


Abstract
Initiation of bacterial DNA replication at oriC is mediated by primosomal proteins that act cooperatively to melt an AT-rich region where the replicative helicase is loaded prior to the assembly of the replication fork. In Bacillus subtilis, the dnaD, dnaB and dnaI genes are essential for initiation of DNA replication. We established that their mRNAs are maintained in fast growing asynchronous cultures. DnaB is truncated at its C-terminus in a growth phase-dependent manner. Proteolysis is confined to cytosolic, not to membrane-associated DnaB, and affects oligomerization. Truncated DnaB is depleted at the oriC relative to the native protein. We propose that DNA-induced oligomerization is essential for its action at oriC and proteolysis regulates its localization at oriC. We show that DnaB has two separate ssDNA-binding sites one located within residues 1–300 and another between residues 365–428, and a dsDNA-binding site within residues 365–428. Tetramerization of DnaB is mediated within residues 1–300, and DNA-dependent oligomerization within residues 365–428. Finally, we show that association of DnaB with the oriC is asymmetric and extensive. It encompasses an area from the middle of dnaA to the end of yaaA that includes the AT-rich region melted during the initiation stage of DNA replication.
INTRODUCTION
Bacterial DNA replication initiates at oriC with the recruitment of two replicative helicases and associated primases around which two replisomes assemble to start copying the parental genome. The replication initiator protein DnaA is ubiquitous but replication initiation mechanisms and their regulatory processes vary between bacteria (1), probably reflecting the different architectures of their replication origins (2,3). DnaA binds to specific sites within oriC to assist local melting in an AT-rich region for loading of the replicative helicase (4–6). During the elongation phase of DNA replication, replication forks are frequently challenged by mutations, nicks, base modifications and other factors, resulting in fork arrest and collapse. Recombination-based and direct repair mechanisms re-initiate replication from stalled sites (7). In Escherichia coli two restart mechanisms, PriA- and PriC-mediated, differ in their ability to recognize nascent DNA at stalled forks (8). PriA, like DnaA, is ubiquitous but other primosomal proteins are not. For example, Bacillus subtilis does not have homologues of E. coli PriB, PriC and DnaT. Instead, two B. subtilis proteins DnaB and DnaD are required for initiation of DNA replication at oriC and stalled replication fork sites (9–12). No homologues of these proteins are found in gram negative bacteria implying that their replication initiation mechanisms are different.
Mechanistic and regulatory details of B. subtilis replication initiation are not clear at present. DnaD interacts with DnaA (13) and PriA (14) and is likely to act in the early stages of primosomal assembly while DnaB acts at a later stage. In fact DnaB is believed to be recruited to the oriC–DnaA–DnaD complex by an interaction with DnaD. DnaD has DNA remodelling activity on supercoiled DNA (15–17) and counteracts the lateral DNA compaction activity of DnaB (18). The DNA remodelling activity of DnaD is the sum of oligomerization and DNA-binding activities on two separate domains. An N-terminal domain mediates scaffold formation (19,20) and a C-terminal domain binds to DNA and has a second DNA-induced oligomerization interface (19). DnaB is multifunctional; it interacts with DnaD on SSB-coated DNA (21), cooperates with DnaI (the helicase loader homologous to E. coli DnaC) as ‘co-loader’ of DnaC (the replicative helicase homologous to E. coli DnaB) (22,23), acts as an origin membrane attachment protein (24–28), regulates the recruitment of DnaD to the membrane attached oriC (29), laterally condenses supercoiled DNA (18) and is essential for regulation of replication initiation synchrony (12).
We found that DnaB is truncated in vivo to smaller polypeptides in middle to late exponential growth and late stationary phases. This truncation takes place in a C-terminal domain, which is responsible for DNA-induced higher-order oligomerization, and is confined to cytosolic, not membrane-associated, DnaB. Using truncated polypeptides we found that DnaB(1–428) forms nucleoprotein complexes similar to those formed by native DnaB, whereas DnaB(1–365) is defective in DNA-induced oligomerization. Tetrameric DnaBN (residues 1–300) binds to ssDNA, not dsDNA, and does not form high-order complexes, while a monomeric DnaBC (residues 301–472) binds to ss and dsDNA and forms higher-order nucleoprotein complexes. Therefore, the dsDNA-binding site of DnaB is located within residues 365–428. Tetramerization of DnaB is mediated within residues 1–300 and DNA-dependent oligomerization within residues 365–428.
ChIP-chip analysis revealed that, relative to full-length DnaB, truncated DnaB is depleted at oriC. There is no significant difference in the mRNA levels of the dnaB, dnaD and dnaI primosomal genes during exponential growth of asynchronous cultures. We propose that DnaB functional regulation in asynchronous cultures is posttranslational and involves proteolytic truncation of its C-terminal domain. Proteolysis of DnaB abolishes high-order DNA-induced oligomerization and regulates its localization at oriC. Truncated DnaB in the stationary phase may bind DNA but will not form higher-order oligomers, and may be essential for functions other than initiation of DNA replication.
MATERIALS AND METHODS
RNA purification and cDNA synthesis
B. subtilis strain 168 was cultured in Luria–Bertani (LB) media. An overnight culture was diluted into LB and incubated at 30 °C. Samples were harvested at OD600 = 0.3 (lag phase, t1), 0.9 (exponential phase, t2), 1.3 (early stationary phase, t3) and overnight (late stationary phase, t4) for RNA and protein isolation. RNA was extracted from 4 ml cell cultures using RNAprotect Bacteria Reagent and Qiagen RNeasy Mini Columns (Qiagen), according to manufacturer’s instructions, except for a 10 s sonication step after the addition of lysozyme and Proteinase K. DNaseI digestion was performed, as described in the Qiagen protocol. RNA was quantified by a NanoDrop ND-1000 (Thermo Scientific), its quality checked by capillary electrophoresis in an Agilent 2100 Bioanalyzer (Agilent Technologies) and stored at −20 °C. cDNA synthesis was carried out with random primers and SuperScript II Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions.
RT–PCR
Primers were designed with Primer Express 3.0 software (PE Applied Biosystems) (Supplementary Figure 1S). RT–PCR reactions were carried out using PowerSYBR green Master Mix on a 7500 Real-Time PCR system (PE Applied Biosystems). Equal amounts of cDNA (5 ng) and 300 nM primers were used in all amplification reactions. PCR conditions were: one cycle of 50°C for 2 min, one cycle of 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 15 s and one cycle of 60°C for 1 min. Product formation was monitored by fluorescence increase from SYBR Green intercalation. The threshold cycle (CT) is the first cycle for which a statistically significance increase in the amount of product is detected. CT values are thus inversely proportional to the cDNA amount in the sample. Relative quantification was used as described before (30) to determine changes in dnaB, dnaD and dnaI expression compared to sigA (ΔCT value), followed by comparisons of the three genes over time (ΔΔCT value). Relative quantification was done using the formula 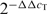 as described previously (30).
as described previously (30).
Protein extraction and western-blot analysis
Cell pellets from 10 ml cultures were suspended in 1 ml 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 1 mM EDTA and sonicated 3 × 10 s. The soluble fraction was separated by centrifugation at 13 000 rpm for 5 min at room temperature. The membrane fraction (pellet) was washed twice with the same buffer before suspension in 8 M urea. Protein was quantified by Bradford assay (31). Extracts were resolved by SDS–PAGE and transferred to nitrocellulose membranes (Hybond-ECL, GE Healthcare) by electrotransfer in Tris–Glycine buffer (12.5 mM Tris pH 9.0, 96 mM glycine and 20% v/v methanol). Membranes were blocked overnight with 10% w/v dried milk in PBS-T (20 mM phosphate buffer, pH 7.2, 150 mM NaCl, 0.05% v/v Tween-20) and washed 3 × 5 min with PBS-T before incubation with relevant antibodies for 60–90 min. After washing with PBS-T for 3 × 5 min membranes were incubated with secondary anti-rabbit IgG HandL Chain Specific Peroxidase Conjugate (Calbiochem) and washed with PBS for 5 min. Detection was performed either by SuperSignal® West Pico Chemiluminescence Substrate (Pierce) using HyperfilmTM ECL films (GE Healthcare) or by the peroxide/DAB method (32). Duplicate gels were run concurrently with the same samples for Coomassie Blue staining. Polyclonal rabbit antibodies were produced by the Medical and Health Sciences support unit at the University of Nottingham and their efficiency was tested by western blots with purified proteins (data not shown). The C-terminal DnaB peptide DLEEQKKKMMEEMQKLKKYSAY was synthesized by Alta Bioscience (Birmingham) and sheep polyclonal antibody against it was made by the Scottish National Blood Transfusion Service. The antibody was purified further by Alta Bioscience using affinity chromatography with a column containing immobilized peptide and supplied in lyophilized 1 mg aliquots. Binding to native DnaB and DnaBC and failure to bind to DnaBN were verified by western blotting (Supplementary Figure 2S).
Protein expression and purification
Native untagged DnaB was expressed from pET28a-dnaB and purified as described before (18). Plasmid constructs for N-terminally His6-DnaB polypeptides were prepared by PCR (Supplementary Figure 1S) using pET28a-dnaB (18) and subcloning into the NdeI/BamHI sites of pET28a (Novagen). DnaD and DnaI were purified as before (33, 15). DnaB polypeptides were expressed at 20°C for 20 h [DnaB and DnaB(1–300)] or 65 h [DnaB(1–356) and DnaB(1–428)] in E. coli BL21(DE3) with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cell pellets were suspended in 20 ml of 50 mM sodium phosphate pH 7.4, 500 mM NaCl, 20 mM imidazole, 1 mM DTT, 10% w/v sucrose, 1 mM phenylmethyl sulfonate and protease inhibitor cocktail (P8849, Sigma; 100 µl/l of culture). Following sonication and clarification by centrifugation (42 000g × 40 min at 4°C) the supernatant was filtered (0.2 µm filter, Sartorius) and loaded onto a Ni2+-charged 5 ml HiTrap chelating column (GE Healthcare) equilibrated in buffer A (50 mM sodium phosphate pH 7.4, 500 mM NaCl, 20 mM imidazole, 1 mM DTT). The column was washed successively with 40 ml of buffer A with 2 M NaCl, buffer A with 1% v/v Triton X-100, dH2O and finally buffer A. Proteins were eluted with a 20–500 mM imidazole gradient over 60 ml. Relevant fractions were pooled and thrombin added (Novagen) at 0.4 units/mg of His6-DnaB(301–472) or half that for other proteins. After digestion for 16 h at 20°C proteins were buffer-exchanged into buffer A and loaded onto the chelating column equilibrated in buffer A. The flow-through was spin-concentrated and loaded onto a Superdex 200 HiLoad 26/60 column (GE Healthcare), or, in the case of DnaB(301–472), a Superdex 75, equilibrated in buffer C (50 mM Tris–HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 100 mM NaCl). Relevant fractions were pooled glycerol added to 10% v/v, proteins quantified with a NanoDrop and stored at −80°C.
Analytical ultracentrifugation
Sedimentation velocity was carried out at 40 000 rpm at 20°C, in a Beckman XL-A. Cells were scanned at 280 nm every 5 min. Samples were loaded into two SEDVEL60K channels (SpinAnalytical, NH, USA). Data were analysed using the maximum entropy, c(s), method of Schuck (34) using the program SEDFIT (34–36). Sedimentation equilibrium was attained at 12 000, 16 000 and 20 000 rpm. Data were processed and fitted using SEDFIT and SEDPHAT (http://www.analyticalultracentrifugation.com).
Gel shifts
Oligonucleotides used in gel shift experiments were gel-purified. Radiolabelling was carried out with [γ32P]ATP (Perkin-Elmer) and T4 polynucleotide kinase (NEB). Probes were further purified in 10 mM Tris–HCl pH 8.0 using Sephacryl S-200 columns (GE healthcare). Annealing conditions for the ds dTA135 probe were determined carefully before preparation of the final substrate. DnaB and its polypeptides were incubated for 20 min at 25°C with 0.46 nM DNA substrates (dT45, dT90, dT135 or dAT135) in 82 mM Tris–HCl pH 7.5, 1.64 mM EDTA, 1.64 mM DTT, 5 mM MgCl2, 100 mM NaCl and 8% v/v glycerol. Samples were resolved by electrophoresis through a chilled (4°C) 18 × 16 cm gel in 0.5 × TBE for 200 min at 200 V. Gels were dried and visualized using a Personal Molecular Imager FX with Quantity-One software (Bio-Rad).
Peptide mass fingerprinting and N-terminal sequencing
Matrix assisted laser desorption ionization-peptide mass fingerprinting (MALDI-PMF) analysis was carried out at the Biopolymer Synthesis and Analysis unit at the University of Nottingham. Following in-gel trypsinization DnaB fragments were excised and eluted from the gel, and peptide spectra were obtained by MALDI. Peptides were identified by Mascot analysis (http://www.matrixscience.com) from databases containing Eubacteria sequences. N-terminal sequencing of intact DnaB fragments excised from gels was carried out by Alta Bioscience (University of Birmingham).
ChIP-chip microarray analysis
Sample preparation for ChIP-chip analysis was carried out as described before (37) with minor modifications. An overnight culture of Bacillus subtilis (strain 168) was diluted into 800 ml LB and incubated at 30°C. At mid-log phase (OD595 = 0.8) 1% v/v formaldehyde was added for 20 min to cross-link protein–DNA complexes and the reaction was quenched by 0.5 M glycine. Cells were harvested and washed with 50 ml 10 mM Tris–HCl pH 7.5, suspended in 2 ml lysis buffer (10 mM Tris–HCl pH 8.0, 20% w/v sucrose, 50 mM NaCl, 10 mM EDTA, 10 mg/ml lysozyme) and incubated at 37°C for 30 min. Immunoprecipitation (IP) buffer (8 ml) was added (50 mM HEPES–KOH pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% v/v Triton X-100, 0.1% w/v sodium deoxycholate, 0.1% w/v sodium dodecyl sulfate and 1 mM phenylmethylsulfonyl fluoride) and the solution sonicated 15 times (20 s bursts, 20 s rest on ice). The mixture was clarified by centrifugation (15 000g, 15 min at 4°C), the supernatant incubated with 300 µl anti-DnaB antibody overnight at 4°C on a rotating wheel and then with 150 µl of Dynabeads® M-280 sheep anti-rabbit IgG (Invitrogen) for 5 h at 4°C. The beads were washed twice with IP buffer, once with wash buffer (10 mM Tris–HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% v/v Nonident-P40 and 0.5% w/v sodium deoxycholate), once with wash buffer plus 0.02 M NaCl and twice with TE buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA). After removing the TE buffer, the beads were suspended in 400 µl elution buffer (50 mM Tris–HCl pH 7.5, 10 mM EDTA, 1% w/v SDS) and incubated at 65°C for 10 min. Cross links were reversed by incubation with 0.8 mg/ml of proteinase K (Thermo Scientific) in 0.5× elution buffer at 42°C for 2 h and 65°C for 6 h. DNA was purified with a PCR purification kit (QIAGEN) and quantified by Nanodrop.
B. subtilis (strain 168) Agilent 4x44K ChIP arrays with AMADID 023001 were prepared by Oxford Gene Technologies (OGT) who carried out array hybridization analysis and provided the final data. Each array comprises 41 770 probes in total, covering 4 185 genes. Each probe is 60 bp and generated using Agilent’s inkjet in-situ synthesis technology. The probes have an average spacing of ∼100 bp with a maximum inter-probe distance of ∼140 bp. The reference sample in the red (Cy5) channel was genomic B. subtilis (strain 168) DNA. Data analysis was carried out with a ChIP browser developed and supplied by OGT.
RESULTS
The mRNAs of the dnaD, dnaB and dnaI genes are maintained throughout asynchronous growth
Regulation of initiation of DNA replication is linked to the balance between DnaA levels and the number of DnaA-binding sites in the cell (38, 39). DnaA is regulated at the transcriptional level (39). The fate of the mRNAs of other primosomal genes during growth is unknown. We investigated the mRNA levels of dnaD, dnaB and dnaI during growth of asynchronous B. subtilis cultures by quantitative RT-PCR. Control experiments to verify direct proportionality between amounts of RNA and PCR signals were carried out and data were normalized against the sigA mRNA (data not shown). SigA codes for σA which is essential for transcription of housekeeping genes. Its mRNA remains constant during exponential growth (40). Comparison of CT values from RT-PCR experiments revealed that mRNA levels of dnaB, dnaI and dnaD were constant throughout growth (Figure 1A). The dnaB and dnaI genes are juxtaposed in the same operon (41) while dnaD is located in a distant operon. CT values indicated that the mRNA levels of dnaB and dnaI were identical and dnaD slightly higher (Figure 1A). Normalization against sigA mRNA and direct comparisons of ΔCT values indicated similar levels for each of the three mRNAs at t1 (early growth-phase) and t2 (middle growth phase) and only a marginal decrease at t3 (late growth phase) that was statistically insignificant. It was not possible to normalize samples from late stationary growth phase relative to each other as control sigA mRNA levels were not constant in these samples (data not shown). Attempts to use a different control, 16S rRNA, were unsuccessful because of growth dependent variability (data not shown). From these data we conclude that the mRNAs of dnaD, dnaB and dnaI are maintained throughout asynchronous growth.
Figure 1.

Growth-dependent expression of dnaD, dnaI and dnaB genes. (A) Growth curve of B. subtilis strain 168 in LB at 30°C over time. Time points t1, t2 and t3 when samples were analysed are indicated. The top bar graph shows levels of dnaB, dnaD, dnaI and sigA as CT values from RT-PCR over time. The bottom bar graph shows changes in gene expression calculated from the formula 2−ΔΔCt for dnaB, dnaD and dnaI, using sigA as control and t1 as calibrator, over time. In both graphs each bar represents the mean of five independent experiments. The error bars indicate standard deviations. (B) Western blot analysis of B. subtilis soluble extracts from overnight cultures. A typical SDS–PAGE gel with decreasing amounts of extracts (72, 36, 18, 9, 4.5, 2.2, 1.1, 0.5 and 0.25 μg) is shown (top left), with western blots for DnaD, DnaI and DnaB, as indicated. DnaB is truncated to smaller versions. Pre-stained molecular weight markers from New England Biolabs are indicated in kDa.
C-terminal proteolytic truncation of DnaB in a growth phase dependent manner
Western blots of B. subtilis extracts from overnight cultures, using polyclonal rabbit antibodies against DnaB, DnaD and DnaI, confirmed the presence of all three proteins (Figure 1B). However, DnaB polypeptides noticeably smaller than the native size (MW 55 171 Da) were apparent (Figure 1B). Truncation of DnaB in vivo was postulated before but no experimental evidence has been published (26). Its nature and biological significance remain a mystery. Heterologous expression of DnaB in E. coli and subsequent purification revealed additional smaller polypeptides (Figure 2A and Supplementary Figure 3S). N-terminal sequencing established that they all had the native DnaB N-terminal sequence (data not shown). Random degradation or cleavage of specific domains would be expected to produce peptides of variable N-termini. However, this was not the case. Western blot analysis of cell lysates from E. coli expressing DnaB showed that the observed truncation is due to ‘in vivo cleavage’ during expression and not due to proteolysis during purification (Figure 2A). Consistent with this, western blot analysis of B. subtilis soluble extracts from early and late exponential growth detected native DnaB (∼55 kDa) in early growth, and native plus truncated (∼40 kDa) DnaB in late growth (Figure 2B). In samples from overnight cultures, a significant fraction of DnaB was truncated suggesting that proteolysis continues into the late stationary growth phase (Figures 1B and 2B). The sizes of fragments did not indicate the appearance of distinct domains that added up to the size of the full-length protein. Instead, the sizes of the fragments were consistent with truncation at its C-terminus and complete digestion of the removed C-terminal fragment.
Figure 2.

Proteolytic truncation of DnaB. (A) Left, SDS–PAGE analysis of DnaB with smaller fragments visible. Truncated fragments are clearly visible in the juxtaposed western blot. Cell extracts from DnaB-expressing E. coli were analysed by western blotting (lane 1, whole cell extract; lane 2, soluble fraction). DnaB fragments were present indicating that truncation was occurring during expression and not during purification. (B) DnaB western blots of B. subtilis soluble extracts from early (lane 1, 20 µg; lane 2, 25 µg) and late (lane 3, 25 µg; lane 4, 20 µg) exponential growth phases, as indicated. DnaB is intact in early growth but a truncated polypeptide (∼40 kDa) appears in late growth. (C) The primary sequence of B. subtilis DnaB (residues 1–472). Predicted α-helices and β-strands (green boxes) are indicated. Secondary-structure prediction was carried out by Jpred 3 (42) and was consistent with several other programs. DnaB residues are colored according to sequence conservation obtained by multiple sequence alignment using Clustal W2 (43) of DnaB proteins from 47 sequenced genomes across the Firmicutes phylum (data not shown); pink (100% identity); red [100% close similarity, score ≥ 2 in the Blosum62 substitution matrix (44)]; orange (100% similarity, Blosum62 score ≥ 0); sky blue (≥90% similarity); dark blue (≥80% similarity). Regions of particular interest are indicated by horizontal lines and are (1) hydrophobic motif (25); (2) putative DNA binding motif (45); (3) 45.7% identity with RTP (46); (4) putative ATP binding motif (25); (5) similarity to Gp49 a putative phage replication initiation protein (29); (6) putative ATP-binding motif (25); (7) trypsin sensitivity (23); and (8) an antibody against a 15 amino acid peptide from this region inhibits binding to the pUB110 plasmid (47). Arrows show cleavage sites in limited proteolysis experiments (23).
MALDI-PMF of truncated DnaB polypeptides purified from E. coli revealed peptides covering residues 1–411 (Supplementary Figure 3S). The smaller polypeptides, labelled 2–5, produced peptides covering residues 1–289 suggesting that proteolysis takes place at the C-terminus (Supplementary Figure 3S). Previous studies identified two domains at each end (residues 1–184 and 297–472) with a small central domain (residues 204–296), (23). Interestingly, the same studies reported proteolytic sensitivity of the C-terminal region, residues 430–472. We constructed two domains of DnaB encompassing residues 1–300 (DnaBN; N-terminal domain plus the central domain, MW 35 157 Da) and 301–472 (DnaBC; C-terminal domain, MW 20 445 Da), (Figure 2C). DnaBC was found to be monomeric by analytical ultracentrifugation (Figure 3A and B) whilst DnaBN was a tetramer at high concentrations and in a dimer-tetramer equilibrium at low concentrations (Kd = 500 nM) (Figure 3C and D). To unequivocally show that the C-terminal region of DnaB was being truncated we raised a sheep polyclonal antibody against a peptide encompassing the last twenty two C-terminal residues (DLEEQKKKMMEEMQKLKKYSAY). We argued that if proteolysis takes place at the C-terminus this antibody will recognize native DnaB but not truncated polypeptides. The antibody was significantly weaker than rabbit anti-DnaB and was not sensitive enough to detect DnaB in B. subtilis extracts (data not shown), but it recognized purified native DnaB, DnaBC and not DnaBN (Supplementary Figure 2S). It was only useful in establishing that over-expressed DnaB in E. coli was truncated from its C-terminus. The combined data show that DnaB is proteolytically truncated in vivo from its C-terminus in a growth-dependent manner and analogous truncation is observed when DnaB is heterologously expressed in E. coli. It is not clear whether E. coli uses the same or different proteases to process DnaB. However, proteolysis of DnaB in E. coli is less efficient probably because of its large over-expression in a short period of time.
Figure 3.

The oligomeric states of the DnaB domains. Sedimentation velocity AUC data for DnaBC (A velocity sedimentation at 40 000 rpm, B: equilibrium sedimentation at 17 000, 24 000 and 29 000 rpm) and DnaBN (C: velocity sedimentation at 40 000 rpm, D: equilibrium sedimentation at 12 000, 16 000 and 20 000 rpm) reveal that DnaBC is a monomer, while DnaBN is tetrameric at higher concentrations but in a dimer–tetramer equilibrium at lower concentrations (Kd = 500 nM). Experiments were carried out at 12.96, 11.25 and 8.07 μM DnaBC and 21.3, 15.6 and 7.8 μM DnaBN.
DnaB has two separate ssDNA binding sites, one in DnaBN and another in DnaBC, and a ssDNA-dependent oligomerization activity in DnaBC
DnaBN and DnaBC did not interact with each other (data not shown) but both bound ssDNA. DnaBN formed defined complexes with oligonucleotides dT45, dT90 and dT135, (Figure 4). The patterns of shifted bands with increasing oligonucleotide sizes suggest that a small fraction of DnaBN binds as a dimer forming a minor band shifted the least with all three substrates, while most DnaBN binds as a tetramer to form one major shifted band with dT45, two with dT90 (two bound tetramers) and three with dT135 (three bound tetramers). Therefore, the apparent DnaBN footprints are ∼45 and 22 nucleotides for the tetramer and dimer, respectively. DnaBC formed mixtures of bigger complexes retarded at the top of the gel higher than it would be anticipated by its monomeric state and size (Figure 4). Native DnaB produced undefined diffuse shifts of decreased mobility with dT45 indicative of mixed higher order oligomers. These oligomers appeared to be more defined with the larger dT90 and dT135 oligonucleotides suggesting that they are more stable with longer ssDNA substrates.
Figure 4.

DnaB–ssDNA interactions. Gel shifts showing binding of DnaB polypeptides to ssDNA (dT45, dT90 or dT135). The concentrations (µM) of proteins in the binding reactions are shown underneath the lanes of the gels. Lanes M are internal controls showing binding reactions with 45 µM DnaBN. The positions of the slowest migrating bands of DnaB(1–365) are marked by asterisks. The equivalent position with native DnaB is also marked by an asterisk in the dT45 gel shift.
To further dissect the interactions with ssDNA we constructed two more truncated polypeptides DnaB(1–365) and DnaB(1–428) (the numbers refer to amino acid residues) (Figure 2C). Both truncated proteins formed stable tetramers and exhibited no signs of significant non-specific aggregation as assessed by sedimentation velocity AUC (Supplementary Figure 4S). DnaB(1–428) formed complexes similar to native DnaB but DnaB(1–365) produced bands that were shifted to a position (marked by an asterisk) much lower than the top of the gel, indicating failure to form higher order oligomers (Figure 4). We conclude that DnaB has two ssDNA binding sites located in DnaBN and DnaBC. It is likely that binding of DnaB(1–365) to ssDNA is mediated by the site located in DnaBN. Given that DnaBC is monomeric in solution we also conclude that binding to ssDNA induces oligomerization of this domain. DnaBC is likely to mediate DNA-induced oligomerization of native DnaB. This oligomerization site is located within residues 365–428 since DnaB(1–365) does not form the big complexes retarded at the top of the gel apparent with native DnaB, DnaBC and DnaB(1–428). Therefore, if the C-terminal truncation of DnaB observed in vivo included residues 365–428 it would abolish the ssDNA-dependent formation of higher order oligomers.
Heparin mimics ssDNA and induces high-order oligomerization of DnaB
To obtain further evidence that DNA binding induces DnaB oligomerization we utilized heparin affinity chromatography combined with size exclusion chromatography. DnaB, DnaBC and DnaB(1–428) were loaded to heparin and eluted with an increasing NaCl gradient (Figure 5). DnaB and DnaB(1–428) were eluted as two overlapping peaks (Figure 5A and B). Subsequent analysis of samples from fractions 1–3 by size exclusion chromatography revealed that fraction 1 contained tetramers, whereas fractions 2 and 3 mixtures of higher oligomers (Figure 5A and B). Velocity sedimentation analysis of fractions 1–3 from native DnaB verified the presence of tetramers in fraction 1 with sedimentation coefficient s* = 6.8 and mixtures of higher oligomers in fractions 2 and 3 with a wide distribution of sedimentation coefficients s* = 10–50 (Supplementary Figure 5S). DnaBC eluted as two separated peaks from the heparin column indicating formation of oligomers. However, subsequent analysis of fractions 1–4 by size exclusion chromatography revealed that they were all monomeric (Figure 5C). The simplest explanation is that DnaBC oligomers are formed in the presence of DNA but are somewhat unstable in the absence of DNA. Therefore, the presence of DnaBN may contribute towards greater stability of higher order oligomers in the absence of DNA. DnaB(1–365) did not bind to heparin (data not shown) indicating that the heparin interaction site is between residues 365–428, likely to coincide with the second ssDNA binding site in DnaBC. Since high salt does not induce oligomerization (data not shown) we conclude that binding to heparin mimics binding to DNA and induces the formation of higher order DnaB oligomers. The ssDNA binding and oligomerization sites in DnaBC are located within residues 365–428.
Figure 5.

Heparin-induced oligomerization. Chromatograms on the left of panels A–C show elution profiles of DnaB (27 µM), DnaB(1–428) (10 µM) or DnaBC (30 µM) from a HiTrap Heparin column (GE Healthcare) with a 0–2 M NaCl gradient over 20 ml. Letters L and E mark loading and elution points, respectively. Fractions (2 ml) were collected and numbered 1–3 or 1–4. The conductivity at each peak is indicated. DnaBN and DnaB(1–365) did not bind to the heparin column under our conditions (data not shown). Gel filtration profiles of the heparin fractions are shown on the right of A–C. Samples (0.5 ml) from heparin fractions (1–3) were loaded onto a Superose 6 (HR 10/30, GE Healthcare), except for DnaBC where Superdex 200 (HR 10/30, GE Healthcare) was used instead. Traces are superimposed and numbered the same as the heparin fractions. Higher order DnaB oligomers are marked accordingly.
DnaB has a dsDNA site within residues 365–428 which also mediates oligomerization
DnaB binds ss and dsDNA (14, 18). Although we showed that DnaB has two ssDNA binding sites within DnaBN and DnaBC and forms tetramers and higher order oligomers via DnaBN and DnaBC interactions, respectively, it is not clear how it interacts with dsDNA. Using our DnaB fragments and carefully prepared ds dTA135 we examined its dsDNA binding properties with gel shift experiments. We discovered that DnaBC binds dsDNA but DnaBN does not (Figure 6). Therefore, there is a dsDNA-binding site within DnaBC whilst DnaBN binds exclusively ssDNA. DnaBC–dsDNA complexes were of very low electrophoretic mobility. Judging from the monomeric state and small size of DnaBC, they are likely to be high-order complexes similar to the ones obtained with ssDNA (Figure 4). To further narrow the location of the dsDNA binding site we examined the DnaB(1–428) and DnaB(1–365) polypeptides (Figure 6). DnaB(1–428) bound to dsDNA forming defined high-order oligomers of very low mobility whereas DnaB(1–365) did not. The dsDNA site is, therefore, located within residues 365–428 coinciding with the area of ssDNA-binding and DNA-induced high-order oligomerization. However, we cannot distinguish whether the ss and dsDNA binding sites in DnaBC are separate, overlapping or identical.
Figure 6.

DnaB–dsDNA interactions. Gel shift assays showing binding to dAT135 of DnaB polypeptides, as indicated. Lanes C show controls with no proteins.
In vivo truncation is specific for DnaB and confined to soluble not to membrane-associated DnaB
To establish whether the observed proteolysis is a feature of DnaB or generally of other B. subtilis primosomal proteins we probed DnaD and DnaI by western blots (Figure 7). DnaB was present in cytosolic and membrane-associated cell extracts from early, middle and late exponential growth, but truncated versions started to become apparent from middle exponential growth only in cytosolic and not in membrane-associated fractions (Figure 7A). The sizes of fragments did not indicate the formation of distinct domains that added up to the size of native DnaB. Furthermore, the fact that DnaB appears to be intact in the early growth phase and in membrane associated fractions rules out premature termination and/or translation of dnaB mRNAs, as there is no reason why such processes should not be apparent in early compared to middle and late growth phases and why only soluble DnaB should be truncated. These data are consistent with our model of C-terminal proteolysis with the separated C-terminal fragment being digested completely. DnaB was mostly truncated in soluble fractions from overnight cultures, compared to membrane-associated fractions where it was somewhat resistant to proteolysis, although some marginal non-specific degradation was also apparent in these samples (Figure 7A). In addition to the 40 kDa fragment, present during exponential growth, another truncated fragment of 46 kDa absent from exponential growth, was apparent in samples from overnight cultures (Figure 7A). DnaD and DnaI were intact (Figure 7B). The former was present in both soluble and membrane-associated fractions and the latter was present only in cytosolic fractions consistent with previous localization studies showing GFP-tagged DnaD and DnaB to be proximal to the membrane and DnaI to be cytosolic (48). We conclude that proteolytic truncation is specific for DnaB and not a general feature of B. subtilis DnaD and DnaI primosomal proteins.
Figure 7.

DnaB, DnaD and DnaI levels in vivo. (A) Western blot of extracts (10 µg) from soluble (s) and pellet (p) fractions at different growth points probed with anti-DnaB. Molecular weight markers are included in lane M. The sizes of DnaB polypeptides are indicated. (B) SDS–PAGE (left) and western blot (right) analyses of protein extracts (10 µg) from soluble (s) and pellet (p) fractions at different growth points probed with anti-DnaD and anti-DnaI as indicated. Molecular weight markers are included in lane M. DnaD and DnaI are intact although some non-specific bands just above DnaD and just below DnaI are visible, possibly as a result of insufficient blocking.
Truncated DnaB is depleted from the oriC
To investigate the biological significance of this truncation, we carried out comparative ChIP-chip analysis of DnaB–DNA cross linked samples, immunoprecipitated from B. subtilis cultures with polyclonal anti-DnaB and anti-DnaBC antibodies. We argued that if there is extensive C-terminal truncation in vivo, then the anti-DnaBC antibody will immunoprecipitate less DnaB compared to the anti-DnaB antibody, and may allow us to differentiate interaction sites of native and truncated DnaB across the B. subtilis genome. We first established that the two antibodies were of comparable strength against native DnaB to allow direct comparison (data not shown). Then B. subtilis cultures were grown at OD595 = 0.8 and treated with formaldehyde. Cross-linked complexes were immunoprecipitated from cell extracts with anti-DnaB and anti-DnaBC antibodies. Following reversal of cross links, genomic DNA fragments were isolated for microarray analysis. Native and truncated DnaB were found to bind with comparable strength to several sites across the entire genome (Figure 8A and data not shown). Detailed analysis of these genomic sites will be presented elsewhere. Here we have analysed binding to oriC and the areas just before and after it (Figure 8B). DnaB was associated with several sites from the middle of dnaA to the end of yaaA giving several signals with G/R > 2 (Figure 8B and C). This region includes one half of the oriC, with the other half located upstream of dnaA (Figure 8D). By comparison, DnaBC samples produced much weaker signals (G/R < 2) in this region (Figure 8 and Supplementary Figure 6s). For every single one of 23 different oligonucleotide probes exhibiting significant signals (G/R > 1.5) in this area, the signals from anti-DnaBC samples were consistently weaker compared to the equivalent signals from anti-DnaB samples, with differences ranging from 50–300% (Supplementary Figure 6s). By comparison, binding signals elsewhere in the genome were similar in both samples (Figure 8A and data not shown), with comparative sets of different oligonucleotide probes within the same area giving signals of variable strengths between the two samples, i.e. sometimes higher for DnaB samples and sometimes higher for DnaBC samples (data not shown). This is highly significant as it eliminates random variations between individual oligonucleotide probes within the oriC region, indicating that the signal difference observed for all the different 23 sites within this area is a real effect. We conclude that DnaB interacts with oriC and that truncated DnaB is depleted at the oriC and not in other sites across the genome. Furthermore, the interaction of DnaB with oriC is asymmetric, confined to the half oriC site downstream of dnaA, that includes the AT-rich region, and involves an extended region further downstream encompassing the dnaN and yaaA genes.
Figure 8.

ChIP-chip microarray analysis. (A) Data from ChIP-chip microarray analysis across the entire B. subtilis genome using anti-DnaB and anti-DnaBC polyclonal antibodies, as indicated, showing that the two sets of data are almost identical. The G/R signal ratio of the green and red dyes (the reference sample was in red) and the genomic positions are shown on the y- and x-axes, respectively. The (+) and (−) DNA strands with the entire complement of genes and their positions are shown underneath. (B) Views of the genomic region upstream of the oriC (nucleotides 4 190 000–0) and the oriC region (nucleotides 0–10 000). Clear association of DnaB with the oriC can be seen. The association of DnaBC with the oriC is clearly less compared to DnaB. (C) Expanded views of the genomic region upstream of the oriC (nucleotides 4 208 000–0) and the oriC-containing region (nucleotides 0–6000), showing clearly reduced association of DnaBC with oriC compared to DnaB. (D) Further expansion of the oriC-containing region (0–5000) showing asymmetric association with the oriC. Binding of DnaB is apparent from positions 1000–3500. The up- and downstream half sites of the oriC on either side of dnaA are indicated. DnaA-binding sites are shown by arrows. Comparative signals in this area using the anti-DnaBC antibody are also shown. Weak association of DnaBC can also be detected by anti-DnaBC but the G/R signal ratios are <2 indicating weaker binding compared to DnaB (G/R > 2). All 23 individual sites within this region where significant binding is detected (sites with G/R > 1.5) the G/R signals from DnaB are significantly higher than those from DnaBC (Supplementary Figure 6S). We conclude that DnaBC is depleted from the oriC.
DISCUSSION
Biological significance of DnaBC
DnaB is proteolytically truncated in a growth-dependent manner. Truncation is apparent from mid-exponential growth and continues into the stationary phase. It is confined to cytosolic DnaB, membrane-associated DnaB is resistant, and involves the C-terminal region which mediates DNA-induced higher order oligomerization. A partially purified membrane fraction of B. subtilis was postulated, as data not shown, to have both native and truncated DnaB, and a cytosolic fraction native DnaB (26). DnaB was proposed to undergo posttranslational modification, involving truncation of a putative hydrophobic N-terminal region (Figure 2C), to become embedded in the cell membrane, although data were never published (26). This, however, is unlikely to be the case as it is the opposite of what we have observed. Our data show that DnaB is intact in early growth and C-terminally truncated in middle-late growth only in cytosolic and not in membrane-associated fractions, although in overnight cultures some non-specific degradation was apparent. It may be that the cytosolic proteases involved have no access to membrane bound DnaB or that the proteolytically sensitive region of DnaB has a different more compact, protease resistant, conformation. The fact that DnaB is intact in early growth and in the membrane associated fraction makes unlikely premature termination and/or translation of dnaB mRNA. A careful inspection of the dnaB sequence does not reveal any potential termination and/or translational slippage signals.
A shorter DnaB version was reported in the dnaB19 mutant (carrying the A379T mutation) at permissive temperatures although again no data were ever published (26). The pKW1 plasmid, indigenous to B. subtilis, did not replicate in dnaB19, whereas it replicated only in dimeric form in dnaB1 (carrying the P113L mutation within DnaBN), suggesting that DnaBC may be involved in resolution of newly replicated plasmids (27). Since membrane-associated DnaB is resistant to proteolysis we propose that, in addition to other functions, it may also be involved in plasmid resolution. It is clear that DnaBC is crucial for initiation of DNA replication and since, as shown here, binds to ss and dsDNA and induces the formation of higher order DnaB oligomers, these activities must be an integral part of its primosomal function.
Linking oriC with the cell membrane
The precise structure/function relationships of the DnaB oligomers remain to be elucidated. Here we can only speculate regarding their structure(s). DNA-induced oligomerization and bead-like structures along the DNA have been visualized by AFM (18). In the same studies intense foci suggested that DnaB is capable of forming ‘piles’ on the DNA with some DnaB molecules inevitably not associated with the DNA in such piles. A speculative suggestion is that this type of oligomerization may be relevant in linking together DnaB associated at the oriC with DnaB bound to the membrane. Replication initiation in B. subtilis likely occurs at the inner face of the cell membrane (24–26) and DnaB may play a role in controlling this process by recruiting the oriC to the inner face of the membrane (29). This may occur by DnaB oligomers forming a cross-bridge with the oriC via interactions with DnaD and with the membrane via interactions with membrane bound DnaB. The C-terminal domain may be instrumental in this process with DNA acting as the initial stimulus to cause cooperative cross-tetramer higher order oligomers. Such cross-bridging oligomers may link oriC with the inner surface of the cell membrane during exponential growth when DNA replication is most active. As growth slows down the link with the membrane may be broken as DnaB is being truncated.
Helicase loading
Higher order oligomers may, alternatively, represent ‘bead-like’ structures of successive DnaB-tetramers sandwiched between two DNA molecules similar to the bead-like structures observed by AFM (18). These bead-like structures laterally compact supercoiled DNA (18) but may also be relevant in helicase loading. Somewhat analogous spiral filamentous nucleoprotein structures, that mediate helicase loading, have been suggested for the E. coli DnaC helicase loader (49). The actual molecular mechanism of this DNA-induced oligomerization is also not known. We speculate that it may involve an element of co-operativity unleased by binding to DNA and possibly involving conformational changes in DnaBC. Certain mutations in DnaBC can affect the functional properties of the protein indicating the potential for structural changes in this domain that could affect the function of the protein. For example, priA suppressors in dnaB have been identified in DnaBC, residues 327, 371 and 404 (10). The mutant DnaB proteins were able to load the replicative DnaC helicase onto ssDNA in a PriA-independent but DnaD- and DnaI-dependent manner. Presumably these mutations within DnaBC altered the conformation of the protein and hence its ability to load the replicative helicase. In a similar manner DNA-driven conformational changes may induce co-operative DnaB oligomerization via DnaBC.
Regulating access to oriC
Truncated DnaB may also be part of a mechanism to regulate access to oriC to gradually switch off replication initiation as growth progressively slows down. This can be achieved by abolishing its interaction with DnaD which is believed to be the mechanism by which DnaB is recruited to oriC (14, 29). The molecular details of this interaction are not known but, interestingly, the DnaB S371P mutation causes a gain in ssDNA affinity (21) and an increase in its interaction with DnaD (29). S371 is within the 365–418 region predicted by our data to bind DNA and to induce DNA-dependent oligomerization. It is, therefore, probable that DnaBC mediates the interaction with DnaD. Truncation of DnaB may thus regulate its interaction with DnaD and hence its recruitment to oriC.
Self-regulation of native DnaB by interfering with oligomerization
Another possibility is that truncated DnaB may regulate functional oligomerization of native DnaB. Truncated DnaB, with an intact DnaBN, will still be able to form mixed tetramers with native DnaB thus interfering with its action. An example of interference with oligomerization by a truncated version of the same protein as a means of regulating its function, can be seen in the case of H-NS. E. coli H-NS is a nucleoid associated protein and a global transcriptional regulator (mainly repressor) of several genes. It has a bipartite structure with a coiled-coil N-terminal domain involved in oligomerization and a DNA-binding C-terminal domain (50). Expression of the N-terminal domain has a dominant inhibitory effect on native H-NS mediated repression by interfering with its oligomerization via their mutual coiled-coil domains (51, 52). Furthermore, a naturally-occurring family of H-NS truncated (H-NST) proteins from enteropathogenic E. coli (EPEC), that lack the DNA-binding domain, can functionally antagonize H-NS mediated repression by interfering with oligomerization (53). It is also possible that truncated DnaB may be required for additional unidentified functions mediated exclusively via DnaBN.
DnaB has multiple DNA-binding sites
DnaB has two ssDNA-binding sites located in DnaBN and DnaBC and a dsDNA-binding site within residues 365–428. This region exhibits similarity to a region of the putative replication initiation phage protein Gp49 (29). The S371P point mutation results in a 50-fold increased affinity for ssDNA (21). However, it is unlikely that a serine residue would be involved in a direct interaction with DNA. Instead, it is likely that a proline in this position has affected local secondary structure making it more favourable to bind ssDNA. At this stage, it is not clear whether the ss and dsDNA-binding sites in DnaBC are separate, overlapping or identical. An antibody against a peptide encompassing residues 444–458 inhibits binding to the pUB110 plasmid (47). This is outside the 365–428 region but the observed DNA binding defect is likely to be due to steric problems when a bulky antibody is bound in the vicinity. Further mutagenesis studies will be required to clarify this.
Asymmetric association of DnaB with oriC
DnaB is a non-specific DNA binding protein recruited to the oriC via a protein–protein interaction with DnaD which in turn is recruited by an interaction with DnaA. Chromatin immunoprecipitation targets DnaB that has been recruited at oriC by the DnaA–DnaD complex. Our ChIP-chip data suggest that truncated DnaB is depleted at oriC and that the association of DnaB with oriC is asymmetric and involves the half site located downstream of dnaA. This is the site that contains the AT-rich sequence thought to melt prior to helicase-loading (54). It makes sense for DnaB to be present in the vicinity of this site if it is to participate directly in loading of the DnaC helicase. There is no association of DnaB with the half oriC site located upstream of dnaA. Instead, DnaB binds to an extended area from the middle of dnaA to the end of yaaA suggesting that remodelling of the oriC during initiation of DNA replication is extensive and involves a wide-area beyond the core oriC sites. Loading of the replicative E. coli DnaB helicase at oriC has also been suggested to involve asymmetric binding of the helicase loader DnaC which through a direct interaction with ATP-charged DnaA recruits a helicase hexamer to one DNA strand, while the second helicase hexamer is recruited to the opposite strand by DnaA alone (49). E. coli DnaC has been postulated to form right-handed spiral oligomers that are likely to reflect the formation of true filaments on the DNA during helicase loading (49). DnaI, the B. subtilis homologue of E. coli DnaC, has an equivalent ATPase C-terminal domain (33, 55) while its co-loader DnaB can also form bead-like structures on DNA (18). It would be interesting to determine whether the asymmetric association of DnaB with oriC reflects loading of the replicative helicase DnaC by the DnaI–DnaB dual loader to one of the DNA strands, equivalent to the DnaC-mediated helicase loading in E. coli.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Biotechnology and Biological Sciences Research Council (BBSRC) grant to PS (BB/E006450/1). Funding for open access charge: Biotechnology and Biological Sciences Research Council (BBSRC).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Charikleia Ioannou for help with preliminary western blots and Ed Bolt for help with oligonucleotide purifications.
REFERENCES
- 1.Mott ML, Berger M. DNA replication initiation: mechanisms and regulation in bacteria. Nat. Rev. Microbiol. 2007;5:343–354. doi: 10.1038/nrmicro1640. [DOI] [PubMed] [Google Scholar]
- 2.Messer W. The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol. Rev. 2002;26:355–374. doi: 10.1111/j.1574-6976.2002.tb00620.x. [DOI] [PubMed] [Google Scholar]
- 3.Mackiewicz P, Zakrewsjak-Crewinska J, Zawilak A, Dudek MR, Cebrat S. Where does bacterial replication start? Rules for predicting the oriC region. Nucleic Acids Res. 2004;32:3781–3791. doi: 10.1093/nar/gkh699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erzberger JP, Pirruccello MM, Berger JM. The structure of bacterial DnaA: implications for general mechanisms underlying DNA replication initiation. EMBO J. 2002;21:4763–4773. doi: 10.1093/emboj/cdf496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham EL, Berger JM. Unraveling the early steps of prokaryotic replication. Curr. Opinion Struct. Biol. 2005;15:68–76. doi: 10.1016/j.sbi.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Kaguni JM. DnaA: controlling the initiation of bacterial DNA replication and more. Annu. Rev. Microbiol. 2006;60:351–371. doi: 10.1146/annurev.micro.60.080805.142111. [DOI] [PubMed] [Google Scholar]
- 7.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 8.Heller RC, Marians KJ. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol. Cell. 2005;17:733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 9.Bruand C, Ehrlich SD, Janniere L. Primosome assembly site in Bacillus subtilis. EMBO J. 1995;14:2642–2650. doi: 10.1002/j.1460-2075.1995.tb07262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruand C, Farache M, McGovern S, Ehrlich SD, Polard P. DnaB, DnaD and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Mol. Microbiol. 2001;42:245–255. doi: 10.1046/j.1365-2958.2001.02631.x. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Kurokawa K, Matsuo M, Fukuhara N, Murakami K, Sekimizu K. Identification of temperature-sensitive dnaD mutants of Staphylococcus aureus that are defective in chromosome DNA replication. Mol. Genet. Genomics. 2004;271:447–457. doi: 10.1007/s00438-004-0996-6. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Kurokawa K, Reutimann L, Mizumura H, Matsuo M, Sekimizu K. DnaB and DnaI temperature-sensitive mutants of Staphylococcus aureus: evidence for involvement of DnaB and DnaI in synchrony regulation of chromosome replication. Microbiology. 2007;153:3370–3379. doi: 10.1099/mic.0.2007/009001-0. [DOI] [PubMed] [Google Scholar]
- 13.Ishigo-Oka D, Ogasawara N, Moriya S. DnaD protein of Bacillus subtilis interacts with DnaA, the initiator protein of replication. J. Bacteriol. 2001;183:2148–2150. doi: 10.1128/JB.183.6.2148-2150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marsin S, McGovern S, Ehrlich SD, Bruand C, Polard P. Early steps of Bacillus subtilis primosome assembly. J. Biol. Chem. 2001;276:45818–45825. doi: 10.1074/jbc.M101996200. [DOI] [PubMed] [Google Scholar]
- 15.Turner IJ, Scott DJ, Allen S, Roberts CJ, Soultanas P. The Bacillus subtilis DnaD protein: a putative link between DNA remodeling and initiation of DNA replication. FEBS Lett. 2004;577:460–464. doi: 10.1016/j.febslet.2004.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Allen S, Roberts CJ, Soultanas P. The Bacillus subtilis primosomal protein DnaD untwists supercoiled DNA. J. Bacteriol. 2006;188:5487–5493. doi: 10.1128/JB.00339-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Machόn C, Orta A, Phillips N, Roberts CJ, Allen S, Soultanas P. Single-molecule atomic force spectroscopy reveals that DnaD forms scaffolds and enhances duplex melting. J. Mol. Biol. 2008;377:706–714. doi: 10.1016/j.jmb.2008.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W, Carneiro M.J.VM, Turner IJ, Allen S, Roberts CJ, Soultanas P. The Bacillus subtilis DnaD and DnaB proteins exhibit different DNA remodeling activities. J. Mol. Biol. 2005;351:66–75. doi: 10.1016/j.jmb.2005.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carneiro MJVM, Zhang W, Ioannou C, Scott DJ, Allen S, Roberts CJ, Soultanas P. The DNA-remodelling activity of DnaD is the sum of oligomerisation and DNA-binding activities on separate domains. Mol. Microbiol. 2006;60:917–924. doi: 10.1111/j.1365-2958.2006.05152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider S, Zhang W, Soultanas P, Paoli M. Structure of the N-terminal oligomerisation domain of DnaD reveals a unique tetramerization motif and provides insights into scaffold formation. J. Mol. Biol. 2008;376:1237–1250. doi: 10.1016/j.jmb.2007.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruand C, Velten M, McGovern S, Marsin S, Serena C, Ehrlich SD, Polard P. Functional interplay between the Bacillus subtilis DnaD and DnaB proteins essential for initiation and re-initiation of DNA replication. Mol. Microbiol. 2005;55:1138–1150. doi: 10.1111/j.1365-2958.2004.04451.x. [DOI] [PubMed] [Google Scholar]
- 22.Velten M, McGovern S, Marsin S, Ehrlich SD, Noirot P, Polard P. A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Mol. Cell. 2003;11:1009–1020. doi: 10.1016/s1097-2765(03)00130-8. [DOI] [PubMed] [Google Scholar]
- 23.Nunez-Ramirez R, Velten M, Rivas G, Polard P, Carazo JM, Donate LE. Loading a ring: Structure of the Bacillus subtilis DnaB protein, a co-loader of the replicative helicase. J. Mol. Biol. 2007;367:764–769. doi: 10.1016/j.jmb.2006.12.075. [DOI] [PubMed] [Google Scholar]
- 24.Winston S, Sueoka N. DNA-membrane association is necessary for initiation of chromosomal and plasmid replication in Bacillus subtilis. Proc. Natl Acad. Sci. USA. 1980;77:2834–2838. doi: 10.1073/pnas.77.5.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoshino T, McKenzie T, Schmidt S, Tanaka T, Sueoka N. Nucleotide sequence of Bacillus subtilis dnaB: a gene essential for DNA replication initiation and membrane attachment. Proc. Natl Acad. Sci. USA. 1987;84:653–657. doi: 10.1073/pnas.84.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sueoka N. Cell membrane and chromosome replication in Bacillus subtilis. Prog. Nucleic Acids Res. 1998;59:34–53. doi: 10.1016/s0079-6603(08)61028-4. [DOI] [PubMed] [Google Scholar]
- 27.Watabe K, Forough R. Effects of temperature sensitive variants of the Bacillus subtilis dnaB gene on the replication of low-copy-number plasmid. J. Bacteriol. 1987;169:4141–4146. doi: 10.1128/jb.169.9.4141-4146.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato Y, McCollum M, McKenzie T, Laffan J, Zuberi A, Sueoka N. In vitro type II binding of chromosomal DNA to membrane in Bacillus subtilis. J. Bacteriol. 1991;173:7732–7735. doi: 10.1128/jb.173.23.7732-7735.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rokop ME, Auchtung JM, Grossman AD. Control of DNA replication initiation by recruitment of an essential initiation protein to the membrane of Bacillus subtilis. Mol. Microbiol. 2004;52:1757–1767. doi: 10.1111/j.1365-2958.2004.04091.x. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Adams JC. Technical considerations on the use of horseradish peroxidase as a neuronal marker. Neuroscience. 1977;2:141–145. doi: 10.1016/0306-4522(77)90074-4. [DOI] [PubMed] [Google Scholar]
- 33.Soultanas P. A functional interaction between the putative primosomal protein DnaI and the main replicative DNA helicase DnaB in Bacillus. Nucleic Acids Res. 2002;30:966–974. doi: 10.1093/nar/30.4.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuck P. Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys J. 1998;75:1503–1512. doi: 10.1016/S0006-3495(98)74069-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 37.Cho BK, Knight EM, Barrett CL, Palsson B.Ø. Genome-wide analysis of Fis binding in Escherichia coli indicates a causative role for A-/AT-tracts. Genome Res. 2008;18:900–910. doi: 10.1101/gr.070276.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herrick J, Kohiyama M, Atlung T, Hansen FG. The initiation mess? Mol. Microbiol. 1996;19:659–666. doi: 10.1046/j.1365-2958.1996.432956.x. [DOI] [PubMed] [Google Scholar]
- 39.Ogura Y, Imai Y, Ogasawara N, Moriya S. Autoregulation of the dnaA-dnaN operon and effects of DnaA protein levels on replication initiation in Bacillus subtilis. J. Bacteriol. 2001;183:3833–3841. doi: 10.1128/JB.183.13.3833-3841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Vries YP, Hornstra LM, de Vos WM, Abee T. Growth and sporulation of Bacillus cereus ATCC 14579 under defined conditions: temporal expression of genes for key sigma factors. Appl. Environ. Microbiol. 2004;70:2514–2519. doi: 10.1128/AEM.70.4.2514-2519.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruand C, Ehrlich SD. The Bacillus subtilis dnaI gene is part of the dnaB operon. Microbiol. 1995;141:1199–1200. doi: 10.1099/13500872-141-5-1199. [DOI] [PubMed] [Google Scholar]
- 42.Cole C, Barber JD, Barton GJ. The Jpred 3 secondaray structure prediction server. Nucleic Acids Res. 2008;36(Web Server issue):W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallacxe IM, Wim A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 44.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogasawara N, Moriya S, Mazza PG. Nucleotide sequence and organization of dnaB gene and neighbouring geneson the Bacillus subtilis chromosome. Nucleic Acids Res. 1986;14:9989–9999. doi: 10.1093/nar/14.24.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kracilek AV, Day AJ, Wake RG, King GF. A sequence similarity between proteins involved in initiation and termination of bacterial chromosome replication. Biochem J. 1991;275(Pt 3):823. doi: 10.1042/bj2750823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watabe K, Forough R. Identification of the product of dnaB gene in Bacillus subtilis. Biochem. Biophys. Res. Commun. 1987;145:861–867. doi: 10.1016/0006-291x(87)91044-8. [DOI] [PubMed] [Google Scholar]
- 48.Meile JC, Wu LJ, Ehrlich SD, Errington J, Noirot P. Systematic localization of proteins fused to the green fluorescent protein in Bacillus subtilis: identification of new proteins at the DNA replication factory. Proteomics. 2006;6:2135–2146. doi: 10.1002/pmic.200500512. [DOI] [PubMed] [Google Scholar]
- 49.Mott ML, Erzberger JP, Coons MM, Berger J. Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell. 2008;135:623–634. doi: 10.1016/j.cell.2008.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dorman CJ, Hinton J.CD, Free A. Domain organization and oligomerisation among H-NS-like nucleoid-associated proteins in bacteria. Trends Microbiol. 1999;7:124–128. doi: 10.1016/s0966-842x(99)01455-9. [DOI] [PubMed] [Google Scholar]
- 51.Williams RM, Rimsky S, Buc H. Probing the structure function and interactions of the Escherichia coli H-NS and StpA proteins by using dominant negative derivatives. J. Bacteriol. 1996;178:4335–4343. doi: 10.1128/jb.178.15.4335-4343.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Free A, Porter ME, Deighan P, Dorman CJ. Requirement for the molecular adapter function of StpA at the Escherichia coli bgl promoter depends upon the level of truncated H-NS protein. Mol. Microbiol. 2001;42:903–918. doi: 10.1046/j.1365-2958.2001.02678.x. [DOI] [PubMed] [Google Scholar]
- 53.Williamson HS, Free A. A truncated H-NS-like protein from enteropathogenic Escherichia coli acts as an H-NS antagonist. Mol. Microbiol. 2005;55:808–827. doi: 10.1111/j.1365-2958.2004.04421.x. [DOI] [PubMed] [Google Scholar]
- 54.Krause M, Ruckert B, Lurz R, Messer W. Complexes at the replication origin of Bacillus subtilis with homologous and heterologous DnaA protein. J. Mol. Biol. 1997;274:365–380. doi: 10.1006/jmbi.1997.1404. [DOI] [PubMed] [Google Scholar]
- 55.Ioannou C, Schaeffer PM, Dixon NE, Soultanas P. Helicase binding to DnaI exposes a cryptic DNA-binding site during helicase loading in Bacillus subtilis. Nucleic Acids Res. 2006;34:5247–5258. doi: 10.1093/nar/gkl690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.