic hypermutation introduces non-templated nucleotide substitutions into the variable region of Ig genes in mature B cells that have been stimulated during immune responses (Di Noia and Neuberger, 2007; Peled et al., 2008) (Figure 2). Antibody variants with higher affinity for the antigen are selected during the germinal center response leading to an overall increase in antibody affinity (Meffre et al., 2000). CSR is a region specific deletional recombination reaction that replaces one antibody constant region for another while maintaining the specificity of the antibody (Stavnezer et al., 2008) (Figure 2). Switched antibody variants have unique effector functions, for example IgA is specialized for secretion into milk. Nearly a decade ago, groups led by Tasuku Honjo and Anne Durandy discovered that activation-induced cytidine deaminase (AID) is essential for both CSR and SHM in mice and humans (Muramatsu et al., 2000; Revy et al., 2000) (Figure 2). Shortly thereafter, nuclear foci of γ-H2AX and Nbs1, which mark sites of DNA damage, were found to accumulate specifically at the IgH locus in an AID-dependent manner (Petersen et al., 2001); this led to the conclusion that AID acts upstream of DSB formation during CSR, and that the canonical cellular DNA damage response is involved in the processing of AID-induced DSBs (Petersen et al., 2001). In addition, AID-dependent damage foci formed in G0/G1, indicating that like RAG1/2 induced DSBs, AID-dependent DSBs may be restricted to this phase of the cell cycle. These cytological observations were later confirmed by ligation-mediated polymerase chain reaction (PCR) assays (Schrader et al., 2007), and by studies showing that DNA repair mutants undergoing CSR accumulate IgH-specific chromosome breaks (rather than S-phase generated chromatid breaks) in mitotic spreads (Franco et al., 2006; Ramiro et al., 2006).
Although these studies did not pinpoint the mechanism by which AID functions during gene rearrangements or translocation, pioneering work by Michael Neuberger and colleagues supported a unifying model in which AID promotes both class switching and somatic hypermutation (Di Noia and Neuberger, 2007; Peled et al., 2008). According to this model, AID acts by deaminating cytosine residues in single-strand DNA (ssDNA) that is exposed during transcription. The resulting uracil:guanine mismatches are then recognized by the uracil DNA glycosylase or the mismatch repair proteins, and processed in an error-prone manner by DNA repair pathways leading either to mutation or DSBs, which are intermediates in class switching (Di Noia and Neuberger, 2007; Stavnezer et al., 2008). This model is consistent with a profound defect in class switching in knockout mice lacking uracil DNA glycosylase and hyper IgM syndrome in humans deficient for uracil DNA glycosylase (Imai et al., 2003; Rada et al., 2002). Furthermore, it is in agreement with the defects in SHM and CSR found in Msh2/6 mutant mice (Di Noia and Neuberger, 2007; Peled et al., 2008; Stavnezer et al., 2008). Finally, this model is also supported by direct biochemical and genetic experiments, showing that cytosine residues in ssDNA, which are exposed during transcription, are the preferred substrate for AID (Di Noia and Neuberger, 2007; Peled et al., 2008; Stavnezer et al., 2008).
AID mutations in non-Ig genes
Unlike RAG1/2, which is a sequence specific endonuclease, AID can deaminate cytosines in nearly any sequence context, but has a preference for RGYW motifs (Di Noia and Neuberger, 2007; Peled et al., 2008). This is a key feature of AID since mutations in a diverse collection of Ig variable regions maximizes the chances of producing somatic variants with higher affinity. An important question that remains to be answered is how AID finds its target genes and avoids producing widespread genomic damage.
An important first step in this direction lies in defining the set of genes that can be mutated by AID. Experiments with human and mouse B cells show that, in addition to Ig, AID can also mutate a number of oncogenes including c-myc, Pim1, Pax5 and RHOH as well as Bcl6, Igα, Igβ and fas (Gordon et al., 2003; Muschen et al., 2000; Pasqualucci et al., 1998; Pasqualucci et al., 2001; Shen et al., 1998). Recently, a broad survey of 118 genes expressed in germinal center B lymphocytes reveals that as many as 25% are mutated by AID, although most of these are hit at a frequency that is 100-fold lower than Ig (Liu et al., 2008). Interestingly, mutations at c-myc, the most frequent translocation partner for IgH in Burkitt's lymphoma, are detected at close to background levels under physiological conditions (Liu et al., 2008; Pasqualucci et al., 2001). However, c-myc accumulates AID-mediated mutations in the absence of base excision and mismatch repair pathways, indicating that some genes that are targeted by AID may be repaired in an error-free manner by the base excision and mismatch-repair machinery (Liu et al., 2008). Consistent with this notion and in contrast to IgH, c-myc does not show DSB repair foci in AID-expressing B cells under physiological conditions (Robbiani et al., 2008).
Although the full extent of genomic damage produced by AID has not yet been defined, deregulated expression of AID in B cells leads to chromatid breaks and translocations involving nearly every chromosome (Robbiani et al., 2009). Thus, AID can produce mutations in many genes other than antibody genes, some of which are rapidly fixed by error-free pathways; others, that are not repaired, can produce DSBs that destabilize the B cell genome.
Translocations initiated by AID
Following the discovery that AID mutates Ig variable and switch regions, it became possible to address the longstanding issue of the origin of DNA breaks in Ig switch regions that lead to oncogenic chromosomal translocations. Given that lesion formation at the IgH locus is co-dependent on AID and uracil DNA glycosylase, it could be predicted that these enzymes might also be required for chromosomal translocations associated with class switching. This hypothesis is borne out by experiments demonstrating that AID is required for c-myc/IgH translocations, which are associated with IL-6 induced mouse plasmacytocis (Ramiro et al., 2004) and pristaine oil injections (Takizawa et al., 2008). Moreover, translocation is dependent on uracil DNA glycosylase (Ramiro et al., 2006), which indicates that processing of U:G mismatches generated by AID is required for translocation (Ramiro et al., 2006).
AID is indirectly implicated in the pathogenesis of human lymphomas because it is expressed in some forms of lymphoma, including diffuse large B cell lymphoma (DLBCL), which are characterized by IgH-associated chromosomal rearrangements (Lenz et al., 2007). Direct evidence that deregulated AID expression leads to B cell lymphoma has only recently been obtained (Robbiani et al., 2009). Several groups produced transgenic mice expressing AID, beginning with Honjo and his colleagues (Okazaki et al., 2003). Some, but not all of these mice are cancer-prone, developing T lymphomas and epithelial tumors that are associated with aberrant SHM (Muto et al., 2006; Okazaki et al., 2003; Shen et al., 2008). However, although AID is almost exclusively expressed in B cells under physiologic circumstances, B cell lymphomas are not found in AID transgenic mice, even when the transgene is expressed exclusively in B cells (Muto et al., 2006). In addition, there are no chromosomal translocations in the tumors that eventually formed in non-B cells (Okazaki et al., 2003). Instead, cancers caused by the de-regulated expression of AID are characterized by point mutations in oncogenes, as well as passenger mutations in other non-tumor promoting genes (Okazaki et al., 2003). This apparent discrepancy between the absence of B cell lymphoma generation in vivo, and frequent AID-mediated c-myc/IgH translocation in vitro has been resolved by the discovery that AID deregulation leads to the rapid onset of mature B cell lymphoma when combined with deletion of p53 (Robbiani et al., 2009). In AID transgenic/p53-deficient mice, lymphomas almost invariably carry reciprocal clonal chromosome translocations. Some of these translocations involve c-myc and IgH, reminiscent of Burkitt's lymphoma, whereas others joined c-myc to the promoter of microRNA-142 (miR-142), a translocation that is also found in human B cell leukemia (Gauwerky et al., 1989). Translocation between c-myc and miR-142 brings c-myc under the control of the mir-142 promoter and deregulates c-myc expression (Robbiani et al., 2009). miR-142 is likely to be involved in these cancer-producing translocations because it too is an AID target gene (Robbiani et al., 2009).
AID-mediated DSBs in non-Ig genes
Relative to the amount of information we have about AID-mediated DSBs at Ig loci, much less is known regarding the role of AID in producing DSBs in non-Ig genes. DSBs are detectable at the IgH as evidenced by the focal accumulation of proteins that mark sites of DSBs (Petersen et al., 2001) (Figure 4, see below). However, these lesions cannot be detected at other loci such as c-myc despite strong evidence that AID produces DSBs in this gene (Robbiani et al., 2008). Genetic experiments that use the restriction enzyme I-Sce1 to create breaks in either IgH, c-myc or both in the presence or absence of AID show that AID is required to produce the DSBs at both loci. However, AID-generated breaks at IgH are more frequent than at c-myc, and the DSBs in c-myc are limiting for translocation (Robbiani et al., 2008; Wang et al., 2009). Which other non-Ig genes besides c-myc are susceptible to AID-mediated DSB formation, and how this compares to the degree to which these genes are somatically mutated has yet to be determined. An indication that DSBs and mutation in non-Ig genes are correlated comes from the recent finding that Igβ, which is a known target for hypermutation by AID, is also a substrate for translocation to IgH in stimulated B cells(Jankovic et al., 2010)[RK2]. Finally, deregulated expression of AID leads to chromatid breaks and translocations involving nearly every chromosome, which suggests that AID can produce DNA breaks in a diverse group of genes throughout the genome (Robbiani et al., 2009). Thus, it is likely that many genes somatically mutated by AID also harbor DNA breaks that can serve as substrates for chromosome translocation.
Figure 4. Evolution of a DNA repair focus.
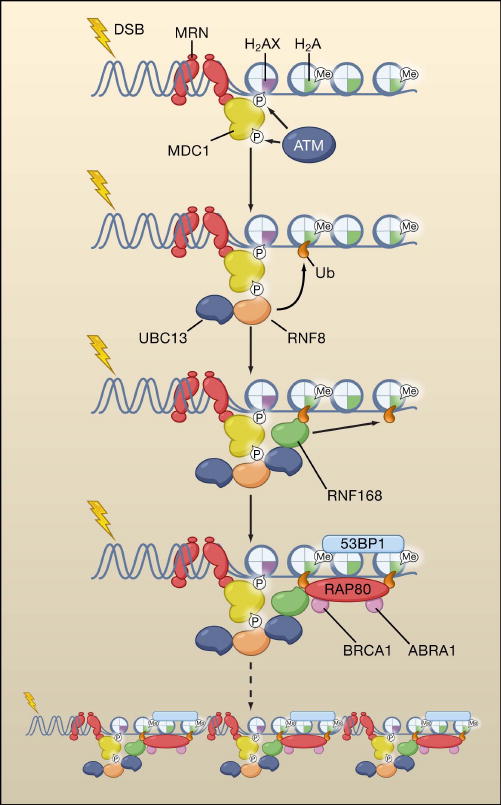
A focus represents large-scale accumulation of factors in the chromatin flanking a double strand break (DSB). MRN recognizes the DSBs, and recruits and activates ATM, a kinase that phosphorylates numerous factors at DSBs including histone H2AX and MDC1. Two E3 ubiquitin ligases RNF8 and RNF168 (coupled with the E2 enzyme UBC13) promote local chromatin ubiquitination, including ubiquitination of H2A histones. RNF8 and RNF168 promote chromatin retention of 53BP1 and Brca1. Modifications of histones may allow exposure of partially occluded histone methyl marks, which facilitates retention of 53BP1. The complex of Rap80/Brca1 interacts directly with ubiquitinated histones. Foci formation stabilizes complexes on chromatin, but is not required for their initial recruitment to damaged sites.
Combined action of RAG1/2 and AID
RAG1/2 and AID are usually expressed in distinct B cell developmental compartments. High levels of RAG1/2 are found in developing B cells in the bone marrow, whereas AID expression is found in mature B cells responding to antigen in the periphery. Assuming that DSBs are rapidly repaired, RAG1/2-induced breaks and AID breaks should not coexist; therefore, they should not be joined in chromosome translocations. However, RAG-induced DSBs can persist, especially in the absence of the ataxia telangiectasia mutated kinase (ATM), an essential DNA damage checkpoint regulator that prevents the long-term persistence of DSBs (Callen et al., 2009a; Callen et al., 2007)[RK3]. Cells that escape this checkpoint can have a DSB at the IgH locus even after RAG expression is extinguished and therefore these lesions could be paired with DSBs created by AID (Callen et al., 2007).
A second mechanism that facilitates translocation between RAG1/2 and AID breaks is the loss of the non-homologous end joining factor XRCC4 (Wang et al., 2009). Conditional deletion of this core component of the non-homologous end joining machinery in mature B cells deficient in p53 leads to B cell lymphomas that carry tumor promoting c-myc/IgH translocations, as well as translocations between the Igλ variable regions and IgH switch regions (Wang et al., 2008). These Igλ-IgH passenger translocations appear to be the result of secondary V(D)J recombination (Wang et al., 2009), also known as receptor editing (Jankovic et al., 2004). This reaction appears to be mediated by delayed extinction of RAG1/2 expression, although the possibility of RAG re-induction cannot be ruled out definitively (Jankovic et al., 2004). The abnormal or delayed repair of RAG1/2-mediated DSBs in XRCC4 deficient cells, or potentially more rarely in normal cells, might promote overlap between the lesions created by the two Ig recombinases; this in turn could produce B cells in which both RAG1/2 and AID breaks coexist (Wang et al., 2008; Wang et al., 2009).
Finally, an analysis of an extensive panel of human chromosome translocations suggests that AID might facilitate off-target DSB formation by RAG1/2. According to this model, the low level AID expression in developing B cells in the bone marrow would create U:G mismatches that lead to the formation of non-B-DNA targets for RAG1/2, in genes like Bcl-2 (Tsai et al., 2008).
In conclusion, both RAG1/2 and AID can produce DSBs in Ig and non-Ig genes. The majority of RAG1/2 breaks are usually found in developing B cells, whereas the AID breaks are usually found in mature activated B cells. However, a number of mechanisms can facilitate the overlap between the two, and thus might account for translocations between RAG1/2 and AID-mediated DSBs.
Repair pathways in Aberrant chromosome joining
Non-homologous end joining and homologous recombination
There are two well-described pathways that mediate DSB repair in mammalian cells: NHEJ and homologous recombination (HR) (Figure 3).
NHEJ directly religates broken DNA, which can result in the loss of genetic material (Weinstock et al., 2006). Although this type of end-joining can be active in any phase of the cell cycle and can compete with HR, it is essential for DSB repair in G0/G1; therefore it plays a major role in V(D)J recombination, class switching and antigen receptor diversification-associated chromosome translocations (Chaudhuri et al., 2007; Jankovic et al., 2007). The absence of any of the core NHEJ components leads to severe combined immunodeficiency, due to the inability to properly repair RAG breaks (de Villartay, 2009; Sleckman, 2005). Most of the core NHEJ factors (Ku70/80, XRCC4, Lig4) are also required for the efficient repair of AID breaks during class switching (Chaudhuri et al., 2007; Jankovic et al., 2007). However, the requirement for core NHEJ factors in switching is not absolute, and alternative end joining pathways (A-NHEJ) that function in the absence of Ku70/80, Lig4, and XRCC4 (Figure 3) can mediate CSR at rates of up to 50% of normal (Boboila et al., 2010a, b; Pan-Hammarstrom et al., 2005; Soulas-Sprauel et al., 2007; Yan et al., 2007).
In contrast to NHEJ, repair of DSBs by HR utilizes the intact homologous chromatid as a complementary template; therefore, HR occurs primarily in S/G2/M (Figure 3). HR requires DSB resection to produce ssDNA substrates for homology search. Specifically, DNA ends must undergo 5′-3′ resection to generate 3′ ssDNA that recruits the RAD51 recombinase which mediates homologous pairing and strand invasion (Takeda et al., 2007) (Figure 3). DSB processing to ssDNA requires the formation of a complex that includes BRCA11, the MRN complex (Mre11/Rad50/Nbs1) and CtIP (Chen et al., 2008; Sartori et al., 2007; Yun and Hiom, 2009) as well as Blooms Helicase and Exo1(Gravel et al., 2008). In this context, Mre11 functions as a nuclease that is required for both DSB resection and HR (Buis et al., 2008; Paull and Gellert, 1998; Sartori et al., 2007). Finally, although DSB resection is essential for HR in the S/G2 phase of the cell cycle, DSB resection also occurs in G0/G1 (Yun and Hiom, 2009) where it may facilitate microhomology based alternative end joining (A-NHEJ) during class switching (Bothmer et al., 2010) and chromosome translocation (Simsek and Jasin).
HR in translocation
Both NHEJ and HR are likely to play important roles in chromosome translocation (Weinstock et al., 2006). Consistent with this notion, there are several rare “chromosomal breakage” diseases that predispose to chromosomal translocation and cancer. Each of the genes mutated in these diseases encode proteins involved in DNA DSB repair. For example, Fanconi anemia, which is associated with bone marrow failure, blood and solid tumors, is caused by mutations in one of 13 complementation groups, FANC A-N, some of which are implicated in HR (Moldovan and D'Andrea, 2009); Nijmegen breakage syndrome and ataxia telangiectasia, which are associated with an increased risk of lymphoma, arise from mutation in Nbs1 and ATM respectively, both of which function in NHEJ and HR (Shiloh, 2003); finally, inherited breast cancer is frequently associated with mutations in BRCA1 or BRCA2, both of which are essential for HR (Venkitaraman, 2002).
In the absence of HR, cells experience an increase in general genomic instability (Sonoda et al., 1998) and a specific increase in the frequency of breaks at so-called fragile sites, which are large genomic regions prone to damage during replication stress (Arlt et al., 2006). In addition to DNA breakage, loss of BRCA1, BRCA2, BLM, or other HR proteins leads to the accumulation of radial and quasi-radial structures, in which sister chromatids from different (homologous or non-homologous) chromosomes fuse together (Venkitaraman, 2002). If the resulting radial type structure contains more than one centromere, then dicentric chromosome rupture during DNA replication could result in breakage-bridge fusion cycles that may eventually lead to the propagation of a “stable” chromosomal translocation. Chromatid breaks that accumulate in Brca1 deficient cells are aberrantly joined by a Lig4 dependent mechanism leading to the formation of radial structures (Bunting et al., 2010).
NHEJ and translocation
Although classical NHEJ (C-NHEJ) core proteins are essential for intra-chromosomal joining during V(D)J recombination and play an important role in class switching, their role in translocations appears to lie in suppressing rather than mediating these aberrant joining events. As was first shown in fibroblasts, loss of Ku80 and Lig4 leads to a massive increase in the frequency of spontaneous DNA breaks and chromosomal translocations (Difilippantonio et al., 2000; Ferguson et al., 2000; Karanjawala et al., 1999). Consistent with these results, translocations induced by RAG1/2, I-Sce1, or AID do not require Ku70, Ku80, DNA-PKcs, or XRCC4 (Boboila et al., 2010b; Callen et al., 2009b; Ramiro et al., 2006; Wang et al., 2009; Weinstock et al., 2007). On the contrary, the absence of Ku, XRCC4, Lig4, Artemis, or DNA-PKcs leads to the accumulation of AID-dependent DNA breaks and translocations in stimulated B cells (Boboila et al., 2010b; Franco et al., 2008; Yan et al., 2007). The absence of C-NHEJ factors may promote abnormal joining by increasing the number of DSB translocation substrates due to inefficient repair in cis, making it more probable that microhomology based A-NHEJ pathways fuse the ends together (see below).
A-NHEJ pathways are implicated in chromosomal translocations on the basis of microhomology at the translocation breakpoints of some human cancers including non-lymphoid cancers (Weinstock et al., 2006). In mice, A-NHEJ is thought to mediate fusion-bridge-fusion translocations arising in B cell lymphomas in C-NHEJ/p53 double-deficient mice because the translocation junctions show microhomology (Difilippantonio et al., 2002; Zhu et al., 2002). In addition, reciprocal chromosome translocations between c-myc and IgH are C-NHEJ independent (Boboila et al., 2010b; Ramiro et al., 2006; Yan et al., 2007). Thus, A-NHEJ appears to be particularly active in chromosome translocations.
The A-NHEJ pathway was first described in mammalian cells by Roth and Wilson (Roth and Wilson, 1986). Although work from a number of laboratories and in different systems indicates that this is a robust pathway (Boboila et al., 2010a; Pan-Hammarstrom et al., 2005; Soulas-Sprauel et al., 2007; Yan et al., 2007), very little is known about the molecular components that mediate A-NHEJ. A-NHEJ appears to be conserved in yeast (McVey and Lee, 2008), may be kinetically slower than classical NHEJ (Boboila et al., 2010a; Han and Yu, 2008; Xie et al., 2009), and is biased towards microhomology mediated joins (typically 5-25 bp homologous sequences at the DSB junctions) (Haber, 2008; Zha et al., 2009). In contrast, classical NHEJ pathways anneal blunt ends or short (<4 bp) homologous sequences (Haber, 2008; Zha et al., 2009).
Among the repair factors implicated in A-NHEJ are Mre11 and CtIP, which form a complex that regulates the initial DNA end resection at breaks, thereby exposing homologous ssDNA sequences for annealing (Bennardo et al., 2008; Deriano et al., 2009; Dinkelmann et al., 2009; Rass et al., 2009; Sartori et al., 2007; Xie et al., 2009; Yun and Hiom, 2009) (Figure 3). Loss of Mre11 decreases the joining efficiency between two I-SceI DSBs (Rass et al., 2009) and over-expression increases end resection and A-NHEJ suggesting a role for Mre11 in DNA end resection and alternative end-joining. Interestingly, another member of the complex that is essential for resection, BRCA1, is required for HR but is dispensable for A-NHEJ (Yun and Hiom, 2009). Other repair factors that may function in A-NHEJ and also as backup repair factors to C-NHEJ include the single-strand break repair proteins DNA Ligase III (Lig3), XRCC1, and Poly (ADP-Ribose) Polymerase I (PARPI) (Robert et al., 2009; Wang et al., 2006) (Figure 3). These proteins normally function in ssDNA repair, but might also act upon processed DSBs during A-NHEJ (Haber, 2008; Zha et al., 2009).
Alternative end joining may also mediate chromosomal fusions that occur in response to telomere erosion (Palm and de Lange, 2008). Loss of telomeric sequences results in the failure of DNA end protection, and chromosomal end fusions ensue. Like translocation, fusion of short telomeres occurs independently of the end joining factors DNA-PKcs, ATM, and Lig4 (Maser et al., 2007; Wong et al., 2003). In fission yeast, chromosomal fusions induced by telomere attrition are also independent of NHEJ, but require the homologous recombination proteins Rad52 and ERCC1, which participate in single strand annealing (Wang and Baumann, 2008). Single strand annealing is a conserved pathway that repairs DSBs arising between long (>30 nt) flanking direct repeats via a mechanism involving annealing of the homologous ssDNA sequences. Single strand annealing requires DSB processing and is dependent on Brca1 and CtIP (Bennardo et al., 2008; Stark et al., 2004). In summary, the repair pathways involved in translocation are unknown, but may involve single-strand DNA formation.
Although A-NHEJ is utilized when C-NHEJ is compromised, the extent to which it is active under physiological conditions and how the choice is made between the two pathways remains unclear. For example, a recent study analyzed the junctions of c-myc/IgH translocations produced in wild-type B cells (Robbiani et al., 2008). Half of the translocation junctions are blunt and the other half show only 1-3 bp homology, which would be consistent with the known properties of C-NHEJ (Robbiani et al., 2008). This distribution and lack of extensive microhomology is similar to junctions formed during normal class switch recombination (Yan et al., 2007). This contrasts with translocation breakpoints found in the absence of C-NHEJ or by I-Sce1-induced DSBs in AID deficient B cells (Robbiani et al., 2008) or embryonic stem (ES) cells (Weinstock et al., 2007), which involve more extensive processing and micro-homology-mediated joining. End processing is inhibited by 53BP1 (Bothmer et al., 2010; Bunting et al., 2010). Loss of this factor promotes HR (Bunting et al., 2010) and microhomology mediated joining (Bothmer et al., 2010), and greatly alleviates replication associated genetic instability in Brca1 deficient cells (Bunting et al., 2010).
Focus forming factors
Several DNA damage response proteins such as Nbs1, ATM and 53BP1 accumulate in the chromatin region surrounding a DSB, forming what are called DNA damage repair ‘foci’ (Fernandez-Capetillo et al., 2003) (Figure 4). Others, like the classical NHEJ proteins, seem to bind directly to DSBs but do not accumulate in regions distal to the break sites (Bekker-Jensen et al., 2006). For example, foci forming proteins Nbs1, phosphorylated histone H2AX (γ-H2AX) and 53BP1 accumulate on the IgH locus in response to AID-mediated breaks during class switching (Petersen et al., 2001) and on antigen receptor genes undergoing RAG1/2 dependent V(D)J recombination in thymocytes (Chen et al., 2000; Savic et al., 2009). The ability of proteins to form foci is dependent on prior phosphorylation of histone H2AX (γ-H2AX) (Celeste et al., 2002), a chromatin mark that spreads over a region spanning thousands to millions of base pairs of DNA (Rogakou et al., 1998). γ-H2AX is believed to serve as an anchor for stabilizing the binding of other focus forming factors to chromatin (Bassing and Alt, 2004; Fernandez-Capetillo et al., 2003), including the recently discovered histone ubiquitin E3 ligase RNF8 (Figure 4).
In addition to accumulating in foci at sites of antigen receptor gene recombination, Nbs1, γ-H2AX, ATM, RNF8, and 53BP1 are also required for the efficient completion of normal class switch recombination (Jankovic et al., 2007; Ramachandran et al., 2010; Santos et al., 2010), and some of these factors also contribute to V(D)J recombination (Bassing et al., 2003; Bredemeyer et al., 2006; Celeste et al., 2003; Difilippantonio et al., 2008; Helmink et al., 2009). However, their role in these reactions is not completely understood. For example, the loss of 53BP1 in B cells undergoing CSR reduces the frequency of long-range joining between different switch regions; however, short-range intra-switch joints appear to be increased (Reina-San-Martin et al., 2007), in part because of increased resection (Bothmer et al., 2010; Bunting et al., 2010) which facilitates microhomology mediated A-NHEJ between repetitive switch sequences (Bothmer et al., 2010). Similarly, 53BP1 appears to be essential for repairing a subset of V(D)J recombination-induced ends, specifically those separated by long distances (Difilippantonio et al., 2008). RNF8 is required for 53BP1 foci formation, yet the CSR defect in RNF8 knockout mice is milder than that observed in the absence of 53BP1 (Ramachandran et al., 2010; Santos et al., 2010). Thus, in both CSR and V(D)J joining, loss of focus forming factors affects only a subset of joints and not end ligation per se. There are several models that could explain these phenomena, including the possibility that 53BP1 stabilizes or promotes long-range chromosomal interactions (Difilippantonio et al., 2008), increases chromatin mobility (Dimitrova et al., 2008) or alters the repair pathway choice between C-NHEJ and A-NHEJ (Bothmer et al., 2010).
Many of the focus forming factors are also essential to maintaining genomic stability and in their absence cells suffer an increase in chromosomal translocations. For example, the loss of either Nbs1, γ-H2AX, ATM, or 53BP1 leads to high levels of chromosomal aberrations during class switching, which are divided among general chromosomal aberrations and those occurring specifically at antigen receptor loci (Jankovic et al., 2007). Furthermore, mutant mice that fail to express these factors are predisposed to cancer (Jankovic et al., 2007). Finally, the combined loss of ATM and DNA-PKcs leads to a synergistic increase in genomic instability as well as additive defects in class-switching, indicating that these proteins act redundantly in lymphocytes (Callen et al., 2009b). In summary, like NHEJ and HR proteins, focus-forming factors are not essential for inter-chromosome exchanges. On the contrary, their absence appears to result in aberrant or inefficient DSB repair in G0/G1, which may increase the availability of DSB substrates for translocation. In contrast, absence of 53BP1 reduces chromosomal instability in S phase and cancer predisposition in Brca1 deficient cells (Bunting et al., 2010; Cao et al., 2009).
Translocation between c-myc and IgH
c-myc/IgH translocations belong to a special subset of translocations that trigger oncogenic stress (Collado et al., 2007). Deregulated expression of oncogenes causes activation of the tumor suppressors ARF and ATM, which in turn trigger p53 dependent senescence or cell death (Collado et al., 2007). This provides a barrier that protects cells against propagating the oncogenic translocation. Consistent with this notion, loss of Nbs1 (ATM activator), ATM, ARF, and p53 increases the frequency of c-myc/IgH but not Igβ/IgH translocations in primary B cells (Jankovic et al., 2010; Ramiro et al., 2006; Robbiani et al., 2009); mutations in ARF/p53 promote myc-induced lymphomagenesis (Eischen et al., 1999; Schmitt et al., 1999); deregulated AID expression causes cancer when p53 is disrupted (Robbiani et al., 2009); and B cell lymphomas almost invariably carry mutations in these proteins in addition to translocations (Kuppers, 2005). Such secondary tumor-suppressor gene mutations may be necessary to avoid pathways that trigger cell death; they appear to play a critical role in many tumors besides those of lymphoid origin (Halazonetis et al., 2008).
Interestingly, and in contrast to ATM, disruption of H2AX or 53BP1 does not affect the rate of c-myc/IgH translocation. However, their loss markedly increases IgH-associated translocations to targets other than c-myc (Franco et al., 2006; Ramiro et al., 2006). One potential explanation for this paradox is that c-myc/IgH triggers an oncogenic stress signal that eliminates cells that carry this translocation, whereas Ig translocations to other regions may not be toxic. Support for this notion comes from the observation that the rate of translocation between IgH and Igβ (another AID target) is not altered by loss of p53 in the same cells that suffer increased c-myc/IgH translocations (Jankovic et al., 2010). Instead, alternative pathways that include BAFF, Bcl-XL, and PKC-δ protect cells against DNA damage in the absence of oncogenic stress (Jankovic et al., 2010).
In summary, the loss of either NHEJ or HR or focus-forming factors leads to an increase in the frequency of translocation and cancer predisposition. We speculate that the loss of these factors increases translocation because bona fide DSB repair is less efficient, resulting in an increase in untethered ends that are available for intermingling, DSB processing and aberrant joining.
The role of nuclear positioning in translocation
In addition to being organized into nucleosomes, chromosomes are found in defined spatially segregated nuclear territories that are in part correlated with transcriptional activity (Lieberman-Aiden et al., 2009). Physical proximity between translocated loci in the nucleus is an additional factor that may impact the frequency of translocation between chromosomes. For example, c-myc and IgH are frequently colocalized in primary B cells but not in other cell types (Osborne et al., 2007; Wang et al., 2009). Importantly, this co-localization is found in resting B cells prior to AID-induced damage at c-myc and IgH, indicating that these loci are spatially proximal before DSBs are formed (Wang et al., 2009). The mechanisms that bring disparate loci together in the nucleus are poorly understood; however, it has been proposed that loci that share transcriptional regulation are more likely to be found together (Spilianakis et al., 2005). However, accessibility for translocation does not appear to require transcription, given that even transcriptionally silent c-myc is in an accessible nuclear compartment that permits c-myc/IgH translocation (Robbiani et al., 2008).
Quantitative high resolution studies find that DSBs exhibit limited mobility similar to the random movement of undamaged chromatin (Kruhlak et al., 2006; Soutoglou et al., 2007). In contrast, a recent study of the dynamics of telomere fusions demonstrates that 53BP1 promotes end joining of dysfunctional telomeres by increasing chromatin mobility (Dimitrova et al., 2008). They provide evidence that uncapped telomeres are more mobile and sample larger territories than normal telomeres in a manner dependent on 53BP1(Dimitrova et al., 2008). The implication is that 53BP1 changes the dynamic behavior of chromatin to facilitate NHEJ reactions involving distant sites (Dimitrova et al., 2008). The potential role of 53BP1 or other factors in influencing chromatin dynamics, position, and translocation is an important topic that is actively being investigated.
Conclusions
Paired DSBs are obligate intermediates for chromosomal translocation. Antigen receptor diversification reactions in lymphoid cells require the formation of DSBs that can serve as substrates for translocations. Importantly, the enzymes that produce DSBs in antigen receptor loci, RAG1/2 and AID, can also produce DNA lesions in non-antigen receptor genes, including oncogenes that can be paired with breaks in antigen receptor loci to form translocations.
Over the last decade, considerable progress has been made in understanding the way in which cells maintain genomic stability and avoid chromosomal translocations in the face of continual DNA damage produced during normal cellular metabolism including antigen receptor gene recombination. Many of the factors that mediate DNA repair by HR and NHEJ also suppress translocation in normal cells. In the absence of these factors, mice and humans are prone to cancer-inducing chromosomal translocations. An understanding of the mechanisms involved in the creation, signaling, and repair of DSBs will help target the activity of tumor-promoting oncogenes, as well as that of caretaker and gatekeeper genes whose loss contributes to tumor initiation and progression.
Acknowledgments
We thank E. Callen for drawing the figures, and Drs. A. Bothmer, S. Bunting, J. Danial, E. Callen, S. Deroubaix, M. Di Virgilio, N. Feldhan, A. Gazumyan, M. Jankovic, I. Klein, K. McBride, R. Pavri, A. Ramiro, B. Reina San-Martin, and D. Robbiani for comments on the manuscript. M.C.N. is an HHMI investigator and is supported by NIH grant AI037526. A.N. is supported by the Intramural Research program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst) 2006;5:1126–1135. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Alt FW. Cell cycle. Vol. 3. Georgetown, Tex: 2004. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity; pp. 149–153. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS genetics. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Jankovic M, Yan CT, Wang JH, Wesemann DR, Zhang T, Fazeli A, Feldman L, Nussenzweig A, Nussenzweig MC, et al. Alternative end-joining catalyzes class switch recombination in the absence of both ligase 4 adn Ku70. The Journal of experimental medicine. 2010a doi: 10.1084/jem.20092449. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Jankovic M, Yan CT, Wang JH, Wesemann DR, Zhang T, Fazeli A, Feldman L, Nussenzweig A, Nussenzweig MC, et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 adn Ku70. Proceedings of the National Academy of Sciences of the United States of America. 2010b doi: 10.1073/pnas.0915067107. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switching. 2010 doi: 10.1084/jem.20100244. unpublished data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- Buis J, Wu Y, Deng Y, Leddon J, Westfield G, Eckersdorff M, Sekiguchi JM, Chang S, Ferguson DO. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting S, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010 doi: 10.1016/j.cell.2010.03.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Croce CM. Chromosomal rearrangements and microRNAs: a new cancer link with clinical implications. J Clin Invest. 2007;117:2059–2066. doi: 10.1172/JCI32577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Bunting S, Huang CY, Difilippantonio MJ, Wong N, Khor B, Mahowald G, Kruhlak MJ, Ried T, Sleckman BP, et al. Cell cycle. Vol. 8. Georgetown, Tex: 2009a. Chimeric IgH-TCRalpha/delta translocations in T lymphocytes mediated by RAG; pp. 2408–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, Pellegrini M, McBride K, Wangsa D, Bredemeyer AL, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130:63–75. doi: 10.1016/j.cell.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Callen E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A, et al. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Molecular cell. 2009b;34:285–297. doi: 10.1016/j.molcel.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Molecular cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Science. Vol. 296. New York, NY: 2002. Genomic instability in mice lacking histone H2AX; pp. 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, et al. Science. Vol. 290. New York, NY: 2000. Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX; pp. 1962–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Curry JD, Schulz D, Guidos CJ, Danska JS, Nutter L, Nussenzweig A, Schlissel MS. Chromosomal reinsertion of broken RSS ends during T cell development. The Journal of experimental medicine. 2007;204:2293–2303. doi: 10.1084/jem.20070583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villartay JP. V(D)J recombination deficiencies. Advances in experimental medicine and biology. 2009;650:46–58. doi: 10.1007/978-1-4419-0296-2_4. [DOI] [PubMed] [Google Scholar]
- Deriano L, Stracker TH, Baker A, Petrini JH, Roth DB. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Molecular cell. 2009;34:13–25. doi: 10.1016/j.molcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, Ried T, Nussenzweig A. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. The Journal of experimental medicine. 2002;196:469–480. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, Ried T, Nussenzweig A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nature structural & molecular biology. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes & development. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, DePinho RA, Alt FW. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6630–6633. doi: 10.1073/pnas.110152897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Celeste A, Nussenzweig A. Cell cycle. Vol. 2. Georgetown, Tex: 2003. Focusing on foci: H2AX and the recruitment of DNA-damage response factors; pp. 426–427. [PubMed] [Google Scholar]
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Molecular cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Franco S, Murphy MM, Li G, Borjeson T, Boboila C, Alt FW. DNA-PKcs and Artemis function in the end-joining phase of immunoglobulin heavy chain class switch recombination. The Journal of experimental medicine. 2008;205:557–564. doi: 10.1084/jem.20080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauwerky CE, Huebner K, Isobe M, Nowell PC, Croce CM. Activation of MYC in a masked t(8;17) translocation results in an aggressive B-cell leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:8867–8871. doi: 10.1073/pnas.86.22.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MS, Kanegai CM, Doerr JR, Wall R. Somatic hypermutation of the B cell receptor genes B29 (Igbeta, CD79b) and mb1 (Igalpha, CD79a) Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4126–4131. doi: 10.1073/pnas.0735266100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Yan CT, Bianco JM, Cogne M, Pinaud E, Alt FW. Long-range oncogenic activation of Igh-c-myc translocations by the Igh 3′ regulatory region. Nature. 2009;462:803–807. doi: 10.1038/nature08633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes & development. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, Danska JS. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes & development. 1996;10:2038–2054. doi: 10.1101/gad.10.16.2038. [DOI] [PubMed] [Google Scholar]
- Haber JE. Alternative endings. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:405–406. doi: 10.1073/pnas.0711334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. Science. Vol. 319. New York, NY: 2008. An oncogene-induced DNA damage model for cancer development; pp. 1352–1355. [DOI] [PubMed] [Google Scholar]
- Han L, Yu K. Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV--deficient B cells. The Journal of experimental medicine. 2008;205:2745–2753. doi: 10.1084/jem.20081623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Bredemeyer AL, Lee BS, Huang CY, Sharma GG, Walker LM, Bednarski JJ, Lee WL, Pandita TK, Bassing CH, et al. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. The Journal of experimental medicine. 2009;206:669–679. doi: 10.1084/jem.20081326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nature immunology. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- Jankovic M, Casellas R, Yannoutsos N, Wardemann H, Nussenzweig MC. RAGs and regulation of autoantibodies. Annual review of immunology. 2004;22:485–501. doi: 10.1146/annurev.immunol.22.012703.104707. [DOI] [PubMed] [Google Scholar]
- Jankovic M, Nussenzweig A, Nussenzweig MC. Antigen receptor diversification and chromosome translocations. Nature immunology. 2007;8:801–808. doi: 10.1038/ni1498. [DOI] [PubMed] [Google Scholar]
- Jankovic M, Robbiani DF, Dorsett Y, Eisenreich T, Xu Y, Tarakhovsky A, Nussenzweig A, Nussenzweig MC. Role of the translocation partner in protection against AID-dependent chromosomal translocations. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:187–192. doi: 10.1073/pnas.0908946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JM, Gellert M. The taming of a transposon: V(D)J recombination and the immune system. Immunol Rev. 2004;200:233–248. doi: 10.1111/j.0105-2896.2004.00168.x. [DOI] [PubMed] [Google Scholar]
- Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr Biol. 1999;9:1501–1504. doi: 10.1016/s0960-9822(00)80123-2. [DOI] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nature reviews. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nature reviews. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- Lenz G, Nagel I, Siebert R, Roschke AV, Sanger W, Wright GW, Dave SS, Tan B, Zhao H, Rosenwald A, et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. The Journal of experimental medicine. 2007;204:633–643. doi: 10.1084/jem.20062041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Science. Vol. 326. New York, NY: 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome; pp. 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- Maser RS, Wong KK, Sahin E, Xia H, Naylor M, Hedberg HM, Artandi SE, DePinho RA. DNA-dependent protein kinase catalytic subunit is not required for dysfunctional telomere fusion and checkpoint response in the telomerase-deficient mouse. Molecular and cellular biology. 2007;27:2253–2265. doi: 10.1128/MCB.01354-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffre E, Casellas R, Nussenzweig MC. Antibody regulation of B cell development. Nature immunology. 2000;1:379–385. doi: 10.1038/80816. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nature reviews. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- Moldovan GL, D'Andrea AD. How the Fanconi Anemia Pathway Guards the Genome. Annual review of genetics. 2009 doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Muschen M, Re D, Jungnickel B, Diehl V, Rajewsky K, Kuppers R. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. The Journal of experimental medicine. 2000;192:1833–1840. doi: 10.1084/jem.192.12.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto T, Okazaki IM, Yamada S, Tanaka Y, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. Negative regulation of activation-induced cytidine deaminase in B cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2752–2757. doi: 10.1073/pnas.0510970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. The Journal of experimental medicine. 2003;197:1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annual review of genetics. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Pan-Hammarstrom Q, Jones AM, Lahdesmaki A, Zhou W, Gatti RA, Hammarstrom L, Gennery AR, Ehrenstein MR. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. The Journal of experimental medicine. 2005;201:189–194. doi: 10.1084/jem.20040772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, Chaganti RS, Klein U, Kuppers R, Rajewsky K, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11816–11821. doi: 10.1073/pnas.95.20.11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, Dalla-Favera R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Molecular cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annual review of immunology. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- Raghavan SC, Swanson PC, Wu X, Hsieh CL, Lieber MR. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428:88–93. doi: 10.1038/nature02355. [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Chahwan RN, Frieder RM, Panier D, Roa S, Zaheen S, Durocher A, Scharff MD, Martin A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2010 doi: 10.1073/pnas.0913790107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, Eisenreich TR, Chen J, Dickins RA, Lowe SW, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006;440:105–109. doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nature structural & molecular biology. 2009;16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- Reina-San-Martin B, Chen J, Nussenzweig A, Nussenzweig MC. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1-/- B cells. Eur J Immunol. 2007;37:235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, M KM, Klein IA, Stone G, Eisenreich TR, et al. AID produces DNA double strand breaks in non-Ig genes and mature B cell lymphomas with recipricol chromosome translocations. Molecular cell. 2009 doi: 10.1016/j.molcel.2009.11.007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert I, Dantzer F, Reina-San-Martin B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. The Journal of experimental medicine. 2009;206:1047–1056. doi: 10.1084/jem.20082468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Roth DB, Wilson JH. Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Molecular and cellular biology. 1986;6:4295–4304. doi: 10.1128/mcb.6.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Besmer E, Villa A, Bozzi F, Allingham JS, Sobacchi C, Haniford DB, Vezzoni P, Nussenzweig MC, Pan ZQ, et al. The RAG1/RAG2 complex constitutes a 3′ flap endonuclease: implications for junctional diversity in V(D)J and transpositional recombination. Molecular cell. 1999;4:935–947. doi: 10.1016/s1097-2765(00)80223-3. [DOI] [PubMed] [Google Scholar]
- Santos MA, Huen MSY, Lopez-Contreras J, Klein IA, Jankovic M, Chen HT, Wong N, Barbancho JLR, Fernandez-Capetillo O, Nussenzweig MC, et al. RNF8-dependent histone ubiquitylation contributes to class switch recombination. 2010 unpublished data. [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic V, Yin B, Maas NL, Bredemeyer AL, Carpenter AC, Helmink BA, Yang-Iott KS, Sleckman BP, Bassing CH. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Molecular cell. 2009;34:298–310. doi: 10.1016/j.molcel.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz DG, Spanopoulou E. Biochemistry of V(D)J recombination. Curr Top Microbiol Immunol. 2005;290:49–85. doi: 10.1007/3-540-26363-2_4. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes & development. 1999;13:2670–2677. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol. 2007;179:6064–6071. doi: 10.4049/jimmunol.179.9.6064. [DOI] [PubMed] [Google Scholar]
- Shen HM, Bozek G, Pinkert CA, McBride K, Wang L, Kenter A, Storb U. Expression of AID transgene is regulated in activated B cells but not in resting B cells and kidney. Mol Immunol. 2008;45:1883–1892. doi: 10.1016/j.molimm.2007.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Peters A, Baron B, Zhu X, Storb U. Science. Vol. 280. New York, NY: 1998. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes; pp. 1750–1752. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature reviews. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nature structural & molecular biology. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleckman BP. Lymphocyte antigen receptor gene assembly: multiple layers of regulation. Immunologic research. 2005;32:253–258. doi: 10.1385/IR:32:1-3:253. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. Embo J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulas-Sprauel P, Le Guyader G, Rivera-Munoz P, Abramowski V, Olivier-Martin C, Goujet-Zalc C, Charneau P, de Villartay JP. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. The Journal of experimental medicine. 2007;204:1717–1727. doi: 10.1084/jem.20070255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Molecular and cellular biology. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annual review of immunology. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Nakamura K, Taniguchi Y, Paull TT. Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Molecular cell. 2007;28:351–352. doi: 10.1016/j.molcel.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Takizawa M, Tolarova H, Li Z, Dubois W, Lim S, Callen E, Franco S, Mosaico M, Feigenbaum L, Alt FW, et al. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. The Journal of experimental medicine. 2008;205:1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Wang JH, Alt FW, Gostissa M, Datta A, Murphy M, Alimzhanov MB, Coakley KM, Rajewsky K, Manis JP, Yan CT. Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. The Journal of experimental medicine. 2008;205:3079–3090. doi: 10.1084/jem.20082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Gostissa M, Yan CT, Goff P, Hickernell T, Hansen E, Difilippantonio S, Wesemann DR, Zarrin AA, Rajewsky K, et al. Mechanisms promoting tranlocations in editing and switching peripheral B cells. Nature. 2009 doi: 10.1038/nature08159. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Baumann P. Chromosome fusions following telomere loss are mediated by single-strand annealing. Molecular cell. 2008;31:463–473. doi: 10.1016/j.molcel.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Weinstock DM, Brunet E, Jasin M. Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat Cell Biol. 2007;9:978–981. doi: 10.1038/ncb1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock DM, Richardson CA, Elliott B, Jasin M. Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair (Amst) 2006;5:1065–1074. doi: 10.1016/j.dnarep.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nature structural & molecular biology. 2009;16:814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Boboila C, Alt FW. Mre11: roles in DNA repair beyond homologous recombination. Nature structural & molecular biology. 2009;16:798–800. doi: 10.1038/nsmb0809-798. [DOI] [PubMed] [Google Scholar]
- Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–821. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]