

Abstract
Mice lacking Kv1.1 Shaker-like potassium channels encoded by the Kcna1 gene exhibit severe seizures and die prematurely. The channel is widely expressed in brain but only minimally, if at all, in mouse myocardium. To test whether Kv1.1-potassium deficiency could underlie primary neurogenic cardiac dysfunction, we performed simultaneous video EEG–ECG recordings and found that Kcna1-null mice display potentially malignant interictal cardiac abnormalities, including a fivefold increase in atrioventricular (AV) conduction blocks, as well as bradycardia and premature ventricular contractions. During seizures the occurrence of AV conduction blocks increased, predisposing Kv1.1-deficient mice to sudden unexplained death in epilepsy (SUDEP), which we recorded fortuitously in one animal. To determine whether the interictal AV conduction blocks were of cardiac or neural origin, we examined their response to selective pharmacological blockade of the autonomic nervous system. Simultaneous administration of atropine and propranolol to block parasympathetic and sympathetic branches, respectively, eliminated conduction blocks. When administered separately, only atropine ameliorated AV conduction blocks, indicating that excessive parasympathetic tone contributes to the neurocardiac defect. We found no changes in Kv1.1-deficient cardiac structure, but extensive Kv1.1 expression in juxtaparanodes of the wild-type vagus nerve, the primary source of parasympathetic input to the heart, suggesting a novel site of action leading to Kv1.1-associated cardiac bradyarrhythmias. Together, our data suggest that Kv1.1 deficiency leads to impaired neural control of cardiac rhythmicity due in part to aberrant parasympathetic neurotransmission, making Kcna1 a strong candidate gene for human SUDEP.
Introduction
People with epilepsy who are otherwise healthy die unexpectedly for unknown pathological reasons at a rate up to 24 times greater than the general population (Ficker et al., 1998). Such deaths are classified as SUDEP, short for sudden unexplained death in epilepsy. A leading explanation for SUDEP, which affects up to 17% of those with idiopathic epilepsy, is that seizures initiate pathogenic neural signaling between the brain and heart, leading to lethal cardiac arrhythmias (Jehi and Najm, 2008; Surges et al., 2009). Genetically, ion channels coexpressed in brain and heart are logical candidates for SUDEP because defects in intrinsic membrane excitability could underlie both epilepsy and cardiac arrhythmias that precipitate death (Nashef et al., 2007). Recently, the heart–brain potassium channel gene, Kcnq1, was identified as the first ion channel gene for SUDEP when it was found that mice bearing human disease-linked KCNQ1 mutations associated with the most common form of cardiac long QT syndrome (LQT1) exhibit spontaneous, unprovoked seizures that can lead to lethal cardiac arrhythmias (Goldman et al., 2009).
Genes with neural-specific expression that can impact cardiac function extrinsically via the autonomic nervous system represent a second potential class of SUDEP candidate genes. The vagus nerve is the primary conduit for parasympathetic signaling from the brainstem, and is balanced by the spinal sympathetic outflow (Berthoud and Neuhuber, 2000). Excess vagal activity causes bradycardia and asystole, and these patterns occur during seizures in up to 21% of drug-resistant epilepsy cases (Rugg-Gunn et al., 2004). Kv1.1 potassium channels, encoded by the Kcna1 gene, may be capable of mediating a potentially lethal cardiac effect via this indirect neural mechanism. Kv1.1 subunits are widely expressed in brain, where they regulate action potential propagation and shape, neuronal repetitive firing properties, and neurotransmitter release (Wang et al., 1994; Dodson and Forsythe, 2004), but are essentially undetectable in myocardial tissue (Nerbonne and Kass, 2005). Mice bearing Kv1.1 channel mutations exhibit hippocampal and peripheral nerve hyperexcitability, severe epilepsy characterized by partial and generalized tonic–clonic seizures, and premature death (Smart et al., 1998; Chiu et al., 1999; Lopantsev et al., 2003; Glasscock et al., 2007). In Kv1.1-deficient mice, the onset of epilepsy at 2–3 weeks of age coincides with the appearance of early lethality in ∼25% of homozygotes, suggesting a link between seizures and sudden death (Smart et al., 1998; Glasscock et al., 2007). Kcna1 loss-of-function gene mutations also cause human excitability phenotypes, including epilepsy, episodic ataxia, and myokymia (Adelman et al., 1995; Zuberi et al., 1999; Liguori et al., 2001). Although some evidence exists for low levels of Kv1.1 transcripts in heart, especially nodal pacemaker cells, the channels have no known contribution to myocardial repolarizing K+ currents or pacemaking, and their deficiency has never been associated with a cardiac defect (Leoni et al., 2005; Marionneau et al., 2005; Nerbonne and Kass, 2005; Harrell et al., 2007). Here we localize Kv1.1 channels to vagal axonal juxtaparanodes and examine the hypothesis that Kv1.1-deficient mice exhibit cardiac dysfunction associated with epileptic activity, which may predispose them to sudden neurocardiogenic death.
Materials and Methods
Animals.
The Kcna1 −/− mice carry a null mutation of the Kcna1 gene on chromosome 6 as a result of gene targeted deletion, as previously described (Smart et al., 1998). Mice were housed at 22°C, fed ad libitum, and submitted to a 12 h light/dark cycle. We performed all procedures in accordance with the guidelines of the National Institutes of Health, as approved by the Animal Care and Use Committee of Baylor College of Medicine.
Genotyping.
We isolated genomic DNA from tail clips using DirectPCR Lysis Reagent (Viagen Biotech). We determined the genotypes of Kcna1 mice using PCR amplification of specific alleles as done previously (Glasscock et al., 2007). We included three primers in the PCR: a mutant-specific primer (5′-CCTTCTATCGCCTTCTTGACG-3′), a wild-type-specific primer (5′-GCCTCTGACAGTGACCTCAGC-3′), and a common primer (5′-GCTTCAGGTTCGCCACTCCCC-3′). The PCR yielded PCR products of ∼337 bp for the wild-type allele and ∼475 bp for the mutant allele.
Simultaneous video EEG–ECG recordings.
Kcna1-null mice and wild-type controls between 1 and 2 months of age were anesthetized with an intraperitoneal injection of 0.02 ml · g−1 Avertin and surgically implanted with bilateral silver wire electrodes (0.005 inch diameter) attached to a microminiature connector. EEG electrodes were inserted into the subdural space through cranial burr holes overlying the temporal cortex. For ECG, two thoracic electrodes were tunneled subcutaneously on either side and sutured in place to record cardiac activity. Mice were allowed to recover for 24 h before measuring simultaneous EEG–ECG activity of freely moving animals using a digital EEG/video monitoring system with Harmonie software, version 6.1c (Stellate Systems). For EEG signals, we used a sampling rate of 250 Hz and filtered using a 0.3 Hz high-pass filter, 70 Hz low-pass filter, and 60 Hz notch filter. For ECG signals, we used a sampling rate of 2 kHz with a 3 Hz high-pass filter.
ECG waveform analysis.
Definitions of ECG intervals and durations are illustrated in supplemental Figure S1 (available at www.jneurosci.org as supplemental material). P duration was manually measured as the time from the beginning of the upstroke of the P wave until its return to the isoelectric baseline; QRS duration was measured from the beginning of the Q wave to the peak amplitude of the downward deflection of the S wave; PR interval from the beginning of the upstroke of the P wave until the maximal amplitude of the R wave; RR interval as the time between consecutive R wave peaks; and QT interval from the beginning of the Q wave until the T wave returns to the isoelectric baseline. Since the QT interval covaries with the RR interval, we calculated a rate-corrected QT interval (QTc) using the following formula: QTc = QT/(RR/100)½ (Mitchell et al., 1998). To calculate average ECG intervals and durations, we analyzed 30 s epochs from a 24 h monitoring session, sampled four times daily at 12:00 A.M., 6:00 A.M., 12:00 P.M., and 6:00 P.M. Each 30 s epoch comprised between 250 and 400 heart beats, depending on the heart rate, which were then averaged together using pClamp 10 software (Molecular Devices) to generate a composite ECG waveform. We then measured each ECG characteristic from the average waveform, except for the RR interval, which was determined for each individual RR interval during the 30 s epoch and averaged. To calculate the average rate of interictal heart conduction blocks per hour for each genotype, we counted all non-seizure-associated second-degree AV blocks during the entire 24 h recording session. We defined a second-degree AV block as a nonconducted P wave in which the RR interval of the pause was at least 1.5 times the RR interval of the previously conducted P wave. To count as more than one event, we required AV blocks to be separated by at least 500 ms. To calculate the average ictal heart conduction blocks per hour, we counted second-degree AV blocks occurring during seizures, divided them by the total seizure duration, and extrapolated to an hourly basis for comparison. Only mice exhibiting at least three seizures were used for ictal cardiac analysis. We defined bradycardia as ≥15% decrease in heart rate compared to the overall heart rate.
Cardiac magnetic resonance imaging.
Short-axis cardiac magnetic resonance imaging (MRI) images were acquired using a Bruker BioSpin MRI PharmaScan 70/16-7.0 T scanner at the Mouse Phenotyping Core (Baylor College of Medicine). During the procedure, mice were anesthetized with isoflurane, and heart rate, respiratory rate, and body temperature were continuously monitored. Sets of 11 images were acquired for each cardiac cycle. Images were acquired with a field of view of 4 cm, slice thickness of 1 mm, and an in-plane resolution of ∼313 μm. Approximately 9–10 image sets were required to cover both ventricles. Images were analyzed off-line using ImageJ software (NIH). Right and left ventricular endocardial areas were measured during systole and diastole and summed to calculate the overall ventricular systolic and diastolic volumes. Ejection fraction (EF) was calculated from the end diastolic volume (EDV) and the end systolic volume (ESV) according to the following formula: EF = (EDV − ESV)/EDV.
Blood pressure measurements.
Systolic blood pressures were measured at the Mouse Phenotyping Core (Baylor College of Medicine) using a noninvasive tail-cuff blood pressure system (IITC Life Science). Two mice (2–3 months old) per genotype were allowed to acclimate to the testing conditions, and then multiple blood pressure measurements were taken daily over the course of 3 d for each mouse. These data were pooled and averaged to calculate the average systolic blood pressure for each mouse.
Pharmacology.
To achieve complete autonomic blockade, we administered propranolol (4 mg · kg−1) and atropine (1 mg · kg−1; Sigma-Aldrich) using concentrations previously shown to be effective in mice (Shusterman et al., 2002; Ieda et al., 2007). For selective parasympathetic or sympathetic blockade, we administered atropine (1 mg · kg−1) or propranolol (4 mg · kg−1) alone, respectively. Drugs were dissolved in 0.9% NaCl and injected intraperitoneally at a concentration of 10 ml · kg−1. For each drug challenge experiment, Kcna1-null mice (4–6 weeks old) were recorded by simultaneous video EEG–ECG for 2 h immediately before drug administration to establish the baseline rate of second-degree AV blocks, injected with drug, and then recorded for another 2 h to determine the drug's effect on the rate of AV blocks.
Immunohistochemistry.
C57BL/6J mice (2–3 months old) were perfused intracardially with PBS and fixed with 4% paraformaldehyde in PBS. Vagus nerves were removed, cryoprotected for 1–2 d at 4°C in 30% sucrose in PBS, frozen in embedding medium, and cut into 10 μm sections using a cryostat maintained at −20°C. Sections were directly mounted on slides and allowed to thaw at room temperature for ∼30 min immediately before processing. Tissue sections were rinsed three times in PBS and incubated for 1 h in antibody vehicle (10% BSA, 0.3% Triton X-100 in PBS). Next, the sections were incubated overnight (15–20 h) at room temperature with rabbit polyclonal anti-Kv1.1 antibody (1:50 dilution in vehicle; antibody courtesy of Dr. J. Trimmer, University of California, Davis, CA) and/or mouse monoclonal anti-Caspr (K65/35) antibody (1:500 dilution in vehicle; UC Davis/NIH NeuroMab Facility). Subsequently, sections were washed three times in antibody vehicle and incubated for 1 h in Alexa Fluor 488 goat anti-rabbit F(ab′)2 secondary antibody and/or Alexa Fluor 555 goat anti-mouse F(ab′)2 secondary antibody (1:1000 dilution in vehicle, Invitrogen). Finally, sections were rinsed once in vehicle and twice in PBS and then air dried at room temperature for 30 min. Once dry, the slides were coverslipped and mounted using ProLong Gold anti-fade reagent with DAPI (Invitrogen). Images were captured using an Olympus IX71 microscope (Olympus America) and adjusted for brightness and contrast using Adobe Photoshop Elements software (Adobe Systems). Control experiments performed by incubating slices with secondary antibodies only, as well as staining tissue sections from Kcna1-null mice with anti-Kv1.1 antibody showed an absence of background staining. For heart immunohistochemistry, mice (129 strain, age 2–3 months) were transcardially perfused with PBS and heart nodal regions dissected. Frozen heart sections (10 μm) were then cut and processed as described above, but without fixation.
Heart tissue preparation.
To obtain region-specific heart tissue for transcript and protein expression analysis, wild-type mice (129 strain; 2–3 months old) mice were transcardially perfused with PBS to remove blood. Dissections were then performed in ice-cold PBS using a microscope. For atria, we cut left and right atrial appendages. For ventricular tissue, we collected left and right atrial walls. For sinoatrial node, we isolated tissue including the intercaval region of the right atrium bounded by the crista terminalis, the superior and inferior vena cavae, and the atrial septum. For the atrioventricular node, we obtained tissue including Koch's triangle bounded by the tricuspid valve, the membranous septum, and the coronary sinus. Tissue was flash frozen using liquid nitrogen or placement at −80°C. Care was taken to remove adjoining fatty tissue as completely as possible.
Reverse transcriptase PCR.
Tissue from whole heart and from regional dissections of brain (cortex, cerebellum, hippocampus, olfactory bulb, spinal cord, and brainstem) and heart [atria, ventricle, sinoatrial (SA) node, and AV node] was homogenized in Trizol, and a phenol chloroform extraction of mRNA was performed. After treatment with DNase, the reverse transcriptase PCR (RT-PCR) was performed using SuperScriptIII RT-PCR System (Invitrogen) with polydT primers as per the provided protocol. PCR amplification of Kcna1 transcripts was performed using primers (forward, 5′-GCATCGACAACACCACAGTC-3′; reverse, 5′-CGGCGGCTGAGGTCACTGTCAGAGGCTAAGT-3′) targeting a 710 bp region in the coding exon of the mouse Kcna1 transcript (accession #NM_010595). PCR amplifications used 35 cycles with a 45 s extension time. To control for the small size of the starting heart tissues, GAPDH amplification (982 bp band; forward, 5′-TGAAGGTCGGTGTGAACGGATTTGGC-3′; reverse, 5′-ATGTAGGCCATGAGGTCCACCAC-3′) of cDNA from all tissues were performed.
Western blotting and immunoprecipitation.
Whole mouse brain and heart were extracted, flash frozen in liquid nitrogen, and subsequently homogenized on ice with a Tissue Tearor in lysis buffer containing the following (in mm): 20 Tris, pH 7.5, 138 NaCl, 3 KCl, 1% Triton X-100, 1 EGTA, 2 EDTA, 1 benzamidine, 1 phenylmethylsulfonylfluoride, 1 dithiothreitol, and 5 μg · ml−1 each of aprotinin, leupeptin, and pepstatin A. Total protein concentration of the brain and heart tissue lysates were determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories). One hundred or three hundred micrograms of heart protein lysate were separated on 8% Tris–glycine–SDS polyacrylamide gels, analyzed by Western immunoblot (IB) using rabbit polyclonal anti-Kv1.1 antibody (5 μg · ml−1 in vehicle; antibody courtesy of Dr. J. Trimmer) and HRP-tagged goat anti-rabbit IgG secondary antibody (1:10,000 dilution in vehicle, Santa Cruz Biotechnology), and subsequently detected using a commercial chemiluminescent substrate (SuperSignal; Pierce Chemical). For immunoprecipitation experiments, heart and brain protein lysates were diluted to 1 mg · ml−1, and each 1 ml sample was precleared for 1 h with 30 μl of protein A–Sepharose (GE Healthcare) and incubated overnight with 5 μg of mouse monoclonal anti-Kv1.1 (K20/78) antibody (UC Davis/NIH NeuroMab Facility). All incubations were performed at 4°C with constant agitation. Antibody-bound protein complexes were captured by the addition of 30 μl of protein A–Sepharose and incubated for another 2 h. Protein A–Sepharose was pelleted by centrifugation, and the immunoprecipitated protein complexes were eluted using SDS-PAGE sample buffer before SDS-PAGE and Western immunoblotting as described above.
Statistical analysis.
All data are presented as mean ± SEM. Statistical analysis was performed using Microsoft Excel 2007 Analysis ToolPak. For comparisons of cardiac data between Kv1.1-null mice and wild-type controls, we used two-tailed t tests. To assess drug effectiveness in pharmacology experiments, we calculated the symmetrized percentage change in the number of AV blocks following drug administration and then tested whether the symmetrized percentage change in baseline significantly differed from zero (μ = 0) using a one-sample t test. This method has greater statistical power to account for small numbers at baseline (Berry and Ayers, 2006). Pharmacology data points were excluded if no AV blocks were observed during the baseline period.
Results
Kv1.1-deficient mice exhibit interictal cardiac abnormalities
Since Kcna1 −/− mice develop epilepsy and die prematurely beginning at 2–3 weeks of age, we hypothesized that Kcna1-deficient mice may possess potentially lethal cardiac defects predisposing them to early death. To identify cardiac abnormalities, we obtained simultaneous video EEG–ECG recordings from freely moving Kcna1 −/− mice and wild-type controls (n = 4 per genotype) over a continuous 24 h period. We found that Kcna1 −/− mice displayed multiple patterns of functional cardiac rhythm disturbances not seen in control mice, including frequent AV conduction blocks, prolonged bradycardia, and excessive premature ventricular contractions (PVCs). The most common AV block observed was a type 1, second-degree block (Wenckebach) characterized by a PR interval that progressively lengthened until the QRS complex was dropped (Fig. 1 A). Less often, we saw type 2, second-degree blocks in which the heart skipped one or more beats without obvious PR lengthening (Fig. 1 B). AV blocks indicate defective electrical signal conduction between the atria and ventricles, often at the level of the AV node (Da Costa et al., 2002). We quantified the number of second-degree AV blocks in mutants and controls and found that Kv1.1-deficient mice displayed approximately five AV blocks per hour during interictal periods, a significant fivefold increase compared to wild-type littermates (p = 0.00034), which exhibited approximately one second-degree AV block per hour (Fig. 2 A). In Kcna1 −/− mutants, 96% (92/96) of the recorded hours contained at least one conduction block, compared to only 47% (45/96) in control mice (Fig. 2 B).
Figure 1.
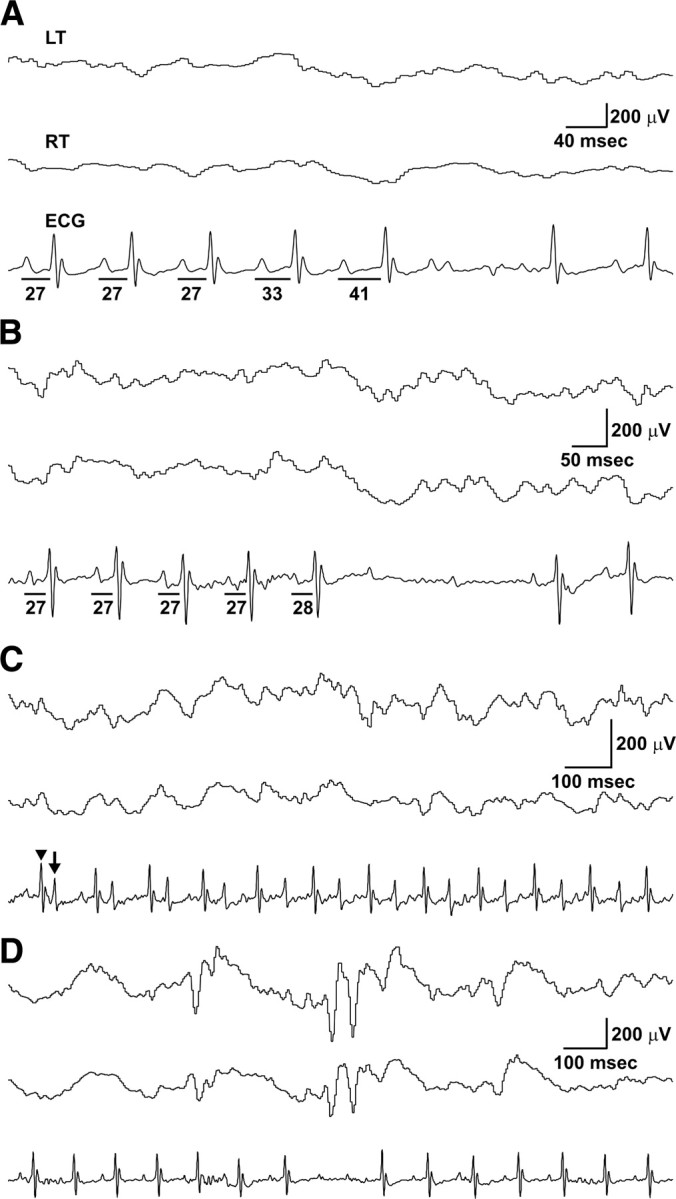
Simultaneous EEG–ECG reveals interictal cardiac abnormalities, including AV blocks and premature ventricular contractions, in Kcna1 −/− mice. A, Example of a type 1, second-degree AV block (Wenckebach) in a Kcna1 −/− mouse demonstrating the characteristic progressive lengthening of the PR interval (denoted by lines with time interval in milliseconds underneath) until the QRS complex is dropped. B, Example of a type 2, second-degree AV block in a Kcna1 −/− mouse characterized by a skipped beat (in this case 2) preceded by normal length PR intervals. C, In at least two null mice, PVCs appeared in a bigeminy pattern composed of normal sinus rhythm (sharper, higher amplitude waves; arrowhead) alternating with abnormal PVCs (broader, lower amplitude waves; arrow). D, Occasionally, AV blocks corresponded with interictal cortical discharges in Kcna1-null mice, but these instances were rare. LT, Left temporal EEG; RT, right temporal EEG.
Figure 2.
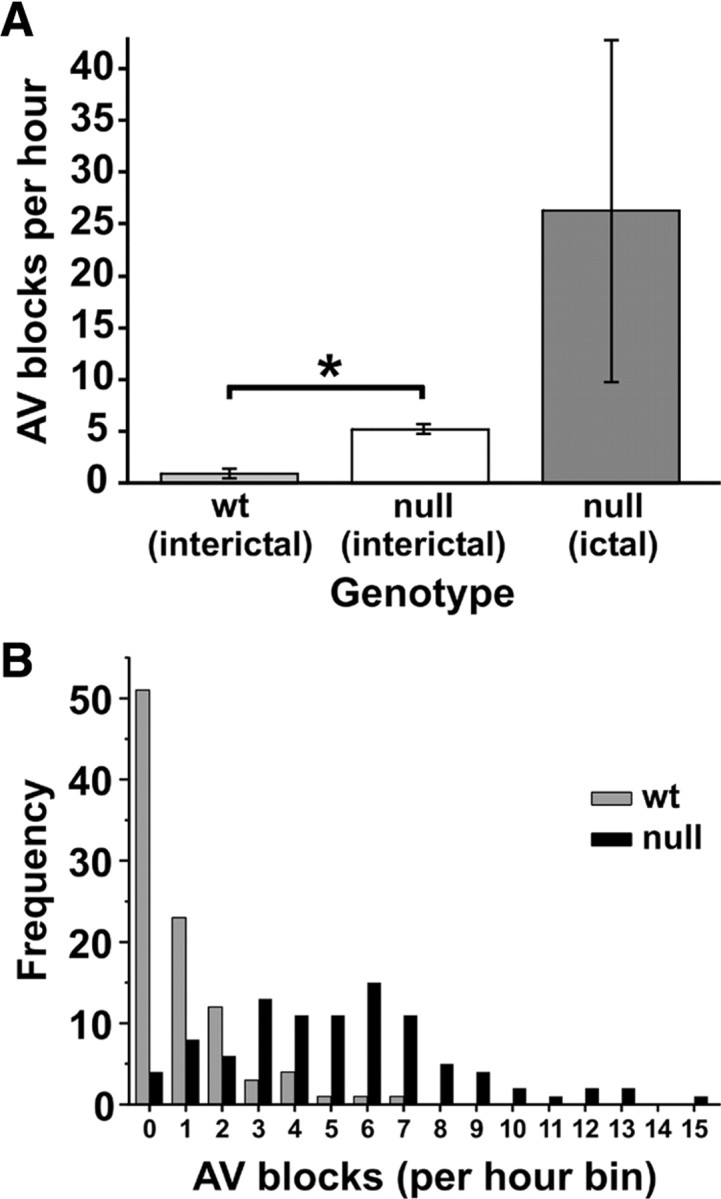
Kcna1 −/− mice exhibit a fivefold increase in basal AV blocks, a difference that is exacerbated by seizure activity. A, Comparison of mean number of AV blocks per hour during interictal periods in wild-type and null mice (n = 4 per genotype) showed a fivefold increase in mutants. During seizures in null mice, the frequency of AV blocks increased another fivefold to 25 times the wild-type rate. B, Histogram distribution of interictal AV blocks per hour in wild-type and null mice during 24 h recording sessions (n = 4 mice per genotype) shows that wild-type mice usually exhibited only 0–2 conduction blocks per hour compared to null mice, which exhibited conduction blocks with much higher frequency, up to 15 or more per hour. At least one AV block was observed in 96% of recording hours in null mice, whereas less than half of the recording hours showed a conduction block in controls. *p < 0.001; two-tailed t test.
In addition to AV blocks, Kcna1 −/− mice were also prone to bradycardia and PVCs. We observed extended periods of sinus bradycardia [heart rate (HR) <540 beats per minute (bpm)] lasting many minutes in Kv1.1-deficient mice. These episodes often corresponded to instances of behavioral myoclonus but without associated cortical EEG discharges, when the mice would assume a “Buddha-like” posture, sitting upright on the hindlimbs while the forelimbs exhibited clonic movements (see supplemental video, available at www.jneurosci.org as supplemental material). Despite these episodes of bradycardia, Kcna1 −/− mice displayed an average overall basal heart rate of 634 ± 44 bpm, which did not differ significantly from their wild-type counterparts (664 ± 16 bpm; p = 0.54). However, the heart rate of Kv1.1-deficient mice was more variable than controls. For example, the minimum–maximum range of heart rate values measured for Kcna1 −/− animals was 491–795 bpm, whereas wild-type mice exhibited a smaller range (615–743 bpm). Only 50% of HR measurements from Kv1.1-deficient mice fell within the wild-type range of observed heart rates. In addition to bradycardia, recordings in at least two mutant mice showed abnormal PVCs, sometimes occurring in a bigeminy pattern characterized by runs of PVCs alternating with normal beats (Fig. 1 C). Apart from the electrical arrhythmia, further analysis of the ECG waveform, blood pressure, cardiac MRI, and microscopic necropsies did not reveal any additional obvious functional or structural pathology in Kcna1 −/− mice that might indicate intrinsic heart disease (Table 1; supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Table 1.
ECG waveform characteristics of Kcna1 wild-type and null mice
| WT | Null | |
|---|---|---|
| RR (ms) | 90.5 ± 2.1 | 96.0 ± 6.8 |
| P (ms) | 14.8 ± 0.4 | 15.6 ± 1.2 |
| PR (ms) | 30.6 ± 0.2 | 30.6 ± 0.5 |
| QRS (ms) | 10.3 ± 0.3 | 9.7 ± 0.1 |
| QT (ms) | 55.4 ± 1.6 | 53.9 ± 4.4 |
| QTc (ms) | 58.2 ± 1.2 | 55.0 ± 3.8 |
| Systolic blood pressure (mmHg) | 127 ± 11 | 114 ± 7 |
Data are given as mean ± SEM and represent n = 4 per genotype, except for blood pressure measurements, which represent n = 2 per genotype. No statistically significant differences were found between genotypes. See Materials and Methods section and supplemental Figure S1 (available at www.jneurosci.org as supplemental material) for detailed description of data collection and analysis.
Seizures exacerbate cardiac abnormalities in Kv1.1-deficient mice
To evaluate whether cardiac abnormalities in Kv1.1-deficient mice correlated with abnormal brain activity, we searched for instances in which interictal EEG discharges and cardiac events occurred in close temporal proximity (within 200 ms). Solitary cortical spikes occur frequently in Kcna1 −/− mutant mice, sometimes on the order of hundreds per hour. AV blocks in Kcna1 −/− mice were occasionally associated with interictal spikes (Fig. 1 D), but these instances were rare (<2%). We next examined the relationship of seizures and cardiac activity in mice by sampling at least three seizure episodes in each animal. Ictal AV blocks were present in four of seven Kcna1 −/− mice, and in ∼30% of the seizures overall. Relative to interictal periods, there was a notable fivefold increase in the rate of AV blocks during seizures, but this trend did not reach statistical significance (p = 0.37) (Fig. 2 A). In addition to conduction blocks, episodes of bradycardia and asystole were associated with seizures.
On one occasion, we captured a SUDEP event in a Kv1.1-deficient mouse during simultaneous video EEG–ECG monitoring. The mouse exhibited generalized tonic–clonic seizures lasting ≤100 s each and occurring approximately once per hour, similar to other Kcna1 −/− mice. However these typical, milder seizures abruptly transitioned into a series of five prolonged (124–277 s), severe seizures, which occurred once an hour for 5 consecutive hours and from which the mouse did not recover. Before the first of these seizures, the mouse showed a stable heart rate of ∼650 bpm, but with each successive seizure, the postictal heart rate progressively decreased, eventually dropping below 400 bpm, where it remained until death (Table 2). The middle three seizures in the series produced severe cardiac instability marked by profound bradycardia (heart rate of 90–210 bpm) lasting up to 20 s, including 1–3 s intervals of asystole (Fig. 3 A–C). Shortly after the fourth seizure, the mouse ceased ambulatory movement, and following ∼2 h of electrocortical silence, ECG activity ceased (Fig. 3 D).
Table 2.
Series of severe seizures precipitating SUDEP in a Kcna1 −/− mouse
| Time of day | Seizure duration (s) | Postictal heart rate (bpm) | ECG abnormalities |
|---|---|---|---|
| 5:41 P.M. | 150 | 630 | First extended seizure in series; no obvious ECG abnormalities during seizure |
| 7:00 P.M. | 124 | 560 | During 2nd seizure, severe bradycardia/asystole with heart rate decreasing to 100 bpm for 9 s (see Fig. 3 A) |
| 7:52 P.M. | 184 | 490 | During 3rd seizure, severe bradycardia/asystole with heart rate decreasing to 90 bpm for 20 s (see Fig. 3 B) |
| 8:49 P.M. | 231 | 400 | During 4th seizure, severe bradycardia/asystole with heart rate decreasing to 210 bpm for 8 s; shortly afterwards, ambulatory movement ceased (see Fig. 3 C) |
| 9:57 P.M. | 277 | 380 | During 5th seizure, one AV block; afterwards, ECG showed variable bradyarrhythmias (∼180–360 bpm) and asystoles until electrical silence |
Figure 3.
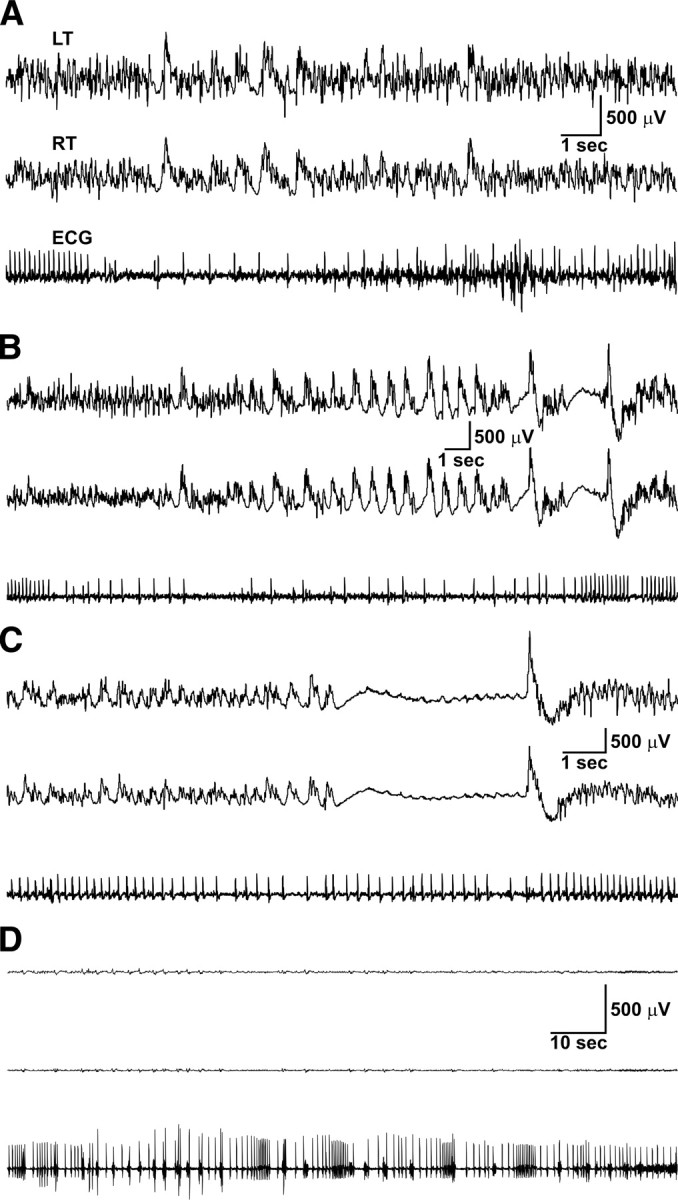
SUDEP in a Kcna1 −/− mouse was preceded by ictal cardiac abnormalities including severe bradycardia and asystole. A–C, Simultaneous EEG–ECG recordings showing cortical and cardiac activity during the second, third, and fourth seizures, respectively, in the series of five severe seizures culminating with death. Note the regular cardiac rhythm in A and B, with an abrupt transition to profound bradycardia (∼100 bpm) interspersed with episodes of asystole lasting up to 20 s before resuming normal rhythm. The postictal heart rate was progressively depressed by each successive seizure as shown in Table 2. D, Simultaneous EEG–ECG recording showing electrocerebral silence coupled with severe bradycardia, asystole, and arrhythmias shortly preceding death. LT, Left temporal EEG; RT, right temporal EEG.
Autonomic blockade indicates a major parasympathetic influence on cardiac dysrhythmias in Kv1.1-deficient mice
Heart conduction abnormalities in Kv1.1-deficient mice could stem from intrinsic heart excitability defects and/or autonomic dysfunction. To determine whether cardiac or neural effects were the predominant cause of interictal AV blocks, we examined their response to pharmacological blockade of the autonomic nervous system using atropine and propranolol to selectively inhibit parasympathetic and sympathetic activity, respectively. Simultaneous blockade of both autonomic branches by administering atropine and propranolol together resulted in nearly complete elimination of AV blocks in Kcna1 −/− mice, suggesting that the conduction defects were primarily mediated by the descending autonomic innervations (p = 0.00017) (Fig. 4). We then administered the drugs separately to determine which branch of the autonomic nervous system contributed most significantly to the conduction blocks. When given atropine alone to selectively inhibit the parasympathetic branch, AV blocks in Kcna1 −/− mice were almost completely ameliorated (p = 0.0016), whereas blocking sympathetic activity alone with propranolol had no significant effect (p = 0.39) (Fig. 4). The striking cessation of AV blocks mediated by selective parasympathetic inhibition suggests that the observed cardiac conduction defects depend on excessive parasympathetic tone.
Figure 4.

Selective pharmacological blockade of the autonomic nervous system suggests parasympathetic exacerbation of AV blocks in Kcna1 −/− mice. The first three panels are scatter plots of raw data showing the number of interictal AV blocks per hour before (Pre) and after (Post) drug administration. First panel, The elimination of interictal AV blocks in Kv1.1-deficient mice following administration of atropine and propranolol together (Atr + Pro) implicates autonomic transmission in arrhythmogenesis (n = 6). Second panel, Interictal AV blocks disappeared with atropine alone, suggesting predominant effect of parasympathetic tone (n = 4). Third panel, In contrast, injection of propranolol alone to inhibit sympathetic activity did not result in a significant change in the rate of AV conduction blocks (n = 6). Fourth panel, Summary of the percentage change in interictal AV blocks (symmetrized; mean ± SEM) following each drug treatment. Both complete autonomic blockade (Atr + Pro) and selective parasympathetic blockade (Atr) significantly decreased the average number of AV blocks per hour, but sympathetic blockade (Pro) had no significant effect. *p < 0.01; one-sample t test with μ = 0. NS, Not statistically significant.
Kv1.1 subunit expression in vagus nerve and heart
The primary source of parasympathetic input to the heart is the vagus nerve, which contains motor efferents originating from neurons in the dorsal motor nucleus of the vagus and the nucleus ambiguus in the medulla. Since cardiac conduction defects in Kv1.1-deficient mice were strongly associated with parasympathetic output, we used a specific Kv1.1 antibody to analyze the expression of Kv1.1 protein subunits within this pathway in wild-type mice. We detected Kv1.1 subunits in a juxtaparanodal pattern along vagal axons in both the cervical/thoracic portions of the nerve and the cardiac branch (Fig. 5 A,B), and identical to that observed at juxtaparanodes in peripheral axons (Mi et al., 1995), consistent with a role in modulating parasympathetic neurotransmission. We confirmed that the Kv1.1 staining pattern in vagus nerve corresponded to juxtaparanodes by double labeling with antibody to the paranodal protein Caspr. The Kv1.1 subunits always clustered directly adjacent to Caspr-positive paranodes in a flanking pattern, as would be expected for a juxtaparanodal protein (Fig. 5 C).
Figure 5.
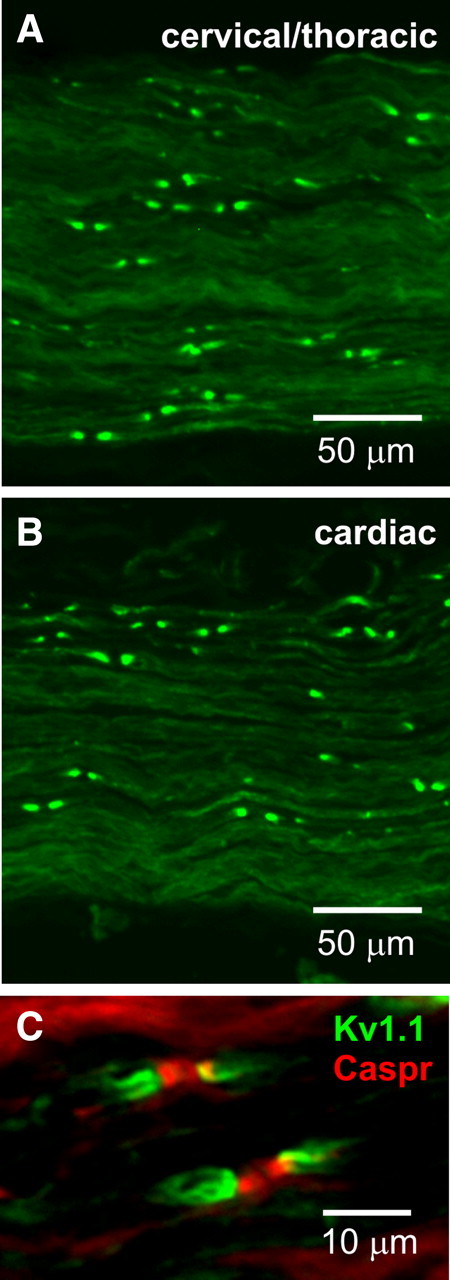
Kv1.1 channels localize to juxtaparanodes of vagus nerve axons. Both the cervical/thoracic portion of the vagus nerve (A) and the cardiac branch (B) exhibited strong Kv1.1 immunoreactivity in a juxtaparanodal staining pattern. C, Double labeling with antibodies to Kv1.1 subunits (green) and to the paranodal protein Caspr (red) confirms that the Kv1.1 staining pattern in vagus nerve corresponds to juxtaparanodes.
Finally, while Kv1.1 channels have been long regarded as predominantly neural-specific with no known expression of transcripts or protein in the heart (Dixon and McKinnon, 1994; Gaborit et al., 2007), recent work in mouse heart has reported the presence of low levels of Kv1.1 mRNA in the atria and ventricles and slightly higher levels in the sinoatrial and atrioventricular nodal pacemaking regions (Leoni et al., 2005; Marionneau et al., 2005; Harrell et al., 2007). To help clarify the localization of Kv1.1 channels in the mouse heart, we used RT-PCR to detect Kv1.1 mRNA in atria, ventricles, sinoatrial node, atrioventricular node, and whole heart (Fig. 6 A), as well as regional brain tissue as a positive control (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Kv1.1 mRNA was not detected in cardiac tissue from Kv1.1-deficient mice (Fig. 6 B). Using Western immunoblotting, we detected Kv1.1 protein as an immunoreactive band with an apparent molecular weight of ∼58 kDa in 300 μg, but not in 100 μg of heart protein lysate (Fig. 6 C). This band was not detected in 300 μg of heart protein lysate from Kv1.1-deficient mice (Fig. 6 C). Using immunoprecipitation and Western immunoblotting of 1 ml samples of protein lysates diluted to 1 mg · ml−1, we detected the same ∼58 kDa band faintly in heart and much more robustly in brain (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). The apparent molecular weight of ∼58 kDa for Kv1.1 protein is consistent with the value previously reported in mouse cortex protein lysate (Cheong et al., 2001). However, we could not detect Kv1.1 protein by immunoblotting regionally dissected heart tissue, even after pooling samples from four mice. Similarly, light microscopic immunohistochemistry of heart sections at multiple levels failed to demonstrate substantial Kv1.1 immunoreactivity.
Figure 6.

Kv1.1 transcript and protein expression in mouse heart. A, RT-PCR using wild-type tissue reveals Kv1.1 transcripts (710 bp) in atria, ventricle, SA node, AV node, and whole heart tissue. RT-PCR of GAPDH mRNA (982 bp) as a positive control showed adequate tissue and transcript levels of the preparations. Omitting template during either the reverse transcriptase (RT control) or PCR (PCR control) steps to control for DNA contamination resulted in no unwanted amplification. B, As expected, RT-PCR of cardiac ventricular tissue from a Kcna1 −/− mouse as a negative control failed to amplify any Kv1.1 mRNA. C, A Kv1.1-immunoreactive band with an apparent molecular weight of ∼58 kDa was detected in 300 μg but not 100 μg of wild-type heart protein lysate following SDS-PAGE and immunoblotting using anti-Kv1.1 polyclonal antibody. As expected, the immunoreactive band was not detected in 300 μg of heart protein lysate from a Kcna1 −/− mouse.
Discussion
A leading pathogenic explanation for SUDEP is that seizures themselves lead to cardiac arrest in at-risk individuals; however, a molecular mechanism for this malignant brain–heart relationship has remained elusive until the recent discovery of arrhythmogenic mutations of ion channels encoding long QT syndrome. The first candidate gene identified for SUDEP, KCNQ1 (KvLQT1), is the most common member of the LQT gene family, all of which are coexpressed in brain and heart. KCNQ1 encodes the Kv7.1 channel that mediates I Ks, a slow delayed rectifying K+ current that is linked to spontaneous seizures and cardiac arrhythmia in mice and humans (Goldman et al., 2009; Johnson et al., 2009). Here we find that Kcna1, a delayed rectifier channel abundant in brain but present at extremely low levels in heart, initiates lethal neurogenic cardiac dysfunction in mice by altering autonomic regulation of the heart. Kcna1 is not considered an LQT gene, and therefore represents a novel class of SUDEP gene candidates, namely ion channels with a primarily neural-restricted expression pattern.
Neurogenic cardiac dysfunction in Kcna1 −/− mice
The atropine-sensitive conduction blocks and ictal bradyarrhythmias in Kcna1-null mice combined with the predominantly neural expression indicate that Kv1.1 deficiency initiates cardiac dysfunction by neurogenic mechanisms, but the locus of the signaling defect remains to be defined. Kv1.1 channels are present in specific brain limbic centers, such as the hippocampus, dentate gyrus, and amygdala, which can modify autonomic outflow via descending pathways (Wang et al., 1994; Finnegan et al., 2006). Kcna1 deletion causes network hyperexcitability defects in these areas, which manifest as epilepsy in mice and humans (Smart et al., 1998; Zuberi et al., 1999; Liguori et al., 2001; Lopantsev et al., 2003; Glasscock et al., 2007). Early studies in monkeys demonstrated that stimulation of limbic regions induces vagally mediated bradycardia that is abolished by atropine or vagotomy (Reis and Oliphant, 1964). In humans, both spontaneous and stimulation-induced hippocampal epileptic activity can trigger cardiac bradyarrhythmias, such as AV blocks, reflecting recruitment of central autonomic centers (Altenmüller et al., 2004). Similarly, seizures in Kv1.1-deficient mice, which tend to originate in the hippocampus (Wenzel et al., 2007), also elicit bradycardia and AV blocks, indicating excessive cardiac parasympathetic tone. Thus, seizure-related vagal imbalance in Kcna1 −/− mice may stem at least in part from hyperexcitability in upstream limbic pathways.
Here we also show that Kv1.1 channels localize to juxtaparanodes of axons in vagus nerve, the primary source of parasympathetic cardiac input from the brain, suggesting that descending brain impulses during seizures may augment latent hyperexcitability already present in Kv1.1-deficient vagus nerve. Juxtaparanodal Kv1.1 channels are critical regulators of excitability and burst firing (Chiu et al., 1999), and the absence of Kv1.1 subunits in mouse sciatic nerve causes prolonged depolarization of the compound action potential following stimulation (Smart et al., 1998). In mouse phrenic nerve, Kv1.1 deletion causes repetitive neuronal activity resulting from both spontaneous and stimulus-evoked nerve-backfiring at preterminal axon transition zones where axons change from myelinated to nonmyelinated (Zhou et al., 1998, 1999). Computer simulations of myelinated nerve terminals corroborate this mechanism, showing that a lack of juxtaparanodal Kv1.1 channels leads to reentrant excitation of nodes due to nerve backfiring at axon transition zones (Zhou et al., 1999). Interestingly, preterminal hyperexcitability leading to backfiring shows developmental regulation, peaking at P17, which corresponds to the time (P14–P21) when Kv1.1 channels first exhibit fully developed axonal localization in hippocampus (Prüss et al., 2010) and when Kcna1-null mice begin to show seizures and sudden death (Rho et al., 1999; Zhou et al., 1999; Glasscock et al., 2007). Similarly, the absence of Kv1.1 channels in vagus nerve would be expected to confer vagal hyperexcitability, predisposing Kcna1-null mice to abnormal brain–heart cardiac regulation.
Although pharmacology and expression data suggest a predominantly extrinsic, neural origin for cardiac defects in Kv1.1-deficient mice, we do not exclude the possibility that Kv1.1 channels make a minor contribution to intrinsic heart rhythms, since we detect low levels of cardiac Kv1.1 expression in mouse. While the analysis of Kv1.1 channels in heart has produced mixed results depending on the organism and technique, Kv1.1 protein in heart has not been previously reported (supplemental Table S1, available at www.jneurosci.org as supplemental material). In mouse, Northern blotting reveals faint expression of Kv1.1 transcripts in whole-heart RNA, but this was attributed to contamination by cells other than myocytes (London et al., 2001). More sensitive RT-PCR-based detection methods identify Kv1.1 mRNA in low-to-high abundance in mouse ventricles and atria and higher transcript levels in nodal regions (Marionneau et al., 2005; Harrell et al., 2007). Cardiac channels undergo significant activity-induced remodeling, and chronic bradycardia in mice leads to compensatory upregulation of Kv1.1 mRNA expression in sinoatrial node, suggesting a potential role in cardiac pacemaking (Leoni et al., 2005). Our work extends these findings by demonstrating the presence of Kv1.1 protein in mouse heart. Since we detected Kv1.1 protein by immunoblotting large (300 μg), but not smaller (100 μg) heart protein lysate samples, it is possible that the previous lack of detection of cardiac Kv1.1 protein was due to insufficient sample loading, along with differences in antibody sensitivity. Immunoblotting regionally dissected heart tissue and immunofluorescence of nodal and myocardial heart sections did not reveal significant Kv1.1 immunoreactivity. The most likely source of Kv1.1 protein in our experiments is either the nodal pacemaking regions where moderately high levels of Kv1.1 mRNA have been detected or intrinsic cardiac neurons, which were occasionally dimly visualized in immunofluorescent sections. Together, the low-level cardiac Kv1.1 expression, the abundant neural Kv1.1 expression, and our pharmacological data suggest that Kv1.1 channels do not play a prominent role in generating intrinsic aberrant heart rhythms, but rather influence cardiac function extrinsically via the autonomic nervous system.
Kv1.1 deficiency as a model of human SUDEP
The neurocardiac features of the Kcna1-null phenotype make it a useful model for studying the relationship between epilepsy and sudden death. The AV conduction blocks and bradycardias observed during seizures in Kcna1-null mice are consistent with ictal cardiac bradyarrhythmias present in humans, where they have been reported in up to 21% of epilepsy patients (Rugg-Gunn et al., 2004; Leung et al., 2006) and >60 cases have been described with potentially life-threatening AV block, bradycardia, and asystole associated with seizures (Wilder-Smith, 1992; Reeves et al., 1996; Devinsky et al., 1997; Lim et al., 2000; Tinuper et al., 2001; Tigaran et al., 2002; Zubair et al., 2009). At least one report describes an instance of seizure-provoked asystole that actually progressed to SUDEP (Dasheiff and Dickinson, 1986). While simple AV conduction blocks were the most common finding during seizures in Kcna1-deficient mice, in the recorded SUDEP event we observed a series of seizures that initiated sustained ictal bradycardia and asystole, similar to the human SUDEP case.
In addition to bradyarrhythmias, Kv1.1 deficiency accurately recapitulates other aspects of human SUDEP pathology. Key risk factors for human SUDEP include a history of generalized tonic–clonic seizures, high seizure frequency, and young age at onset of epilepsy (Nashef et al., 2007). The seizure phenotype of Kv1.1-deficient mice accurately models all of these characteristics: frequent tonic–clonic seizures (up to 20 times daily) that begin in young mice between 2 and 3 weeks of age, and early lethality in ∼75% of the homozygotes (Smart et al., 1998; Glasscock et al., 2007). The criteria for SUDEP also include the absence of structural cardiac defects upon postmortem examination (Nashef et al., 2007), and we failed to identify any cardiac MRI or histopathology in Kcna1-null mice. In humans, SUDEP usually occurs in close temporal association with a seizure (Langan et al., 2000), and in our Kcna1 mouse, cardiac dysfunction and death were closely preceded by a series of severe seizures. Interestingly, in the only reported case of human SUDEP to occur during intracranial EEG monitoring, the patient experienced four closely spaced seizures followed by a fifth and final seizure, which ended with complete cessation of brain activity despite the presence of a regular pulse for another 2 min (Bird et al., 1997). A similar pattern of terminal progression from repeated seizures to electrocerebral silence followed by cardiac arrest was observed during the lethal event recorded in our Kcna1 −/− mouse.
Complex multigene interactions may also influence susceptibility to SUDEP, given the ability of genetic variants to modify the severity of epilepsy and cardiac arrhythmias independently (Viswanathan et al., 2003; Glasscock et al., 2007; Nashef et al., 2007). Interestingly, we found that the same P/Q-type Ca2+ channel mutation that rescues premature death and decreases seizure activity in Kcna1-null mice (Glasscock et al., 2007) also reduced the occurrence of interictal AV conduction blocks by ∼50% (data not shown). P/Q channels also strongly reduce release at both afferent and efferent vagal axons (Hong and Chang, 1995; Kawada et al., 2006; Pamidimukkala et al., 2006; Ohba et al., 2009). This complexity suggests that multiple ion channel gene variants may be important combinatorial modifiers of both brain and heart phenotypes, providing a novel explanation for the sporadic nature of human SUDEP. Although the contribution of Kv1.1 deficiency to human SUDEP cases remains to be investigated, Kcna1 −/− mice establish a new class of SUDEP candidate genes that induce lethal cardiac dysfunction by autonomic neural mechanisms independent of heart expression.
Footnotes
This work was supported by National Institutes of Health Grant NS29709 (J.L.N.) and an American Heart Association postdoctoral fellowship (E.G.).
References
- Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Altenmüller DM, Zehender M, Schulze-Bonhage A. High-grade atrioventricular block triggered by spontaneous and stimulation-induced epileptic activity in the left temporal lobe. Epilepsia. 2004;45:1640–1644. doi: 10.1111/j.0013-9580.2004.34403.x. [DOI] [PubMed] [Google Scholar]
- Berry D, Ayers G. Symmetrized percent change for treatment comparisons. Am Stat. 2006;60:27–31. [Google Scholar]
- Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci. 2000;85:1–17. doi: 10.1016/S1566-0702(00)00215-0. [DOI] [PubMed] [Google Scholar]
- Bird JM, Dembny KAT, Sandeman D, Butler S. Sudden unexplained death in epilepsy: an intracranially monitored case. Epilepsia. 1997;38(Suppl 11):S52–S56. [Google Scholar]
- Cheong A, Dedman AM, Xu SZ, Beech DJ. K(V)alpha1 channels in murine arterioles: differential cellular expression and regulation of diameter. Am J Physiol Heart Circ Physiol. 2001;281:H1057–H1065. doi: 10.1152/ajpheart.2001.281.3.H1057. [DOI] [PubMed] [Google Scholar]
- Chiu SY, Zhou L, Zhang CL, Messing A. Analysis of potassium channel functions in mammalian axons by gene knockouts. J Neurocytol. 1999;28:349–364. doi: 10.1023/a:1007013731231. [DOI] [PubMed] [Google Scholar]
- Da Costa D, Brady WJ, Edhouse J. Bradycardias and atrioventricular conduction block. BMJ. 2002;324:535–538. doi: 10.1136/bmj.324.7336.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasheiff RM, Dickinson LJ. Sudden unexpected death of epileptic patient due to cardiac arrhythmia after seizure. Arch Neurol. 1986;43:194–196. doi: 10.1001/archneur.1986.00520020080028. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Pacia S, Tatambhotla G. Bradycardia and asystole induced by partial seizures: a case report and literature review. Neurology. 1997;48:1712–1714. doi: 10.1212/wnl.48.6.1712. [DOI] [PubMed] [Google Scholar]
- Dixon JE, McKinnon D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circ Res. 1994;75:252–260. doi: 10.1161/01.res.75.2.252. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Forsythe ID. Presynaptic K+ channels: electrifying regulators of synaptic terminal excitability. Trends Neurosci. 2004;27:210–217. doi: 10.1016/j.tins.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Ficker DM, So EL, Shen WK, Annegers JF, O'Brien PC, Cascino GD, Belau PG. Population-based study of the incidence of sudden unexplained death in epilepsy. Neurology. 1998;51:1270–1274. doi: 10.1212/wnl.51.5.1270. [DOI] [PubMed] [Google Scholar]
- Finnegan TF, Chen SR, Pan HL. Mu opioid receptor activation inhibits GABAergic inputs to basolateral amygdala neurons through Kv1.1/1.2 channels. J Neurophysiol. 2006;95:2032–2041. doi: 10.1152/jn.01004.2005. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582:675–693. doi: 10.1113/jphysiol.2006.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell MD, Harbi S, Hoffman JF, Zavadil J, Coetzee WA. Large-scale analysis of ion channel gene expression in the mouse heart during perinatal development. Physiol Genomics. 2007;28:273–283. doi: 10.1152/physiolgenomics.00163.2006. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Chang CC. Calcium channel subtypes for the sympathetic and parasympathetic nerves of guinea-pig atria. Br J Pharmacol. 1995;116:1577–1582. doi: 10.1111/j.1476-5381.1995.tb16375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Kanazawa H, Kimura K, Hattori F, Ieda Y, Taniguchi M, Lee JK, Matsumura K, Tomita Y, Miyoshi S, Shimoda K, Makino S, Sano M, Kodama I, Ogawa S, Fukuda K. Sema3a maintains normal heart rhythm through sympathetic innervation patterning. Nat Med. 2007;13:604–612. doi: 10.1038/nm1570. [DOI] [PubMed] [Google Scholar]
- Jehi L, Najm IM. Sudden unexpected death in epilepsy: impact, mechanisms, and prevention. Cleve Clin J Med. 2008;75(Suppl 2):S66–S70. doi: 10.3949/ccjm.75.suppl_2.s66. [DOI] [PubMed] [Google Scholar]
- Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA, Ackerman MJ. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. 2009;72:224–231. doi: 10.1212/01.wnl.0000335760.02995.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada T, Yamazaki T, Akiyama T, Uemura K, Kamiya A, Shishido T, Mori H, Sugimachi M. Effects of Ca2+ channel antagonists on nerve stimulation-induced and ischemia-induced myocardial interstitial acetylcholine release in cats. Am J Physiol Heart Circ Physiol. 2006;291:H2187–H2191. doi: 10.1152/ajpheart.00175.2006. [DOI] [PubMed] [Google Scholar]
- Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211–213. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni AL, Marionneau C, Demolombe S, Le Bouter S, Mangoni ME, Escande D, Charpentier F. Chronic heart rate reduction remodels ion channel transcripts in the mouse sinoatrial node but not in the ventricle. Physiol Genomics. 2005;24:4–12. doi: 10.1152/physiolgenomics.00161.2005. [DOI] [PubMed] [Google Scholar]
- Leung H, Kwan P, Elger CE. Finding the missing link between ictal bradyarrhythmia, ictal asystole, and sudden unexpected death in epilepsy. Epilepsy Behav. 2006;9:19–30. doi: 10.1016/j.yebeh.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Liguori R, Avoni P, Baruzzi A, Di Stasi V, Montagna P. Familial continuous motor unit activity and epilepsy. Muscle Nerve. 2001;24:630–633. doi: 10.1002/mus.1048. [DOI] [PubMed] [Google Scholar]
- Lim EC, Lim SH, Wilder-Smith E. Brain seizes, heart ceases: a case of ictal asystole. J Neurol Neurosurg Psychiatry. 2000;69:557–559. doi: 10.1136/jnnp.69.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Guo W, Pan X, Lee JS, Shusterman V, Rocco CJ, Logothetis DA, Nerbonne JM, Hill JA. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of I(K,slow) and resistance to drug-induced qt prolongation. Circ Res. 2001;88:940–946. doi: 10.1161/hh0901.090929. [DOI] [PubMed] [Google Scholar]
- Lopantsev V, Tempel BL, Schwartzkroin PA. Hyperexcitability of CA3 pyramidal cells in mice lacking the potassium channel subunit Kv1.1. Epilepsia. 2003;44:1506–1512. doi: 10.1111/j.0013-9580.2003.44602.x. [DOI] [PubMed] [Google Scholar]
- Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, Lei M, Escande D, Demolombe S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol. 2005;562:223–234. doi: 10.1113/jphysiol.2004.074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Deerinck TJ, Ellisman MH, Schwarz TL. Differential distribution of closely related potassium channels in rat Schwann cells. J Neurosci. 1995;15:3761–3774. doi: 10.1523/JNEUROSCI.15-05-03761.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998;274:H747–H751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- Nashef L, Hindocha N, Makoff A. Risk factors in sudden death in epilepsy (SUDEP): the quest for mechanisms. Epilepsia. 2007;48:859–871. doi: 10.1111/j.1528-1167.2007.01082.x. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Ohba T, Takahashi E, Murakami M. Modified autonomic regulation in mice with a P/Q-type calcium channel mutation. Biochem Biophys Res Commun. 2009;381:27–32. doi: 10.1016/j.bbrc.2009.01.184. [DOI] [PubMed] [Google Scholar]
- Pamidimukkala J, Habibi S, Hay M. Frequency-dependent depression of exocytosis and the role of voltage-gated calcium channels. Brain Res. 2006;1078:1–8. doi: 10.1016/j.brainres.2006.01.044. [DOI] [PubMed] [Google Scholar]
- Prüss H, Grosse G, Brunk I, Veh RW, Ahnert-Hilger G. Age-dependent axonal expression of potassium channel proteins during development in mouse hippocampus. Histochem Cell Biol. 2010;133:301–312. doi: 10.1007/s00418-009-0668-z. [DOI] [PubMed] [Google Scholar]
- Reeves AL, Nollet KE, Klass DW, Sharbrough FW, So EL. The ictal bradycardia syndrome. Epilepsia. 1996;37:983–987. doi: 10.1111/j.1528-1157.1996.tb00536.x. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Oliphant MC. Bradycardia and tachycardia following electrical stimulation of the amygdaloid region in monkey. J Neurophysiol. 1964;27:893–912. doi: 10.1152/jn.1964.27.5.893. [DOI] [PubMed] [Google Scholar]
- Rho JM, Szot P, Tempel BL, Schwartzkroin PA. Developmental seizure susceptibility of kv1.1 potassium channel knockout mice. Dev Neurosci. 1999;21:320–327. doi: 10.1159/000017381. [DOI] [PubMed] [Google Scholar]
- Rugg-Gunn FJ, Simister RJ, Squirrell M, Holdright DR, Duncan JS. Cardiac arrhythmias in focal epilepsy: a prospective long-term study. Lancet. 2004;364:2212–2219. doi: 10.1016/S0140-6736(04)17594-6. [DOI] [PubMed] [Google Scholar]
- Shusterman V, Usiene I, Harrigal C, Lee JS, Kubota T, Feldman AM, London B. Strain-specific patterns of autonomic nervous system activity and heart failure susceptibility in mice. Am J Physiol Heart Circ Physiol. 2002;282:H2076–H2083. doi: 10.1152/ajpheart.00917.2001. [DOI] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- Surges R, Thijs RD, Tan HL, Sander JW. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol. 2009;5:492–504. doi: 10.1038/nrneurol.2009.118. [DOI] [PubMed] [Google Scholar]
- Tigaran S, Mølgaard H, Dam M. Atrio-ventricular block: a possible explanation of sudden unexpected death in epilepsy. Acta Neurol Scand. 2002;106:229–233. doi: 10.1034/j.1600-0404.2002.02017.x. [DOI] [PubMed] [Google Scholar]
- Tinuper P, Bisulli F, Cerullo A, Carcangiu R, Marini C, Pierangeli G, Cortelli P. Ictal bradycardia in partial epileptic seizures: autonomic investigation in three cases and literature review. Brain. 2001;124:2361–2371. doi: 10.1093/brain/124.12.2361. [DOI] [PubMed] [Google Scholar]
- Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J Clin Invest. 2003;111:341–346. doi: 10.1172/JCI16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. 1994;14:4588–4599. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Vacher H, Clark E, Trimmer JS, Lee AL, Sapolsky RM, Tempel BL, Schwartzkroin PA. Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia. 2007;48:2023–2046. doi: 10.1111/j.1528-1167.2007.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder-Smith E. Complete atrio-ventricular conduction block during complex partial seizure. J Neurol Neurosurg Psychiatry. 1992;55:734–736. doi: 10.1136/jnnp.55.8.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhang CL, Messing A, Chiu SY. Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: role of potassium channels under the myelin sheath in young nerves. J Neurosci. 1998;18:7200–7215. doi: 10.1523/JNEUROSCI.18-18-07200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Messing A, Chiu SY. Determinants of excitability at transition zones in Kv1.1-deficient myelinated nerves. J Neurosci. 1999;19:5768–5781. doi: 10.1523/JNEUROSCI.19-14-05768.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubair S, Arshad AB, Saeed B, Luqman S, Oommen KJ. Ictal asystole—late manifestation of partial epilepsy and importance of cardiac pacemaker. Seizure. 2009;18:457–461. doi: 10.1016/j.seizure.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, McWilliam RC, Stephenson JBP, Kullmann DM, Hanna MG. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 1999;122:817–825. doi: 10.1093/brain/122.5.817. [DOI] [PubMed] [Google Scholar]