


Abstract
Recombinase-mediated cassette exchange (RMCE) exploits the possibility to unidirectionally exchange any genetic material flanked by heterotypic recombinase recognition sites (RRS) with target sites in the genome. Due to a limited number of available pre-fabricated target sites, RMCE in mouse embryonic stem (ES) cells has not been tapped to its full potential to date. Here, we introduce a universal system, which allows the targeted insertion of any given transcriptional unit into 85 742 previously annotated retroviral conditional gene trap insertions, representing 7013 independent genes in mouse ES cells, by RMCE. This system can be used to express any given cDNA under the control of endogenous trapped promoters in vivo, as well as for the generation of transposon ‘launch pads’ for chromosomal region-specific ‘Sleeping Beauty’ insertional mutagenesis. Moreover, transcription of the gene-of-interest is only activated upon Cre-recombinase activity, a feature that adds conditionality to this expression system, which is demonstrated in vivo. The use of the RMCE system presented in this work requires one single-cloning step followed by one overnight gateway clonase reaction and subsequent cassette exchange in ES cells with efficiencies of 40% in average.
INTRODUCTION
A number of different strategies for the modification of the mouse genome have been used, most of which involve homologous recombination (HR) techniques to introduce genetic material into desired genetic locations. However, HR efficiency is often dependent on the nature of the genomic target site itself as well as on the design of the targeting vector. The use of site-specific recombinases (SSR) allows secondary modifications of previously inserted cassettes (1,2). Recognition motifs of SSRs usually consist of two palindromic sequences separated by an asymmetric spacer, which determines their orientation. Engineered target sites with defined base pair changes in the spacer or inverted repeats were also shown to recombine efficiently with each other. Generally, SSRs will recombine identical (homotypic) recognition targets (for a review, see 3,4). Although originally derived from Bacteriophage P1 or S. cerevisiae respectively, Cre (causes recombination in the phage P1 genome) and FLP (flippase; named for its ability to flip DNA segments in S. cerevisae) recombinases have been demonstrated to function efficiently in human cell lines and mouse embryonic stem (ES) cells (2,5–7). Recently, the German Gene Trap Consortium (GGTC) introduced the use of multipurpose cassettes, which are delivered randomly to the genome by retroviral gene trapping and allow post-entrapment modifications (8,9). We used the so-called FlEx (FLP-excision; 10) technology in order to conditionally inactivate or reactivate gene trap mutations by using two independent FlEx arrays, which each allow unidirectional inversions of trapping cassettes. The FlEx method involves two subsequent recombination events, the first of which is a FLP inversion, and the second is a FLP excision. The result is an inverted gene trap vector cassette flanked by two heterotypic FLP recombination target sites (FRT and F3) that can be re-inverted in a cell-specific manner by Cre recombinase but could serve as well as a genomic docking-site for FLP recombinase-mediated cassette exchange (RMCE; 11,12). By representing a decrease in entropy, the reversal of excision (i.e. the integration of DNA cassettes) occurs against thermodynamic and kinetic barriers and is therefore highly inefficient transiently (13). However, as we demonstrate, the combination of an alternative selection marker and a codon-optimized, thermostable FLP recombinase (FLPo; 14) resulted in efficient exchange of DNA cassettes, with ‘FlExed’ β-galactosidase/neomycin-resistance (β-geo) cassettes.
The power of this exchange system lies within the size of the GGTC and Eucomm sublibraries, which were established using FlEx-containing gene trap vectors. With a total number of 7013 independent genes containing FlEx vector insertions that were characterized molecularly by genomic splinkerette (splk) sequence tags (15; http://www.genetrap.de; http://www.eucomm.org), the libraries represent catalogues of mouse ES cells, with the potential to establish mouse lines for each clone. RMCE in FlEx containing gene trap clones facilitates the placement of any cDNA construct under the control of previously trapped endogenous promoters of choice, which will usually recapitulate their expression in vivo, if the interaction of promoters with other transcriptional elements is not influenced by the insertion.
Therefore, in order to design a powerful RMCE vector system, compatible with the established GGTC FlEx library, we developed a simple procedure that involves a single-cloning step into a Gateway Entry vector (Invitrogen) followed by an overnight Gateway reaction to end up with the final RMCE vector. This circular vector is co-transfected along with FLP recombinase into FlEx clones, followed by hygromycin selection for 9 days. Since exchange efficiencies typically range around 40% on average, few clones need to be isolated and PCR—screened using generic primers in order to distinguish correctly exchanged alleles from randomly inserted cassettes. Even more easily, any cDNA, which is already situated between the attL 1/2 sites of a pEntry vector, can be introduced directly into the exchange vector described here without further cloning steps.
MATERIALS AND METHODS
Vectors
Exchange vectors pEX-Dest and pEx-FLP
The pre-Gateway pEX-FLP backbone, designated as pEx-Dest was synthesized (http://www.geneart.com) and consists of an adenoviral splice acceptor sequence (in the following referred to as SA), followed by unique XhoI, SalI and NotI restriction sites, flanked by two identically oriented loxP sites and finally by inversely oriented 5′-FRT and 3′-F3 sites and served as a backbone for further cloning.
The hygromycin-coding sequence followed by a phosphoglyceratekinase (PGK)-polyA signal was PCR-amplified using a set of primers with a 5′-overhang containing the NotI restriction sites and AT-spacer nucleotides: 5′-atgcggccgcgccaccatgaaaaagcctga-3′, 5′-atgcggccgcaagcttctgatggaattaga-3′, using pPGK-Hygromycin-pA as a template (provided by Ralf Kühn). After T/A-cloning in pCR II-Topo vector, the fragment was cloned into the NotI site of pEX-Dest.
Between the 3′ loxP and the F3 site, a unique PmeI restriction site was used to insert a blunt-end Gateway cassette A (Invitrogen) containing an attR 1/2 flanked ccdB cassette (16) to finalize pEx-Dest (Figure 1).
Figure 1.

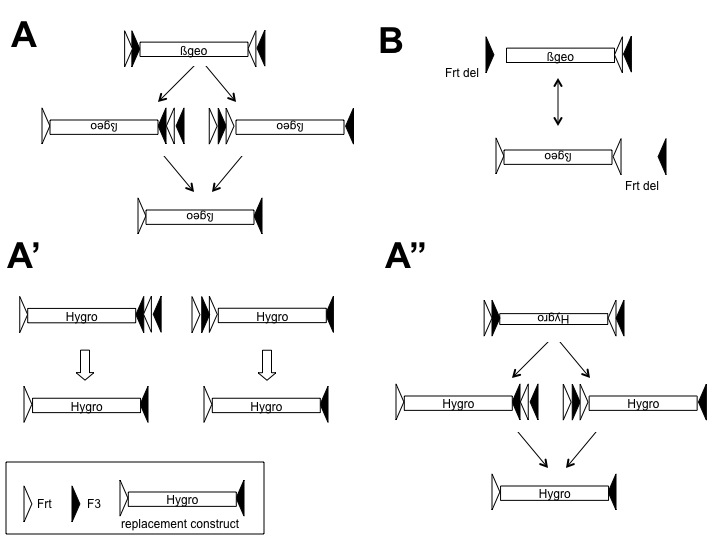
(A) Schematic outline of the procedure for individual modification and use of pEX-FLP. Any DNA sequence, which is cloned between the attL1/2 recombination sites in the pENTR-EX, can be exchanged with an attR1/2 flanked cat ccdb cassette of pEX-Dest in vitro. (B) Schematic illustration of the FLPo RMCE in ES cells. pEX-FLP harbors two heterospecific flippase recombination sites, therefore cotransfection of FLPo and pEX-FLP to any FlEx gene trap ES cell clone allows replacement of β-geo cassette. (C) Analysis of successfully exchanged locus with pEX-FLP-dsRed containing a adSA-dsRed-BGHpA sequence flanked by IR/DR of Sleeping Beauty transposase. A representative PCR screen of seven hygromycin resistant clones (E307D01 derivates, A01–A07) concerning orientation of the genetrap vector and successful exchange (left: primers B045, B048 and B050; right: primers SR, TP). A01 and A06 are inverted β-geo insertions and show an 800-bp band on the left and no band on the right. A02–A05 are successfully exchanged clones and show an 839-bp band on the left and the small 239-bp band on the right. Clone A07 carries the original β-geo insertion and gives rise to the 631-bp band on the left and the 516-bp band on the right.
Entry vector pENTR-EX
The basis for pENTR-EX was a pENTR4 insert (Invitrogen) which contains attL1/2 sites flanking the ccdB cassette (Figure 1A). For pENTREx-FLP-dsRed, the coding sequence of the red fluorescence protein (dsRed, from Discosoma sp.) was PCR-amplified from DsRed2-N1 (Clontech, 17) using the oligonucleotides: 5′-ataagcttaccatggcctcctccgaggac-3′ (fwd), 5′-atgagctcctacaggaacaggtggtggcg-3′ (rev) and cloned into a pCRII Topo plasmid.
Next, the pCRII Topo-dsRed HindIII/SacI fragment was ligated to HindIII/SalI and SacI/SalI fragments of the β-geo gene trap vector (adenoviral splice acceptor, β-galactosidase/neomycin phosphotransferase fusion gene, bovine growth hormone polyadenylation sequence, a gift of Phil Soriano to T. F.) to yield an SA-dsRed-BGHpA harbouring vector. To generate pENTREx-FLP-dsRed, the pENTR4 insert and a plasmid containing a ‘Sleeping Beauty’ (SB) transposon (PGK-Neomycin-pA flanked by SB inverted and direct repeats (IR/DR), the transposase recognition sites) were cut each by EcoRI/SalI and ligated. Then, the SalI and the NotI restriction sites of the ligation product were removed, by digesting with the respective enzymes, overhangs were blunted by T4 DNA polymerase and the vector was religated.
The insert flanked by the IR/DR sequences was obtained after HindIII digest to ligate the resulting cassette with a HindIII fragment of a pEx-FLP-synthetic backbone (http://www.geneart.com), containing a SalI site, followed by two unique oriented lox5171 sites, which flank unique NotI and AscI restriction sites.
Next, an SA-dsRed-pA sequence was released by XhoI and ligated to the attL1/2 sites of the pENTR-EX vector, linearized with SalI.
A hyperactive SB transposase expression plasmid (Caggs-SB100X-pA) is under the control of a chicken-beta actin-CMV enhancer (Caggs) promoter (18). A codon-optimized FLP recombinase (PGK-FLPo-pA; 14) was a gift from Phil Soriano to F.S. Caggs-CRE-IRES-Puro plasmid was described (4).
To generate pENTREx-FLP-hTDP-43(A315T), the SacI fragment of SA-dsRed-BGHpA (see above) was cloned into pBluescriptII-KS followed by the isolation of a BamHI/SalI fragment, which was then cloned to the BamHI/XhoI sites of pENTR4.
A full-length cDNA clone containing TDP-43(A315T) with 5′-HindIII and 3′-SacI overhangs was ligated as a HindIII/SacI fragment to the ade2-SA and BGH-pA of a SA-dsRed-BGHpA cassette in pENTR4, from which the dsRed coding sequence had been removed by HindIII/SacI.
Gateway reaction
The final exchange vectors pEx-FLP-hTDP-43(A315T) and pExFLP-dsRed were established from pENTR-Ex vectors and pEX-Dest in a Gateway reaction using clonase and following standard protocols (Invitrogen).
Gene trap insertions
With the exception of Tardbp and Gtf2ird insertions (Figure 2), gene trap clones with insertions of rFLPRosabetageo or rsFRosabetageo were chosen randomly in either 0, +1 or +2 reading frames. rsFRosabetageo consists of a splice acceptor α-geo polyA cassette, flanked on each side by FRT, F3, loxP and lox5171 or lox511, in head-to-head orientation (FlEx array, Figure 1B). For further details, see http://www.genetrap.de.
Figure 2.

Schematic overview of the efficiency of FLPo-mediated RMCE in FlEx gene trap ES cell clones. Shown are all transfected gene trap clone insertions and the corresponding rounded efficiency of exchange reaction of pEx-FLP-dsRed in three different reading frames (0 upper, +1 middle and +2 lowest values in percent, red values indicating the frame matching the gene trap insertion). For the Tardbp insertion, the exchange vector pEx-FLP-hTDP-43(A315T) was used. All gene trap clones used had insertions in an intron of the respective gene, and the ratio on the right indicates in which intron out of how many total introns the insertion is located. On the left, the size of the genes is indicated in kilobases, and the total size of the genes is not drawn in scale, with omitted DNA indicated. Untranslated and translated exons according to Ensembl. MGI Gene Identifiers and absolute expression levels in ES cells are listed in Supplementary Table S1.
ES cell culture
Gene trap clones
Transfections were performed using the FlEx conditional GGTC gene trap clones (for further details see http://www.genetrap.de):
E079H11, E068C09, E311D09, E224B05, D045A10, E287F07, E326E05, E307D01, E326E12, E288B02, E224H09, E326E04 and E284H06.
Insertions were determined by splinkerette (splk)-PCR (15) and confirmed by genomic PCR using the following primers:
E311D09 (Etl4): 5′-gccggaagagatgctgagtc-3′, 5′-tacccgtgtatccaataaaccc-3′
E068C09 (Etl4): 5′-aaactggttttcattggggatca-3′, 5′-tacccgtgtatccaataaaccc-3′
E326E05 (Msi2): 5′-tcccccatgtttctgtaattgg-3′, 5′-gccaaacctacaggtgggtcttt-3′
E307D01 (Msi2): see hygromycin excision.
E287F07 (Fnbp1): β-geo insertion not confirmed.
E079H11 (Gtf2ird1): 5′-atcgaatgtagcccaggatg-3′, 5′-gccaaacctacaggtgggtcttt-3′
E326C04 (Ahdc1): 5′-catcttgaacctcaagtttgccttt-3′, 5′-tacccgtgtatccaataaaccc-3′
E284H06 (Ahdc1): 5′-ctggcttcctcccacttgtgtt-3′, 5′-tacccgtgtatccaataaaccc-3′
E224H09 (Ahdc1): 5′-agaggtgaccctgctggaaatg-3′, 5′-tacccgtgtatccaataaaccc-3′
E288B02(Scpep1): 5′-ccaaggtgggaaagatgaggtg-3′, 5′-gccaaacctacaggtgggtcttt-3′
E326E12 (Scpep1): 5′-cacatggtgaccttcagagcag-3′, 5′-gccaaacctacaggtgggtcttt-3′
D045A10 (Tardbp): 5′-ccaagtcccatggtgacaac-3′, 5′-cgagtgttgcttcggagag-3′
Recombinase-mediated cassette exchange
All clones were derived from feeder independent E14 Tg2A.4 cells (19; a gift from Kent Lloyd to T. F.) gene trap lines, with the exception of D045A10, which was derived from a TBV2 cell line (20) and was cultured on mouse embryonic fibroblast feeder layer. ES cell lines were grown under standard culture conditions (http://www.genetrap.de). Cells were co-electroporated with 30 µg supercoiled plasmid DNA of pEX-FLP and 70 µg of FLPo and selected for hygromycin resistance after 48 h (150 µg/ml, Sigma-Aldrich) for at least 9 days.
About 107 cells were electroporated per experiment with an EPI 2500 Elektroporations-Impulsgenerator 0–2500 V (Fischer), cells were pulsed for 2 ms at 300 V in 0.4-cm cuvettes in 700 µl phosphate-buffered saline (PBS). After electroporation, ES cells were plated on gelatine-coated culture dishes (5 × 106 cells/100 mm).
Hygromycin resistant colonies were transferred to 96-well dishes and expanded to three replicates of 48-wells. One plate was frozen as a stock, one was used to determine β-gal activity and one to isolate genomic DNA for further analysis.
Cre transfection
One exchange clone (E307D01 derivative) was transfected with 50 µg of supercoiled Caggs-CRE-IRES-Puro plasmid. After electroporation, 50% of electroporated cells were plated on a 5 × 100-mm gelatine-coated dishes and cultured for 48 h without selection. Pooled genomic DNA served as a template for PCR analysis. The remaining cells of the electroporation were plated on a 1 × 100-mm gelatine-coated dish and puromycin-selected for 5 days (1 µg/ml, Sigma). After expansion, total RNA was extracted (Trizol) and subsequent RT-PCR was performed.
SB transfection
One exchanged clone (E307D01 derivative) was transfected with variable amounts of supercoiled SB plasmid (0, 10 and 70 µg). After plating each electroporation on 1 × 100-mm gelatine-coated dishes, cells were cultured for 48 h, and genomic DNA was extracted of each pooled dish for PCR analysis.
Analysis of exchange clones
5′ FRT site PCR
Before cassette exchanges, the integrity of 5′ FRT sites was determined by PCR using the oligonucleotides: SR 5′-gccaaacctacaggtggggtcttt-3′ and B034 5′-tgtaaaacgacgggatccgcc-3′ (9). Loss of a 5′ FRT site yields a 548-bp product instead of a 673-bp product.
X-gal staining
Initially β-Gal-positive gene trap clones become β-Gal-negative after successful cassette exchange or gene trap inversion, therefore by β-gal activity false positive clones can be excluded. X-Gal staining was performed as described (21).
Southern blot analysis
To evaluate copy number and possible random insertions after RMCE, genomic DNA of exchanged clones was analysed in a Southern blot analysis using a 800-bp neomycin (Pst I fragment of pPKJ; 22) and a 727-bp hygromycin (EcoR I/Sca I fragment of pEX-FLP) probe. DNA from 151 independent clones (90xE224B05, 12xE288B02 and 10xE326E12 derivates) was digested overnight at 37°C by HindIII for both probes and run on 0.8% agarose gel in 1xTAE. Gels were blotted on a nylon membrane (Hybond N) and hybridized following standard procedures.
FLP inversion PCR
Conditional gene trap insertions are flanked by FlEx cassettes. Therefore, FLPo also inverted the β-geo gene trap vector, which resulted in false negative β-gal activity. To analyse all RMCE clones, a multiplex PCR was performed, which yielded either the inversion band of about 800 bp or the original band of 630 bp using following oligonucleotides: 5′-ctccgcctcctcttcctccat-3′ (B045), 5′-cctcccccgtgccttccttgac-3′ (B048) and 5′-tttgaggggacgacgacagtat-3′ (B050). Correctly exchanged clones produce a 839-bp fragment in this assay for pExFLP-dsRed (Figure 1C) and 448 bp for pEx-FLP-hTDP-43(A315T; Figure 4C).
Figure 4.

In vivo excision of the hygromycin-resistance cassette. (A) The gene trap vector rFLPRosabetageo, which was inserted in intron 1 of the mouse Tardbp gene (clone D045A10 in Fig. 2), was replaced by pEx-FLP-hTDP-43(A315T). A mutant mouse line with this genomic modification was established and crossed with Rosa26-Cre deleter mice. (B) The RMCE allele in the mouse Tardbp intron 1 after Cre-mediated excision of the hygromycin-resistance cassette. (C) Left: total protein was extracted from embryonic heads (lanes 1–3), after mating to Cre-deleter mice at E17.5 or HEK293 cells (positive control, lane 4). Three western blots were done in parallel using antibodies against human Tdp-43 (hTDP-43), human and mouse TDP-43 (mTDP-43) and β-actin. TDP-43 runs at ∼45 kDa, β-actin runs at 42 kDa. Right: DNA for genotyping was obtained from tails. PCR was performed using the primers SR/TP or B048/B045 as depicted in (A). The presence of Cre was detected using primers specific for Cre-recombinase. Exc: excision (of the hygromycin-resistance cassette); Ret: retention (of the hygromycin-resistance cassette). LTR: long terminal repeat, SA: splicing acceptor, PGK-pA: phosphoglycerate kinase poly A signal; BGHpA: bovine growth hormone polyA signal; attB1/2: gateway clonase recognition sites.
Exchange PCR
Clones were screened for successful exchange by using an internal oligonucleotide in the splice acceptor of pEX-FLP-dsRed and one in the 5′-LTR: 5′-gccaaacctacaggtggggtcttt-3′ (SR) and 5′-atcaaggaaaccctggactactg-3′ (TP). Using these primers, positive exchange resulted in a 239-bp band, no exchange yielded a 631-bp PCR product. For pEx-FLP-hTDP-43(A315T), a positive exchange produced a 262-bp product.
Further analysis
Hygromycin excision by Cre
This experiment was first performed in vitro, using the exchange clone A03, a derivative from gene trap clone E307D01. To analyse Cre-mediated excision, DNA of five pooled transfected dishes (50 µg Caggs-Cre-IRES-Puro) compared to the original exchange clone was extracted and screened by PCR for hygromycin excision using following oligonucleotides: 5′-atcaaggaaaccctggactactg-3′ (TP) and 5′-gccaaacctacaggtggggtcttt-3′ (SR). Undeleted hygromycin cassette led to a 239-bp product, after successful deletion a 619-bp product was amplified.
To analyse the resulting dsRed expression after Cre-mediated deletion under control of the endogenous promoter, RNA of three individual clones and the original exchange clone was extracted and analysed in a RT-PCR followed in a triplex PCR. Three oligonucleotides were designed, one situated in Msi2 exon II (5′-aatgtttatcggtggactgagc-3′), one in Msi2 exon III (5′-cgtttcgttgtgggatctct-3′) and an internal primer in the dsRed cassette (5′-gtgcttcacgtacaccttggag-3′). This PCR yielded a 427-bp product for the wild-type genomic sequence, a 550-bp fragment after successful Cre excision and a 94-bp fragment of the wild-type transcript.
In vivo excision of hygromycin by Cre was done using Rosa26Cre transgenic mice (Taconic; 006467-T-F Heterozygous C57BL/6NTac-Gt(ROSA)26Sortm16(cre)Arte). Transgenic Cre mice were mated to pEx-FLP-hTDP-43(A315T) mutants, and embryos were sacrificed at E17.5. Genomic DNA was isolated from tails, and genotyping was done using SR/TP and B048/B045 primer combinations and Cre-specific primers pCre1 5′-atgcccaagaagaagaggaaggt-3′ and pCre2 5′-gaaatcagtgcgttcgaacgctaga-3′. Undeleted hygromycin produced a band of 262 bp, and deletion of hygromycin led to a slightly larger fragment of 321 bp. The product for Cre-specific primers was 447 bp. For pEx-FLP-hTDP-43(A315T), the SR/TP and B048/B045 PCR product sizes after hygromycin deletion differed in size from all other clones tested. The reason for this was the absence of SB-transposase IR/DR recognition sites from the pEx-FLP-hTDP-43(A315T) vector (Figure 4).
Western blot analysis
Isolated proteins (RIPA buffer) were run on Nu–PAGE 10% Bis–Tris gel (Invitrogen) and transferred onto PVDF membrane (Pall Corporation). ES cell protein was probed with a polyclonal rabbit anti RFP antibody (Abcam, 1:5000 dilution) or a monoclonal mouse anti-beta-actin (Biozol, 1:5000). The secondary antibody was peroxidase-conjugated goat anti-rabbit (Jackson Immuno Research Laboratories, Inc., 1:10 000 dilution) or goat anti-mouse (Jackson Immuno Research Laboratories, Inc., 1:10 000). Mouse tissue protein was probed with TARDBP polyclonal antibody: 10782-2-AP; Proteintech Group, Inc.: Purified rabbit anti human TARDBP polyclonal Antibody (dilution: 1:1500) and TARDBP monoclonal antibody: anti-human TARDBP antibody (ab57105; ABCAM; 1.25 µg/ml). For signal detection, ECL Detection Reagents I + II (GE Healthcare UK Limited) was used in conjunction with Amersham Hyperfilm ECL.
Transposase remobilization by SB100
This experiment was performed using exchange clone A03 (E307D01 derivative), which was transfected with different amounts of SB plasmid (0, 10 and 70 µg). To analyse mobilization of the dsRed cassette by the SB100 transposase, two PCRs with one 3′ external reverse oligonucleotide (B045) and two different forward primers, either in the BGHpA of the dsRed cassette (B048) or in the hygromycin coding sequence (H) were performed. Mobilization events led to a 1009-bp product using the H/B045 primer combination in addition to the 839-bp product (B048/B045). Oligonucleotide sequences are as follows: 5′-caagctctgatagagttggtcaag-3′ (H), 5′-cctcccccgtgccttccttgac-3′ (B048) and 5′-ctccgcctcctcttcctccat-3′ (B045).
RESULTS
The vectors
Any given DNA can be introduced into pEX-Dest via the Gateway (Invitrogen) system; therefore, the RMCE system introduced here can be applied universally. For this purpose, it was designed as a two-component vector system, consisting of pEX-Dest, harboring an in vitro selection marker and all necessary recombinase recognition sites (RRS) and the shuttle vector pENTR-EX. Both components harbour the corresponding attL/R sites to insert any sequence of interest to establish the final pEX-FLP in a Gateway reaction. Instead of pENTR-EX, any Gateway-compatible entry vector could be used (Figure 1A).
pENTR-EX
The vector pENTR4 (Invitrogen) served as a source for attL1/2 recombination sites in pEX-FLP. The cat/ccdb cassette was replaced by the IR/DR flanked adSA-dsRed-BGHpA-lox5171-lox5171 fragment (Figure 1C). The features of the inserted cassette are (i) inverted and direct repeats (IR/DR), which are recognized by the SB transposase; (ii) an adenoviral splice acceptor sequence (SA) followed by the promoterless coding sequence of the red fluorescence protein (dsRed); (iii) the Bovine Growth Hormone poly(A) sequence (BGHpA); (iv) two identically oriented lox5171 sites which flank unique cloning sites, for later removal of an additional cassette, if needed. Only in the case of D045A10 (insertion in Tardbp), the cat/ccdb cassette was replaced by an ade-2 SA-flanked human cDNA (kindly provided by Manuela Neumann) carrying an A315T mutation (23) followed by BGHpA (Figure 4).
pEX-Dest
The vector was designed for FLP-mediated RMCE with FlEx gene trap vectors (Figure 1A; 8). The features of the pEX-Dest are (i) the face-to-face oriented 5′ FRT and 3′ F3 recombination sites, which flank all other functional parts; (ii) the adSA followed by the hygromycin resistance gene; (iii) two head-to-tail oriented loxP sites flanking the hygromycin selection cassette for Cre-mediated excision in vitro or in vivo; (iv) the PGK poly(A) signal; (v) the clonase recombination sites attR1/2 to enable the insertion of the pENTR-EX insert.
RMCE using FlEx gene trap clones
All gene trap clones presented in this study had insertions of a retroviral SA-β-geo-pA vector, flanked by RRS in FlEx configuration (8; see Figure 1B). In brief, the cassette consists of a combination of inversely oriented original and mutant SSRs for FLP and Cre recombinases. This configuration allows unidirectional inversion of the β-geo cassette by the respective recombinase. Since the pEX-FLP insert was flanked with one set of heterotypic oppositely oriented SSRs for FLP recombinase, transient co-transfection of pEX-FLP and FLPo into FlEx gene trap clones allows recombination between the homotypic RRS. The RMCE strategy is outlined in Figure 1B.
Different outcomes after co-transfection of pEX-FLP and FLPo and hygromycin selection are possible: (i) recombination of pEX-FLP and β-geo in identical orientation in the event of recombination between the outer FRT/F3 sites; (ii) recombination of pEX-FLP in inverse orientation, resulting in hygromycin sensitivity in the event of recombination between the inner FRT/F3 sites. Inverse orientation was not selected, since it may be followed by another FlEx inversion and excision event, leading to correctly exchanged clones; (iii) inversion of the β-geo cassette combined with a random insertion of the pEX-FLP within a transcriptionally active gene, leading to false positive clones (see Supplementary Figure S1 for a comprehensive description of theoretical events before and after RMCE).
Different PCR strategies were used to identify successfully exchanged clones. Hygromycin resistant clones were first analysed for β-gal activity. β-Gal-negative clones were further screened by ‘exchange PCR’ using a 5′ primer in the LTR (SR) and a nested primer in the splice acceptor (TP), which yielded either the hygromycin band of 239 bp (A02–A05), the β-geo band of 516 bp (A07) or no band after β-geo inversion (A01; Figure 1C right). To distinguish clones with inverted β-geo cassettes and random insertions of pEX-FLP and to identify false positive clones as described earlier, a triplex PCR using primers B045, B048 and B050 was performed, which yielded different product sizes depending on the orientation of the gene trap vector (8). DNA from RMCE clones yielded a slightly larger product (839 bp; A02–A05) as compared to the β-geo inversion (800 bp; A01, A06), whereas original β-geo insertions led to a 631-bp product (A07; Figure 1C, left). An assortment of clones was additionally screened in Southern blot analyses using lacZ, neomycin and hygromycin probes to detect multiple or possible random insertions. More than 90% of successfully exchanged clones showed an expected a loss of lacZ and neomycin resistance (data not shown).
A total of 13 different conditional gene trap clones with insertions in eight independent genes were co-transfected with pEX-FLP in three reading frames (see Figure 2). The Scpep1 clones were electroporated twice and results were averaged.
Insertions in the same gene were either chosen for different gene trap vector reading frames or different introns (Ahdc1, Msi2, Etl4 and Scpep1) to determine possible differences in exchange efficiencies between 5′ and 3′ insertions in the same gene and between different reading frames of pEX-FLP. Cell number and DNA amount per electroporation were identical in all cases to obtain comparable results. The efficiencies of RMCE were calculated by relating the numbers of correctly exchanged clones to the total number clones isolated per electroporation. Of the gene trap insertions used for exchange reactions, seven were in frame 0, four in frame +1 and two in frame +2, ten in more 5′ introns and three in more 3′ introns of the respective gene (Figure 2).
Total clone number of all 38 electroporations varied from less than 10 clones in 17 cases up to several hundred clones per plate, the average clone number per electroporation was 124. The exchange efficiency of the matching frame varied from 0 to 93% and was 40% on average. The efficiency of both nonmatching frames per clone varied between 0 and 100%, and the average efficiency was 25%. In 8 of the 13 different electroporated gene trap clones, the matching frame was most efficient, while in five cases a nonmatching frame led to successful exchange.
In five cases (Etl4 3′, Fnbp1, Scpep1 5′ and both Msi2 insertions), only one electroporated pEX-FLP frame led to hygromycin resistant clones, and in four out of these, it represented a frame matching the β-geo insertion. The only exception was the Msi2 insertion in intron 2, where only frame 0 yielded positive clones, while the β-geo insertion was in frame +1 (Figure 2).
In vitro excision of selection marker
Further screens were performed using the successfully exchanged clone A03 (Figure 1C) derived from the E307D01 (Msi2, intron 4) gene trap clone.
To minimize secondary effects, the selection marker of pEX-FLP vector was designed to be removable by Cre recombinase in vitro or in vivo. To demonstrate excision in vitro, clone A03 was transiently transfected with Caggs-Cre-IRES-Puro plasmid with and without subsequent puromycin selection. Genomic DNA of plates without selection were pooled and screened by PCR with primers located within the splice acceptor sequence (hygromycin and dsRed) and in the LTR. This PCR yielded different-sized products before (239 bp) or after (619 bp) excision of the selection marker (Figure 3A). Lanes marked 1–5 (pooled transfected plates) show both band sizes, whereas untransfected A03 only shows the smaller product. Without puromycin selection, Cre excision occurred only partially. To ensure the splicing to the dsRed cassette after Cre excision, the same transfection was performed followed by puromycin selection and cDNA of individual clones was screened by PCR with primers located in Msi2 exon II and an internal primer in the dsRed coding sequence (Figure 3B). This primer set yielded a 550-bp product in three independent clones (lanes 1–3), which was sequence-verified. As an internal control, a third primer located in Msi2 exon III was added, which yielded a 94-bp product of the wild-type transcript. As further controls, genomic DNA of two gene trap clones (E307D01 and E326E12) and the plasmid DNA of pEX-FLP were used. Gene trap genomic DNA templates yielded the wild-type genomic product (427 bp) including the intron 3/4.
Figure 3.

(A) Removal of hygromycin resistance upon Cre activity. Cre-transfected exchange clone A03 (E307D01/pExFLP-dsRed derivative) was analysed using primers located within both the hygromycin/dsRed cassettes (TP) and the LTR of the original gene trap vector (SR). PCR on total genomic DNA of five pooled transfected dishes (lanes 1–5) compared to genomic DNA of clone A03 before Cre transfection (A03 −Cre). The internal primer (TP) gives rise to the larger product (619 bp) after successful excision of the hygromycin cassette. Before hygromycin excision, primer TP amplifies a smaller product of 239 bp. (B) RT-PCR and triple PCR analysis to determine splicing to the dsRed after hygromycin excision by using external primers in exon II (5) of the insertion locus (Msi2) and an internal dsRed primer (I) and as internal wild-type control a primer located in exon III (3) of the Msi2 gene. cDNA of individual clones derived from Cre-transfected clone A03 after puromycin selection (lanes labelled 1–3) compared to cDNA of clone A03 before Cre transfection (lane A03 −Cre). As control genomic DNA of two gene trap clones (E307D01 and E326E12) and pEX-FLP-dsRed plasmid was used. (C) Western blot analysis of protein extracted from ES cells probed with a polyclonal RFP antibody. As controls wild-type ES cells either untransfected (WT) or transiently transfected with two different dsRed expression plasmids (WT + RFPI + II) were used. Exchange clones A03 and D04 were analysed prior (−CRE) and after (+CRE) transient CRE transfection. DsRed protein positive samples show a 27-kDa band. Lower blot shows loading control with beta actin monoclonal antibody (42 kDa).
After Cre excision of the selection marker, subsequent expression of the dsRed gene was validated in a western blot analysis. Wild-type ES cells, two different positive exchange clones (A03 and D04, E307D01-derived) before Cre transfection and two puromycin resistant clones after transient Cre transfection (A03 and D04 derived) were screened. As positive control, wild-type ES cells were transiently transfected with two different dsRed expression plasmids (WC150-DsRed2N1 and WC156-DsRed2N1), expanded for 3 days and total protein was extracted. No RFP was detectable before Cre transfection (A03 −Cre and D04 −Cre) or in wild-type protein (Figure 3C).
Germline transmission and excision of hygromycin in vivo
In order to analyse the germline potential of exchange clones, one successfully exchanged clone (Tardbp; clone 5/10) was injected into 73 C57Bl/6 host blastocysts after superovulation. Injected embryos were transferred to four pseudopregnant CD1 females and yielded 16 pups of which five were high-percentage agouti colour male chimeras and one was a low-percentage female. Three of the five male chimeras transmitted the mutation into the germline.
In the exchange vector for the Tardbp insertion, the gene encoding TDP-43 (24), the cDNA of interest was a human mutant form of TDP-43 carrying an A315T mutation, which was recently found in familial ALS patients (23). In order to activate the expression of the human isoform, mice were bred to Rosa26-Cre expressing animals (Tac-Gt(ROSA)26Sortm16(cre)Arte; Taconic), and total protein was isolated from embryos at E17.5. As we have shown in Figure 4, embryos carrying both the Cre recombinase and the exchange vector pEx-FLP-TDP-43 (A315T) exhibit an excision of the hygromycin cassette and initiate the expression of the human mutant isoform. The human isoform was detected by a monoclonal antibody (ABCAM), which does not crossreact with the mouse protein. A polyclonal antibody (Proteintech) which recognizes both the mouse and human TDP-43, served as a control along with an antibody for β-actin.
Overall, seven RMCE clones were injected into host blastocysts of which two went germline and four are in the chimera stage with a chimaerism over 80%, as based on agouti colour. One injected clone did not yield chimeras.
Mobilization of dsRed by SB transposase
Clone A03 (E307D01 derivate, Msi2 insertion in intron 4) was transiently transfected with different amounts (0, 10 and 70 µg) of a SB expression vector. Pooled genomic DNA of transfected plates was screened by PCR. SB100-mediated excision was analysed by using two primer combinations. Primers were located either in the hygromycin coding sequence (H) or in the BGHpA of the dsRed (B048) cassette (which would be mobilized by SB100) and with an external primer located 5′ of the F3 site (B045). Templates of plates transfected with 10 and 70 µg SB100 showed both bands, the SB-excision band with primers H/B048 of 1009 bp and the unexcised band of 839 bp with primers B048/B045, whereas the controls 0 µg and A03 without transfection (A03-SB) only yielded the 839-bp product. As further control, the gene trap clone E307D01 was used as template and yielded only a 631-bp band with primers B048/B045 (Figure 5). All PCR products were sequence-verified.
Figure 5.

PCR analysis of SB100 transfected exchange clone to determine mobilization of IR/DR flanked cassette. Genomic DNA of pooled SB100 transfected dishes (0, 10 and 70-µg SB100 plasmid) of exchange clone A03 (E307D01/pExFLP-dsRed derivative) was screened with primers located 5′ of the F3 site (B045), in the pA of dsRed (B048) and in the hygromycin-resistance cassette (H). DNA was screened with either primer combination H/B045 or B048/B045, as controls genomic DNA of clone A03 (A03 −SB) and E307D01 was used. Primers H/B045 yielded a 1-kb product after successful excision of the IR/DR flanked cassette. Without mobilization, a 839-bp band was amplified by primers B048/B045, whereas the original gene trap insertion led to a 631-bp band.
DISCUSSION
A number of studies have recently demonstrated the advantages of RMCE systems in order to generate mice carrying different knock-in and even ‘humanized’ alleles. However, these were entirely based on one-gene-at-a-time approaches including all the tedious vector design and gene-targeting efforts (25–29).
As we demonstrate in this work, RMCE with existing FlEx promoter traps allows the modification of an entire ES cell library in a highly efficient and straightforward manner. Typical exchange efficiencies ranged around 40% in average with a preference towards the first introns of genes, which is in agreement with the overall insertion preferences of splice-acceptor containing gene trap vectors in large-scale screens (30,31).
As we determined, overall exchange efficiencies were directly related to the amount of FLPo recombinase plasmid. At otherwise identical conditions, amounts of FLPo plasmid below 50 µg resulted in few correct exchange events. Only FLPo plasmid amounts of 70 µg resulted in significant exchange efficiencies (data not shown). The required high-FLPo amounts may also reflect the fact that a number of different events are possible when exchanging for FlEx gene traps, namely FLP-mediated inversions and excisions before the exchange reaction (Supplementary Figure S1). Nevertheless, the relatively high-FLPo recombinase plasmid amounts applied here raised some concern about unspecific secondary integrations of pEx-FLP. In three cases (two Ahdc1—clones; Figure 2), 500–2080 clones were selected as hygromycin-resistant after electroporation at identical conditions, as compared to only 50–100 clones in the majority of experiments, underlining initially the theoretical possibility of secondary pEx-FLP insertions in transcriptionally active genes. Therefore, 96 hygromycin-resistant Ahdc1 exchange clones were analysed by Southern blotting using a hygromycin probe. We found that only 2% of successfully exchanged clones carried secondary hygromycin insertions (not shown), and the overall success rate was comparable to other clones. We therefore suspected that differences in endogenous gene expression levels may account for the differing total clone numbers after RMCE. Therefore, expression levels of the trapped genes in Figure 2 were determined according to the absolute gene expression values using a recently published Affymetrix Chip Array data set, providing quantitative information on the expression levels of 7435 ENSEMBL genes in an undifferentiated E14 ESCs (32). However, no correlation with exchange efficiencies was determined (Supplementary Table 1).
Correctly exchanged clones are identified in a generic PCR using primers located within the hygromycin resistance cassette as well as in the LTR of the original gene trap vector, which is retained in the locus after exchange (Figure 1B). By Southern blot, we found more than 90% of successfully exchanged clones with an expected loss of both lacZ and neomycin (data not shown).
In order to express the cDNA of interest, the loxP-flanked hygromycin resistance cassette needs to be removed. As we show for the clones E307D01 and D045A10 (Figure 3), hygromycin was excised upon Cre activity in vitro. As we demonstrate for DsRed in Figure 3C, the gene-of-interest becomes active only ‘after’ Cre activity, a feature that adds conditionality to this RMCE system if tissue-specific Cre recombinases are used in vivo.
Cre removal may be performed in vivo in order to avoid higher passages of ES cell clones, which may result in reduced germline rates. As we demonstrate, mating of a mouse line carrying a human mutant form of Tardbp to a ubiquitously expressing Cre deleter line resulted in solid expression of the human isoform in mouse embryos after hygromycin excision (Figure 4). These mutants represent potential mouse models for familial amytrophic lateral sclerosis and will be published elsewhere.
Although a successful exchange reaction is expected to occur in a frame-dependent manner, we show that matching frames become less important towards the 5′ end of genes. From the GGTC library, the existence of larger numbers of β-geo gene traps, selected using vectors with Kozak consensus translational start sequences in different reading frames inserted in identical introns of several genes, support our view: for this analysis, only clones with a clear genomic tag were chosen. Of a total of 5220 introns trapped, 3472 were trapped in only one, 1083 in two and 665 in all three reading frames. Of the clones, which were trapped in only one frame, 63% were either in the first or second intron (referred to as 5′ insertion), while 84 and 87% of hits with multiple frames were in the 5′-end of genes. As likely reasons for this, we suspect initiation of translation starting at the Kozak site of the hygromycin resistance cassette, especially in cases where the first exon is untranslated (e.g. Tardbp clone; Figure 2) as well as alternative splicing. In addition, we do not rule out the presence of alternative promoters, which were recently predicted for 40–50% of all human and mouse genes (33). Therefore, fusions with endogenous peptides are possible, but cannot be predicted. In case fusions are not desired, the use of T2A cassettes will ensure the production of two independent peptides (34).
A seemingly similar RMCE system with even slightly higher efficiencies (75%) of their exchange vector pEXCH1 was recently introduced by Cobellis et al. (35). The exchange vector pEXCH2, however, which was designed to carry the transgenic cargo, uses a non-removable TK promoter-driven antibiotic resistance cassette, which selected higher numbers of random insertions and therefore had exchange efficiencies of a lower rate (7%) when compared to pEx-FLP. In addition, pEXCH vectors are Gateway-incompatible and do not allow for conditional gene expression.
The first-generation of conditional gene trap vectors (8) bears a 50% risk of losing a 5′ Frt site, which most likely occurs during reverse transcriptase activity in packaging cells. This risk was reduced to 10% after exchange of the spacer between 5′ Frt and 5′ F3 site (unpublished results). The loss of the 5′ Frt site may lead to continuous F3/F3 recombination and therefore rotation of the original trap or—after RMCE—the replacement vector, until transient FLP recombinase expression is lost. RMCE events on inverted β-geo FlEx vectors with deletions of the 5′ Frt site result in, depending on the presence of FLP recombinase, rotating hygromycin cassettes, of which only correctly orientations are selected by the antibiotic. After sequencing of 5′ FlEx arrays, we identified one clone (Tardbp; Figure 2), which lacked the 5′ Frt site. However, also in this case, overall exchange efficiencies were 40–50%.
In summary, the RMCE system presented here extends the possibilities for further conditional or non-conditional gene expression, restricted only by the availability of FlEx gene trap clones. First, it represents an alternative to conventional transgenic technology with entire control over copy number and endogenous expression already in vitro. This should circumvent, e.g., epigenetic inactivation as a result of high-copy number followed by cosuppression of transgenes (36). Second, FlEx-RMCE facilitates genetic pathway dissection by allowing e.g. replacement of presumed upstream transcriptional regulators by their potential targets. Third, this system provides a simple method in order to replace given mouse genes with the human counterpart on the transcriptional level. As we demonstrate, it is especially suited for the expression of human disease-causing genes containing point mutations in the corresponding cells and tissues of the mouse. The combined GGTC and EUCOMM FlEx libraries currently represent more than 791 independent confirmed or candidate genes for human genetic diseases. Fourth, this system will be particularly of interest to further extend the catalogue of Cre-expressing mouse lines.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: NGFN (01KW0106, 01KW9948 and 0313435B to W.W., Z.I. and T.F.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Phil Soriano, Peter W. Chang and Ralf Kühn for probes and constructs. Mutant Tardbp cDNA was cloned and kindly provided by Manuela Neumann and Kathrin Dittmar. They thank Jordi Guimera for scientific discussion and critical reading of the manuscript. They appreciate the expert technical assistence of Denise Herold, Helga Grunert and Irina Rodionova. All gene trap sequence tags are deposited in the Ensembl and NCBI genomic databases.
REFERENCES
- 1.Seibler J, Bode J. Double-reciprocal crossover mediated by FLP-recombinase: a concept and an assay. Biochemistry. 1997;36:1740–1747. doi: 10.1021/bi962443e. [DOI] [PubMed] [Google Scholar]
- 2.Seibler J, Schübeler D, Fiering S, Groudine M, Bode J. DNA cassette exchange in ES cells mediated by FLP recombinase: an efficient strategy for repeated modification of tagged loci by marker-free constructs. Biochemistry. 1998;37:6229–6234. doi: 10.1021/bi980288t. [DOI] [PubMed] [Google Scholar]
- 3.Branda CS, Dymecki SM. Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev. Cell. 2004;6:7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- 4.Schnütgen F, Stewart AF, von Melchner H, Anastassiadis K. Engineering embryonic stem cells with recombinase systems. Methods Enzymol. 2006;420:100–136. doi: 10.1016/S0076-6879(06)20007-7. [DOI] [PubMed] [Google Scholar]
- 5.O’Gorman S, Fox DT, Wahl GM. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science. 1991;251:1351–1355. doi: 10.1126/science.1900642. [DOI] [PubMed] [Google Scholar]
- 6.Dymecki SM. FLP recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice. Proc. Natl Acad. Sci. USA. 1996;93:6191–6196. doi: 10.1073/pnas.93.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bethke B, Sauer B. Segmental genomic replacement by Cre-mediated recombination: genotoxic stress activation of the p53 promoter in single-copy transformants. Nucleic Acids Res. 1997;25:2828–2834. doi: 10.1093/nar/25.14.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnütgen F, De-Zolt S, Van Sloun P, Hollatz M, Floss T, Hansen J, Altschmied J, Seisenberger C, Ghyselinck NB, Ruiz P, et al. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc. Natl Acad. Sci. USA. 2005;102:7221–7226. doi: 10.1073/pnas.0502273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Floss T, Schnütgen F. Conditional gene trapping using the FLEx system. Methods Mol. Biol. 2008;435:127–138. doi: 10.1007/978-1-59745-232-8_9. [DOI] [PubMed] [Google Scholar]
- 10.Schnütgen F, Doerflinger N, Calleja C, Wendling O, Chambon P, Ghyselinck NB. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol. 2003;21:562–565. doi: 10.1038/nbt811. [DOI] [PubMed] [Google Scholar]
- 11.Schlake T, Bode J. Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci. Biochemistry. 1994;33:12746–12751. doi: 10.1021/bi00209a003. [DOI] [PubMed] [Google Scholar]
- 12.Oumard A, Qiao J, Jostock T, Li J, Bode J. Recommended method for chromosome exploitation: RMCE-based cassette-exchange systems in animal cell biotechnology. Cytotechnology. 2006;50:93–108. doi: 10.1007/s10616-006-6550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baer A, Bode J. Coping with kinetic and thermodynamic barriers: RMCE, an efficient strategy for the targeted integration of transgenes. J. Curr. Opin. Biotechnol. 2001;12:473–480. doi: 10.1016/s0958-1669(00)00248-2. [DOI] [PubMed] [Google Scholar]
- 14.Raymond CS, Soriano P. High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE. 2007;2:e162. doi: 10.1371/journal.pone.0000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horn C, Hansen J, Schnütgen F, Seisenberger C, Floss T, Irgang M, De-Zolt S, Wurst W, von Melchner H, Noppinger PR. Splinkerette PCR for more efficient characterization of gene trap events. Nat. Genet. 2007;39:933–934. doi: 10.1038/ng0807-933. Erratum in: Nat Genet., 2007, 39, 1528. [DOI] [PubMed] [Google Scholar]
- 16.Bahassi EM, Salmon MA, Van Melderen L, Bernard P, Couturier M. F plasmid CcdB killer protein: ccdB gene mutants coding for non-cytotoxic proteins which retain their regulatory functions. Mol. Microbiol. 1995;15:1031–1037. doi: 10.1111/j.1365-2958.1995.tb02278.x. [DOI] [PubMed] [Google Scholar]
- 17.Yarbrough D, Wachter RM, Kallio K, Matz MV, Remington SJ. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-A resolution. Proc. Natl Acad. Sci. USA. 2001;98:462–467. doi: 10.1073/pnas.98.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mátés L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009;41:753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- 19.Skarnes WC. Gene trapping methods for the identification and functional analysis of cell surface proteins in mice. Methods Enzymol. 2000;328:592–615. doi: 10.1016/s0076-6879(00)28420-6. [DOI] [PubMed] [Google Scholar]
- 20.Hill PD, Wurst W. Methods in Enzymology. Vol. 225. New York: 1993. Screening of novel pattern formation of genes using gene trap approaches; pp. 664–681. [DOI] [PubMed] [Google Scholar]
- 21.Uez N, Lickert H, Kohlhase J, de Angelis MH, Kühn R, Wurst W, Floss T. Sal4 isoforms act during proximal-distal and anterior-posterior axis formation in the mouse embryo. Genesis. 2008;46:463–477. doi: 10.1002/dvg.20421. [DOI] [PubMed] [Google Scholar]
- 22.Adra CN, Boer PH, McBurney M. Cloning and expression of the mouse pgk-1 gene and the nucleotide sequence of its promoter. Gene. 1987;60:65–74. doi: 10.1016/0378-1119(87)90214-9. [DOI] [PubMed] [Google Scholar]
- 23.Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White C.L., III, Bigio EH, Caselli R, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cesari F, Rennekampff V, Vintersten K, Vuong LG, Seibler J, Bode J, Wiebel FF, Nordheim A. Elk-1 knock-out mice engineered by FLP recombinase-mediated cassette exchange. Genesis. 2004;38:87–92. doi: 10.1002/gene.20003. [DOI] [PubMed] [Google Scholar]
- 26.Liu K, Hipkens S, Yang T, Abraham R, Zhang W, Chopra N, Knollmann B, Magnuson MA, Roden DM. Recombinase-mediated cassette exchange to rapidly and efficiently generate mice with human cardiac sodium channels. Genesis. 2006;44:556–564. doi: 10.1002/dvg.20247. [DOI] [PubMed] [Google Scholar]
- 27.Roebroek AJ, Reekmans S, Lauwers A, Feyaerts N, Smeijers L, Hartmann D. Mutant Lrp1 knock-in mice generated by recombinase-mediated cassette exchange reveal differential importance of the NPXY motifs in the intracellular domain of LRP1 for normal fetal development. Mol. Cell Biol. 2006;26:605–616. doi: 10.1128/MCB.26.2.605-616.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jägle U, Gasser JA, Müller M, Kinzel B. Conditional transgene expression mediated by the mouse beta-actin locus. Genesis. 2007;45:659–666. doi: 10.1002/dvg.20342. [DOI] [PubMed] [Google Scholar]
- 29.Sato T, Kawamura Y, Asai R, Amano T, Uchijima Y, Dettlaff-Swiercz DA, Offermanns S, Kurihara Y, Kurihara H. Recombinase-mediated cassette exchange reveals the selective use of Gq/G11-dependent and -independent endothelin 1/endothelin type A receptor signaling in pharyngeal arch development. Development. 2008;135:755–765. doi: 10.1242/dev.012708. [DOI] [PubMed] [Google Scholar]
- 30.Floss T, Wurst W. Functional genomics by gene-trapping in embryonic stem cells. Methods Mol. Biol. 2002;185:347–379. doi: 10.1385/1-59259-241-4:347. [DOI] [PubMed] [Google Scholar]
- 31.Hansen J, Floss T, Van Sloun P, Fuchtbauer EM, Vauti F, Arnold HH, Schnütgen F, Wurst W, von Melchner H, Ruiz P. A large-scale, gene-driven mutagenesis approach for the functional analysis of the mouse genome. Proc. Natl Acad. Sci. USA. 2003;100:9918–9922. doi: 10.1073/pnas.1633296100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nord AS, Vranizan K, Tingley W, Zambon AC, Hanspers K, Fong LG, Hu Y, Bacchetti P, Ferrin TE, Babbitt PC, et al. Modeling insertional mutagenesis using gene length and expression in murine embryonic stem cells. PLoS ONE. 2007;2:e617. doi: 10.1371/journal.pone.0000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baek D, Davis C, Ewing B, Gordon D, Green P. Characterization and predictive discovery of evolutionarily conserved mammalian alternative promoters. Genome Res. 2007;17:145–155. doi: 10.1101/gr.5872707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat. Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 35.Cobellis G, Nicolaus G, Iovino M, Romito A, Marra E, Barbarisi M, Sardiello M, Di Giorgio FP, Iovino N, Zollo M, et al. Tagging genes with cassette-exchange sites. Nucleic Acids Res. 2005;33:e44. doi: 10.1093/nar/gni045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bingham PM. Cosuppression comes to the animals. Cell. 1997;90:385–387. doi: 10.1016/s0092-8674(00)80496-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}