


Abstract
In April 2009, a new influenza A (H1N1 2009) virus emerged that rapidly spread around the world. While current variants of this virus have caused widespread disease, particularly in vulnerable groups, there remains the possibility that future variants may cause increased virulence, drug resistance or vaccine escape. Early detection of these virus variants may offer the chance for increased containment and potentially prevention of the virus spread. We have developed and field-tested a resequencing kit that is capable of interrogating all eight segments of the 2009 influenza A(H1N1) virus genome and its variants, with added focus on critical regions such as drug-binding sites, structural components and mutation hotspots. The accompanying base-calling software (EvolSTAR) introduces novel methods that utilize neighbourhood hybridization intensity profiles and substitution bias of probes on the microarray for mutation confirmation and recovery of ambiguous base queries. Our results demonstrate that EvolSTAR is highly accurate and has a much improved call rate. The high throughput and short turn-around time from sample to sequence and analysis results (30 h for 24 samples) makes this kit an efficient large-scale evolutionary biosurveillance tool.
BACKGROUND
While the current influenza A H1N1 2009 virus is known to be sensitive to neuraminidase-inhibitor chemoprophylaxis, it has limited diversity at neutralizing antibody binding sites and overall mortality rates comparable with seasonal influenza (1); variations at numerous sites within the influenza A genome are predicted to alter these characteristics, with potentially important consequences for healthcare provision.
In order to enable large-scale identification of variations of H1N1(2009) viruses from multiple patient samples, it is necessary to develop a low-cost method for rapidly whole-genome sequencing the H1N1 samples. Historically, sequencing of viral genomes is performed using standard dye termination technologies. These conventional sequencing technologies produce accurate data but are too slow, costly and labour-intensive to be practical for large-scale epidemiologic or evolutionary investigations in viral outbreaks. Oligonucleotide resequencing microarrays that are capable of identifying nucleotide sequence variants may offer an alternative solution (2,3) and in recent years, have been used for detecting and subtyping influenza viruses (4,5). By analysing sequences generated from tiling probes across targeted regions of various strains of the influenza virus [e.g. partial fragments of the haemagglutinin (HA) and neuraminidase (NA) genes], important information such as viral subtypes, lineages and sequence variants can be determined. Apart from influenza, resequencing microarrays have also been used to obtain whole-genome primary sequences for orthopoxviruses (6), biothreat viruses (7) and SARS (8). The reported studies mainly use platform accompanying software that employs probabilistic base-calling algorithms such as ABACUS (3) and Nimblescan PBC (8). Although statistically sound, these methods are susceptible to hybridization noise caused by factors such as poor probe quality, poor amplification or mutations. This results in numerous ambiguous and false positive base calls that may affect the accuracy of downstream evolutionary analysis. Efforts have been made to improve the call rates and accuracies of existing probabilistic base-calling algorithms. For example, Model-P uses probe and sequence features to build intensity-prediction models that compute maximum likelihood scores for base-calling (9). Another approach filters low-confidence base calls from problematic regions (e.g. regions with high mutation rates or repeats), thereby reducing the number of false-positive base calls (10). Depending on the stringencies of the filters used, call rates may suffer as a result.
To address if these arrays can be used as a practical, large-scale re-sequencing tool, we have developed a system comprising customized sequence amplification primers, a 12-plex DNA resequencing array and an automated base-calling and variant analysis software (EvolSTAR). We demonstrate that the sequences obtained from the array are highly reproducible with ≥99.99% accuracy and 99.02 ± 0.82% genome coverage. The short turnaround time from sample to sequence and analysis results (∼30 h for 24 samples) makes this kit an efficient large-scale evolutionary surveillance tool.
This article describes the development of the various genetic analysis components, and their validation using clinical samples. Accession numbers for 84 complete H1N1(2009) genomes generated are listed in Supplementary Data File 1.
MATERIALS AND METHODS
RNA isolation and amplification of patient isolates
Viral RNA from the diagnostic swabs or RNA extracted from MDCK cell cultures was extracted using the DNA minikit (Qiagen, Inc, Valencia, CA, USA) according to manufacturer’s instructions. RNA was reverse-transcribed to cDNA using customized random primers designed using LOMA (11) and then amplified by PCR using proprietary H1N1(2009) specific primers. The presence of H1N1(2009) in the samples was confirmed using a separate real-time PCR assay based on the published primer sequences from the Centre for Disease Control and Prevention (CDC), USA.
Design of probes in mutation hotspots
We found 36 mutation hotspots in the alignments where mutations occurred near one another (within 20 bp). A perfect match (PM) probe residing in a mutation hotspot may contain mismatches that will have a detrimental effect on its hybridization intensity. To avoid this problem, we designed additional PM probes that contain all possible combinations of mutations found in each mutation hotspot. Thus, if two mutations are found within 20 bp of each other in the alignments, then we need in total four (22) PM probes to encode them. In general, 2x PM probes are needed to completely encode a cluster of x mutations that occur within 20 bp of one another in the alignments.
Determination of neighbourhood hybridization intensity profile types
We have identified five distinct types of neighbourhood hybridization intensity profile belonging to true non-mutations (wild-type), true mutations, isolated errors/‘N’s, long consecutive errors/‘N’s, and unknown errors/‘N’s, respectively. For each non-high-confidence query base, we determine the type of its neighbourhood hybridization signal intensity profile (NHIP) by the following criteria:
True-non-mutation—The PM probe (of both strands) of the query base must be a high-confidence call [i.e. it has hybridization intensity ≥ 1.4-fold that of its mismatch (MM) probes]. Neighbourhood PM probes are also high-confidence calls. Let the mean hybridization intensity of the three nearest PM probes to the immediate left of the mutation base (at position −1, −2 and −3), denoted as μ{−1,−2,−3}, the mean hybridization intensity of the three PM probes to the far left of the mutation base (at position −4, −5 and −6), denoted as μ{−4,−5,−6}, the mean hybridization intensity of the three nearest PM probes to the immediate right of the mutation base (at position 1, 2 and 3), denoted as μ{1,2,3}, and the mean hybridization intensity of the three PM probes to the far right of the mutation base (at position 4, 5 and 6), denoted as μ{4,5,6}. We impose that μ{−1,−2,−3} ≈ μ{−4,−5,−6} and μ{1,2,3} ≈ μ{4,5,6}
True-mutation—The PM probe (of both strands) of the query base must have hybridization intensity ≥ 1.4-fold that of its MM probes. To detect the characteristic dip, we check four mean hybridization intensities: the mean hybridization intensity of the three nearest PM probes to the immediate left of the mutation base (at position −1, −2 and −3), denoted as μ{−1,−2,−3}, the mean hybridization intensity of the three PM probes to the far left of the mutation base (at position −4, −5 and −6), denoted as μ{−4,−5,−6}, the mean hybridization intensity of the three nearest PM probes to the immediate right of the mutation base (at position 1, 2 and 3), denoted as μ{1,2,3}, and the mean hybridization intensity of the three PM probes to the far right of the mutation base (at position 4, 5 and 6), denoted as μ{4,5,6}. If μ{−1,−2,−3} < μ{−4,−5,−6} and μ{1,2,3} < μ{4,5,6}, we say this is a dip pattern and the query base is likely to be mutated.
Isolated error/‘N’—The PM probe (of both strands) of the query base has hybridization intensity < 1.4-fold that of its MM probes. Neighbourhood PM probes are high-confidence calls.
Long consecutive errors/‘N’s—The PM probe (of both strands) of the query base has hybridization intensity < 1.4-fold that of its MM probes. A majority of neighbourhood PM probes are non-high-confidence calls.
Unknown error/‘N’—All other neighbourhood hybridization profile patterns that do not fall under the previous categories.
Computing the likelihood in nucleotide substitution bias analysis
We define that a probe encodes the base b if b is located in the centre-most position of the probe and is the base to be interrogated. For a given query base, suppose the PM probe encodes b1 while the MM probes encode b2, b3 and b4, respectively, where {b1, b2, b3, b4} = {A, C, G, T} and the hybridization intensity reduction order is b1b2b3b4. To validate if the observed PM probe encoding b1 is indeed the true PM probe of the sample sequence, we compute the likelihood ratio of fobs and frand, where fobs is probability of observing the hybridization intensity reduction order b1b2b3b4 given that the PM probe encodes b1 and frand is the probability of observing the hybridization intensity reduction order b1b2b3b4 by chance. Precisely,
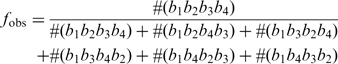 |
and
where #(wxyz) is the number of observed hybridization intensity reduction orders from high-confidence base-calls and t is the total number of hybridization intensity reduction orders excluding b1b2b3b4 obtained from high-confidence base-calls. If the likelihood ratio >2, we expect that the observed PM probe encoding b1 is indeed the true PM probe of the sample sequence.
EvolSTAR two-step process
EvolSTAR employs a two-step process for base-calling (Figure 1). In the first step, each base query is scrutinized for signs of hybridization intensity abnormalities. If the gain-of-signal of the query base is strong and has no mutation, the base is called. In the second step, EvolSTAR then tries to recover base queries that have any hybridization intensity abnormalities with two analysis methods, namely neighbourhood hybridization intensity profile analysis and nucleotide substitution bias analysis.
Figure 1.

FlowChart of EvolSTAR. FlowChart of EvolSTAR. Bold arrows are ‘Yes’ paths, while normal arrows are ‘No’ paths. In the first step, each base query is scrutinized for signs of hybridization intensity abnormalities. Base queries with hybridization intensity abnormalities are passed to step 2 for further analysis.
Step 1: Identification of base queries with ambiguity
On our array platform, the hybridization intensity of each probe is given by the mean and standard deviation of the fluorescence intensities of nine individually scanned pixels. Hence, we define the signal-to-noise ratio (SNR) of a probe as the ratio of the mean to the standard deviation of the intensities of the nine pixels associated with the probe. In our experiments, we found that >95% of all probes had SNR less than TSNR (TSNR = μSNR + 2σSNR, where μSNR and σSNR are the mean and standard deviation of SNR of all probes on the array). The remaining 5% of probes with SNR ≥ TSNR are unreliable. Hence, base queries with one or more probes with SNR ≥ TSNR are analysed further in step 2. Furthermore, all base queries whose PM probe in the forward strand and PM probe in the reverse strand are non-complementary, or have weak PM/MM hybridization intensity differentiation (<1.4-fold) are also passed to step 2. Lastly, we also pass all putative mutation calls to step 2 for confirmation.
Step 2: Mutation confirmation and base query recovery
A high-confidence mutation call may be a result of coincidental non-specific hybridization of the same MM probe in both strands. As such, it may be inadequate to discern true mutations based solely on differences in the hybridization intensities of PM and MM probes. From our analysis of true-mutation calls made by PBC, we have found that true mutations have a signature NHIP type as per described in Figure 2b. Thus, query bases that result in a mutation call must have this signature NHIP. Finally, to confirm the mutation, we perform nucleotide substitution bias analysis on these query bases. For each of the query bases with NHIP of type described in Figure 2b, we compute the likelihood ℓ that the observed PM probe (representing the mutation) is indeed the true PM probe of the sample sequence given the hybridization intensity-based ordering of its MM probes (see ‘Materials and methods’ section). If ℓ > 2, the query base results in a strong mutation call (represented by upper case base calls ‘A’, ‘C’, ‘G’ or ‘T’). If ℓ > 1, the query base results in a mutation call with weak support (represented by lower case base calls ‘a’, ‘c’, ‘g’ or ‘t’). Otherwise, they are re-assigned an unknown ‘N’ call.
Figure 2.

Summary of characteristics of neighbourhood hybridization intensity profiles for different type of calls. Summary of the characteristics of the NHIP for five types of call: (a) true-non-mutation, (b) true-mutation, (c) isolated error or ‘N’, (d) long chains of consecutive errors or ‘N’, (e) unknown error or ‘N’) based on their respective observed neighbourhood hybridization intensity profiles. The PM probe (red circle) of query base is at position 0 while neighbourhood PM probes (black circles) are numbered according to their distance away from the query base. A PM probe is significantly differentiated from its MM probes if its hybridization intensity is at least t-fold that of all its MM probes.
For query bases that results in a mutation call but have NHIP of type described in Figure 2c, they are most likely isolated errors caused by poor PM probe quality. Hence, we correct the base-calls of these query bases to their respective reference bases (but represented by lower case base calls ‘a’, ‘c’, ‘g’ or ‘t’) in the reference sequences. We also perform the same correction to non-high-confidence query bases with NHIP of type described in Figure 2c.
We try to recover the remaining query bases that have NHIP of type described in Figure 2d or 2e by analysing the substitution bias from their PM and MM probes in the forward and reverse strands separately. Similar to how a mutation is confirmed, we compute the likelihood ℓf that the observed PM probe (representing the unsure base call) is indeed the true PM probe of the sample sequence given the hybridization intensity-based ordering of its MM probes in the forward strand. We also compute a similar likelihood ℓr for the PM probe in the reverse strand. If the PM probes in both strands are complementary and ℓf, ℓr > 2, the query base results in a strong base call (represented by upper case base calls ‘A’, ‘C’, ‘G’ or ‘T’). However, in many cases, the PM probes in both strands are not complementary due to non-specific hybridization of MM probes in one or both strands. For such query bases, we make base calls based on ℓf and ℓr: if ℓf > ℓr and ℓf > 2, a base call with weak support (represented by lower case base calls ‘a’, ‘c’, ‘g’ or ‘t’) is made from the PM probe in the forward strand. Else, if ℓr > ℓf and ℓr > 2, a base call with weak support is made from the PM probe in the reverse strand. Otherwise, they are assigned an unknown ‘N’ call.
Note that since nucleotide substitution biases may vary depending on the experimental conditions, experimental reagents or input samples, for each experiment, we obtain a set of high-confidence base-calls and use them to infer the hybridization intensity reduction orders for each PM probe encoding. This is then used to compute likelihood scores for base-calling non-high-confidence query bases and mutation confirmation.
RESULTS
Design of resequencing array
We generated a consensus sequence for each segment of the H1N1(2009) virus by aligning all 1715 complete and partial sequences available from the NCBI H1N1 flu resources database (http://www.ncbi.nlm.nih.gov/genomes/FLU/SwineFlu.html) as of 11 June 2009 using MAFFT (12) with high-accuracy option.
Tiling probes spanning the entire genome segments on both the forward and reverse strands were created at one base resolution (8). Analysis of the sequence alignments revealed that there were no deletions, insertions or recombination. However, we found 36 mutation hotspots in the alignments where mutations occurred near one another (within 20 bp). Thus, we added additional probes that represent all possible combinations of mutations in these mutation hotspots onto the array. To ensure that we have accurate sequence of the drug binding pocket targeted by NA inhibitors (13) such as oseltamivir (Tamiflu®) and zanamivir (Relenza®) in the NA gene of the H1N1(2009) virus, additional probes were added. In total, the array contains 8236 control probes and 121 928 H1N1(2009) probes, which provides 2 × coverage of the entire H1N1(2009) genome, and up to 8 × coverage of the regions comprising the 36 mutation hotspots and 10 drug-binding sites (Supplementary Data File 2).
Optimization of RT–PCR primers and conditions
Due to the small amount of virus present in samples relative to human or cell-line total RNA, it was necessary to amplify the viral RNA through PCR. We employed a combination of sequence-specific and random PCR approaches using LOMA-optimized primers as previously described (11). The addition of random primers ensured complete genome amplification, even if mutations were present at the specific-primer binding sites. PCR conditions were optimized by conducting five duplicate hybridizations of the same virus sample cultured from a patient sample under different PCR conditions. The optimized method was then tested on RNA isolated directly from nasal swabs obtained from the same patient and from virus grown in cell culture. Microarray sequences generated from these replicate experiments were compared with capillary sequencing to estimate sequencing accuracy.
Algorithm for sequence determination
Following PCR product labelling, hybridization and scanning, signal intensities for each probe was generated using Genepix 4.0 software, and annotated using Nimblescan 2.5 software. Initially, the standard Nimblescan software which employs a gain-of-signal approach [PBC algorithm (8)], was used to determine the viral sequence. The PBC algorithm assumes that the signal intensity of the PM probe (which matches exactly to the sequence in the sample) will be significantly higher than that of the MM probes. While this approach sufficed for ∼90% of base queries, we observed that the discrimination between the PM and MM signals was not clear for the remaining probes.
These ambiguous signals were caused by the presence of multiple mutations in the probe sequence, homopolymers and hybridization artefacts. We developed a novel algorithm, Evolution Surveillance and Tracking Algorithm for Resequencing arrays (EvolSTAR), to resolve this problem. EvolSTAR improves upon PBC by adding an analysis of the NHIP and nucleotide substitution bias (described below).
NHIP
Due to the use of tiling probes in resequencing arrays, a single nucleotide mutation at a particular query base could cause a dramatic reduction in the hybridization intensities of neighbouring PM probes up to six bases away (14). This effect can be measured by studying the NHIP of each query base. We defined the NHIP of each query base as the observed pattern of hybridization intensities of its PM and MM probes and neighbouring (±6 bases from query base) PM and MM probes. To study the effects of sequence variation (mutation) and noise on the NHIP of a query base, we sequenced RNA from H1N1(2009) patient 380 by capillary sequencing and on duplicate microarrays. We compared sequence calls generated using by Nimblescan or by capillary sequencing and compiled a list of true (correct) calls, error calls and ‘N’ (unknown) calls. In total, of the expected 13 588 bases of the H1N1 virus (based on genome described at http://www.ncbi.nlm.nih.gov/genomes/taxg.cgi?tax=211044) the microarray called 13 449 bases while capillary sequence was able to call 12 832 bases.
Figure 3 shows the NHIPs of a representative set of 40 randomly selected query bases that result in true-non-mutation calls (wild-type calls). We observed that in these NHIPs, the PM probe of the query base together with neighbouring PM probes, have hybridization intensities significantly higher (>1.4-fold) than that of their MM probes in general. We also identified 10 mutations using capillary sequencing in the patient sample. The NHIPs of these 10 true-mutation calls (Figure 4) are very different from NHIPs of wild-type calls. The presence of a mutation at the query base created a MM in neighbouring PM probes and caused a drop in their hybridization intensities. The closer this mutation is to the centre of a neighbouring PM probe, the bigger the drop in hybridization intensity. This results in a distinctive dip to the immediate left and right of the centre of the NHIP where the mutation is.
Figure 3.

Observed neighbourhood hybridization intensity profiles for true-non-mutation calls. A representative set of observed NHIPs for true-non-mutation calls from patient sample 380. This representative set consists of five true-non-mutation calls randomly selected from each segment. Each line represents the NHIP (±6 bp from query base position) of a true-non-mutation call.
Figure 4.

Observed neighbourhood hybridization intensity profiles for true-mutation calls. The observed NHIPs for all 10 identified true-mutation calls from patient sample 380.
Unlike the NHIPs of wildtype and true-mutation calls, the NHIPs of most errors and ‘N’ calls appear haphazard (Figure 5). However, when we traced the locations of these errors and ‘N’ calls on the genome, we found that some are isolated among good calls while others are conjugated in a small locality of the genome. We investigated the NHIPs of isolated errors and ‘N’ calls that occurred among good calls and found that in these NHIPs, only the PM probe of the query base that is an error or ‘N’ call has poor hybridization differentiation with its MM probes while other PM probes have hybridization intensities significantly higher than that of their MM probes in general (Figure 6). This suggests that for such calls, only the PM and MM probes of the query base are noisy while neighbouring PM and MM probes are unaffected. In addition, we also found that long chains of consecutive error and ‘N’ calls (especially at the 5′- and 3′-end of the sample sequences) often have NHIPs where the PM probe of the query base together with neighbouring PM probes, have poor hybridization differentiation with their MM probes (Figure 7). These error and ‘N’ calls usually occur at the ends of the genome segments. In summary, NHIP analysis showed that all true mutation calls had a characteristic profile (Figure 2b) that differed from wild-type sequence calls (Figure 2a). Ambiguous calls arising from different causes, such as homopolymers, isolated errors and hybridization artifacts also have profiles that are distinct from true mutation profiles (Figure 2). See ‘Materials and Methods’ section for details.
Figure 5.

Observed neighbourhood hybridization intensity profiles for unknown error/‘N’ calls. A representative set of observed NHIPs for unknown error/‘N’ calls from patient sample 380. This representative set consists of two unknown error/‘N’ calls randomly selected from each segment.
Figure 6.

Observed neighbourhood hybridization intensity profiles for isolated error/‘N’ calls. The observed NHIPs for all three identified isolated error/‘N’ calls from patient sample 380. These errors are flanked by true (correct) calls.
Figure 7.

Observed neighbourhood hybridization intensity profiles for long consecutive error/‘N’ calls. The observed NHIPs for five regions where there are long consecutive (≥5) error/‘N’ calls from patient sample 380.
Nucleotide substitution bias
The presence of nucleotide substitution bias in Nimblegen resequencing arrays has been previously described (15). However, this knowledge has so far been used only to improve probe design. In this article, we propose a novel method that makes use of nucleotide substitution bias in the array to improve base-calling accuracy and call rate. The key idea is to build a likelihood model of the substitution bias among the probes of non-ambiguous calls on the array; then use this to call bases with ambiguous signals.
To build the likelihood model, we first determined the substitution bias on our platform by comparing the PM and MM probes (of both strands) of 25 028 true calls made by PBC from the two replicate microarray experiments of patient sample 380 mentioned in the previous section. For each true call, we generated a hybridization intensity reduction order by ranking the PM and MM probes of a particular strand in decreasing order of hybridization intensity and recording their respective frequencies (Table 1). Table 1 shows that for each PM probe encoding, certain hybridization intensity reduction orders occur much more frequently than others. For example, if the PM probe encoding is ‘A’ (regardless of strand), then it is most likely that the hybridization intensity reduction order is ‘TGC’ or ‘GTC’. Thus, by matching the hybridization intensity reduction orders of its PM/MM probes with that in Table 1, we can compute the likelihood that the putative base call for a query base with ambiguous signals is correct (see ‘Materials and Methods’ section). In this way, we can recover base calls of ambiguous query bases exceeding a reasonably high likelihood threshold and achieve better accuracy and call rate than PBC.
Table 1.
Hybridization intensity reduction orders found in two replicated hybridization experiments of patient sample 380
| PM probe encoding | Hybridization intensity reduction order | Forward strand | Reverse strand |
|---|---|---|---|
| Frequency | Frequency | ||
| A | CGT | 547 | 246 |
| CTG | 558 | 237 | |
| GCT | 957 | 367 | |
| GTC | 2215 | 1407 | |
| TCG | 1049 | 611 | |
| TGC | 3015 | 2873 | |
| C | AGT | 2035 | 2712 |
| ATG | 1752 | 2400 | |
| GAT | 382 | 341 | |
| GTA | 159 | 134 | |
| TAG | 360 | 377 | |
| TGA | 165 | 129 | |
| G | ACT | 1474 | 1043 |
| ATC | 976 | 624 | |
| CAT | 1639 | 1534 | |
| CTA | 868 | 788 | |
| TAC | 594 | 410 | |
| TCA | 542 | 454 | |
| T | ACG | 432 | 529 |
| AGC | 562 | 636 | |
| CAG | 623 | 841 | |
| CGA | 1066 | 1616 | |
| GAC | 1421 | 1878 | |
| GCA | 1637 | 2841 |
Hybridization intensity reduction orders found in 25 028 true calls from two replicated hybridization experiments of patient sample 380. For each true call, for each strand, we rank the PM probe and its MM probes based on their hybridization intensities in decreasing order. We count the frequency of each hybridization intensity reduction order.
Grading the quality of the sequence calls
EvolSTAR employs a two-step process for base-calling (details in ‘Materials and methods’ section). First, it determines if the gain-of-signal from the PM probe is strong. If strong, then the base sequence is called and annotated as ‘high quality’. If the gain-of-signal is ambiguous, or if the base called is different from the expected sequence, then NHIP and nucleotide substitution bias is deployed to verify the sequence call. From empirical experiments and comparison with capillary sequence data, we observed that high quality sequence calls are made when the signal from the PM probe for both forward and reverse strands are at least 40% higher than that of the MM probes.
The second-step processes the ambiguous bases. For bases confirmed to be mutations through NHIP analysis and satisfy the substitution bias rule, they are graded as high quality sequence calls and denoted in the FASTA sequence as UPPER CASE characters. For bases confirmed to be an isolated error through NHIP analysis and also satisfy the substitution bias rule, they are graded as low confidence sequence calls and denoted in the FASTA sequence in lower case characters. The rest of the bases are called ‘N’ in the FASTA sequence. The flowchart of EvolSTAR is shown in Figure 1.
Performance of EvolSTAR
To validate the software, we hybridized 14 patient samples in duplicate onto the microarray. The microarrays were analysed in parallel using Nimblescan (PBC algorithm) and EvolSTAR, and the sequences obtained were compared to Sanger capillary sequencing. We counted the number of true-non-mutation calls, true-mutation calls, error calls and ambiguous (‘N’) calls for both methods (Table 2; Supplementary Data File 3). We also confirmed that the substitution bias in all 14 duplicate hybridization experiments (Table 3) were consistent with that found in Table 1. Compared with the available capillary sequences for the 14 samples, EvolSTAR had an average error rate of 0.0029% and 12 ambiguous calls per sample (346 in total). This is far superior than Nimblescan PBC, where we obtained an average error rate of 0.083% and 158 ambiguous calls per sample (4434 in total). Furthermore, EvolSTAR called all true mutations correctly. The genome coverage attained by EvolSTAR (99.02 ± 0.82%) is also much higher than that of Nimblegen PBC (94.3 ± 6.06%).
Table 2.
Comparison of calls made by EvolSTAR and PBC for 14 samples
| Sample | Program | Rep. | Total sites verified by capillary | Mutations (verified by capillary) | True-non- mutation calls | True mutation calls | Missed mutations | Error calls |
|---|---|---|---|---|---|---|---|---|
| 129 | EvolSTAR | 1 | 4767 | 6 | 4737 | 6 | 0 | 0 |
| PBC | 1 | 4767 | 6 | 4500 | 6 | 0 | 3 | |
| EvolSTAR | 2 | 4767 | 6 | 4737 | 6 | 0 | 0 | |
| PBC | 2 | 4767 | 6 | 4474 | 6 | 0 | 6 | |
| 141 | EvolSTAR | 1 | 4051 | 6 | 4026 | 6 | 0 | 0 |
| PBC | 1 | 4051 | 6 | 3832 | 6 | 0 | 10 | |
| EvolSTAR | 2 | 4051 | 6 | 4021 | 6 | 0 | 0 | |
| PBC | 2 | 4051 | 6 | 3808 | 6 | 0 | 4 | |
| 279 | EvolSTAR | 1 | 693 | 2 | 670 | 2 | 0 | 0 |
| PBC | 1 | 693 | 2 | 358 | 1 | 1 | 8 | |
| EvolSTAR | 2 | 693 | 2 | 682 | 2 | 0 | 0 | |
| PBC | 2 | 693 | 2 | 645 | 2 | 0 | 0 | |
| 354 | EvolSTAR | 1 | 8950 | 9 | 8942 | 9 | 0 | 0 |
| PBC | 1 | 8950 | 9 | 8802 | 9 | 0 | 1 | |
| EvolSTAR | 2 | 8950 | 9 | 8944 | 9 | 0 | 0 | |
| PBC | 2 | 8950 | 9 | 8851 | 9 | 0 | 0 | |
| 380 | EvolSTAR | 1 | 12 832 | 10 | 12 803 | 10 | 0 | 0 |
| PBC | 1 | 12 832 | 10 | 12 466 | 10 | 0 | 6 | |
| EvolSTAR | 2 | 12 832 | 10 | 12 816 | 10 | 0 | 0 | |
| PBC | 2 | 12 832 | 10 | 12 542 | 10 | 0 | 4 | |
| 384 | EvolSTAR | 1 | 6002 | 6 | 5992 | 6 | 0 | 0 |
| PBC | 1 | 6002 | 6 | 5888 | 6 | 0 | 0 | |
| EvolSTAR | 2 | 6002 | 6 | 5993 | 6 | 0 | 0 | |
| PBC | 2 | 6002 | 6 | 5895 | 6 | 0 | 1 | |
| 507 | EvolSTAR | 1 | 3921 | 8 | 3913 | 8 | 0 | 0 |
| PBC | 1 | 3921 | 8 | 3736 | 8 | 0 | 3 | |
| EvolSTAR | 2 | 3921 | 8 | 3916 | 8 | 0 | 0 | |
| PBC | 2 | 3921 | 8 | 3758 | 8 | 0 | 2 | |
| 581 | EvolSTAR | 1 | 8574 | 10 | 8567 | 10 | 0 | 0 |
| PBC | 1 | 8574 | 10 | 8458 | 10 | 0 | 2 | |
| EvolSTAR | 2 | 8574 | 10 | 8566 | 10 | 0 | 0 | |
| PBC | 2 | 8574 | 10 | 8461 | 10 | 0 | 5 | |
| 582 | EvolSTAR | 1 | 3057 | 4 | 3051 | 4 | 0 | 0 |
| PBC | 1 | 3057 | 4 | 2986 | 4 | 0 | 0 | |
| EvolSTAR | 2 | 3057 | 4 | 3053 | 4 | 0 | 0 | |
| PBC | 2 | 3057 | 4 | 3001 | 4 | 0 | 0 | |
| 593 | EvolSTAR | 1 | 3054 | 3 | 3053 | 3 | 0 | 0 |
| PBC | 1 | 3054 | 3 | 3007 | 2 | 1 | 0 | |
| EvolSTAR | 2 | 3054 | 3 | 3053 | 3 | 0 | 0 | |
| PBC | 2 | 3054 | 3 | 2992 | 2 | 1 | 0 | |
| 9 061 364 | EvolSTAR | 1 | 5129 | 5 | 5123 | 5 | 0 | 0 |
| PBC | 1 | 5129 | 5 | 5064 | 5 | 0 | 0 | |
| EvolSTAR | 2 | 5129 | 5 | 5122 | 5 | 0 | 0 | |
| PBC | 2 | 5129 | 5 | 5042 | 5 | 0 | 0 | |
| 9 061 365 | EvolSTAR | 1 | 3000 | 3 | 2993 | 3 | 0 | 0 |
| PBC | 1 | 3000 | 3 | 2956 | 3 | 0 | 1 | |
| EvolSTAR | 2 | 3000 | 3 | 2991 | 3 | 0 | 0 | |
| PBC | 2 | 3000 | 3 | 2941 | 3 | 0 | 0 | |
| 9 061 366 | EvolSTAR | 1 | 1683 | 3 | 1683 | 3 | 0 | 0 |
| PBC | 1 | 1683 | 3 | 1649 | 3 | 0 | 1 | |
| EvolSTAR | 2 | 1683 | 3 | 1682 | 3 | 0 | 1 | |
| PBC | 2 | 1683 | 3 | 1636 | 3 | 0 | 1 | |
| 923 | EvolSTAR | 1 | 4373 | 5 | 4365 | 5 | 0 | 0 |
| PBC | 1 | 4373 | 5 | 4187 | 5 | 0 | 1 | |
| EvolSTAR | 2 | 4373 | 5 | 4330 | 5 | 0 | 1 | |
| PBC | 2 | 4373 | 5 | 3738 | 5 | 0 | 6 |
Types of calls and their frequencies generated by EvolSTAR and PBC in replicated microarray hybridizations of 14 patient samples. Partial or complete capillary sequences were generated for each sample and used to verify the calls made by EvolSTAR and PBC on each replicate. We then count the frequency of true-non-mutation, true-mutation, error and ‘N’ calls in each replicate.
Table 3.
Hybridization intensity reduction orders found in 14 hybridization experiments
| PM probe encoding | Hybridization intensity reduction order | Forward strand | Reverse strand |
|---|---|---|---|
| Frequency | Frequency | ||
| A | CGT | 2618 | 1030 |
| CTG | 2347 | 975 | |
| GCT | 4848 | 1870 | |
| GTC | 12 571 | 8889 | |
| TCG | 4417 | 2624 | |
| TGC | 16 805 | 16 692 | |
| C | AGT | 10 843 | 14 309 |
| ATG | 10 606 | 14 473 | |
| GAT | 1777 | 1567 | |
| GTA | 748 | 618 | |
| TAG | 2006 | 1784 | |
| TGA | 790 | 623 | |
| G | ACT | 9114 | 7403 |
| ATC | 5490 | 3647 | |
| CAT | 9369 | 8811 | |
| CTA | 4104 | 3143 | |
| TAC | 2839 | 1976 | |
| TCA | 2458 | 1790 | |
| T | ACG | 1926 | 2080 |
| AGC | 2489 | 2524 | |
| CAG | 3211 | 3721 | |
| CGA | 6191 | 8656 | |
| GAC | 7550 | 9533 | |
| GCA | 10 713 | 17 092 |
Hybridization intensity reduction orders found in 135 830 true calls from 14 hybridization experiments. For each true call, for each strand, we rank the PM probe and its MM probes based on their hybridization intensities in decreasing order. We count the frequency of each hybridization intensity reduction order.
We wondered if, and by how much, incorporating NHIP and substitution biases analysis to the PBC results would improve the performance of the PBC algorithm. We observed that more than 70% of the 65 error calls (false mutation calls) made by PBC did not have the characteristic NHIP of a true-mutation shown in Figure 2b. The remaining 30% of the error calls had a NHIP reminiscent of a true-mutation NHIP but did not satisfy the substitution bias rule. Using NHIP and substitution biases analysis together, we were able to reduce the number of false mutation calls to only two. Most of the 4434 ‘N’ calls made by PBC were due to conflicting base calls from the forward and reverse strand. By analysing the NHIP and hybridization intensity reduction order of the query base in the forward and reverse strand individually, we were able to identify the noisy strand and hence, make the base call only from the non-noisy strand. We were able to recover 92% of the ‘N’ calls made by PBC using this approach.
In addition, we evaluate the robustness and reproducibility of EvolSTAR by employing six pairs of replicate experiments consisting of one pair nasal swab and five pairs of cell culture isolates, belonging to the same patient sample 305 (Supplementary Data File 4). Of the experiments, two pairs of replicates (305_nasal and 305_cell_cond1) were amplified under the same optimal experimental conditions while each of the other pairs (305_cell_cond2, 305_cell_cond3, 305_cell_cond4, 305_cell_cond5) were amplified under different sub-optimal experimental conditions (simulating experimental volatility). Compared with the available capillary sequences for sample 305, EvolSTAR had an average error rate of 0.0012% and 28 ambiguous calls per sample (338 in total). On the other hand, Nimblescan PBC obtained a relatively higher average error rate of 0.169% and 237 ambiguous calls per sample (2855 in total). Our results showed that EvolSTAR is robust and performs well when samples are prepared under sub-optimal conditions. Even for nasal swab samples that tend to have much less concentration of virus RNA than cell cultures, EvolSTAR suffered only a slight drop in performance compared to Nimblescan PBC.
In conclusion, we have shown that EvolSTAR is robust and generates sequence calls of high accuracy and reproducibility in this pilot study consisting of 40 microarray experiments. Meanwhile, efforts will be put in to continually evaluate EvolSTAR with more samples and update it on a regular basis as the H1N1(2009) influenza virus evolves.
Visualization of sequence calls
Besides a FASTA output of the virus sequence, EvolSTAR generates a visualization map of the sequence calls using a heat map based on the percentage identity of the called sequence to the reference sequence measured at 50 bp windows (Figure 8). The map template consists of all eight segments of the 2009 influenza A(H1N1) virus and the locations of known drug binding sites (marked with green lines) on the NA gene. Locations of all mutation calls are denoted by red triangles beneath the heat map bar. Sequences that are of low coverage (<90%) are automatically flagged, and the overall PM/MM discrimination ratio for each segment is displayed. The heat map bar allows the technician to rapidly assess the quality of the sequence data obtained from the microarray and identify regions where PCR did not work well, or presence of potential recombination/reassortment events. Mutations, especially those in close proximity to drug binding sites, can be quickly visualized. Other details such as coverage, number of base calls successfully made, number of mutations and number of ‘N’ calls for each sequence call are also shown on the visualization map.
Figure 8.

Visualization map of EvolSTAR. Visualization map of all eight segments of the 2009 influenza A(H1N1) virus and the locations of known drug binding sites (marked with green lines) on the neuraminidase (NA) gene (segment 6). A heat map bar is used to represent the quality and coverage of its sequence calls. The locations of all mutation calls made by EvolSTAR are represented by red triangles beneath the heat map bar. Sequences with coverage <90% are automatically flagged as ‘low coverage’. Other details such as coverage: percentage of base calls successfully made, match: number of base calls that match the reference sequence i.e. non-mutation base calls, strong mismatch: number of high confidence base calls that do not match the reference sequence i.e. mutation base calls, weak mismatch: number of low-confidence base-calls that do not match the reference sequence i.e. mutation base calls and Ns: number of ‘N’ calls, for each sequence call are also shown on the visualization map.
DISCUSSION
Traditional statistical and probabilistic sequence-calling techniques ascertain that a base call is of high confidence if they exceed pre-defined significance or probability thresholds. This approach works well for high-confidence base-calls but is inadequate to extract sufficient information from noisy base-calls. It is also difficult to determine the validity of a mutation call purely based on the distribution of hybridization intensities of its PM and MM probes. In this work, we have described two new hybridization intensity analysis methods that enable us to confidently identify true mutations and recover some noisy base calls. Compared to PBC, EvolSTAR has achieved superior call rates and accuracies, especially in low-concentration samples with high CT values. The robustness of the base calls enables our approach to be a practical large-scale evolutionary surveillance tool.
Although we are confident that our resequencing array can successfully generate complete sequences for the H1N1(2009) virus and its variants at the current stage, we cannot rule out the possibility of reassortments between the H1N1(2009) virus and other influenza viruses. Clearly, our resequencing array cannot fully sequence such events and will generate sequences with poor quality and coverage of the reassorted segments. To investigate the effects of a reassortment event on our array, we independently amplified segments 1, 2, 3, 5, 6 and 7 of the 2009 influenza A(H1N1) virus and segment 4 of a H3N2 influenza A virus, and hybridized them onto our array. The visualization map of this experiment is shown in Figure 9. As expected, the sequence call for segment 4 [based on PM/MM probes from the segment 4 consensus of the 2009 influenza A(H1N1) virus] is poor in quality and coverage. However, we observed that we were able to get good base calls from region 1150–1547. This region turns out to be the only significantly similar (70% matched) region between the segment 4 consensus of the 2009 influenza A(H1N1) virus and segment 4 of a H3N2 virus (CY039087). This shows that identifying regions of high similarity between the 2009 influenza A(H1N1) virus with other influenza viruses and checking if these regions have good sequence calls may be a plausible way of detecting reassortments. The drawback of this approach is that it will fail to detect reassortment of certain segments where there are no regions of high similarity between the H1N1(2009) virus and the parental influenza virus. It is also difficult to annotate and differentiate every region that the H1N1(2009) virus and all other influenza viruses share similarity with. We propose an alternative approach to detect reassortments. By analysing the PM/MM hybridization intensity fold-change of high confidence calls of all eight segments, we found that the average PM/MM hybridization intensity fold-change of high confidence calls in segments 1, 2, 3, 5, 6 and 7 belonging to the 2009 influenza A(H1N1) virus is ∼4.5 while the average PM/MM hybridization intensity fold-change of high confidence calls in segment 4 belonging to the H3N2 influenza A virus is only 1.9. The most likely reason for this huge drop in the average PM/MM hybridization intensity fold-change of high confidence calls is that the signal gained by most of the segment 4 PM probes on our array are through cross-hybridization to the segment 4 sequence of the H3N2 influenza A virus, and thus much lower than signal gained from true specific binding. Thus, by computing and comparing the average PM/MM hybridization intensity fold-change of high confidence calls in each segment, we can identify potential reassortments in a given H1N1(2009) virus sample. Virus samples with possible reassortments can then be sequenced using capillary sequencing or customized reassortment resequencing arrays.
Figure 9.

Visualization map of a 2009 influenza A(H1N1) virus with artificial reassortment of H3N2 segment 4. Visualization map of a 2009 influenza A(H1N1) virus with artificial reassortment of H3N2 segment 4. We independently amplified segments 1, 2, 3, 5, 6 and 7 of the 2009 influenza A(H1N1) virus and segment 4 of a H3N2 influenza A virus, and hybridized them onto our array. As expected, the sequence call for segment 4 [based on PM/MM probes from the segment 4 consensus of the 2009 influenza A(H1N1) virus] is poor in quality and coverage.
So far, the sequence diversity of H1N1 2009 influenza virus has been rather limited. From our analysis, it would be possible to resequence all the published isolates using this resequencing approach. However, as antigenic drift is expected to occur, it is likely that the resequencing array would need to be updated at least annually. Updating the array requires only bioinformatics input, and does not require any other additional manufacturing costs. Thus, this combination of sample amplification primers, low-cost multiplex array and robust interpretation software allows sustainable, rapid, large-scale biosurveillance of the influenza H1N1(2009) virus.
From a broader perspective, this study has highlighted the feasibility of using resequencing microarrays for high-throughput full genome sequencing of viruses. In our application, resequencing microarrays are relatively low-cost, costing only a 10th that of a 454 run, and equivalent to that of a traditional capillary sequencing run. However, through multiplexing, our system can generate full genomes of 24 different H1N1(2009) samples in 30 h. In comparison, capillary sequencing and next-generation technologies such as 454 may obtain full genomes of only one or two different samples in the same time-frame. In practice, capillary sequencing is labour-intensive and thus impractical for large-scale full genome sequencing of viruses in an outbreak. In our experience with the 454 system, much of the amplified material is still human (as the bulk of the patient sample material is human RNA with very little influenza RNA), requiring very deep sequencing to obtain a complete flu genome sequence, with one compartment of a run not yielding sufficient viral information. Furthermore, assembly of the sequence fragments is required before any analysis can be done. Any abnormalities or gaps in the assembly would then require additional runs of 454, incurring more cost and time. Hence, our approach based on resequencing microarrays presents a cost-effective and efficient solution for high-throughput full genome sequencing of viruses.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for this work was provided by the Genome Institute of Singapore and Exploit Technologies Pte Ltd, Agency for Science, Technology and Research (A*STAR).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Sebastian Maurer-Stroh of the Bioinformatics Institute of Singapore for his expert advice on 2009 H1N1 Influenza A drug binding pocket targeted by NA inhibitors. Samples were from Ministry of Health of Singapore (MOH) and DSO National Laboratories (Singapore).
REFERENCES
- 1.Barclay L. WHO issues guidelines for antiviral treatment for H1N1 and other Influenza. Medscape Med. News. 2009 http://www.medscape.com/viewarticle/707922. [Google Scholar]
- 2.Warrington JA, Shah NA, Chen X, Janis M, Liu C, Kondapalli S, Reyes V, Savage MP, Zhang Z, Watts R, et al. New developments in high-throughput resequencing and variation detection using high density microarrays. Hum. Mutat. 2002;19:402–409. doi: 10.1002/humu.10075. [DOI] [PubMed] [Google Scholar]
- 3.Cutler DJ, Zwick ME, Carrasquillo MM, Yohn CT, Tobin KP, Kashuk C, Mathews DJ, Shah NA, Eichler EE, Warrington JA, et al. High-throughput variation detection and genotyping using microarrays. Genome Res. 2001;11:1913–1925. doi: 10.1101/gr.197201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Daum LT, Vora GJ, Metzgar D, Walter EA, Canas LC, Malanoski AP, Lin B, Stenger DA. Identifying influenza viruses with resequencing microarrays. Emerg. Infect. Dis. 2006;12:638–646. doi: 10.3201/eid1204.051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin B, Malanoski AP, Wang Z, Blaney KM, Long NC, Meador CE, Metzgar D, Myers CA, Yingst SL, Monteville MR, et al. Universal detection and identification of avian influenza virus by use of resequencing microarrays. J. Clin. Microbiol. 2009;47:988–993. doi: 10.1128/JCM.01346-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sulaiman IM, Sammons SA, Wohlhueter RM. Smallpox virus resequencing GeneChips can also rapidly ascertain species status for some zoonotic non-variola orthopoxviruses. J. Clin. Microbiol. 2008;46:1507–1509. doi: 10.1128/JCM.00158-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leski TA, Lin B, Malanoski AP, Wang Z, Long NC, Meador CE, Barrows B, Ibrahim S, Hardick JP, Aitichou M, et al. Testing and validation of high density resequencing microarray for broad range biothreat agents detection. PLoS ONE. 2009;4:e6569. doi: 10.1371/journal.pone.0006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong CW, Albert TJ, Vega VB, Norton JE, Cutler DJ, Richmond TA, Stanton LW, Liu ET, Miller LD. Tracking the evolution of the SARS coronavirus using high-throughput, high-density resequencing arrays. Genome Res. 2004;14:398–405. doi: 10.1101/gr.2141004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhan Y, Kulp D. Model-P: a basecalling method for resequencing microarrays of diploid samples. Bioinformatics. 2005;21:182–189. doi: 10.1093/bioinformatics/bti1129. [DOI] [PubMed] [Google Scholar]
- 10.Pandya GA, Holmes MH, Sunkara S, Sparks A, Bai Y, Verratti K, Saeed K, Venepally P, Jarrahi B, Fleischmann RD, et al. A bioinformatic filter for improved base-call accuracy and polymorphism detection using the Affymetrix GeneChip® whole-genome resequencing platform. Nucleic Acids Res. 2007;35:e148. doi: 10.1093/nar/gkm918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee WH, Wong CW, Leong WY, Miller LD, Sung WK. LOMA: a fast method to generate efficient tagged-random primers despite amplification bias of random PCR on pathogens. BMC Bioinformatics. 2008;9:368. doi: 10.1186/1471-2105-9-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toh K. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinformatics. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 13.Maurer-Stroh S, Ma J, Lee RT, Sirota FL, Eisenhaber F. Mapping the sequence mutations of the 2009 H1N1 influenza A virus neuraminidase relative to drug and antibody binding sites. Biol. Direct. 2009;4:18. doi: 10.1186/1745-6150-4-18. discussion 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng J, Moorhead M, Weng L, Siddiqui F, Carlton VE, Ireland JS, Lee L, Peterson J, Wilkins J, Lin S, et al. High-throughput, high accuracy array-base resequencing. Proc. Natl Acad. Sci. USA. 2009;106:6712–6717. doi: 10.1073/pnas.0901902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seringhaus M, Rozowsky J, Royce T, Nagalakshmi U, Jee J, Snyder M, Gerstein M. Mismatch oligonucleotides in human and yeast: guidelines for probe design on tiling microarrays. BMC Genomics. 2008;9:635. doi: 10.1186/1471-2164-9-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.