


Abstract
Mammalian inosine triphosphatase encoded by ITPA gene hydrolyzes ITP and dITP to monophosphates, avoiding their deleterious effects. Itpa− mice exhibited perinatal lethality, and significantly higher levels of inosine in cellular RNA and deoxyinosine in nuclear DNA were detected in Itpa− embryos than in wild-type embryos. Therefore, we examined the effects of ITPA deficiency on mouse embryonic fibroblasts (MEFs). Itpa− primary MEFs lacking ITP-hydrolyzing activity exhibited a prolonged doubling time, increased chromosome abnormalities and accumulation of single-strand breaks in nuclear DNA, compared with primary MEFs prepared from wild-type embryos. However, immortalized Itpa− MEFs had neither of these phenotypes and had a significantly higher ITP/IDP-hydrolyzing activity than Itpa− embryos or primary MEFs. Mammalian NUDT16 proteins exhibit strong dIDP/IDP-hydrolyzing activity and similarly low levels of Nudt16 mRNA and protein were detected in primary MEFs derived from both wild-type and Itpa− embryos. However, immortalized Itpa− MEFs expressed significantly higher levels of Nudt16 than the wild type. Moreover, introduction of silencing RNAs against Nudt16 into immortalized Itpa− MEFs reproduced ITPA-deficient phenotypes. We thus conclude that NUDT16 and ITPA play a dual protective role for eliminating dIDP/IDP and dITP/ITP from nucleotide pools in mammals.
INTRODUCTION
The accumulation of modified or damaged bases in genomic DNA is a major threat for the alteration of genetic information as a result of mutagenesis or even for programmed cell death. It has been established that such damaged bases in genomic DNA arise from two independent pathways: one is a consequence of the direct modification of the normal bases in the DNA and the other is that of the incorporation of modified nucleotides generated in resident nucleotide pools (1,2).
To control the quality of the nucleotide pools, organisms possess a number of nucleoside triphosphatases, which degrade non-canonical nucleoside triphosphates to the corresponding monophosphates. We had identified and characterized three mammalian enzymes: (i) oxidized purine nucleoside triphosphatase encoded by MTH1 gene for 8-oxo-2′-deoxyguanosine triphosphate (8-oxo-dGTP), 8-oxoGTP, 2-hydroxy-2′-deoxyadenosine triphosphate (2-OH-dATP) and 2-OH-ATP (3,4); (ii) inosine triphosphatase encoded by ITPA gene for deaminated purine nucleoside triphosphates such as 2′-deoxyinosine triphosphate (dITP), ITP and 2′-deoxyxanthosine triphosphate (dXTP) and XTP (5,6); and (iii) a newly discovered enzyme, dCTP pyrophosphatase encoded by DCTPP1 gene for halogenated dCTPs such as 5-iodo-2′-deoxycytidine triphosphate (7).
To clarify the biological significance of the damaged nucleotides and the enzymes that eliminate them, we had previously produced and analyzed knockout mice lacking MTH1 or ITPA. Mth1−/– mice are viable and survive normally but exhibit an increased incidence of spontaneous tumorigenesis in the liver, stomach and lung (8), while Itpa−/– mice die before weaning with features of growth retardation and heart failure (6).
ATP, the most abundant of the nucleotides, plays a fundamental role in a wide variety of cellular processes, including energy transfer, signal transduction, RNA synthesis, cytoskeleton remodeling and muscle contraction. Deamination of adenine at C-6 converts ATP to ITP; such modification is catalyzed by enzymes such as adenosine or AMP deaminase (9), or induced chemically under oxidative stress (7). Because ITP retains a molecular structure similar to that of ATP, it can act as an aberrant substrate replacing ATP in some biological processes (10,11). In the case of cardiac function, a number of sarcomere proteins require ATP for their normal activities. It is likely that during cardiac development in Itpa−/– mice the accumulated ITP competes with ATP, which is required for actomyosin function in the sarcomere, thus causing heart failure (6).
Bradshaw and Kuzminov (12) reported that an Escherichia coli mutant of rdgB gene-encoding inosine triphosphatase has no obvious phenotype; however, the mutant exhibits synergistic lethality in the presence of recA or recBC mutations. They concluded that RdgB acts to avoid incorporation of 2′-deoxyinosine (dI) in DNA and thereby blocks chromosome fragmentation by hydrolyzing dITP in E. coli. These observations strongly suggest that ITPA deficiency in mouse cells also causes chromosomal abnormalities.
In the present study, we examined mouse embryonic fibroblasts (MEFs) prepared from wild-type (Itpa+/+), Itpa+/− and Itpa−/– embryos to explore the cellular dysfunction caused by ITPA deficiency. We found that Itpa−/– embryos accumulated more than eight times higher levels of dI in nuclear DNA than did wild-type embryos. Moreover, Itpa−/– primary MEFs with no ITP-hydrolyzing activity exhibited prolonged doubling times, increased chromosome aberrations and accumulation of single-strand breaks in nuclear DNA. Surprisingly, these phenotypes all disappeared following immortalization of Itpa−/– MEFs, with a significant increase in IDP-hydrolyzing activity accompanied by a decreased accumulation of dI in nuclear DNA. We have thus identified a novel enzyme which constitutes a dual enzyme system for eliminating dITP/ITP and dIDP/IDP from nucleotide pools together with ITPA in mammals.
MATERIALS AND METHODS
Nucleotides
Nucleotides used as substrates for enzyme assay were purchased from Sigma-Aldrich (St Louis, MO, USA), or Jena Bioscience GmbH (Jena, Germany). Separation and purification of nucleotides were performed on a Waters Alliance 2690 HPLC separation module (Waters Corp., Milford, MA, USA) equipped with a Model 996 photodiode array detector and a Wakopak Handy ODS column (4.6 × 250 mm) using 100 mM triethyl ammonium hydrogen carbonate solution (pH 7.4) (Wako Pure Chemicals, Osaka, Japan) as the mobile phase. Purified nucleotides were lyophilized five times with solubilization in distilled water.
Itpa gene knockout mice
Itpa gene knockout mice were established as described (6). Genotypes were analyzed using tail DNA. PCR primers used to detect the wild-type and Itpa mutant alleles were P46 and P47, or P29 and LNEO1, respectively (Supplementary Table S2). Heterozygous male (Itpa+/−) were backcrossed with C57BL/6J female (Itpa+/+) (Clea Japan, Tokyo, Japan) for more than five generations (N > 5). All animals were maintained in an air-conditioned, light/time-controlled, specific-pathogen-free room. All studies were approved by the Animal Care and Use Committee, Medical Institute of Bioregulation, Kyushu University.
Preparation of primary and immortalized MEFs
Twenty embryonic gestation day (E)13.5 and E14.5 embryos were obtained by intercross mating of inbred N10 or N11 Itpa+/− mice (three pairs). Their genotypes were Itpa+/+, Itpa+/− and Itpa−/– at a ratio of 5:10:5. Skin fibroblasts were aseptically isolated from these embryos and at least four independent embryos were used for each genotype (Itpa+/+, Itpa+/−, Itpa−/–). These primary MEFs were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 units/ml penicillin and 100 µg/ml streptomycin at 37°C under 5% CO2 in air. Primary MEFs in culture were harvested by treatment with 0.15% trypsin–0.08% EDTA in PBS and replated for further passage. Those from Passage 2 were stocked as primary MEFs. Spontaneously immortalized MEFs were established after single colony isolation during 30–40 passages. Their genotypes were determined by genomic polymerase chain reaction (PCR) amplification and three lines of immortalized MEFs were independently established from three embryos for each genotype.
Cell proliferation assays
Primary or immortalized MEFs were seeded at 1 × 105 cells (Figures 1C and 4A) or 0.5 × 105 cells (Figure 7C) per well in six-well plates (Nalge Nunc International K.K., Tokyo, Japan). Cells were harvested every 2 days or every day, respectively, and the numbers of cells were counted using a hemocytometer.
Figure 1.

ITPA-deficient primary MEFs exhibit various cellular dysfunctions. (A) ITPA deficiency caused a significantly increased accumulation of inosine in cellular RNA. Inosine level was determined by LC–MS/MS analysis of cellular RNA prepared from embryos (N3). Result of non-repeated measures ANOVA (two-tailed), P = 1.69 × 10−7. Student–Newman–Keuls (SNK) post hoc test, **P < 0.01 (versus Itpa+/+ and Itpa+/−). Data are shown as the mean ± SD (n = 3 independent embryos). (B) ITPA deficiency caused a significantly increased accumulation of deoxyinosine (dI) in nuclear DNA. Deoxyinosine (dI) level was determined by LC–MS/MS analysis of nuclear DNA prepared from embryos (N3). Result of non-repeated measures ANOVA (two-tailed), P = 0.00038. SNK post hoc test, **P < 0.01 (versus Itpa+/+ and Itpa+/−). Data are shown as the mean ± SD (n = 3 independent embryos). (C) ITPA deficiency impairs normal cell proliferation. Primary MEFs (Passage 2) isolated from four separate Itpa−/– embryos showed significant prolonged doubling time in comparison to those from Itpa+/+ and Itpa+/− embryos. Result of repeated measures ANOVA (two-tailed), P = 0.0005. Bonferroni/Dunn post-hoc test, *P < 0.05 (versus Itpa+/−), **P < 0.01 (versus Itpa+/+). Data are shown as the mean ± SD (n = 4 independent MEFs). (D) ITPA deficiency causes G2/M arrest. Primary MEFs (Passage 5) were subjected to flow cytometry analysis and the percentages of Cell-cycle phases in each MEF set were determined. Result of non-repeated measures ANOVA (two-tailed), P = 1.74 × 10−8. Bonferroni post hoc test, **P < 0.01 (versus Itpa+/+ and Itpa+/−). Data are shown as the mean ± SD (n = 3 independent isolates).
Figure 4.

ITPA-deficient phenotypes are reversed during immortalization. (A) Immortalized Itpa−/– MEFs (triangle) showed the same proliferation rate as did immortalized Itpa+/+ (circle) and Itpa+/− (square) MEFs. Error bars represent the SD (n = 3 independent isolates). (B) The frequency of chromosomal abnormalities was decreased in immortalized Itpa−/– MEFs to the levels seen in immortalized Itpa+/+ MEFs. Data are shown as the mean ± SD (n = 3 independent isolates).
Figure 7.

Knockdown of Nudt16 mRNA suppressed ITPA-deficient phenotypes in immortalized Itpa−/– MEFs. (A) Expression of Nudt16 mRNA. To knock down the expression of Nudt16, two different siRNAs (80, 82 or 80 + 82) against Nudt16 mRNA or control siRNA were introduced into immortalized Itpa−/– MEFs. Forty-eight hours after the introduction, total RNA was prepared and quantitative real-time RT–PCR was performed. Levels of Nudt16 mRNA were normalized to those of Gapdh mRNA and their relative values are shown. Data are shown in a bar graph (mean ± SD, n = 3). (B) Expression of NUDT16 protein. Western blotting was performed for crude cell extracts (40 µg) prepared from immortalized Itpa−/– MEFs treated with two Nudt16 siRNAs (80 + 82) or control siRNA, using anti-NUDT16. GAPDH was detected as an internal control. (C) Knockdown of Nudt16 mRNA suppressed proliferation of immortalized Itpa−/– MEFs. Expression of Nudt16 mRNA was blocked using a mix of two different Nudt16 siRNAs and cell proliferation was examined. Circles, Itpa+/+; triangles, Itpa−/–; open marks, control siRNA; closed marks, Nudt16 siRNAs. Result of repeated measures ANOVA, two-tailed, P < 0.0001; Bonferroni/Dunn post hoc test, **P < 0.01 (versus other three measures). Error bars represent the SD (n = 4). (D) Knockdown of Nudt16 mRNA significantly increased the accumulation of inosine in RNA of immortalized Itpa−/– MEFs. Level of inosine in RNA was determined by LC–MS/MS analysis of RNA prepared from immortalized Itpa−/– MEFs treated with control or Nudt16 siRNAs. Result of unpaired Student’s t-test (two-tailed), **P < 0.01. Data are shown as a bar graph with the mean ± SD (n = 3). (E) Knockdown of Nudt16 mRNA significantly increased the accumulation of dI in nuclear DNA of immortalized Itpa−/– MEFs. Level of dI in nuclear DNA was determined by LC–MS/MS analysis of nuclear DNA prepared from immortalized Itpa−/– MEFs treated with control or Nudt16 siRNAs. Result of unpaired Student’s t-test (two-tailed), **P < 0.01. Data are shown as a bar graph with the mean ± SD (n = 3). (F) Increased immunoreactivity against ssDNA in immortalized Itpa−/– MEFs after Nudt16 knockdown. Immunofluorescence microscopy with anti-ssDNA antibody revealed significantly increased immunoreactivity after Nudt16 knockdown. Result of unpaired Student’s t-test (two-tailed), **P < 0.01. Data are shown as a bar graph with the mean ± SD (n = 4). (G) Knockdown of Nudt16 mRNA increased chromosomal abnormalities in immortalized Itpa−/– MEFs. The frequency of chromosomal abnormalities was increased significantly in immortalized Itpa−/– MEFs after Nudt16 knockdown. Result of unpaired Student’s t-test (two-tailed), P < 0.05 (versus control siRNA). Data are shown as pie charts with the mean ± SD (n = 4).
Cell-cycle analysis
Flow cytometric analysis of the cell cycle was performed as described (13). Cells (1 × 106 cells per assay) were centrifuged, washed with phosphate buffered saline (PBS) and suspended in PBS containing 0.2% Triton X-100. We then added 5 µl of RNase A (1 mg/ml) and 50 µl of propidium iodide (PI, 1 mg/ml). DNA content and cell numbers were analyzed with an LSR flow cytometer (Becton Dickinson, San Jose, CA, USA). The data were analyzed using CellQuest and ModFit software (Becton Dickinson).
Karyotype analysis
A 50% confluent culture of MEFs was treated with 0.1 µg/ml colcemid (Nacalai Tesque Inc., Kyoto, Japan) for 30 min. After hypotonic treatment of harvested cells in 75 mM KCl, cells were fixed in freshly prepared Carnoy’s fixative (methanol:acetic acid 3:1), and the cell suspension was dropped onto a glass slide, air-dried and immediately stained with freshly prepared Giemsa staining solution (Merck KGaA, Darmstadt, Germany cat. no. 1.09204.0509, 25× diluted in PBS) for 20 min. After rinsing the slide in PBS twice and in distilled water twice, air-dried slides were cover-slipped using Permount (Fisher Scientific, Waltham, MA, USA; SP15-100). The slide was observed under an Axioscope 2 plus microscope equipped with AxioCam and AxioVision software (Carl Zeiss MicroImaging Japan, Tokyo, Japan). A total 30 cells in metaphase was examined for each preparation.
Quantification of deoxyinosine or inosine by liquid chromatography coupled with tandem mass spectrometry
The preparation and digestion of nuclear DNA samples were as described (14), except that 10 mM 2, 2, 6, 6-tetramethylpiperidine-N-oxyl (TEMPO, Wako Pure Chemicals) and 20 µM 2′-deoxycoformycin (a kind gift from the Chemo-Sero-Therapeutic Research Institute, Kumamoto, Japan), an adenosine deaminase inhibitor, was added at all stages of manipulation, as described by Taghizadeh et al. (15). RNA was prepared using RNeasy Mini Kits (Qiagen Inc., Valencia, CA, USA) according to the manufacturer’s instructions in the presence of 20 mM TEMPO and 20 µM 2′-deoxycoformycin. DNA or RNA samples were digested with nuclease P1 (Yamasa, Chiba, Japan) and alkaline phosphatase (Sigma-Aldrich, P-5521) and digested samples were subjected to LC–MS/MS analysis using the Shimadzu VP-10 HPLC system (SHIMADZU CORPORATION, Kyoto Japan) connected to the API3000 MS/MS system (PE-SCIEX, Applied Biosystems, Foster City, CA, USA), as described (14).
Immunostaining
To detect single-stranded (ss) DNA, the slides were incubated with anti-ssDNA (IBL, Takasaki, Japan; code number 18 731, 1/100 dilution) in combination with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, Carlsbad, CA. USA), as described (16). Nuclei were counterstained with 4′, 6-diamino-2-phenylindole (DAPI, 50 ng/ml; Vector, Burlingame, CA, USA). A cover slide was mounted onto the slide with Vectashield (Vector). The slide was observed using an Axioskop 2 plus microscope equipped with AxioCam and AxioVision software (Carl Zeiss MicroImaging Japan). A total of 100 cells were examined for each preparation.
Inosine triphosphatase and Inosine diphosphatase assays
Embryo samples or pellets of immortalized MEFs (1 × 107 cells) were washed twice with PBS and quickly frozen in liquid nitrogen. Frozen samples in 100 µl of lysis buffer containing 50 mM Tris–HCl (pH 8.0), 50 mM NaCl, 1 mM dithiothreitol and protease inhibitor cocktail (Nacalai Tesque), were sonicated at 4°C. The lysate was centrifuged at 17 360 × g for 60 min and the supernatant was collected as a crude cell extract. The protein concentration was determined with a Protein Assay system (Bio-Rad, Hercules, CA, USA) using bovine serum albumin (Thermo Fisher Scientific Inc., Waltham, MA, USA) as a standard. Inosine triphosphatase or Inosine diphosphatase activities were assayed by measuring the hydrolysis of ITP or IDP to IMP. The reaction mixture contained 50 mM Tris–HCl (pH 8.5), 50 mM MgCl2, 1 mM DTT, 0.2 mM ITP or IDP and 1–10 µg of the crude-cell extract to be examined. The reaction was run at 30°C for 20 min and stopped by adding 150 mM EDTA. The reaction mixture was applied to HPLC analysis as described (6). Separation and quantification of nucleotides were performed by HPLC using a Waters Alliance 2690 separation module equipped with a Model 996 photodiode array detector. A buffer consisting of 75 mM sodium phosphate (pH 6.4), 0.4 mM EDTA, with 20% acetonitrile was used as the mobile phase in a TSK-GEL DEAE-2SW column, 4.6 × 250 mm (Tosoh Corp., Tokyo, Japan).
Quantitative real-time reverse transcription polymerase chain reaction
MEFs were seeded at 1 × 105 cells per well with 500 µl of medium in 24-well plates and cultured to 70–80% confluency, or for 3 days. RNA was extracted from the harvested cells using an Isogen kit (Nippon Gene Inc., Tokyo, Japan). Totally 2 µg of total RNA was subjected to RNase-free DNase I treatment and cDNA synthesis using random decamers and a Cells-to-cDNA II kit (Ambion), according to the manufacturer’s instructions. Quantitative real-time PCR was performed using an ABI Prism 7000 sequence detection system with 10 ng cDNA, a set of Nudt16 primers (FmNud3RT, RmNud3RT; 200 nM) or a set of Gapdh primers (F-Gapdh, R-Gapdh; 50 nM) and Power SYBR Green PCR Master Mix (Applied Biosystems) in a total volume of 25 µl. The PCR reaction was performed as follows: a single cycle of 50°C for 2 min, a single cycle of 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The primers were designed using PRIMER EXPRESS software (Applied Biosystems) and their sequences are shown in Supplementary Table S2. Specificity of the PCR products was established by dissociating curve analysis and by running the products on a 2% agarose gel to verify their size. The Nudt16 mRNA level is expressed relative to the Gapdh mRNA level. Serially diluted cDNA was used to obtain a standard curve for each transcript.
Western blotting
MEFs were seeded at 5 × 105 cells per dish in 10 ml medium in a 90 mm Petri dish (Nalge Nunc International K.K.) and were cultured to 70–80% confluency or for 3 days. Cells were washed twice with PBS and harvested using 2× SDS sample buffer [125 mM Tris–HCl (pH 6.8), 4% SDS, 10% glycerol, 4% 2-mercaptoethanol]. The protein concentration was determined using a Protein Assay system as above. Protein samples were separated by SDS–PAGE and transferred to 0.45 µm Immobilon-P membrane (Millipore Inc., Madison, WI, USA) and western blot analysis using anti-hNUDT16 (1 µg/ml) or anti-ITPA antiserum (1/500 dilution) (5) with horseradish peroxidase-conjugated protein A and an ECL-Plus kit (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) was performed as described (17). The same membrane was treated with WB stripping solution (Nacalai Tesque) and reprobed with anti-GAPDH (Millipore, Inc., Billerica, MA, USA; MAB374, 105× diluted) and HRP-anti-mouse IgG (BD Biosciences, San Jose, CA, USA).
Expression of recombinant mouse NUDT16 protein
An expression vector for the mouse NUDT16 protein was constructed by inserting the NdeI-HindIII fragment of pET28a(+):mNudt16 into the NdeI-HindIII region of pET32a(+) (Merck KGaA), thus Trx·Tag-His·Tag-S·Tag sequences were removed. Escherichia coli BL21 cells were transfected with pET32a(+) vector or pET32a:mNudt16 using a Cell-porator (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's; instructions. Transformants were selected on LB-agar plates in the presence of 30 µg/ml ampicillin. Established transformants were cultured until the OD600 reached 0.6 and then incubated with 1 mM isopropyl β-d-thiogalactoside for a further 3 h. Cells were harvested by centrifugation and resuspended in 1 ml of 2× SDS sample buffer. Samples were subjected to 12.5% SDS–PAGE and the expression of mouse NUDT16 protein without tag was confirmed by western blotting.
Introduction of silencing RNA into immortalized MEFs
Nudt16 siRNA (Ambion/Applied Biosystems, Austin, TX, USA; Silencer Select; s93780, s93782, 25 µM) or control siRNA (Ambion/Applied Biosystems, Silencer Select Negative Control #1 siRNA, cat. no. 4390844) was introduced into immortalized MEFs (Itpa+/+, Itpa−/–) by electroporation using a MicroPorator-mini (Digital Bio Technology, Seoul, Korea, MP-100, 1100 V, 10 ms for 2 pulses) and cells were replated appropriately into six-well plates 1 day after electroporation for further analysis.
Statistical analysis
Statistical analysis was performed using Stat View 5.0 (SAS Institute Inc., Cary, NC, USA). The statistical significance between two groups was determined with Student’s t-test, and that among more than three groups was determined with non-repeated or repeated measures ANOVA with an appropriate correction for multiple comparisons as described in each figure legend. P-values <0.05 are considered statistically significant.
RESULTS
ITPA deficient primary MEFs exhibit various cellular dysfunctions
Itpa−/– mice with a 129·C57BL/6J mixed genetic background exhibited incomplete embryonic lethality and the surviving pups die about 2 weeks after birth with growth retardation and heart failure (6). After backcrossing the heterozygotes (Itpa+/−) to C57BL/6J mice for more than five generations (N5), intercrosses of the obtained Itpa+/− mice yielded Itpa−/– embryos in uterus in accordance with Mendel’s laws until embryonic gestation day (E) 18, but there were few newborn pups (Supplementary Table S1), indicating that ITPA deficiency causes perinatal lethality in a C57BL/6J genetic background.
We confirmed significantly increased accumulation of inosine (567.3 ± 41.4 residues per 106 guanosine) in cellular RNA prepared from Itpa−/– embryos (N14) in comparison to those from Itpa+/+ (10.5 ± 1.50) and Itpa+/− (11.4 ± 1.07) embryos by liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) analysis (Figure 1A), as previously observed in various tissues of surviving Itpa−/– pups (6).
Furthermore, LC–MS/MS analysis of nuclear DNA prepared from embryos revealed that Itpa−/– embryos (E14.5) obtained from intercrosses of Itpa+/− mice (N3) contained significantly more dI in their nuclear DNA (20.1 ± 4.8 residues per 106 nucleosides): more than eight times that measured in Itpa+/+ embryos (2.34 ± 0.76) (Figure 1B). The increased dI levels were also confirmed in Itpa−/– embryos after further backcrossing to C57BL/6J mice (N14, E13.5, 24.5 ± 2.24 dI residues per 106 nucleosides).
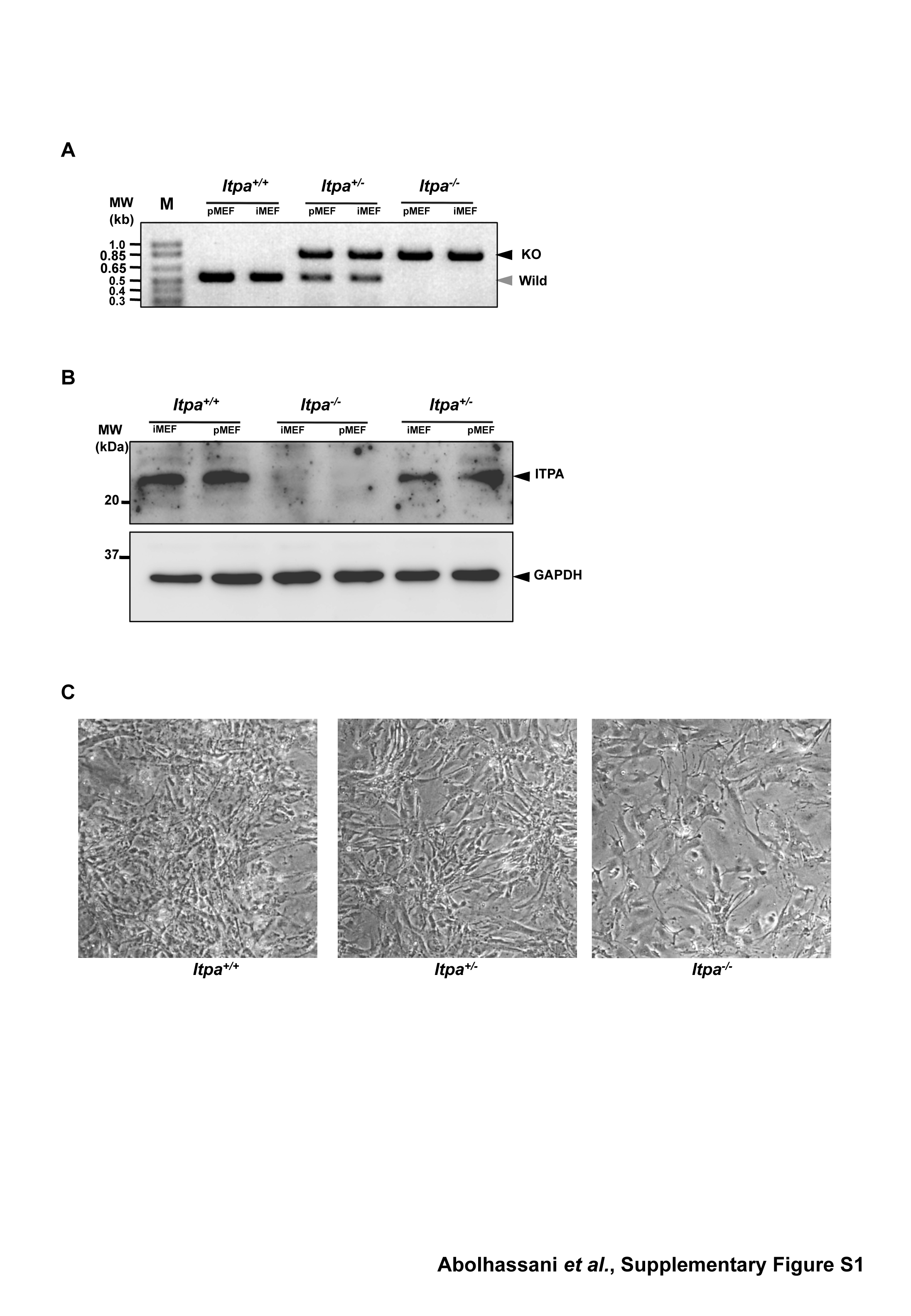
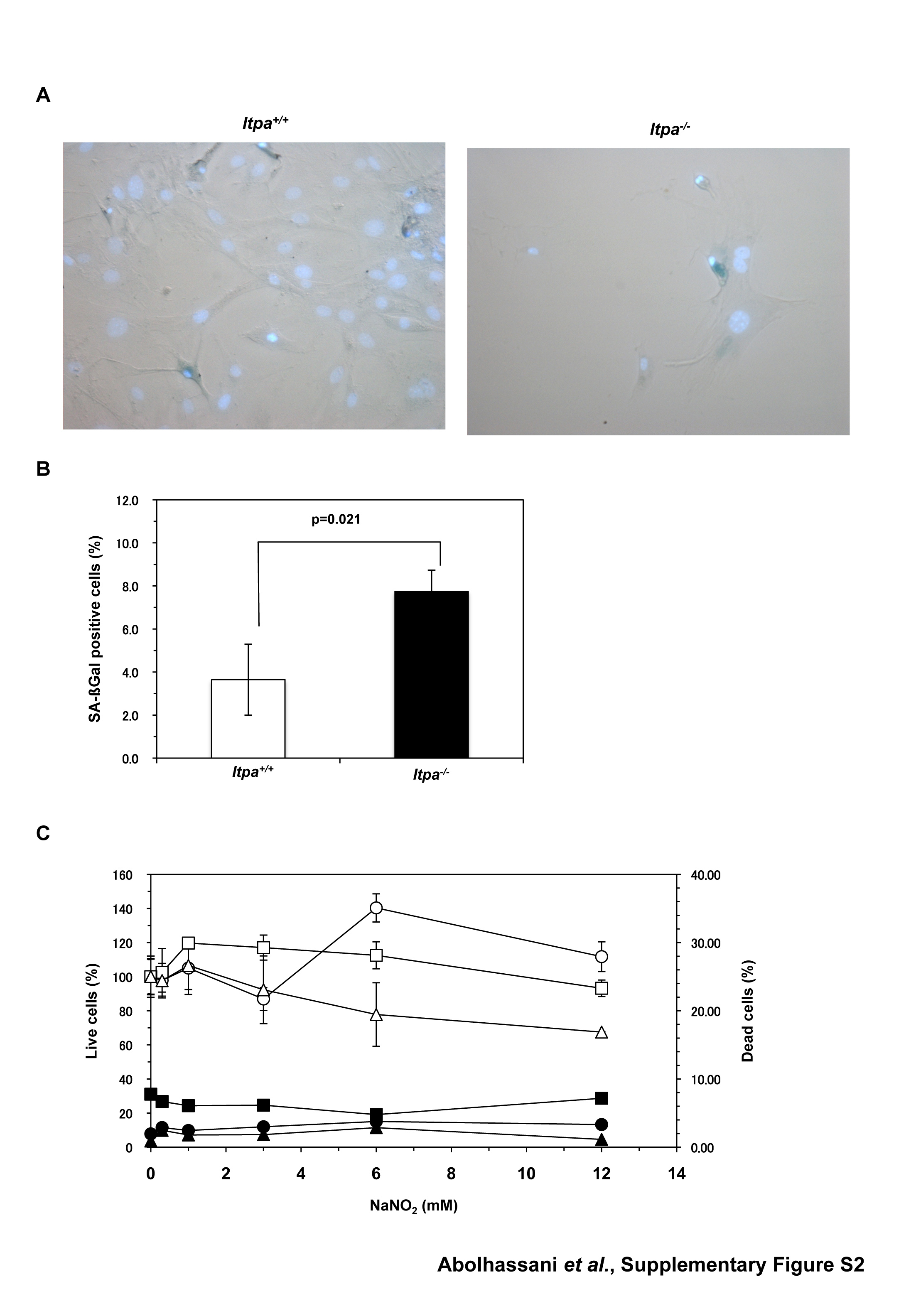
To examine the nature of the cellular dysfunction caused by ITPA deficiency, we isolated embryos (at E13.5 and E14.5) from intercrosses of Itpa+/− littermates (N10, one pair; N11, two pairs) and determined their genotypes (Supplementary Table S1). Among 20 embryos, five were found to be Itpa−/–, 10 were Itpa+/− and the remaining five were Itpa+/+. Then we isolated primary MEFs independently from four embryos of each genotype and their genotypes and the expression levels of ITPA protein were confirmed (Supplementary Figure S1A and B). All Itpa−/– primary MEFs at the second passage showed significantly longer doubling time (132.0 ± 14.6 h) than those from Itpa+/+ (78.6 ± 10.1 h) and Itpa+/− (87.5 ± 14.3 h) embryos (Figure 1C). There was no obvious difference in their morphology under phase contrast microscopy (Supplementary Figure S1C). In Passage 5, we observed essentially the same proliferation deficiency in Itpa−/– MEFs and slightly increased numbers of senescence-associated β-galactosidase (SA-β-Gal)-positive cells in Itpa−/– MEFs (Itpa+/+ versus Itpa−/–, 3.64% versus 7.74%; Supplementary Figure S2A and B). Flow cytometry analysis of the cell cycle revealed that Itpa−/– MEFs exhibited a significant increase in the G2/M phase (Itpa+/+ versus Itpa−/–, 28.4% versus 77.4%; Figure 1D) and a slight increase in the sub G1 fraction (Itpa+/+ versus Itpa−/–, 1.61% versus 5.51%; fraction M1 in Figure 2A). There was an apparent increase in cells with an abnormally increased DNA content in Itpa−/– MEFs compared with Itpa+/+ MEFs (fraction M3 in Figure 2A), indicating that the G2/M phase shown in Figure 1D might have contained some tetraploid cells at the G1 phase (Figure 2B). There was no increase in dead cells detected as propidium iodide (PI)/Hoechst-double positive cells (Supplementary Figure S2C). These results indicate that ITPA deficiency caused delay or arrest in cell-cycle progression. We further observed that exposure of Itpa−/–, but not Itpa+/+, Itpa+/− MEFs, to sodium nitrite (NaNO2), which causes predominant deamination of purine bases (18), resulted in growth suppression without inducing cell death (Supplementary Figure S2C).
Figure 2.

Increased DNA content in ITPA deficient primary MEFs. (A) Flow cytometric analysis of the cell cycle was performed and the sub G1 fraction (M1), diploid fraction (M2) and a fraction with an increased DNA content (M3) were determined. (B) ITPA deficiency increased chromosomal ploidy in primary MEFs. Percentages of diploid, tetraploid and others are shown in pie charts with the mean ± SD (three independent isolates). The frequency of tetraploidy was increased significantly in Itpa−/– MEFs. Results show non-repeated measures ANOVA (two-tailed): P = 0.094. P-value is shown following a Bonferroni post hoc test.
We next examined for chromosomal abnormalities in mitotic cells (Figure 3A). As shown in Figure 3B, chromosomal structural abnormalities were more frequently observed in Itpa−/– MEFs than in Itpa+/+ MEFs, especially premature centromere separation (3.33 times more common), chromatid gaps (2.04 times) and chromatid breakages (1.52 times). Moreover, the percentage of cells with abnormal chromosomes in Itpa−/– primary MEFs was significantly higher than among Itpa+/+ and Itpa+/− MEFs. There was an increase in ploidy abnormalities among Itpa−/– primary MEFs (Figure 2B): thus 41.2% in the mitotic fraction exhibited tetraploidy, while about 25% of mitotic fractions in Itpa+/+ and Itpa+/− MEFs were detected as tetraploids. This confirmed the increase in ploidy among Itpa−/– MEFs.
Figure 3.

ITPA deficiency increases chromosome abnormalities with increased accumulation of SSBs in nuclear DNA. (A) Various chromosome structural abnormalities were observed in Itpa−/– primary MEFs (Passage 2). (B) ITPA deficiency increases chromosome abnormalities. The frequency of chromosomal abnormality was significantly increased in Itpa−/– MEFs in comparison to Itpa+/+ and Itpa+/− MEFs. Result of non-repeated measures ANOVA (two-tailed), P = 0.0149. Bonferroni post hoc test, P < 0.01. Data are shown as pie charts with the mean ± SD (n = 4 independent isolates). (C) Detection of immunoreactivity against ssDNA in Itpa−/– primary MEFs. Immunofluorescence microscopy with anti-ssDNA antibody (green) revealed significantly increased ssDNA immunoreactivity in nuclei (DAPI, blue) of Itpa−/– primary MEFs (Passage 2) compared with the wild-type (Itpa+/+). (D) Significant Increase in the ssDNA-positive population in Itpa−/– primary MEF tested by unpaired Student’s t-test (two-tailed): **P < 0.01. Data are shown in a bar graph (mean ± SD, n = 4 independent isolates).
Because chromatid gaps or breakages are most likely to be caused by the accumulation of single- or double-strand breaks in DNA, we examined levels of single-strand breaks (SSBs) in DNA using an antibody against single-stranded (ss) DNA (anti-ssDNA) (Figure 3C) (16). Immunofluorescence microscopy with anti-ssDNA revealed that the percentages of immunoreactive nuclei in Itpa−/– primary MEFs were significantly higher than in Itpa+/+ MEFs (Figure 3D). Thus, more SSBs had accumulated in the DNA of Itpa−/– primary MEFs. Furthermore, the nuclei were heterogeneous in Itpa−/– MEFs and both the small and large nuclei exhibited ssDNA immunoreactivity (Figure 3C).
Immortalization of Itpa−/– MEFs reversed the ITPA-deficient phenotypes with increased ITP/IDP-hydrolyzing activity
To obtain established cell lines with ITPA deficiency, spontaneously-immortalized MEFs were isolated after 30–40 passages of each primary MEF line. We noticed that each immortalized MEF population showed the same proliferation rate, irrespective of their genotype (Figure 4A). The extent of chromosomal abnormalities (Figure 4B), and both levels of dI (2.49 ± 0.24 dI residues per 106 nucleosides) and ssDNA (positive in 6.17 ± 0.68% of cells) in nuclear DNA (Figure 7E and F) were similarly decreased in immortalized Itpa−/– MEFs. Furthermore, the G1 phase fraction increased significantly in immortalized Itpa−/– MEFs (Figure 5A) compared with primary MEFs (Figure 2A). Because the percentages of tetraploids were significantly higher in both primary and immortalized Itpa−/– MEFs than Itpa+/+ or Itpa+/− MEFs (Figures 2B and 5B), the G2/M fraction in immortalized Itpa−/– MEFs was apparently less than in primary Itpa−/– MEFs.
Figure 5.

Increased DNA content in ITPA deficient immortalized MEFs. (A) Flow cytometric analysis of the cell cycle was performed and the sub G1 fraction (M1), diploid fraction (M2) and a fraction with an increased DNA content (M3) were determined. (B) ITPA deficiency increased chromosomal ploidy in immortalized MEFs. Percentages of diploid, tetraploid and others are shown in pie charts with the mean ± SD (three independent isolates). The frequency of tetraploidy was increased significantly in Itpa−/– MEFs. Results show non-repeated measures ANOVA (two-tailed): P = 0.00015. P-values are shown following a Bonferroni post hoc test.
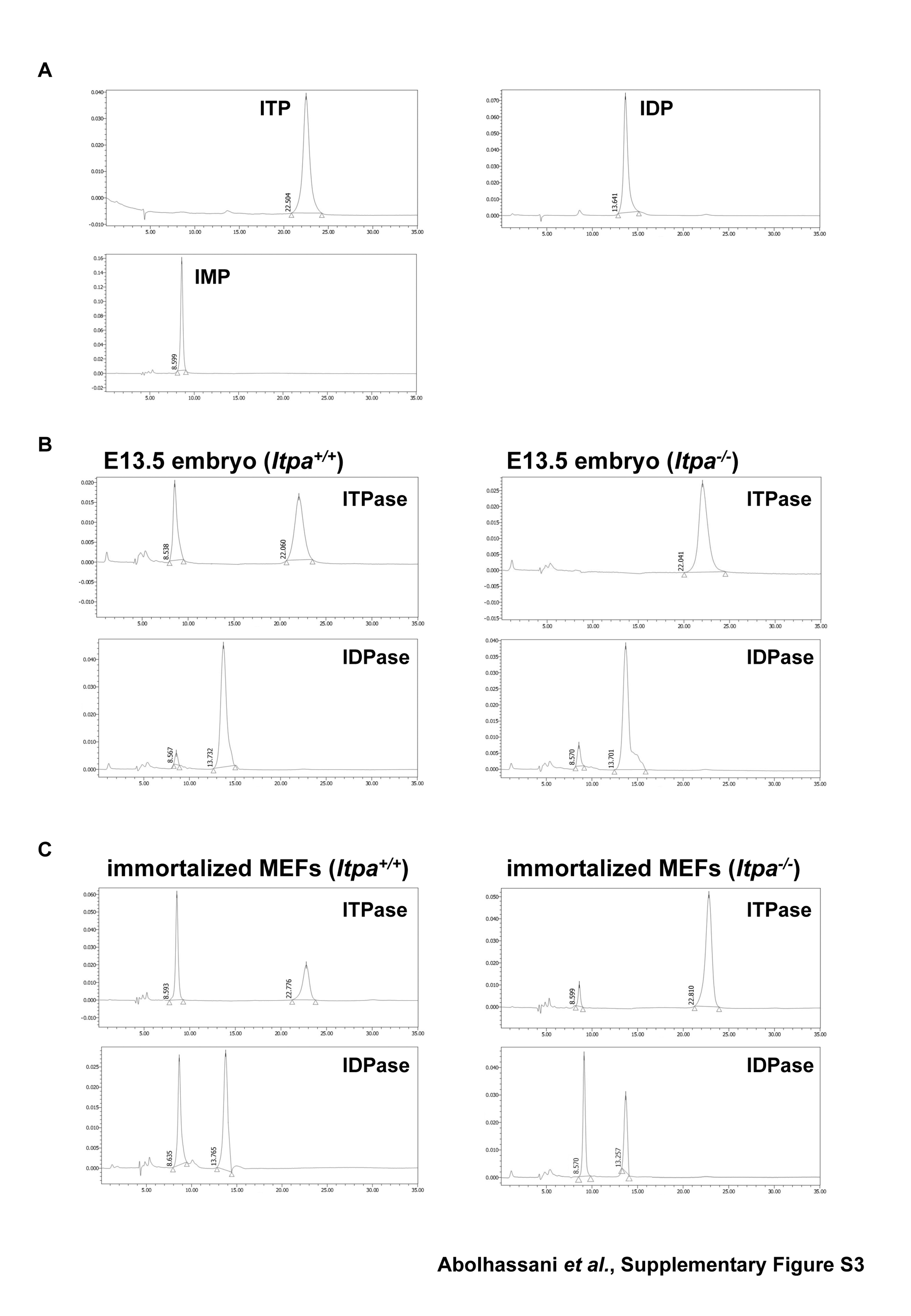
To test whether any backup enzyme for ITPA deficiency might exist in the immortalized Itpa−/– MEFs, we measured ITP-hydrolyzing activity in extracts prepared from embryos and immortalized MEFs (Table 1, Supplementary Figure S3). There was no detectable ITP-hydrolyzing activity in extracts prepared from Itpa−/– embryos, but a significantly high rate was detected in Itpa+/+ embryos, thus confirming ITPA deficiency in Itpa−/– embryos. We also confirmed that Itpa−/– primary MEFs (Passage 3) did not generate IMP from ITP. On the other hand, we detected substantial activity in extracts from immortalized Itpa−/– MEFs, although the levels were less than 20% of that detected in immortalized Itpa+/+ MEFs or embryos. We then measured IDP-hydrolyzing activity in these extracts (Table 1, Supplementary Figure S3). Levels were increased both in Itpa+/+ and Itpa−/– MEFs during immortalization and the activity was more significantly increased in immortalized Itpa−/– MEFs than in immortalized Itpa+/+ MEFs. These results strongly suggest that an increased expression of IDP or ITP-hydrolyzing enzyme(s) blocked or reversed the ITPA-deficient phenotypes observed in primary Itpa−/– MEFs.
Table 1.
Specific ITP/IDP hydrolysis activity in crude-cell extracts prepared from embryos and immortalized MEFs
| Isolate no. 1 |
Isolate no. 2 |
||||
|---|---|---|---|---|---|
| Cell extract | Substrate | Specific activity (u/µg) | SD | Specific activity (u/µg) | SD |
| Itpa+/+ embryo | ITP | 6.34 | 0.8 | 4.08 | 0.7 |
| Itpa+/+ embryo | IDP | 0.84 | 0.2 | 1.26 | 0.4 |
| Itpa+/+ iMEF | ITP | 6.42 | 0.9 | 7.24 | 1.0 |
| Itpa+/+ iMEF | IDP | 3.49 | 0.5 | 3.68 | 0.4 |
| Itpa−/– embryo | ITP | ND | ND | ||
| Itpa−/– embryo | IDP | 0.86 | 0.3 | 1.85 | 0.4 |
| Itpa−/– iMEF | ITP | 1.09 | 0.1 | 1.36 | 0.3 |
| Itpa−/– iMEF | IDP | 5.90 | 0.5 | 7.85 | 1.5 |
As a measure,1 unit (u) was defined as the level of activity producing 1 pmol of IMP at 30°C for 1 min with 0.2 mM ITP/IDP, pH = 8.0, n = 3. Key: ND, not detected; iMEFs, immortalized MEFs.
NUDT16 with strong (deoxy)inosine diphosphatase activity is responsible for cancellation of ITPA-deficient phenotypes and an increase in ITP/IDP-hydrolyzing activity during immortalization
We recently found that the human NUDT16 (nudix [nucleoside diphosphate linked moiety X]-type motif 16) protein, identified as an ITP/XTP/GTP binding protein, has strong IDP and 2′-deoxy-IDP (dIDP)-hydrolyzing activities with weak ITP/dITP-hydrolyzing activities. We also confirmed that mouse NUDT16 protein has essentially the same activities as the human protein (Iyama et al., in preparation). Therefore, we compared Nudt16 mRNA levels among primary and immortalized Itpa+/+ or Itpa−/– MEFs (Figure 6A). Quantitative real-time reverse transcription polymerase chain reaction (RT–PCR) analysis revealed that the expression levels of Nudt16 mRNA were similar between primary MEFs derived from Itpa+/+ and Itpa−/– embryos. In contrast, the expression levels of Nudt16 mRNA in immortalized Itpa−/– MEFs were more than 3-fold higher than those of primary Itpa−/– MEFs and more than 2-fold higher than those in immortalized Itpa+/+ MEFs (Figure 6A). Significantly increased expression of NUDT16 protein in immortalized Itpa−/– MEFs was confirmed by western blotting analysis of crude cell extracts prepared from these MEFs (Figure 6B). We thus conclude that the increased expression of Nudt16 in immortalized Itpa−/– MEFs is responsible for the increase in ITP/IDP-hydrolyzing activity compared with immortalized Itpa+/+ MEFs.
Figure 6.

NUDT16 with strong dIDP/IDP-hydrolyzing activity is the back-up enzyme responsible for the cancellation of ITPA-deficient phenotypes during immortalization. (A) Expression of Nudt16 mRNA. Quantitative real-time RT–PCR was performed to compare Nudt16 mRNA levels between primary (passage 3) and immortalized MEFs. Levels of Nudt16 mRNA were normalized to those of Gapdh mRNA. Values relative to the highest level of Nudt16 mRNA in immortalized Itpa−/– MEFs (isolate No. 3) are shown. Unpaired Student’s t-test (two-tailed) showed P < 0.01 between primary and immortalized Itpa−/– MEFs. Data are shown in a bar graph (mean ± SD), with fold changes between primary and immortalized MEFs (n = 3 independent isolates). Open and black bars show primary MEFs; shaded bars indicate immortalized MEFs. (B) Expression of NUDT16 protein. Western blotting was performed for crude cell extracts (40 µg) prepared from primary (Passage 2) and immortalized MEFs, using anti-NUDT16 antibody (top). GAPDH was detected as an internal control (middle). Membranes were stained with Gel Code Blue in order to confirm the loading and transfer (bottom). Intensities of NUDT16 bands were measured and relative levels normalized to GAPDH are shown in a bar graph. Open and black bars show primary MEFs; shaded bars indicate immortalized MEFs. Result of non-repeated measures ANOVA (two-tailed), P < 0.019. Bonferroni post hoc test, **P < 0.01 (versus others). Error bars represent the SD (n = 3 independent isolates).
To examine the contribution of NUDT16 to the reversal of ITPA-deficient phenotypes in immortalized Itpa−/– MEFs, knockdown of Nudt16 mRNA expression was performed using a mixture of two different Nudt16 silencing (si)RNAs. This treatment caused an efficient reduction of both Nudt16 mRNA and protein levels to less than 20% of the levels seen in controls (Figure 7A and B). Knockdown of Nudt16 expression in immortalized Itpa−/– but not in Itpa+/+ MEFs caused a significant reduction in proliferation rate (Figure 7C), as observed in primary Itpa−/– MEFs (Figure 1C).
We next measured inosine levels in cellular RNA with or without Nudt16 siRNAs. Immortalized Itpa−/– MEFs contained 516.8 ± 22.3 inosine residues per 106 guanosine residues of RNA in the presence of control siRNA which was slightly lower than that in Itpa−/– embryos (567.3 ± 41.4 residues per 106 guanosine), and Nudt16 expression knockdown increased the level to 648.5 ± 01.7 residues per 106 guanosine residues of RNA (Figure 7D). As shown in Figure 7E, immortalized Itpa−/– MEFs contained 2.49 ± 0.24 dI residues per 106 nucleosides in nuclear DNA in the presence of control siRNA which was equivalent to that in immortalized Itpa+/+ MEFs (2.74 ± 0.17 dI residues per 106 nucleosides). Nudt16 expression knockdown in immortalized Itpa−/– MEFs significantly increased the level more than 5-fold (12.73 ± 0.99 residues per 106 nucleosides). In contrast, knockdown of Nudt16 expression in immortalized Itpa+/+ MEFs did not affect the level of dI accumulation in nuclear DNA (2.96 ± 0.3 dI residues per 106 nucleosides).
Knockdown of Nudt16 expression significantly increased the immunoreactivity against ssDNA in immortalized Itpa−/– MEFs (Figure 7F). Moreover, karyotyping of immortalized Itpa−/– MEFs after Nudt16 expression knockdown revealed a significant increase in chromosome structural abnormalities, such as chromatid gaps, premature separation and triradial forms compared with controls (Figure 7G).
Thus, increased expression of Nudt16 was responsible for reversal of the ITPA-deficient phenotypes with reduction of dI accumulation in nuclear DNA but not inosine in cellular RNA.
DISCUSSION
ITPA deficiency increases the accumulation of deoxyinosine in nuclear DNA resulting in severe cellular dysfunction
Here, we showed for the first time that ITPA deficiency caused a significant accumulation of dI in the nuclear DNA of mouse embryos, most of which are likely to die around the time of birth. In wild-type embryos, fewer than three residues of dI per 106 nucleosides, corresponding to about 20 000 residues in a whole cell, were detected in nuclear DNA, whereas about 20 residues of dI per 106 nucleosides reaching more than 105 residues per cell accumulated in Itpa−/– embryos. These results indicate that both spontaneous generation of dITP and incorporation of dITP into DNA occurred at significantly high frequencies. Thus, ITPA deficiency caused severe cellular dysfunction resulting in perinatal lethality. Indeed, we demonstrated that this deficiency in primary MEFs increased the accumulation of SSBs in nuclear DNA detected as ssDNA immunoreactivity and as chromosomal abnormalities such as chromatid/chromosome gaps or breaks. There was also premature centromere separation. All these chromosomal anomalies are likely to cause G2/M arrest thus suppressing cell proliferation, as observed in primary Itpa−/– MEFs.
In E. coli, the lethality of rdgB recA or rdgB recBC double mutants, the former-encoding inosine triphosphatase, is suppressed by the inactivation of endonuclease V (EndoV) (12), which cleaves at the second phosphodiester bond at 3′ to dI and initiates nucleotide excision repair (19). It is likely that an inosine triphosphatase deficiency in E. coli results in the accumulation of its substrate nucleotides, dITP in the nucleotide pools, thus causing an increased accumulation of dI into DNA. Further excision repair initiated by EndoV leads to chromosomal fragmentation in recA or recBC mutants.
In mammals, there are at least two enzymes that might be involved in excision repair of dI: the mammalian homolog of EndoV and alkyladenine DNA glycosylase (AAG) which can excise hypoxanthine, a deaminated adenine base (20–24). Because mammalian cells are likely to be less efficient in recombination repair than E. coli (25), ITPA deficiency itself thus causes severe phenotypes with massive generation of SSBs in DNA, likely caused by efficient excision repair of dI or hypoxanthine.
In primary Itpa−/– MEFs, increased frequency of chromosomal abnormalities such as chromatid/chromosome gaps or breaks might result from increased SSB accumulation in nuclear DNA, which is likely to be caused by excision repair of accumulated dI in nuclear DNA. It is noteworthy that exposure of peripheral lymphocytes to ITP or IDP in culture were reported to cause chromosome aberrations such as chromatid breaks and gaps (26) and sister chromatid exchange (27). Although the precise mechanism inducing chromosome aberration by ITP or IDP is not known, exposure to a high concentration of ITP or IDP might also result in an increase of dI in nuclear DNA, thus increasing SSBs and chromosome abnormalities as observed in Itpa−/– primary MEFs. Moreover, both ITPA deficiency and exposure to ITP or IDP increase premature centromere separation (26). Increased inosine levels in cellular RNA prepared from Itpa−/– embryos indicated that the ITP level was also significantly increased in the absence of ITPA. Therefore, these chromosomal abnormalities might be caused by the increased level of ITP in nucleotide pool. Because sister chromatid cohesion is established by a cohesin complex composed of Rad21, Smc1α, Smc3 and two Scc3 orthologs, SA1 and SA2 (28,29) and whose reaction requires ATP, ITP might compete with ATP to disrupt sister chromatid cohesion, thus resulting in premature centromere separation and inappropriate chromatid separation.
Increased expression of Nudt16 suppresses the ITPA-deficient phenotype during immortalization
During immortalization of Itpa−/– MEFs, most of the ITPA-deficient phenotype characteristics, such as prolonged doubling time, G2/M arrest, SSBs accumulation, chromosome abnormalities were canceled efficiently. This was accompanied by a significant reduction of dI accumulation in nuclear DNA. The inosine level in cellular RNA was still high in immortalized Itpa−/– MEFs (516.8 ± 22.3 inosine residues per 106 guanosine residues) as much as seen in Itpa−/– embryos (567.3 ± 41.4 inosine residues per 106 guanosines), thereby indicating that the ITPA-deficient phenotypes are most likely to be attributed to the increased accumulation of dI in nuclear DNA.
We identified the human NUDT16 protein (Iyama et al., manuscript in preparation) as a dIDP/IDP-hydrolyzing enzyme, which can bind ITP/XTP/GTP and efficiently hydrolyzes dIDP/IDP, and to a lesser extent dITP/ITP, to dIMP/IMP. In the present study, we found that the levels of mouse Nudt16 mRNA and NUDT16 protein were significantly higher in immortalized Itpa−/– MEFs than in immortalized wild-type MEFs or primary Itpa−/– MEFs. Because knockdown of Nudt16 expression efficiently reproduced the ITPA-deficient phenotypes accompanied by significant increases of dI in nuclear DNA, and to a lesser extent of inosine in cellular RNA, we conclude that NUDT16 and ITPA play a dual protective role for eliminating dITP/ITP and dIDP/IDP from nucleotide pools in mammals.
ITP can be hydrolyzed slowly to IDP by ATPase or other nucleoside triphosphatases (11,30), so increased dIDP hydrolysis in immortalized Itpa−/– MEFs is likely to be sufficient to eliminate dITP from the nucleotide pools. Moreover, ITP can be generated in a variety of tissue extracts as well as in erythrocytes (31). We reported previously that ITP accumulated in erythrocytes but not in tissues including the heart and liver derived from Itpa−/– mice, whereas IMP accumulated markedly in RNA prepared from the latter (6). Because both RNA and DNA synthesis takes place in the latter tissues, DNA and RNA polymerases are likely to utilize ITP and dITP efficiently as nucleotide precursors, thereby consuming most of the ITP or dITP that accumulates in the nucleotide pools in the absence of ITPA.
Considering the likely source of ITP or dITP in the nucleotide pools, IMP generated from AMP by AMP deamination must be the most relevant precursor, because most cells can synthesize IDP or ITP from IMP (31) and IDP may be converted to dIDP by ribonucleotide reductase, thus generating dITP (21). To minimize accumulation of ITP or dITP in the nucleotide pools, hydrolysis of IDP or dIDP to the corresponding monophosphates catalyzed by NUDT16 is likely to be as critical as is any hydrolysis of ITP or dITP to the corresponding monophosphates.
In the present study, we showed that increased expression of NUDT16 in Itpa−/– immortalized MEFs is sufficient to cancel the ITPA-deficient phenotypes observed in Itpa−/– embryos or primary MEFs, suggesting that the lower expression level of NUDT16 in normal tissues may be why any ITPA deficiency causes such severe phenotypes. On the other hand, ITPA deficiency in humans is likely to be related to azathioprine intolerance in patients with inflammatory bowel disease, but does not cause any severe phenotype (32–34), compared with the Itpa−/– mice. It is possible that human NUDT16 expression might be higher than that in mouse, thus compensating for any ITPA deficiency.
Identification of NUDT16 as a backup enzyme for ITPA deficiency in mice will shed light on the mechanisms that enable humans to be resistant to ITPA deficiency. Towards this goal, it is important to know the relative expression of ITPA and NUDT16 in human cells and organs, and to characterize the enzymatic properties of NUDT16.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Education, Culture, Sports, Science and Technology of Japan [20013034 to Y.N., 20012038 to K.S.]; the Japan Society for the Promotion of Science [19390114 to D.T., 08J03650 to T.I.]; Kyushu University Global COE program [YN]. Funding for open access charges: Ministry of Education, Culture, Sports, Science and Technology of Japan; Kyushu University Global COE program.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank M. Ohtsu in the Laboratory for Technical Support of our institute and N. Adachi, A. Matsuyama, K. Hayashi, K. Nakabeppu and K. Asakawa for technical assistance.
REFERENCES
- 1.Nakabeppu Y, Tsuchimoto D, Furuichi M, Sakumi K. The defense mechanisms in mammalian cells against oxidative damage in nucleic acids and their involvement in the suppression of mutagenesis and cell death. Free Radic. Res. 2004;38:423–429. doi: 10.1080/10715760410001688348. [DOI] [PubMed] [Google Scholar]
- 2.Nakabeppu Y, Behmanesh M, Yamaguchi H, Yoshimura D, Sakumi K. In: Oxidative Damage to Nucleic Acids. Evans MD, Cooke MS, editors. TX/New York: Landes Bioscience/Springer, Austin; 2007. pp. 40–53. [Google Scholar]
- 3.Nakabeppu Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat. Res. 2001;477:59–70. doi: 10.1016/s0027-5107(01)00096-3. [DOI] [PubMed] [Google Scholar]
- 4.Nakabeppu Y, Kajitani K, Sakamoto K, Yamaguchi H, Tsuchimoto D. MTH1, an oxidized purine nucleoside triphosphatase, prevents the cytotoxicity and neurotoxicity of oxidized purine nucleotides. DNA Rep. 2006;5:761–772. doi: 10.1016/j.dnarep.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Behmanesh M, Sakumi K, Tsuchimoto D, Torisu K, Ohnishi-Honda Y, Rancourt DE, Nakabeppu Y. Characterization of the structure and expression of mouse Itpa gene and its related sequences in the mouse genome. DNA Res. 2005;12:39–51. doi: 10.1093/dnares/12.1.39. [DOI] [PubMed] [Google Scholar]
- 6.Behmanesh M, Sakumi K, Abolhassani N, Toyokuni S, Oka S, Ohnishi YN, Tsuchimoto D, Nakabeppu Y. ITPase-deficient mice show growth retardation and die before weaning. Cell Death Differ. 2009;16:1315–1322. doi: 10.1038/cdd.2009.53. [DOI] [PubMed] [Google Scholar]
- 7.Nonaka M, Tsuchimoto D, Sakumi K, Nakabeppu Y. Mouse RS21-C6 is a mammalian 2′-deoxycytidine 5′-triphosphate pyrophosphohydrolase that prefers 5-iodocytosine. FEBS J. 2009;276:1654–1666. doi: 10.1111/j.1742-4658.2009.06898.x. [DOI] [PubMed] [Google Scholar]
- 8.Tsuzuki T, Egashira A, Igarashi H, Iwakuma T, Nakatsuru Y, Tominaga Y, Kawate H, Nakao K, Nakamura K, Ide F, et al. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl Acad. Sci. USA. 2001;98:11456–11461. doi: 10.1073/pnas.191086798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shenoy TS, Clifford AJ. Adenine nucleotide metabolism in relation to purine enzymes in liver, erythrocytes and cultured fibroblasts. Biochim. Biophys. Acta. 1975;411:133–143. doi: 10.1016/0304-4165(75)90292-5. [DOI] [PubMed] [Google Scholar]
- 10.Gower WR, Jr, Carr MC, Ives DH. Deoxyguanosine kinase. Distinct molecular forms in mitochondria and cytosol. J. Biol. Chem. 1979;254:2180–2183. [PubMed] [Google Scholar]
- 11.Burton K, White H, Sleep J. Kinetics of muscle contraction and actomyosin NTP hydrolysis from rabbit using a series of metal-nucleotide substrates. J. Physiol. 2005;563:689–711. doi: 10.1113/jphysiol.2004.078907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradshaw JS, Kuzminov A. RdgB acts to avoid chromosome fragmentation in Escherichia coli. Mol. Microbiol. 2003;48:1711–1725. doi: 10.1046/j.1365-2958.2003.03540.x. [DOI] [PubMed] [Google Scholar]
- 13.Ide Y, Tsuchimoto D, Tominaga Y, Nakashima M, Watanabe T, Sakumi K, Ohno M, Nakabeppu Y. Growth retardation and dyslymphopoiesis accompanied by G2/M arrest in APEX2-null mice. Blood. 2004;104:4097–4103. doi: 10.1182/blood-2004-04-1476. [DOI] [PubMed] [Google Scholar]
- 14.Tsuruya K, Furuichi M, Tominaga Y, Shinozaki M, Tokumoto M, Yoshimitsu T, Fukuda K, Kanai H, Hirakata H, Iida M, et al. Accumulation of 8-oxoguanine in the cellular DNA and the alteration of the OGG1 expression during ischemia-reperfusion injury in the rat kidney. DNA Rep. 2003;2:211–229. doi: 10.1016/s1568-7864(02)00214-8. [DOI] [PubMed] [Google Scholar]
- 15.Taghizadeh K, McFaline JL, Pang B, Sullivan M, Dong M, Plummer E, Dedon PC. Quantification of DNA damage products resulting from deamination, oxidation and reaction with products of lipid peroxidation by liquid chromatography isotope dilution tandem mass spectrometry. Nat. Protoc. 2008;3:1287–1298. doi: 10.1038/nprot.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oka S, Ohno M, Tsuchimoto D, Sakumi K, Furuichi M, Nakabeppu Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008;27:421–432. doi: 10.1038/sj.emboj.7601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuchimoto D, Sakai Y, Sakumi K, Nishioka K, Sasaki M, Fujiwara T, Nakabeppu Y. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001;29:2349–2360. doi: 10.1093/nar/29.11.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kow YW. Repair of deaminated bases in DNA. Free Radic. Biol. Med. 2002;33:886–893. doi: 10.1016/s0891-5849(02)00902-4. [DOI] [PubMed] [Google Scholar]
- 19.Dalhus B, Arvai AS, Rosnes I, Olsen Ø., Backe PH, Alseth I, Gao H, Cao W, Tainer JA, Bjørås M. Structures of endonuclease V with DNA reveal initiation of deaminated adenine repair. Nat. Struct. Mol. Biol. 2009;16:138–143. doi: 10.1038/nsmb.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karran P, Lindahl T. Enzymatic excision of free hypoxanthine from polydeoxynucleotides and DNA containing deoxyinosine monophosphate residues. J. Biol. Chem. 1978;253:5877–5879. [PubMed] [Google Scholar]
- 21.Myrnes B, Guddal PH, Krokan H. Metabolism of dITP in HeLa cell extracts, incorporation into DNA by isolated nuclei and release of hypoxanthine from DNA by a hypoxanthine-DNA glycosylase activity. Nucleic Acids Res. 1982;10:3693–3701. doi: 10.1093/nar/10.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saparbaev M, Laval J. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc. Natl Acad. Sci. USA. 1994;91:5873–5877. doi: 10.1073/pnas.91.13.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moe A, Ringvoll J, Nordstrand LM, Eide L, Bjørås M, Seeberg E, Rognes T, Klungland A. Incision at hypoxanthine residues in DNA by a mammalian homologue of the Escherichia coli antimutator enzyme endonuclease V. Nucleic Acids Res. 2003;31:3893–3900. doi: 10.1093/nar/gkg472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallur AC, Maher RL, Bloom LB. The efficiency of hypoxanthine excision by alkyladenine DNA glycosylase is altered by changes in nearest neighbor bases. DNA Rep. 2005;4:1088–1098. doi: 10.1016/j.dnarep.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Pardo B, Gómez-González B, Aguilera A. DNA double-strand break repair: how to fix a broken relationship. Cell. Mol. Life Sci. 2009;66:1039–1056. doi: 10.1007/s00018-009-8740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Auclair C, Gouyette A, Levy A, Emerit I. Clastogenic inosine nucleotide as components of the chromosome breakage factor in scleroderma patients. Arch. Biochem. Biophys. 1990;278:238–244. doi: 10.1016/0003-9861(90)90253-u. [DOI] [PubMed] [Google Scholar]
- 27.Vormittag W, Brannath W. As to the clastogenic-, sister-chromatid exchange inducing-and cytotoxic activity of inosine triphosphate in cultures of human peripheral lymphocytes. Mutat. Res. 2001;476:71–81. doi: 10.1016/s0027-5107(01)00085-9. [DOI] [PubMed] [Google Scholar]
- 28.Watrin E, Peters JM. Cohesin and DNA damage repair. Exp. Cell Res. 2006;312:2687–2693. doi: 10.1016/j.yexcr.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 29.Uhlmann F. A matter of choice: the establishment of sister chromatid cohesion. EMBO Rep. 2009;10:1095–1102. doi: 10.1038/embor.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanderheiden BS. ITP pyrophosphohydrolase and IDP phosphohydrolase in rat tissue. J. Cell. Physiol. 1975;86:167–175. doi: 10.1002/jcp.1040860118. [DOI] [PubMed] [Google Scholar]
- 31.Vanderheiden BS. Inosine di- and triphosphate synthesis in erythrocytes and cell extracts. J. Cell. Physiol. 1979;99:287–301. doi: 10.1002/jcp.1040990303. [DOI] [PubMed] [Google Scholar]
- 32.Sumi S, Marinaki AM, Arenas M, Fairbanks L, Shobowale-Bakre M, Rees DC, Thein SL, Ansari A, Sanderson J, De Abreu RA, et al. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum. Genet. 2002;111:360–367. doi: 10.1007/s00439-002-0798-z. [DOI] [PubMed] [Google Scholar]
- 33.Marinaki AM, Duley JA, Arenas M, Ansari A, Sumi S, Lewis CM, Shobowale-Bakre M, Fairbanks LD, Sanderson J. Mutation in the ITPA gene predicts intolerance to azathioprine. Nucleosides Nucleotides Nucleic Acids. 2004;23:1393–1397. doi: 10.1081/NCN-200027639. [DOI] [PubMed] [Google Scholar]
- 34.Seela F, Xu K. Pyrazolo[3,4-d]pyrimidine ribonucleosides related to 2-aminoadenosine and isoguanosine: synthesis, deamination and tautomerism. Org. Biomol. Chem. 2007;5:3034–3045. doi: 10.1039/b708736e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}