

Abstract
Cellular properties are influenced by complex factors inherent to their microenvironments. While oxygen deprivation (hypoxia) occurs in tumours because of rapid cell proliferation and aberrant blood vessel formation, embryonic cells develop in a naturally occurring hypoxic environment. Cells respond to hypoxia by stabilizing hypoxia-inducible factors (HIFs), which are traditionally viewed to function by altering cellular metabolism and blood vessel architecture. Recently, HIFs have been shown to modulate specific stem cell effectors, such as Notch, Wnt and Oct4 that control stem cell proliferation, differentiation and pluripotency. Direct molecular links have also been established between HIFs and critical cell signalling pathways such as cMyc and p53. These novel links suggest a new role for HIFs in stem cell and tumour regulation.
Keywords: hypoxia-inducible factors (HIF-1, HIF-2); stem cell signalling; cancer pathways
Introduction
From metabolic adaptation to stem cell signalling
HIF-1α regulates Notch effects on cellular differentiation
Stem cell pluripotency requires HIF target gene function
O2 regulation of Wnt signalling is differentiation-stage specific
HIFs, stem cell pathways and disease
HIF and cMyc
HIF and p53
Introduction
Oxygen is a key requirement for survival, serving as the primary electron acceptor in a multitude of biochemical reactions, and fulfilling cellular energy demands by generating ATP through aerobic metabolism. This makes physiologically low O2 levels, or hypoxia, one of the most relevant cellular stresses. Hypoxia plays a central role in normal development, and is also an important aspect of variety of pathological conditions, including stroke, tissue ischaemia, inflammation and the growth of solid tumours.
Hypoxia was first correlated with tumour progression in the early 20th century, when researchers demonstrated increased tumour cell glycolysis in the presence of high O2 levels and the existence of a direct link between cellular hypoxia and radioresistance [1] (see timeline in [1]). More recently, a connection between mammalian embryogenesis and O2 levels was revealed in the 1970s, when Morriss and New demonstrated that successful development of the neural fold by ex utero mouse embryos required culture conditions with low O2 levels [2].
Cells respond to hypoxia through coordinated changes in gene expression. The transcription factors primarily responsible for these changes are hypoxia-inducible factors (HIFs). HIF is composed of two subunits, an α subunit that is oxygen labile, but which is rapidly stabilized in response to low O2 conditions, and a β subunit (also known as ARNT, the aryl hydrocarbon receptor nuclear translocator) that is constitutively expressed [3]. The stabilized α subunit subsequently heterodimerizes with the β subunit to form a potent transcription factor which recognizes specific promoter elements known as hypoxia response elements and activates target genes essential for adaptive responses.
Exposure to low O2 levels initiates a HIF response in almost all vertebrate cells which ranges from rapid changes at the cellular level, such as altered carbohydrate metabolism, to systemic changes including erythropoiesis and angiogenesis. HIF responses are also triggered in embryonic stem cells. Prior to the establishment of the circulatory system, the early embryo develops in a naturally occurring hypoxic environment (<3% O2, 20 mm Hg) [4, 5] compared to the physiological range of adult tissues (2–9% O2, 14.4–64.8 mm Hg), or ambient air (21% O2, 150 mm Hg). Therefore, the lack of established vasculature in the early embryo, or aberrant vasculature in a rapidly proliferating tumour, which typically produces regions of severe hypoxia or anoxia, offers an environment conducive for the stabilization and function of HIF proteins.
Genetic analyses of HIFs in multiple species have confirmed these O2 sensors as critical regulators of ontogeny. Targeted disruption of Hif-1α, and Arnt in mice is embryonic lethal, and the embryos die primarily because of cardiac and vascular defects [6–8]. Targeted disruption of Hif-2α results in distinct phenotypes in different mouse strains, including embryonic lethality because of bradycardia and vascular defects [9], perinatal lethality because of impaired lung maturation [10] and postnatal lethality because of multiorgan failure and defective mitochondrial metabolism [11]. These studies also revealed the importance of HIF-mediated processes, including angiogenesis, invasion and metastasis, in the natural course of diseases such as solid tumours [12].
Initially recognized for their ability to promote metabolic adaptation, recent work has uncovered a new role for HIFs, that of stem cell regulation. It is increasingly appreciated that adult tissues maintain localized domains where O2 levels are low. Importantly, stem cells are known to occupy specialized microenvironments or ‘niches’, and, are regulated by factors inherent to their microenvironments. In vitro studies employing low O2 culture conditions (≤5% O2) have revealed regulatory links between O2 availability and cell proliferation, survival and differentiation of stem and progenitor cells. Among the defined stem and progenitor cell responses to hypoxia are stimulation of the proliferation of central nervous system precursor cells [13] and neural crest stem cells [14], enhanced survival of the chondrocyte growth plate [15] and the inhibition of adipocyte differentiation [16]. This raises the exciting possibility of stem cell regulation by hypoxia.
In this review, we discuss the role of O2 availability and HIFs in the regulation of cell signalling pathways and their influence on stem cell behaviour. In addition, we describe HIF regulation of critical cellular pathways (e.g. cMyc and p53) as it relates to tumorigenesis.
From metabolic adaptation to stem cell signalling
That tissue stem cells reside within specific anatomical locations termed ‘niches’ was proposed nearly four decades ago from studies on transplanted haematopoietic progenitors [17]. These analyses put forth the concept of microenvironment factors present in the niche which regulate stem cell properties such as the ability to self-renew and differentiate into specialized lineages. In recent years niches, and the intracellular interactions within have been delineated with increasing precision. For example, it is now known that in the Drosophila ovarioles and testis, specialized adherens junctions between the niche cells and germ stem cells anchor the stem cells to the niche [18], and promote asymmetric division [19, 20]. Microenvironmental cues also promote asymmetric cell division in the C. elegans zygote [21], and embryonic neuroblasts [22]. In mammals, stem cell niches have been described in multiple tissues, including the gonad [23], skin [24–26], intestine [27], bone marrow (BM) [28, 29] and brain [30, 31].
Genetic, molecular and 3-Dimensional culture analyses [32] have identified a number of conserved signal transduction pathways that are provided by niche cells and promote stem cell maintenance. These microenvironmental cues include signals from the BMP, Notch, Wnt, JAK-STAT and Sonic hedgehog pathways, which individually, or through integration with other signals, regulate stem cell properties [33–35]. Characterization of multiple niches, however, has also revealed variations in the niche anatomy, and novel regulatory factors are constantly emerging. One such factor is hypoxia.
It is generally believed that the BM, which is the primary site for adult haematopoiesis, maintains a significantly lower O2 tension compared to other organs and tissues [36]. Though haematopoietic stem cells (HSCs) have been identified within spatially distinct and multiple, functionally distinct niches (with evidence for both osteoblast association and sinusoidal perivascular associations, or both ([37, 38]), HSC’s have long been speculated to align along an oxygen gradient of the BM [39]. Chow et al. employed mathematical modelling of pO2 distribution in the BM to predict strong probability of HSC association with almost anoxic regions [40]. Similar results were obtained with in vivo dye perfusion studies [41]. Furthermore, our work suggests a similar link between hypoxia and the dentate gyrus, a site for adult neurogenesis, which seems to be regulated by low O2 levels (Mazumdar J. and Simon M.C., unpublished observation). That stem cells may occupy hypoxic niches and be regulated by low O2 gradients is also supported by in vitro low O2 culture condition studies [36, 42] which offer a demonstration of the direct influence of local O2 concentrations on stem cell self-renewal and differentiation. Danet et al. demonstrated that culturing human BM HSCs under hypoxic conditions (1.5% O2) increased the expansion of Lin−CD34+DC38− cells, a subpopulation of primitive progenitors and stem cells, and enhanced the ability of these enriched cells to engraft and repopulate the haematopoietic compartment of immunodeficient NOD/SCID mice [42]. Low O2 levels also positively influenced haematopoietic progenitor cell numbers isolated from embryonic yolk sacs or generated from ES cells grown in three-dimensional embryoid bodies in vitro[43, 44].
Low O2 regulation of stem cells extends beyond the haematopoietic lineages for instance, culturing neural crest stem cells or neuronal stem cells under hypoxic conditions (5% O2) increased their proliferation and skewed cellular differentiation towards specific fates [13, 14]. Moreover, hypoxia directly influences differentiation of human placental cytotrophoblast [45], and promotes functional cartilage formation from human embryonic stem cells [46]. Finally, culturing neuroblastoma and breast cancer cells under low O2 conditions confers stem cell-like properties and promotes dedifferentiation in these hypoxic neoplastic cell lines [47, 48]. Together, these findings suggest that hypoxia, which promotes stem cell quiescence and shields cells from oxidative damage, may be a strategic requirement for the maintenance of stem cell pools, especially in long-lived animals that cannot afford premature stem cell depletion.
Genetic ablation of HIF proteins phenocopies many of the hypoxic effects on stem cells, therefore indicating that stem cell responses to hypoxia are likely mediated by HIF proteins. Targeted mutation of the ARNT subunit, which eliminates both HIF-1α and HIF-2α function, results in a decreased number of progenitor cells of all haematopoietic lineages in embryoid body assays [43]. This phenotype is recapitulated in Arnt−/− mouse embryos, which display reduced number of haematopoietic progenitor cells in the embryonic yolk sac, as compared to Arnt replete counterparts [44]. One of the primary abnormalities of Arnt−/– embryos leading to embryonic lethality is a defective placenta, a complex organ whose development is intricately regulated by the hypoxic uterine environment.
Human placental cells proliferate under hypoxic culture conditions, whereas high O2 levels arrest proliferation and promote differentiation [45, 49]. Analysis of placentas from Arnt−/– (or Hif-1α–/–, Hif-2α–/– double) mutant mouse embryos revealed that HIF activity is required for appropriate trophoblast stem (TS) cell fate specification. The presence of HIF activity is essential for the differentiation of TS cells into spongiotrophoblasts which occupy a particularly hypoxic zone, followed by terminal differentiation into trophoblast giant cells which lie close to the O2-rich maternal spiral arteries [50]. Lack of HIF activity, on the other hand results in significantly reduced spongiotrophoblast cell numbers, and skews the differentiation of TS cells towards synciotrophoblasts (fused trophoblast precursor cells) [50, 51]. The effects of HIF activity on trophoblast cell fate determination have also been recapitulated using TS cell lines cultured in vitro. For instance, impaired placental invasion of Arnt−/– or Hif-1α–/– cells in vivo correlates with reduced migration and invasion of Arnt or HIF-1α depleted TS cells [52]. These experiments implicate the HIF proteins in the control of HSC and TS cell function.
Observations from our laboratory suggest a molecular link between HIF-1α and neural stem cell regulation (Mazumdar J. and Simon M.C., unpublished observation), thus indicating the potential influence of hypoxia on stem cells beyond the haematopoietic lineage. Current information, however, does not reveal whether the observed hypoxic effects can be generalized to all stem cell lineages, and entails further analysis. As noted recently by Keith and Simon [53], only a few HIF target genes that could mediate these effects have been identified. VEGF is one such target which accounted for many of the HIF-mediated effects on haematopoietic progenitors [44], but there is little doubt that other factors and signalling pathways are involved. Given that some hypoxic effects on stem cells parallel stem cell regulation by signalling pathways, and because a correlation between hypoxia and signalling pathways has been observed [47, 54], it is possible that altered stem cell behaviour under low O2 involves hypoxic regulation of stem cell pathways. Recent work has identified several such regulatory mechanisms by which HIFs directly modify cellular differentiation and stem cell function (Fig. 1).
Figure 1.
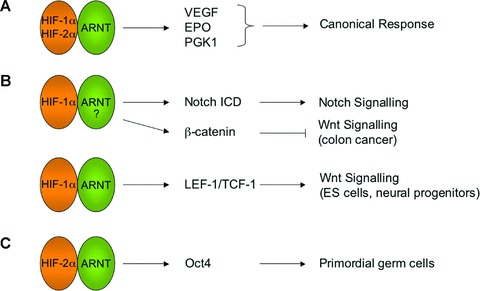
Hypoxia regulates stem cells in a HIF-dependent manner. A schematic diagram of molecular links between HIFs and stem cell pathways is shown. (A) Canonical HIF functions comprise adaptive responses including angiogenesis and altered metabolism mediated by target genes such as VEGF and PGK-1. (B) HIF-1α interacts with Notch-1 ICD, resulting in stabilization of Notch-1 ICD and the activation of target genes. In colon cancer cells, HIF-1α interaction with β-catenin results in reduced β-catenin-LEF/TCF transcriptional complex formation and decreased Wnt/β-catenin signalling. The exact role of ARNT in these interactions is not fully understood. Interestingly, in ES cells and other primary cells, HIF-1α/ARNT positively influences Wnt/β-catenin signalling by directly activating β-catenin transcriptional partners LEF-1 and TCF-1. (C) HIF-2α/ARNT promotes primordial germ cell maintenance through direct activation of Oct4, a pluripotency transcription factor.
HIF-1α regulates Notch effects on cellular differentiation
Previously, global gene profiling experiments indicated putative links between hypoxia and stem cell signalling pathways, for instance Notch [47] and Wnt/β-catenin signalling [54]. An interesting molecular intersection occurred when Gustafsson et al. working with isolated satellite cells (muscle progenitor cells) identified a direct molecular link between HIF-1α and Notch signalling [55]. Subsequently, direct molecular links have been established between HIFs and other key stem cell regulatory proteins including Oct4 [56], cMyc [57] and β-catenin [58].
Notch comprises a relatively simple core-signalling transduction pathway, yet controls cell differentiation in many different tissues and at multiple stages in a given lineage [59]. Signalling is initiated when the Jagged or Delta family of ligands binds a Notch receptor, triggering a two-step receptor proteolysis event [59]. The second proteolytic cleavage, mediated by γ-secretase, results in the release of the Notch intracellular domain (ICD) from the plasma membrane and its transport to the nucleus, where it forms a DNA-binding complex with other coactivators including MAML, CSL and p300, and activates target-gene expression [59]. Gustafsson et al. reported that hypoxia blocked the differentiation of myogenic satellite cells, a myogenic cell line (C2C12), and primary neural stem cells in a Notch-dependent manner [55]. Employing neural stem cells and neurogenic mouse embryonic carcinoma cells, the authors demonstrated that hypoxic treatment increased stabilization of the transcriptionally active Notch ICD, and stimulated Notch target genes Hes-1 and Hey-2. Reversal of cell differentiation after incubation with the γ-secretase inhibitor L-685,458 confirmed the requirement of Notch ICD in this regulation. In a novel molecular link, HIF-1α was demonstrated to mediate Notch ICD stabilization. In immunoprecipitation assays, HIF-1α physically interacted with Notch-1 ICD and accompanied it to Notch responsive promoters to activate target genes. However, a role for HIF’s activation of Notch target genes were only partially investigated: exactly how HIF-1α proteins integrate into the Notch ICD:MAML:CSL complex is not yet understood, nor is it known whether this response modulates the expression of all Notch target genes, or only a subset [55]. Of note, the hypoxic activation of Notch signalling bears remarkable similarity to cross talk between Notch and BMP/TGF-β signalling, which also blocks myogenic differentiation in C2C12 cells in a Notch-dependent manner [60, 61].
The importance of Notch pathway signalling in blocking cellular differentiation has been highlighted in several systems including Drosophila, C. elegans and mammals [59]. For example, Notch signalling is critical for the maintenance of undifferentiated stem and progenitor cell populations in the mammalian intestinal crypt, and also influences differentiation of mature enterocytes [62]. Forced Notch activation in haematopoietic BM or T cell progenitor cells inhibits differentiation and results in T cell acute lymphoblastic leukaemia [63]. That the primary effect of hypoxia, acting through Notch, is to inhibit the differentiation of multiple myogenic lines (see above), therefore offers a striking molecular link. As previously noted by Keith and Simon [53], the findings of Gustafsson et al. make it interesting to investigate whether altered Notch signalling underlies some of the developmental defects observed in HIF-deficient embryos, and in adult cells and tissues (such as the chondrocyte growth plate) which selectively lacks HIF-1α[15]. Given HIF-1α’s demonstrated role in the activation of Notch target genes, a hypoxic niche may prove particularly favourable for the optimal activation of Notch targets that inhibits cell differentiation, thereby contributing to stem cell quiescence and maintenance of homeostasis. Direct analysis of this regulation will require selective inactivation of HIF-α or Notch in specific stem cell populations in vivo. It is important to remember, however, that Notch effects can be complex and context dependent, as overexpression of Notch components rather promotes terminal differentiation in epidermal keratinocytes and certain neural stem and progenitor cells [62, 64, 65].
Stem cell pluripotency requires HIF target gene function
Several lines of study have indicated a potential involvement of low O2 in the maintenance of cell quiescence [41], proliferation [13] and differentiation [14, 16]. It was previously noted that bovine blastocysts produced under reduced O2 (<2% O2) tensions exhibited significantly more inner cell mass (ICM) cells than those that were maintained at higher O2 levels [66]. The ICM and its embryonic stem (ES) cell counterparts are pluripotent. Ezashi et al.[67] demonstrated that human ES cells, when cultured either at 3–5% O2 or 21% O2 proliferate at a similar rate. However, hypoxia substantially reduced the appearance of differentiated regions in these ES cultures, as assessed by morphology, and immunohistochemically (by the loss of stem cell markers such as stage-specific embryonic antigen-4 [SSEA-4], and the transcription factor OCT4 [see below], and by the gain of SSEA-1). These results suggest that hypoxic conditions are required to maintain the full pluripotency of mammalian ES cells.
Our laboratory recently reported that hypoxia regulates stem cell function through direct activation of specific HIF target genes. HIF-1α and HIF-2α are the two major HIFs that mediate responses to hypoxia. While they are known to have their unique targets, they also share targets. To determine the functional redundancy between HIF-1α and HIF-2α in embryonic development, Covello et al. targeted a HIF-2α cDNA into the Hif-1α locus in murine ES cells, thereby replacing HIF-1α expression with HIF-2α[68]. This ‘knock-in’ allele was designed to dissect the overlapping functions and functional redundancies between the two HIFs, and served as a stringent genetic tool to investigate the extent to which expanded HIF-2α expression, under the regulatory control of the Hif-1α locus, could complement a HIF-1α null mutation [56]. Since, HIF-1α-deficient embryos died at E8.5–E10.5, a HIF-2α knock-in strain, if completely incapable of complimenting HIF-1α function, should have displayed a similar embryonic lethality. Surprisingly, embryos with expanded HIF-2α (HIF-2α knock-in homozygotes) were recovered at significantly reduced frequencies at E6.5 – E7.5. The recovered embryos displayed developmental patterning defects and exhibited increased expression of certain genes, including Oct4. Up-regulation of Oct4 was also confirmed in HIF-2α knock-in ES cell-derived embryoid bodies. Subsequent analysis revealed phenotypes in vivo, such as increased mesoderm differentiation, correlated with expanded Oct4 expression, a critical transcriptional regulator controlling ES cell pluripotency. Chromatin immunoprecipitation (ChIP) assays revealed that HIF-2α is a direct upstream regulator of Oct4 and was capable of binding hypoxic regulatory elements in the murine Oct4 promoter. This transcriptional activity was not shared by HIF-1α.
Oct4 occupies promoters for many important developmental regulators in human ES cells [69], and, along with Nanog, forms a transcriptional network, which regulates pluripotency in mouse embryonic stem cells [70]. In vivo, Oct4 protein is abundant in the ICM of early blastocysts and down-regulated in trophoectoderm, whereas it is highly expressed in the nascent primitive endoderm [71]. Oct4 expression is down-regulated in somatic cells around the time of gastrulation, but retained in primordial germ cells and in some adult stem cell populations [72]. Therefore, Oct4 seems to maintain a precisely balanced interactive state with other signalling partners. Deviations from the strict Oct4 expression levels have dramatic effects on ES cell differentiation, for instance a 2-fold increase triggers differentiation into primitive endoderm or mesoderm, whereas a 2-fold decrease in Oct4 expression redirects ES cells to differentiate into trophoectoderm [71]. This is consistent with expanded mesoderm formation in HIF-2α knock-in embryoid bodies, as a consequence of HIF-2α expansion and Oct4 activation. Interestingly, HIF-2α-deficient embryos have severely reduced numbers of primordial germ cells which require Oct4 for survival/renewal [73], consistent with a physiological requirement of HIF-2α in Oct4-dependent stem cell function.
The molecular links between HIFs and key stem cell regulatory proteins such as Notch, and Oct4 confirm the emerging concept that HIFs not only act in metabolic adaptations, but can also regulate critical stem cell phenotypes. They also raise the possibility of cross talk between hypoxia and other stem cell signalling pathways.
O2 regulation of Wnt signalling is differentiation-stage specific
The Wnt signalling pathway has profound effects on cell function in D. melanogaster, Caenorhabditis elegans and mammals [34]. The key cytosolic transducer of the pathway, β-catenin, is stabilized when cell surface receptors (LRP-5/6 and Frizzled family of proteins) are engaged by secreted Wnt ligands. Stabilized β-catenin translocates to the nucleus where it interacts with the LEF/TCF family of transcriptional activators, to activate target genes [34]. Wnt signalling is frequently dysregulated in colon carcinoma, and stimulates proliferation of colorectal tumour cells [74]. On the other hand, hypoxia, a characteristic feature of solid tumours, blocks colorectal tumour cell proliferation. Kaidi et al. reported that hypoxia inhibited the proliferation of colon carcinoma cells in a β-catenin-dependent manner [58]. Hypoxic treatment resulted in increased cell-cycle arrest and down-regulated expression of the Wnt/β-catenin target cMyc, a potent cell-cycle regulator. Hypoxic inhibition of Wnt/β-catenin signalling was mediated by physical interaction of HIF-1α with β-catenin, resulting in reduced formation of β-catenin-TCF-4 complexes. Intriguingly, β-catenin/HIF-1α interaction was found to increase HIF transcriptional activity, which might help cells to adapt to severe hypoxia [58].
We have recently elucidated a divergent aspect of this molecular link in stem cells. We observed a positive influence of hypoxia on Wnt/β-catenin signalling in multiple embryonic cells including murine ES cells and isolated neural stem cells (Mazumdar J. and Simon M.C., unpublished observations). Furthermore, we found that this regulation is differentiation-stage specific, and confirmed previous observations using HCT-116 cells [58]. In vivo studies recapitulated this link in adult hippocampal neurogenesis, a Wnt-dependent process. Deletion of neuronal HIF-1α resulted in significant down-regulation of newborn neurons. The defect was reversible upon administration of a pharmacological inhibitor of glycogen synthase kinase-3, a negative regulator of the pathway. Wnt signalling has been shown to positively influence the properties of haematopoietic [75, 76] and intestinal stem cells [76]. Future work will determine whether other Wnt-dependent stem cells are subject to HIF-1α regulation of Wnt/β-catenin signalling.
HIFs, stem cell pathways and disease
Solid tumours frequently harbour areas with compromised circulation because of structurally disorganized blood vessels. Among other phenotypes such as angiogenesis, invasion and metastasis, low O2 levels are associated with more aggressive tumours [77, 78]. For some cancers, there is now evidence that hypoxia induces an aggressive characteristic by causing spontaneous dedifferentiation of tumour cells. It has been recently shown that low O2 levels can promote dedifferentiation and confer stem cell properties in neuroblastoma, a childhood cancer, and breast cancer [79]. Importantly, in both these tumour forms there is a correlation between low differentiation stage and aggressive phenotype. Neuroblastoma is a childhood cancer that originates from the developing sympathetic nervous system and consists of both neurons and neuroendocrine cells. Jogi et al. showed that hypoxia (1–5% O2) decreased the expression of several neuronal/neuroendocrine marker genes, but induced the expression of markers that were associated with neural crest sympathetic progenitors (such as c-Kit and Notch) in cultured neuroblastoma cells [47]. Similar changes in gene expression were also noted in hypoxic regions of neuroblastoma xenografts grown in immunocompromised mice. The dedifferentiation effect of hypoxia is not restricted to neuroblastomas, as loss of differentiation markers and gain of stem cell characteristics were found to occur also in hypoxic ductal breast carcinoma cells [48]. In addition, O2 concentration has been shown to influence the biological effects of Notch-1 signalling in human tumours including adenocarcinoma of the lung [80], and melanoma development [81]. Notch is also a molecular effector of HIF-1α mediated hypoxic epithelial–mesenchymal transition of tumour cells [82], and development of thymic lymphomas [83]. In another interaction, the putative molecular link between HIF-1α and Wnt/β-catenin signalling also may have ramifications in tumour biology. For example, β-catenin has been implicated in the maintenance of cutaneous cancer stem cells [84], and this link may be influenced by local oxygen concentrations of the solid tumour. Taken together, these findings implicate oxygenation levels as an important aspect of microenvironmental niches, which, along with other niche components such as stromal cell contacts, extracellular matrix proteins, growth factors and temperature, may play an important role in influencing stem or tumour cell behaviour.
HIF and cMyc
Hypoxia is a key component of the tumour microenvironment, and plays an important role in the progression of cancer, particularly in solid tumours [85]. HIF-1α and HIF-2α are the key mediators of the hypoxic response in cells. The HIFs regulate cellular responses to hypoxia by modulating numerous pathways, such as angiogenesis, metabolism, translation, and cell growth. In the context of tumour physiology, hypoxia can enhance glycolysis and promote dedifferentiation, invasion, and metastasis. The broad spectrum of activities influenced by the HIFs overlaps with many of the pathways affected by cMyc [86]. This interaction between HIF and cMyc can influence tumorigenesis (Fig. 2). HIF and cMyc interactions have been documented to play a role in colon cancer [87], clear cell renal cancer [57], breast cancer [88], multiple myeloma [89] and skin cancer [90].
Figure 2.
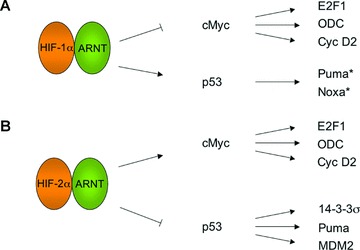
HIF-dependent regulation of hypoxic tumours. A schematic diagram of the molecular links between the HIFs and the cancer associated genes cMyc and p53 is shown. (A) Whereas, HIF-1α interacts with cMyc leading to the inhibition of cMyc activity, HIF-1α effects on p53 result in the activation of p53 activity. (B) HIF-2α interacts with cMyc leading to the activation of cMyc activity. In contrast, HIF-2α opposes p53 activity by promoting intracellular redox homeostasis. The net effect of the interactions of the HIFs with cMyc and p53 is dependent upon the cellular context. *These activities are inferred from the literature.
Initially, it was thought that HIF and cMyc interact indirectly through their overlapping pathways, specifically through their influence on glycolysis [91]. More recently however, it is believed that the interaction might be more direct [57, 87, 92]. Koshiji et al. demonstrated that HIF-1α, even in the absence of hypoxia, can promote cell cycle arrest by countering cMyc in colon cancer cells [87]. Using a HIF-1α construct in which the DNA binding domain was mutated, the authors demonstrated that HIF-1α’s ability to mediate cell cycle arrest is independent of its transcriptional activities. Furthermore, HIF-1α was shown to mediate cell cycle arrest by directly countering cMyc activity, which results in the induction of p21.
HIF-1α inhibition of cMyc can also lead to decreased mitochondrial biogenesis [92]. It has been previously shown that cMyc promotes mitochondrial biogenesis [93]. Upon observing decreased mitochondrial biogenesis in clear cell renal carcinoma cells which constitutively express HIF-1α, Zhang et al. postulated that the decreased mitochondrial biogenesis was most likely because of HIF-1α inhibition of cMyc [92]. Transfecting the cells with a dominant negative HIF-1α resulted in increased mitochondrial biogenesis. Increased expression of cMyc targets upon HIF-1α knockdown confirmed that HIF-1α was acting through cMyc, and that HIF-1α had an inhibitory effect. HIF-1α was found to inhibit cMyc activity either by promoting the transcription of MXI-1, a repressor of cMyc, or by promoting the proteasome-dependent degradation of cMyc.
Whereas HIF-1α appears to counteract cMyc’s effects, HIF-2α promotes cMyc activity, at least in clear cell renal cancer [57]. While HIF-1α and HIF-2α share many common targets like VEGF, they also regulate distinct sets of genes [85]. Furthermore, they are known to have contrasting effects on some targets [94]. Using a clear cell renal cancer cell line that expresses both HIF-1α and HIF-2α, Gordan et al. demonstrated that HIF-1α and HIF-2α have antagonistic effects on cell cycle progression because of their contrasting effects on cMyc [57]. Employing shRNA targeting only one of the HIF-α isoforms at any given time, HIF-1α and HIF-2α was shown to have opposite effects on cell proliferation. RNAi-mediated inhibition of cMyc confirmed HIFs opposing role on cell proliferation as an effect of cMyc.
The relationship between HIF-α and cMyc has great clinical significance. The effects of HIF-1α and HIF-2α on cMyc can be used to classify VHL-deficient clear cell renal carcinoma into two subtypes [95]. VHL-deficient clear cell renal carcinoma can be divided into two groups based on HIF-α expression: one that expresses both HIF-1α and HIF-2α and another that expresses HIF-2α only. The interactions between HIF-α and cMyc define the molecular characteristics of these two groups of tumours. Tumours that express HIF-2α only have increased cMyc activity and thus possess greater proliferative capacity. Because of the antagonistic affects of HIF-1α and HIF-2α on cMyc, tumours that express both HIF-1α and HIF-2α, display neither enhanced nor diminished cMyc activity. Instead, this tumour subgroup shows increased Akt/mTOR and ERK/MAPK activity.
When considering the interaction between HIF and cMyc, the cellular context can become very important [86]. While HIF-1α usually inhibits cMyc, cases have been observed where HIF-1α may promote cMyc activity. It is believed that these discrepancies occur because of the nature of cMyc expression, i.e. whether it is physiological or deregulated. The dosage of cMyc also plays a role in the outcome of its interaction with HIF.
HIF and p53
The interaction between HIF and p53 is also likely to influence tumour development (Fig. 2). An et al. demonstrated that HIF-1α can stabilize wild-type p53 [96]. While hypoxia mimetics like cobalt chloride and desferrioxamine induced p53 in wild type cells, the induction did not occur in cells deficient in HIF-1α activity. Furthermore, HIF-1α interacted with p53 immunoprecipitates from MCF7 breast cancer cells exposed to hypoxia or hypoxia mimetics. It was later discovered that HIF-1α binds p53 via its oxygen-dependent degradation (ODD) domain [97]. The ODD domain of HIF-1α is natively unstructured and threads through p53, tightly binding p53 dimers. While on one hand HIF-1α stabilizes p53, on the other p53 can also promote the degradation of HIF-1α[98]. Wild-type p53 has been suggested to direct the ubiquitination and degradation of HIF-1αvia Mdm2. In many tumours, p53 is mutated and its activity diminished, resulting in HIF-1α stabilization. This in turn leads to greater tumour vascularization during tumorigenesis.
Work by Moeller et al. shed more light on the effect of HIF-1α and p53 interactions [99]. The authors demonstrated that HIF-1α activation of p53 results in increased p53 phosphorylation and p53-induced apoptosis in the presence of γ-radiation. Moreover, γ-radiation of hypoxia-stimulated HCT116 cells resulted in greater levels of apoptosis as measured by caspase 3/7 activation and DNA fragmentation when compared to γ-radiated normoxic HCT116 cells. This increased apoptosis is abrogated by the expression of dominant-negative HIF-α, indicating HIF-1α’s role in the process. The role of p53 in apoptosis was demonstrated by measuring increased p53 phosphorylation levels in radiated/hypoxia treated cells, when compared to the radiated/normoxic cells. In addition, the increased levels of p53 phosphorylation were abolished by HIF-1α inhibition. These results confirm that HIF-1α and p53 interact to bring about increased apoptosis in radiated cells under hypoxic conditions.
Analogous to the situation with cMyc, HIF-1α and HIF-2α appear to have contrary effects on p53 [100]. While HIF-1α promotes the phosphorylation of p53, HIF-2α actually inhibits p53 phosphorylation. Using A498 cells, a renal carcinoma cell line that constitutively express HIF-2α, Bertout et al. demonstrated that HIF-2α inhibits the phosphorylation of p53 upon radiation [100]. Furthermore, in the HIF-2α-deficient cells, radiation-induced p53 phosphorylation resulted in p53 target gene expression and subsequent cell death.
The interactions between HIF-α and p53 have important clinical significance. For instance clear cell renal tumours that express HIF-2α only have markedly less phospho-p53 when compared to tumours that express both HIF-1α and HIF-2α[100]. This decreased phospho-p53 correlated with decreased expression of p53 targets like Puma and 14–3-3σ. Gene expression analysis revealed that because of HIF-2α’s inhibition of the p53 phosphorylation, there is an overall reduction in p53 pathway activity in clear cell renal tumours that express HIF-2α only.
Similar to the case of cMyc, the effects of HIF on p53 can be context-dependent [99,101]. HIF-1α seems to have opposite effects on p53 in endothelial cells when compared to tumour cells.
Summary
Hypoxia plays a key role in normal development, as well as disease progression. The HIFs are the primary mediators of the cellular response to hypoxia. In this review, we examined the effect of the HIFs on multiple pathways. Hypoxia in the stem cell niche promotes stem cell homeostasis by maintaining quiescence and by shielding cells from oxidative damage. These niches also provide other cues that promote stem cell maintenance. Some of these cues include signals from the BMP, Notch, Wnt, JAK-STAT, Oct4 and Sonic hedgehog pathways. The HIFs can interact with many of these pathways and thus play a role in regulating stem cell proliferation, differentiation and pluripotency.
The HIFs also play a critical role in many disease conditions, including stroke, tissue ischaemia, inflammation and the growth of solid tumours. The role of the HIFs in cancer progression has always been appreciated because of their ability to promote angiogenesis. However, it is becoming more evident that this is not the only means by which the HIFs affect tumour growth. It is now clear that the HIFs interact with many oncogenic pathways like cMyc and p53 to modulate cancer progression. What is even more interesting is that HIF-1α and HIF-2α have antagonistic effects on cMyc and p53. Much more work is needed before we fully understand the complex role that hypoxia and the HIFs play in these settings.
Acknowledgments
We thank Simon lab members for useful discussions. This work was supported by the Abramson Family Cancer Research Institute, and the Howard Hughes Medical Institute. We apologize to any colleagues whose work was not cited because of space constraints.
References
- 1.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–75. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morriss GM, New DA. Effect of oxygen concentration on morphogenesis of cranial neural folds and neural crest in cultured rat embryos. J Embryol Exp Morphol. 1979;54:17–35. [PubMed] [Google Scholar]
- 3.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell JA, Yochim JM. Intrauterine oxygen tension during the estrous cycle in the rat: its relation to uterine respiration and vascular activity. Endocrinology. 1968;83:701–5. doi: 10.1210/endo-83-4-701. [DOI] [PubMed] [Google Scholar]
- 5.Rodesch F, Simon P, Donner C, et al. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80:283–5. [PubMed] [Google Scholar]
- 6.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–15. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–62. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maltepe E, Schmidt JV, Baunoch D, et al. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–7. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 9.Peng J, Zhang L, Drysdale L, et al. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci USA. 2000;97:8386–91. doi: 10.1073/pnas.140087397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Compernolle V, Brusselmans K, Acker T, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–10. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 11.Scortegagna M, Ding K, Oktay Y, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet. 2003;35:331–40. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 12.Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 13.Studer L, Csete M, Lee SH, et al. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20:7377–83. doi: 10.1523/JNEUROSCI.20-19-07377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison SJ, Csete M, Groves AK, et al. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci. 2000;20:7370–6. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schipani E, Ryan HE, Didrickson S, et al. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–76. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yun Z, Maecker HL, Johnson RS, et al. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell. 2002;2:331–41. doi: 10.1016/s1534-5807(02)00131-4. [DOI] [PubMed] [Google Scholar]
- 17.Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25. [PubMed] [Google Scholar]
- 18.Song X, Zhu CH, Doan C, et al. Germline stem cells anchored by adherens junctions in the Drosophila ovary niches. Science. 2002;296:1855–7. doi: 10.1126/science.1069871. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita YM, Jones DL, Fuller MT. Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science. 2003;301:1547–50. doi: 10.1126/science.1087795. [DOI] [PubMed] [Google Scholar]
- 20.Yamashita YM, Mahowald AP, Perlin JR, et al. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science. 2007;315:518–21. doi: 10.1126/science.1134910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein B, Hird SN. Specification of the anteroposterior axis in Caenorhabditis elegans. Development. 1996;122:1467–74. doi: 10.1242/dev.122.5.1467. [DOI] [PubMed] [Google Scholar]
- 22.Siegrist SE, Doe CQ. Extrinsic cues orient the cell division axis in Drosophila embryonic neuroblasts. Development. 2006;133:529–36. doi: 10.1242/dev.02211. [DOI] [PubMed] [Google Scholar]
- 23.Seydoux G, Braun RE. Pathway to totipotency: lessons from germ cells. Cell. 2006;127:891–904. doi: 10.1016/j.cell.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 24.Ghazizadeh S, Taichman LB. Multiple classes of stem cells in cutaneous epithelium: a lineage analysis of adult mouse skin. EMBO J. 2001;20:1215–22. doi: 10.1093/emboj/20.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cotsarelis G. Epithelial stem cells: a folliculocentric view. J Invest Dermatol. 2006;126:1459–68. doi: 10.1038/sj.jid.5700376. [DOI] [PubMed] [Google Scholar]
- 26.Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22:339–73. doi: 10.1146/annurev.cellbio.22.010305.104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol. 2004;20:695–723. doi: 10.1146/annurev.cellbio.20.010403.092805. [DOI] [PubMed] [Google Scholar]
- 28.Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006;7:333–7. doi: 10.1038/ni1331. [DOI] [PubMed] [Google Scholar]
- 29.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim DA, Huang YC, Alvarez-Buylla A. The adult neural stem cell niche: lessons for future neural cell replacement strategies. Neurosurg Clin N Am. 2007;18:81–92. doi: 10.1016/j.nec.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Merkle FT, Alvarez-Buylla A. Neural stem cells in mammalian development. Curr Opin Cell Biol. 2006;18:704–9. doi: 10.1016/j.ceb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009;15:701–6. doi: 10.1038/nm.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–17. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 35.Fre S, Vignjevic D, Schoumacher M, et al. Epithelial morphogenesis and intestinal cancer: new insights in signaling mechanisms. Adv Cancer Res. 2008;100:85–111. doi: 10.1016/S0065-230X(08)00003-1. [DOI] [PubMed] [Google Scholar]
- 36.Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82:2031–7. [PubMed] [Google Scholar]
- 37.Kiel MJ, Yilmaz OH, Iwashita T, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 38.Kiel MJ, Yilmaz OH, Morrison SJ. CD150- cells are transiently reconstituting multipotent progenitors with little or no stem cell activity. Blood. 2008;111:4413–4. doi: 10.1182/blood-2007-12-129601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.BI L. The architecture of bone marrow cell populations. OH; 1992. Dayton: [DOI] [PubMed] [Google Scholar]
- 40.Chow DC, Wenning LA, Miller WM, et al. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J. 2001;81:685–96. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parmar K, Mauch P, Vergilio JA, et al. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA. 2007;104:5431–6. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Danet GH, Pan Y, Luongo JL, et al. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. 2003;112:126–35. doi: 10.1172/JCI17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adelman DM, Maltepe E, Simon MC. Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev. 1999;13:2478–83. doi: 10.1101/gad.13.19.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramirez-Bergeron DL, Simon MC. Hypoxia-inducible factor and the development of stem cells of the cardiovascular system. Stem Cells. 2001;19:279–86. doi: 10.1634/stemcells.19-4-279. [DOI] [PubMed] [Google Scholar]
- 45.Genbacev O, Zhou Y, Ludlow JW, et al. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–72. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 46.Koay EJ, Athanasiou KA. Hypoxic chondrogenic differentiation of human embryonic stem cells enhances cartilage protein synthesis and biomechanical functionality. Osteoarthritis Cartilage. 2008;16:1450–6. doi: 10.1016/j.joca.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Jogi A, Ora I, Nilsson H, et al. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci USA. 2002;99:7021–6. doi: 10.1073/pnas.102660199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helczynska K, Kronblad A, Jogi A, et al. Hypoxia promotes a dedifferentiated phenotype in ductal breast carcinoma in situ. Cancer Res. 2003;63:1441–4. [PubMed] [Google Scholar]
- 49.Caniggia I, Mostachfi H, Winter J, et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3) J Clin Invest. 2000;105:577–87. doi: 10.1172/JCI8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cowden Dahl KD, Fryer BH, Mack FA, et al. Hypoxia-inducible factors 1alpha and 2alpha regulate trophoblast differentiation. Mol Cell Biol. 2005;25:10479–91. doi: 10.1128/MCB.25.23.10479-10491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adelman DM, Gertsenstein M, Nagy A, et al. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000;14:3191–203. doi: 10.1101/gad.853700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowden Dahl KD, Robertson SE, Weaver VM, et al. Hypoxia-inducible factor regulates alphavbeta3 integrin cell surface expression. Mol Biol Cell. 2005;16:1901–12. doi: 10.1091/mbc.E04-12-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–72. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu CJ, Wang LY, Chodosh LA, et al. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–74. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gustafsson MV, Zheng X, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–28. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 56.Covello KL, Kehler J, Yu H, et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–70. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gordan JD, Bertout JA, Hu CJ, et al. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9:210–7. doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 59.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 60.Dahlqvist C, Blokzijl A, Chapman G, et al. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development. 2003;130:6089–99. doi: 10.1242/dev.00834. [DOI] [PubMed] [Google Scholar]
- 61.Blokzijl A, Dahlqvist C, Reissmann E, et al. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163:723–8. doi: 10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson A, Radtke F. Multiple functions of Notch signaling in self-renewing organs and cancer. FEBS Lett. 2006;580:2860–8. doi: 10.1016/j.febslet.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 63.Pear WS, Aster JC. T cell acute lymphoblastic leukemia/lymphoma: a human cancer commonly associated with aberrant NOTCH1 signaling. Curr Opin Hematol. 2004;11:426–33. doi: 10.1097/01.moh.0000143965.90813.70. [DOI] [PubMed] [Google Scholar]
- 64.Morrison SJ, Perez SE, Qiao Z, et al. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell. 2000;101:499–510. doi: 10.1016/s0092-8674(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 65.Taylor MK, Yeager K, Morrison SJ. Physiological Notch signaling promotes gliogenesis in the developing peripheral and central nervous systems. Development. 2007;134:2435–47. doi: 10.1242/dev.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harvey AJ, Kind KL, Pantaleon M, et al. Oxygen-regulated gene expression in bovine blastocysts. Biol Reprod. 2004;71:1108–19. doi: 10.1095/biolreprod.104.028639. [DOI] [PubMed] [Google Scholar]
- 67.Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA. 2005;102:4783–8. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Covello KL, Simon MC, Keith B. Targeted replacement of hypoxia-inducible factor-1alpha by a hypoxia-inducible factor-2alpha knock-in allele promotes tumor growth. Cancer Res. 2005;65:2277–86. doi: 10.1158/0008-5472.CAN-04-3246. [DOI] [PubMed] [Google Scholar]
- 69.Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loh YH, Wu Q, Chew JL, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–40. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 71.Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–6. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 72.Tai MH, Chang CC, Kiupel M, et al. Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis. 2005;26:495–502. doi: 10.1093/carcin/bgh321. [DOI] [PubMed] [Google Scholar]
- 73.Kehler J, Tolkunova E, Koschorz B, et al. Oct4 is required for primordial germ cell survival. EMBO Rep. 2004;5:1078–83. doi: 10.1038/sj.embor.7400279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 75.Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 76.De Lau W, Barker N, Clevers H. WNT signaling in the normal intestine and colorectal cancer. Front Biosci. 2007;12:471–91. doi: 10.2741/2076. [DOI] [PubMed] [Google Scholar]
- 77.Vaupel P. Oxygen transport in tumors: characteristics and clinical implications. Adv Exp Med Biol. 1996;388:341–51. [PubMed] [Google Scholar]
- 78.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–84. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 79.Axelson H, Fredlund E, Ovenberger M, et al. Hypoxia-induced dedifferentiation of tumor cells–a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol. 2005;16:554–63. doi: 10.1016/j.semcdb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 80.Chen Y, De Marco MA, Graziani I, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67:7954–9. doi: 10.1158/0008-5472.CAN-07-1229. [DOI] [PubMed] [Google Scholar]
- 81.Bedogni B, Warneke JA, Nickoloff BJ, et al. Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest. 2008;118:3660–70. doi: 10.1172/JCI36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sahlgren C, Gustafsson MV, Jin S, et al. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. 2008;105:6392–7. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bertout JA, Patel SA, Fryer BH, et al. Heterozygosity for hypoxia inducible factor 1alpha decreases the incidence of thymic lymphomas in a p53 mutant mouse model. Cancer Res. 2009;69:3213–20. doi: 10.1158/0008-5472.CAN-08-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Malanchi I, Peinado H, Kassen D, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008;452:650–3. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 85.Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–7. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dang CV, Kim JW, Gao P, et al. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–6. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 87.Koshiji M, Kageyama Y, Pete EA, et al. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–56. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Robey IF, Stephen RM, Brown KS, et al. Regulation of the Warburg effect in early-passage breast cancer cells. Neoplasia. 2008;10:745–56. doi: 10.1593/neo.07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang J, Sattler M, Tonon G, et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res. 2009;69:5082–90. doi: 10.1158/0008-5472.CAN-08-4603. [DOI] [PubMed] [Google Scholar]
- 90.Scortegagna M, Martin RJ, Kladney RD, et al. Hypoxia-inducible factor-1alpha suppresses squamous carcinogenic progression and epithelial-mesenchymal transition. Cancer Res. 2009;69:2638–46. doi: 10.1158/0008-5472.CAN-08-3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dang CV, Lewis BC, Dolde C, et al. Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. J Bioenerg Biomembr. 1997;29:345–54. doi: 10.1023/a:1022446730452. [DOI] [PubMed] [Google Scholar]
- 92.Zhang H, Gao P, Fukuda R, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–20. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 93.Li F, Wang Y, Zeller KI, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–34. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–46. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.An WG, Kanekal M, Simon MC, et al. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392:405–8. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 97.Sanchez-Puig N, Veprintsev DB, Fersht AR. Binding of natively unfolded HIF-1alpha ODD domain to p53. Mol Cell. 2005;17:11–21. doi: 10.1016/j.molcel.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 98.Ravi R, Mookerjee B, Bhujwalla ZM, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 99.Moeller BJ, Dreher MR, Rabbani ZN, et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005;8:99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 100.Bertout JA, Majmundar AJ, Gordan JD, et al. HIF2{alpha} inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci USA. 2009;106:14391–6. doi: 10.1073/pnas.0907357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moeller BJ, Cao Y, Li CY, et al. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–41. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]