

Abstract
Focal epilepsy often develops following traumatic, ischemic, or infectious brain injury. While the electrical activity of the epileptic brain is well characterized, the mechanisms underlying epileptogenesis are poorly understood. We have recently shown that in the rat neocortex, long-lasting breakdown of the blood–brain barrier (BBB) or direct exposure of the neocortex to serum-derived albumin leads to rapid upregulation of the astrocytic marker GFAP (glial fibrillary acidic protein), followed by delayed (within 4–7 d) development of an epileptic focus. We investigated the role of astrocytes in epileptogenesis in the BBB-breakdown and albumin models of epileptogenesis. We found similar, robust changes in astrocytic gene expression in the neocortex within hours following treatment with deoxycholic acid (BBB breakdown) or albumin. These changes predict reduced clearance capacity for both extracellular glutamate and potassium. Electrophysiological recordings in vitro confirmed the reduced clearance of activity-dependent accumulation of both potassium and glutamate 24 h following exposure to albumin. We used a NEURON model to simulate the consequences of reduced astrocytic uptake of potassium and glutamate on EPSPs. The model predicted that the accumulation of glutamate is associated with frequency-dependent (>100 Hz) decreased facilitation of EPSPs, while potassium accumulation leads to frequency-dependent (10–50 Hz) and NMDA-dependent synaptic facilitation. In vitro electrophysiological recordings during epileptogenesis confirmed frequency-dependent synaptic facilitation leading to seizure-like activity. Our data indicate a transcription-mediated astrocytic transformation early during epileptogenesis. We suggest that the resulting reduction in the clearance of extracellular potassium underlies frequency-dependent neuronal hyperexcitability and network synchronization.
Introduction
Epilepsy is one of the most common neurological disorders, affecting 0.5–2% of the population worldwide. While the characteristic electrical activity in the epileptic cortex has been studied extensively, the mechanisms underlying epileptogenesis are poorly understood. Focal neocortical epilepsy often develops following traumatic, ischemic, or infectious brain injury. Under these conditions, vascular damage is common and includes local breakdown of the blood–brain barrier (BBB) (Tomkins et al., 2001; Neuwelt, 2004; Abbott et al., 2006). The BBB has long been recognized as crucial for maintenance of the brain's microenvironment, but it was only recently documented that disruption of the blood–brain barrier plays an important role in the pathogenesis of epilepsy (Seiffert et al., 2004; Marchi et al., 2007; van Vliet et al., 2007). It was found, for example, that in the rat neocortex, long-lasting BBB disruption leads to gradual development (within 4–7 d) of an epileptic focus that persists for weeks (Seiffert et al., 2004; Ivens et al., 2007; Tomkins et al., 2007). Our experiments further indicated that during BBB breakdown, serum-derived albumin diffuses into the brain's extracellular space and is rapidly transported into astrocytes via a specific receptor-mediated mechanism. Albumin uptake by astrocytes was followed by a rapid (within hours) upregulation of the astrocytic marker glial fibrillary acidic protein (GFAP) (Seiffert et al., 2004; Ivens et al., 2007), suggesting that astrocytic dysfunction plays a role in injury-induced epileptogenesis.
Data accumulating from human and animal studies support the notion that glial cells make an important contribution to the control of neuronal function under both normal and pathological conditions [for review, see Araque et al. (2001), Seifert et al. (2006), and Wetherington et al. (2008)]. Studies on epileptic tissue show significant alterations in the expression of astrocytic proteins, including increased expression of GFAP (Bordey and Sontheimer, 1998) and reduced expression of proteins involved in the regulation of extracellular potassium ([K+]o) and glutamate (Hinterkeuser et al., 2000; Schröder et al., 2000). Despite the vast body of data confirming changes in the morphology and function of astrocytes in epileptic tissue, the direct role of these cells in the development of the epileptic network (i.e., in epileptogenesis) remains unclear. In this study, we combined molecular, electrophysiological, and computer modeling approaches to investigate the potential role of vascular-injury-induced and albumin-induced early transformation of astrocytes in altered neuronal excitability and epileptogenesis.
Materials and Methods
Animals were housed and handled according to the directives of the internationally accredited Institutional Animal Care and Use Committees at Charité University Medicine, Berlin, and Ben-Gurion University of the Negev, Beer-Sheva. All experimental procedures were approved by the ethical committees supervising experiments on animals at Charité University Medicine (in vivo approval no.: G0104/05, in vitro: T0228/04) and Ben-Gurion University of the Negev (approval no.: BGU-R-71-2006).
In vivo experiments.
The in vivo experiments were performed as previously described by Seiffert et al. (2004). In brief, adult male Wistar rats (120–250 g) were anesthetized using ketamine and xylazine and placed in a stereotactic frame. A 4-mm-diameter bone window was drilled over the somatosensory cortex, the dura was opened, and the underlying cortex was perfused with artificial CSF (ACSF). For the “treated” rat group, the BBB-disrupting agent deoxycholic acid sodium salt (DOC, 2 mm, Sigma-Aldrich) or bovine serum albumin (0.1 mm, >98% in agarose cell electrophoresis; catalog no. A7906, Sigma Aldrich) was added to the ACSF. Albumin concentrations corresponded to 25% of the normal serum concentration [determined to be 0.4 mm for 10 rats; see also Geursen and Grigor (1987); final osmolarity of 303–305 mOsmol/L]. For the sham-operated control group, the cortex was perfused with ACSF. The composition of the ACSF was as follows (in mm): 129 NaCl, 21 NaHCO3, 1.25 NaH2PO4, 1.8 MgSO4, 1.6 CaCl2, 3 KCl, and 10 glucose. Rats were killed at 7–8, 24, or 48 h following treatment, before the onset of epileptiform activity (>4 d) (Seiffert et al., 2004).
Microarrays.
Total RNA from animals treated with DOC or with albumin was isolated from the somatosensory cortex, directly under the craniotomy area, using the TRIzol reagent (Invitrogen), and prepared using the Affymetrix GeneChip one-cycle target labeling kit. Biotinylated cRNA was then fragmented and hybridized to the GeneChip Rat Genome 230 2.0 Array according to manufacturer's protocols (Affymetrix Technical Manual). All microarray data are available at the Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE12304, a subset of arrays used by Cacheaux et al. (2009). The array data were normalized by using GCRMA (GC Robust Multi-Array Average) or RMA (Robust Multi-Array Average) analysis using the probe annotations distributed by Affymetrix (NetAffx). In addition, we compared these results to those obtained with annotations based on the Unigene build (Dai et al., 2005). For the subset of genes we focused on in this study, we found the expression values obtained from these annotations to be very similar (correlation coefficient of >0.850 for most treatments, p < 0.001, data not shown) and therefore the Affymetrix probe set annotations are used throughout the text. One array was run for each treatment (DOC and albumin) and for every contralateral hemisphere for the following time points: 7/8, 24, and 48 h. The data from a sham-treated animal (24 h) were used to normalize the other arrays. To identify genes involved in astrocytic functions, we used GeneCards (http://www.genecards.org) (Rebhan et al., 1998), querying for “astrocyte.” For comparison of the relative changes in the expression of astrocytic versus neuronal genes, we used gene sets published by Cahoy et al. (2008) of astrocytic and neuronal enriched genes (expressed by S100β+ and S100β−/PDGFRα−/MOG− cells, respectively). Cluster analysis was performed with MATLAB by assessing the expression relationship as the Euclidean distance in N-dimensional space between measurements (N denotes number of gene transcripts). Arrays were then clustered according to distance data, by using the unweighted pair group method with arithmetic mean (Gronau and Moran, 2007).
In vitro astrocytic and neuronal culture preparations.
Primary neuronal cortical cultures were prepared from embryonic day 18 rats as reported previously (Kaufer et al., 2004). Briefly, cells were dissociated with a papain solution for 20 min at 37°C. After the removal of the papain solution, the tissue was resuspended in growth medium [MEM with Earle's salts containing 2.5% B27 supplement, 0.1% mito serum extender, 5% fetal bovine serum (FBS), 20 mm glucose, and 5 mm l-glutamine] and dissociated by mechanical trituration. The cells were plated, and after 4 h in vitro the cell culture medium was replaced with Neurobasal medium supplemented with 2% B27 supplement and 0.5 mm GlutaMAX. The cells were maintained in 5% CO2 at 37°C. After 7 d in vitro, cytosine arabinofuranoside (AraC) (10 μm) was added to the cultures. After 10 d in vitro, the cells were incubated with 0.4 mm albumin for 24 h at 37°C. For astrocytic cultures, astrocytes were isolated from the cerebral cortices of P0 rat pups. Cells were dissociated with papain and mechanical trituration. The cells were cultured in high-glucose DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C and in 5% CO2 (medium was replaced every 3–4 d). After 10 d in vitro, the culture medium was replaced with serum-free high-glucose DMEM (containing 1% penicillin/streptomycin) for 18 h. The cells were then incubated in serum-free medium containing 0.4 mm albumin for 24 h at 37°C. For immunostainings cells were washed with PBS and fixed in 4% paraformaldehyde for 15 min. The cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min and washed in PBS. They were then incubated with 5% normal donkey serum in PBS for 1 h at room temperature followed by overnight incubation at 4°C with either mouse anti-NeuN (1:1000; Millipore Bioscience Research Reagents) or mouse anti-GFAP (1:1000; Cell Signaling Technology). The cells were washed in PBS, incubated with donkey anti-mouse Cy3 (1:1000; Jackson ImmunoResearch) for 1 h at room temperature, and then counterstained with DAPI.
Real-time PCR.
Total RNA was isolated from the somatosensory cortices of animals treated with DOC or albumin (24 h treatment; n = 3) or from primary cultures (astrocytic and neuronal, n = 3 independent experiments). Expression levels were determined by real-time reverse transcriptase-PCR (RT-PCR) with an iQ5 detection system (Bio-Rad) using gene-specific primer pairs. RT-PCR data were analyzed using the PCR Miner program (Zhao and Fernald, 2005), and fold changes in gene expression were represented relative to sham-operated controls (in vivo samples) or serum-deprived controls (in vitro samples). Ribosomal 18S RNA (18S rRNA) was used as an internal control for variations in sample preparation. DNase treatment was performed for all samples, followed by first-strand cDNA synthesis (iScript cDNA Synthesis kit, Bio-Rad). PCRs were performed with iQ SYBR Green Supermix (Bio-Rad). Primer specificity was verified by melt curve analysis. The amplification cycles for 18S, Gja1, GS, SLC1A2 (GLT-1), SLC1A3 (GLAST), and Kcnj10 consisted of 40 cycles of 10 s at 95°C, 30 s at 55°C, and 30 s at 72°C. The amplification cycles for Gjb2 and Gjb6 consisted of 40 cycles of 10 s at 95°C, 30 s at 60°C, and 30 s at 72°C.
Primer sequences (forward, reverse) were as follows: 18S rRNA (GenBank accession number M11188.1, 5′-CCATCCAATCGGTAGTAGCG-3′, 5′-GTAACCCGTTGAACCCCATT-3′); SLC1A3 (GenBank accession number NM_019225.1; 5′-GAGGCCATGGAGACTCTGAC-3′, 5′-CGAAGCACATGGAGAAGACA-3′); GS (GenBank accession number NM_017073.3; 5′-AGCGACATGTACCTCCATCC-3′, 5′-TACAGCTGTGCCTCAGGTTG-3′); Kcnj10 (GenBank accession number X83585.1; 5′-GAGACGACGCAGACAGAGAG-3′, 5′CCACTGCATGTCAATGAAGG-3′); Gjb2 (GenBank accession number NM_001004099.1; 5′-GGTTTGTGATGTGAGCATGG-3′, 5′-CTCAGCACACCAAGGATGAA-3′); Gjb6 (GenBank accession number NM_053388.1; 5′-GCCAAGATGAGTCACAGCAA-3′, 5′-TCAGAGCTGGATCACAATCG-3′); Gja1 (GenBank accession number NM_012567.2; 5′-TCCTTGGTGTCTCTCGCTTT-3′, 5′-TTTGGAGATCCGCAGTCTTT-3′); SLC1A2 (GenBank accession number NM_017215.2; 5′-GGTCAATGTAGTGGGCGATT-3′, 5′-GGACTGCGTCTTGGTCATTT-3′).
In vitro electrophysiological recordings.
For electrophysiological experiments, rats were deeply anesthetized with isoflurane and then decapitated. Brains were quickly removed, and transverse hippocampal-cortical slices (400 μm thick) were prepared using a vibratome (Campden Instruments). Slices were maintained in a humidified, carbogenated (5% CO2 and 95% O2) gas atmosphere at 36 ± 1°C and perfused with ACSF in a standard interface chamber (Seiffert et al., 2004; Ivens et al., 2007). To mimic the altered ionic environment during BBB disruption, recordings were acquired in a serum-adapted electrolyte solution (sACSF) (Seiffert et al., 2004). sACSF was similar in composition to the ACSF except for different concentrations of MgSO4 (0.8 mm), CaCl2 (1.3 mm), KCl (5.7 mm), and glutamine (1 mm). “Treated” slices were incubated with sACSF containing 0.1 mm bovine serum albumin for 2 h before transfer to the perfusion chamber.
Field potential recordings were obtained 6–10 h following perfusion with sACSF. Control slices were treated similarly, using sACSF without albumin. Glass microelectrodes (∼3 MΩ, 154 mm NaCl) were positioned in layer 4 of the neocortex. Slices were stimulated with brief (100 μs) pulses, by using bipolar stimulation electrodes placed at the border between white and gray matter in the same cortical column. Trains of 50 stimuli were applied at 2, 5, 10, 20, 50, and 100 Hz, at 2.5× threshold stimulation intensity. Signals were amplified (SEC-10L; NPI Electronics), filtered at 2 kHz, displayed on an oscilloscope, digitized online (CED-1401micro; Cambridge Electronics Design), and stored for offline analysis. Extracellular potassium concentrations ([K+]o) were measured with ion-sensitive microelectrodes (ISMEs) (Lux and Neher, 1973; Jauch et al., 2002).
In vitro intracellular recordings were obtained from pyramidal neurons (layer 2–3) 23–28 h following the in vivo treatment with albumin or from control rats. Currents were recorded using the whole-cell patch configuration, as described previously (Pavlovsky et al., 2003). In brief, glass pipettes were pulled from capillaries using a vertical puller (Narashige) and filled with a solution comprising the following (in mm): 150 CsCl, 1 MgCl2, 10 HEPES, 4 Na2ATP, 0.1 CaCl2, and 1.1 mm EGTA, pH adjusted to 7.2 with a final osmolarity of 290–310 mOsm. Cells were visualized using infrared differential interference phase contrast video-microscopy. Recordings were performed using AxoPatch 700B (Molecular Devices), digitized at 10 kHz, and recorded using pClamp 9.2 (Molecular Devices). Patch pipette's resistance was 4–5 MΩ. Series resistance was not electronically compensated; however, cells in which series resistance varied by >25% were excluded from the analysis. Stimulation protocols were started at least 5 min following impalement to allow intracellular dialysis with the pipette solution. EPSCs were evoked—using a bipolar stimulating electrode positioned <200 μm from the recorded cell—at 75% of the intensity producing maximal EPSCs. NMDA currents were recorded in the presence of blockers of AMPA/kainate (i.e., 30 μm CNQX) and of GABAA receptors (i.e., 10 μm bicuculline methiodide). Cells were voltage clamped to +40 mV to alleviate NMDA receptor blockade and inactivate fast Na+ currents. In some experiments, dihydrokainic acid (DHK, Tocris Bioscience), 100 μm, was added to the extracellular solution to selectively block the astrocyte-specific glutamate transporter, SLC1A2 (Arriza et al., 1994).
Computer simulations.
A computer model was implemented using the NEURON modeling environment (Hines and Carnevale, 1997) with 20 μs time steps. The model consisted of a multicompartment isolated cell, simulating a layer 2/3 cortical neuron, using only passive membrane properties. Geometric parameters and spatial relationships of the 74 compartments were modeled after Traub et al. (2003). Resting membrane potential was set at −65 mV (determined by Na+ and K+ conductance); membrane capacitance Cm was 0.9 μF/cm2; and the cytoplasmic resistance was set at 250 Ω/cm2. Simulated excitatory inputs consisted of eight synapses on apical dendrites (located 1368 μm from the soma), contributing currents with AMPA and NMDA kinetics modeled after Saftenku (2005) and Kampa et al. (2004), respectively. AMPA to NMDA maximal current ratios were set at 1 (Myme et al., 2003). Synaptic currents were triggered by a surge of “glutamate,” decaying with first-order kinetics (baseline time constant = 1.2 ms). Downregulation of uptake mechanisms was simulated by changing the time constant of the decay function, similar to the effect of the application of dl-threo-β-benzyloxyaspartate (dl-TBOA) (Diamond, 2005). To investigate the effects of altered [K+]o, each compartment was enveloped by a fixed space in which potassium was allowed to accumulate. [K+]o“diffused” either into the bathing solution or into astrocytes with KIR kinetics. Since KIR channel conductance is proportional to [K+]o (Sakmann and Trube, 1984), K+ influx into “astrocytes” was determined by the local potassium gradient ([K+]o − [K+]bath) modulated by KIR conductance (log[K+]o) (adapted from Ciani et al., 1978) as follows:
 |
where IK is the momentary K+ flux (in nA/cm2), IKrest is the resting K+ flux (in nA/cm2), τECS is a time constant for potassium diffusion into the extracellular space, τastrocytic is a time constant for potassium diffusion into astrocytes, F is the Faraday constant, C is the ratio of astrocytic K+ uptake to extracellular diffusion, and V is the radius of enveloping extracellular space, set at 20 nm (Egelman and Montague, 1999; Savtchenko et al., 2000). The ionic flux equation describes first-order potassium clearance by both free diffusion and “astrocytic” uptake (Kager et al., 2000). Lateral diffusion of K+ ions was not taken into account. To simulate a decrease in astrocytic potassium clearance, τastrocytic was increased to mimic a reduction in astrocytic KIR channels. “Resting” ion concentrations were set at the following concentrations (in [mm]): [Na+]o, 145; [Na+]i, 12; [K+]o, 3.5; and [K+]i, 140.
Statistical analysis.
Data are expressed as means ± SEM. Differences between treated and control slices were determined by the Mann–Whitney U test for two independent samples. Statistical tests were performed using SPSS 13.0 for Windows. The level of statistical significance was set at p < 0.05, unless otherwise stated.
Results
Astrocytic transcriptional changes following BBB opening or exposure to albumin
To explore changes in astrocytic gene expression during epileptogenesis, we analyzed gene-array data from DOC- and albumin-treated brains (n = 3 from each treatment) during the first 48 h after treatment and before the development of epileptiform activity (Seiffert et al., 2004; Ivens et al., 2007). When compared with sham-operated controls, the two treatments, at each time point, resulted in similar changes in expression of astrocytic-enriched genes with a correlation coefficients between the different treatments (see Materials and Methods) of r2 = 0.69, 0.82, and 0.85 for 8, 24, and 48 h following treatment, respectively (p < 0.0001) (Fig. 1a). Unsupervised hierarchical cluster analysis revealed further similarities between changes in transcript levels in treated cortices (which cluster according to time after treatment) (Fig. 1b), while transcript changes in the contralateral, untreated hemispheres are relatively dissimilar and cluster together.
Figure 1.
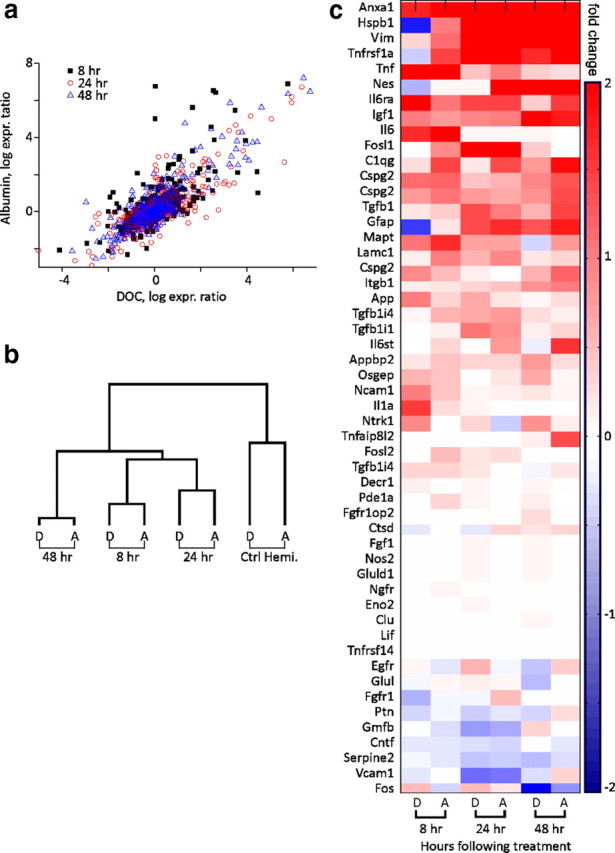
Transcriptional changes in astrocytes following exposure to albumin or BBB disruption. a, Sham-normalized expression levels of mRNA for genes preferentially expressed in astrocytes at 8, 24, and 48 h following treatment with the BBB disrupting agent DOC (D) or albumin (A). b, Hierarchical cluster analysis comparing astrocytic gene expression for DOC-treated and albumin-treated cortices at 8, 24, and 48 h following treatment, and the contralateral, nontreated hemisphere (Ctrl Hemi.). c, Average number of gene transcripts upregulated or downregulated by >150% grouped by cell type across all time points.
In a recent study, Cahoy et al. (2008) created a transcriptome database reflecting cell type-specific, comprehensive mRNA expression levels in astrocytes, neurons and oligodendrocytes. We used these gene lists to classify genes into “astrocytic” or “neuronal” categories. When compared with the “neuronal” category at all examined time points, the “astrocytic” category included a higher average number of genes that underwent a change in expression of more than ±150%. Comparison between the results at 8 and 48 h after treatment showed an increase in the average number of genes that reached 150% change in both groups (34 vs 40 for astrocytes and 21 vs 28 for neurons, 8 and 48 h after treatment, respectively). We also examined the expression levels of genes reported as overexpressed in reactive astrocytes (Ridet et al., 1997) and found a large overlap with overexpressed genes 8, 24, and 48 h following both treatments (Fig. 1c). These results are consistent with the hypothesis that an early and prominent change in astrocytic gene expression is an important early feature of BBB-breakdown or albumin-induced epileptogenesis (see Discussion).
Altered expression of astrocytic potassium- and glutamate-regulating genes
In their pioneering study, Kuffler and Potter (1964) established that astrocytes are crucial for the control of the brain's extracellular environment. Specifically, these cells limit the accumulation of [K+]o and glutamate (Newman et al., 1984; Oliet et al., 2001), thus potentially contributing to the regulation of neuronal excitability. We therefore searched our gene array results for changes in the level of expression of several potassium and glutamate homeostasis-related genes. We found that transcripts coding for the predominantly astrocytic (Kcnj10), but not neuronal (e.g., Kcnj2 or Kcnc1) (Butt and Kalsi, 2006), inward-rectifying K+ channel (KIR) were downregulated. In addition, the mRNA coding for the astrocytic glutamate transporters of the solute carrier family 1, subfamily A members SLC1A2 and SLC1A3 (Su et al., 2003; Chaudhry et al., 1995), but not for SLC1A4, was downregulated. In contrast, SLC1A1 (preferentially expressed in neurons) (Rothstein et al., 1994) did not show significant changes in expression levels. Glutaminase (Gls, Gls2) and glutamine synthetase (GS), both of which are predominantly expressed in astrocytes (Derouiche and Frotscher, 1991) and are responsible for regulating glutamate levels, were also downregulated (Fig. 2a). Furthermore, our gene arrays showed that at most time points there was a significant downregulation of gap junction proteins (Gja1, Gjb2, Gjb6) 24 h following treatment, a finding that indicates reduced spatial buffering capacity (Wallraff et al., 2006). Real-time RT-PCR confirmed the main observations obtained from the gene arrays, i.e., significant upregulation of GFAP, and downregulation of KCNJ10 (KIR 4.1) [data not shown and Ivens et al. (2007), their Fig. 5] as well as KCNJ3 (KIR 3.1), SLC1A2 and SLC1A3, and Gja1, Gjb2, and Gjb6 at all time points (connexins 43, 26, and 30, respectively) (Fig. 2b). In contrast, glutamine synthetase did not show significant downregulation (Fig. 2b).
Figure 2.

Alterations in astrocytic potassium- and glutamate-regulating genes. a, Sham-normalized mRNA expression levels for genes associated with K+ and glutamate homeostasis at 8, 24, and 48 h following in vivo treatment with DOC (D) and albumin (A). b, Sham-normalized mRNA expression levels for selected transcripts (see Results) obtained by real-time RT-PCR 24 h following DOC (gray bars) or albumin (black bars) treatments. c, Astrocyte-enriched cell cultures immunostained for GFAP (red) or NeuN (green). Nuclei visualized with DAPI staining (blue). The graph shows mRNA expression levels in albumin-exposed cultures compared with controls. d, Same as c, for neuron-enriched cultures. *p < 0.05.
Figure 5.

Application of NEURON-based model to determine the effects of glutamate accumulation. a, Somatic EPSP amplitudes for different glutamate time constants. b, Simultaneous glutamate “application” (kinetics represented in the upper trace) at each of the eight synapses elicits AMPA-mediated (middle) and NMDA-mediated (bottom) currents under control conditions (black) and a twofold increase in glutamate decay time constant (blue). c, Ratio of fifth to first EPSP amplitude (P5/P1) at different stimulation frequencies and varying glutamate decay time constants (values related to control). d, Same as in c with GNMDA = 0.
The microarray results hinted at a rapid and robust change in astrocytic gene expression in vivo following BBB breakdown or brain exposure to serum albumin. To further validate the specificity of the astrocytic response to albumin, we exposed cell cultures enriched with either astrocytes or neurons (see Materials and Methods) to albumin for 24 h. Significantly, the astrocytic cultures responded with significant downregulation of the same transcripts found to respond in vivo to albumin (SLC1A3, GS, Gja1, Gjb2, and Gjb6) (Fig. 2c). No significant differences in expression levels of the same transcripts were found in the neuronal-enriched culture (except for downregulation of GS and upregulation of Gjb6 mRNA levels) (Fig. 2d), supporting the notion that the changes observed in vivo do indeed reflect an astrocytic response.
Does epileptogenesis involve reduced glutamate and potassium clearance?
To confirm that the transcriptional changes induced by albumin were associated with altered cellular functions, we investigated the clearance of extracellular glutamate and potassium in cortical slices 24 h following albumin treatment in vivo. To measure synaptic glutamate levels during neuronal activation, we recorded the slowly inactivating (Lester et al., 1990) NMDA currents in cortical neurons by using the whole-cell patch configuration (in the presence of non-NMDA glutamate and GABA receptor blockers; see Materials and Methods). Cells were clamped at +40 mV to prevent a potential confounding effect of postsynaptic depolarization due to the accumulation of synaptic [K+]o. Mean single EPSC rise time and amplitude were similar in both control and albumin-treated groups [14.5 ± 0.5 vs 13.2 ± 0.7 ms and 505 ± 100 vs 492 ± 140 pA, for rise time (data not shown) and amplitude, respectively in treated vs controls (Fig. 3a, inset)], suggesting that no changes in postsynaptic NMDA receptor density or properties at this time point (data not shown). We measured synaptic glutamate elicited by 50 extracellular stimulations at 2, 5, 10, 20, 50, and 100 Hz before and after adding the astrocytic SLC1A2-specific inhibitor, DHK. In neurons from control animals, DHK had no effect on single EPSCs or EPSCs elicited at low stimulation frequencies (<20 Hz). In contrast, stimulation frequencies of >20 Hz resulted in increased NMDA currents (or reduced depression when normalized to the first stimulus) (Fig. 3b,c, left), suggesting that astrocytic glutamate transporters efficiently reduce synaptic glutamate levels at high frequencies of neuronal activation. The same experiments were then repeated 24 h following cortical application of albumin (i.e., during epileptogenesis). In contrast to the control experiments, DHK had no effect on EPSC amplitude in treated slices (Fig. 3b,c, right), supporting reduced expression of the astrocytic transporter SLC1A2. Repetitive stimulation, however, resulted in a stronger depression of EPSC amplitude in treated slices than in controls (see Discussion).
Figure 3.

Electrophysiological evidence for reduced glutamate and potassium buffering during epileptogenesis. a, Single NMDA-mediated EPSCs in control slices and 24 h following albumin treatment in vivo in ACSF (gray) and following DHK (black) application. Inset, Mean EPSC amplitude in ACSF. b, NMDA-mediated EPSCs during train stimulation at 50 Hz in control and treated animals. c, Mean evoked NMDA-mediated EPSC at different stimulation frequencies. d, [K+]o levels in control and treated slices during 20 Hz stimulation. e, Mean [K+]o levels during extracellular stimulation at 2–100 Hz (right). #p < 0.03, *p < 0.001 (n = 6 albumin-treated cells, 5 animals, n = 9 control cells, 7 animals).
To study K+ clearance from the extracellular space, we recorded from control and treated slices (24 h following treatment with albumin) by using ISMEs. We previously reported slower decay kinetics of [K+]o in response to pressure application in BBB-treated animals (Ivens et al., 2007). Here we tested for [K+]o accumulation during neuronal activation at different frequencies of stimulation. In slices from control animals, the increase in [K+]o was limited to 25% of baseline levels (<3.75 mm) at all stimulation frequencies with the used stimulation intensities and number of stimuli. In contrast, in treated slices [K+]o accumulation was significantly higher at frequencies of ≥10 Hz, reaching 6.7 mm (Fig. 3d,e).
Modeling reduced K+ clearance results in frequency-dependent facilitation of EPSPs
To elucidate the possible contribution of astrocytic dysfunction to neuronal excitability, we developed a NEURON-based model of a postsynaptic neuron and an astrocyte. To evaluate the role of increased [K+]o accumulation and glutamate accumulation, we focused on examining changes to excitatory synaptic currents in the postsynaptic neuron (see Materials and Methods). Excitatory synaptic input was simulated by simultaneous application of glutamate at all eight distal dendritic processes (Fig. 4a). In light of our experimental data, we simulated the reduction in K+ clearance by manipulating a [K+]o-regulated potassium removal mechanism (IKIR), while keeping the diffusion component constant. In the absence of neuronal activity, reducing KIR-mediated potassium clearance had no effect on resting [K+]o and thus had a negligible effect on the rising phase and maximal amplitude of a single EPSP (Fig. 4b). Reducing potassium buffering and consequent increased K+ accumulation during repetitive stimulation resulted in enhanced EPSP duration due to slower repolarization (due to a reduced driving force for K+ and a slight increase in NMDA-mediated current) (see below) (Fig. 4b). During repetitive activation, the accumulation of [K+]o near the dendritic compartment reached a maximum of 8.7 (and 16 mm) for reduction to 50% (and 10%) of astrocytic [K+]o buffering capacity, respectively (Fig. 4c). [K+]o accumulation during repetitive stimulation had a differential effect on AMPA- and NMDA-mediated currents: while the AMPA current showed frequency-dependent depression due to receptor desensitization, the NMDA component was strongly facilitated due to membrane depolarization (Fig. 4b). Reducing astrocytic potassium uptake from the extracellular space to 10% of control values resulted in an increase in total charge transfer mediated by the NMDA component of 44, 344, and 84% at 10, 20, and 100 Hz, respectively, while the AMPA charge transfer decreased by 5, 24, and 15%, respectively (Fig. 4d). Overall, there was a frequency-dependent increase in EPSP amplitude (Fig. 4e) associated with longer decay time (Fig. 4f). Repeated simulations with no NMDA conductance (GNMDA = 0, with concomitant increased AMPA conductance, to achieve similar depolarization for a single stimulus) resulted in a much smaller facilitation (Fig. 4, compare g, h).
Figure 4.
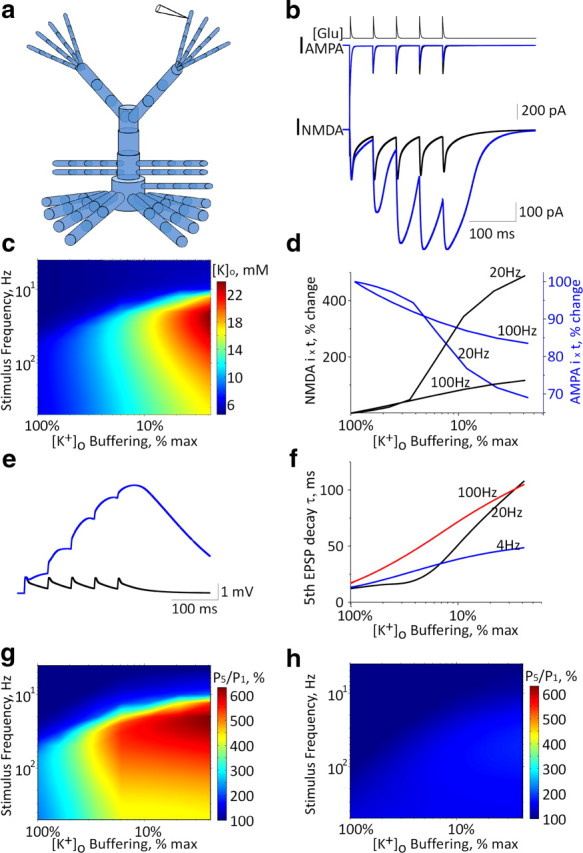
Application of NEURON-based model to determine the effects of [K+]o accumulation. a, Schematic diagram of the modeled layer 2/3 pyramidal neuron containing 74 compartments with eight synapses (with AMPA and NMDA currents), one at each distal dendrite. b, Increasing glutamate levels at each of the eight synapses (top trace representing kinetics of synaptic glutamate level) elicits AMPA-mediated (middle trace) and NMDA-mediated (bottom trace) currents under control conditions (black) and under reduced astrocytic potassium clearance (under a 10-fold decrease of astrocytic K+ clearance, blue trace). c, Maximal K+ concentrations recorded in the vicinity of a distal dendritic compartment during repetitive stimulation as a function of [K+]o clearance and stimulation frequency. d, Percentage change of total charge transfer by NMDA (black) and AMPA (blue) receptors during stimulation at 20 and 100 Hz. e, Somatic EPSPs under control conditions (black) and under reduced astrocytic potassium clearance (10-fold reduction of control levels, blue trace). f, Fifth EPSP (elicited by stimulation at 4, 20, and 100 Hz) decay time constant at different levels of astrocytic potassium clearance rates. g, Ratio of fifth to first EPSP amplitude (P5/P1) at stimulation frequencies of 4–500 Hz at different levels of astrocytic potassium clearance rates. h, Same as in g with GNMDA = 0.
Modeling reduced glutamate clearance results in frequency-dependent depression of EPSPs
We next used the NEURON model and simulation paradigms described above to test the expected effect of reduced glutamate uptake. We simulated the reduction in glutamate uptake by slowing the transmitter's synaptic decay function. A twofold increase in the glutamate decay time constant resulted in a 48% increase in EPSP amplitude (from 25 to 37 mV) at a single postsynaptic dendrite and a 60% increase in the amplitude of the summated somatic EPSP (Fig. 5a). While for a single stimulation both AMPA and NMDA components were increased, with repetitive activation, a marked decrease in the AMPA current (due to receptor desensitization) (Otis et al., 1996) and a strong facilitation of the NMDA current (due to postsynaptic depolarization) (Mayer et al., 1984) (Fig. 5b,c) were measured. Somatic EPSP facilitation (ratio of fifth to first EPSP amplitude) (Fig. 5c) was maximal at 100 Hz with our initial conditions for glutamate clearance. Inhibiting glutamate clearance did not affect EPSP facilitation at low stimulation frequencies (<20 Hz) but reduced it at high stimulation frequencies (>80 Hz). The decreased facilitation was due to a reduced AMPA current through the desensitized receptors, thus keeping the membrane potential below the threshold for NMDA receptor activation. In simulations performed in the absence of NMDA conductance, EPSP facilitation was reduced at most stimulation frequencies, with only a small (<150%) residual facilitation measured at high stimulation frequencies (>100 Hz) (Fig. 5d).
Modeling the concerted effect of reduced potassium and glutamate clearance
The simulations showed that while synaptic glutamate levels mainly affected the first EPSP in the train, an activity-dependent increase in [K+]o mainly enhanced EPSP facilitation in a frequency-dependent manner. Since our molecular data indicated a decrease in both potassium and glutamate buffering mechanisms, we simulated their joint effect on synaptic transmission. Decreasing the clearance of [K+]o led to maximal EPSP facilitation when stimulating at 20 Hz, while a concurrent twofold reduction in glutamate uptake shifted the optimal frequency for maximal facilitation to 10 Hz (Fig. 6a). Concurrent reductions in glutamate and [K+]o clearance led to increases in the duration of the first EPSP, which in turn elicited increased and longer NMDA receptor activation per stimulus. The longer EPSPs allowed for a larger charge transfer with longer interstimulus intervals (i.e., reduced frequency) (Fig. 6b), thus lowering the optimal stimulation frequency. To assess the sensitivity of the synaptic response during repetitive stimulation (at 20 Hz), we used several glutamate decay time constants and varying levels of [K+]o uptake. We plotted the maximal EPSP amplitude as a function of [K+]o; Figure 6c demonstrates that increasing synaptic glutamate led to small increases in the maximal EPSP amplitudes for all levels of [K+]o. However, synaptic facilitation was decreased with reduced glutamate uptake: thus, synaptic [K+]o accumulation to 10 mm was associated with 40% EPSP facilitation (upon the fifth stimulation) under baseline glutamate clearance, but with only 22% facilitation when glutamate decay time was doubled (Fig. 6d).
Figure 6.

Modeling the concerted effect of reduced potassium and glutamate clearance. a, EPSP facilitation (relative to maximal value) for a 10-fold decrease in [K+]o clearance (black), a twofold slowing of glutamate decay time constant (red), and downregulation of both uptake mechanisms (blue) as a function of stimulation frequency. b, EPSP traces for 10 and 20 Hz trains under a 10-fold decrease in astrocytic K+ clearance (gray and blue traces, respectively) and with both uptake mechanisms downregulated (at 10 Hz, black). The dashed line marks the resting potential. c, Maximal EPSP amplitude elicited by a train of five stimuli as a function of maximal [K+]o for different glutamate uptake decay time constants (for 1.2, 2.2, 3.2, and 3.5 ms). d, EPSP facilitation [ratio of fifth to first EPSP amplitude (P5/P1)] for 20 Hz stimulation for different glutamate decay time constants (as in c).
Electrophysiological evidence for frequency-dependent synaptic facilitation during epileptogenesis
Our simulation data predicted maximal EPSP facilitation at 20 Hz when [K+]o clearance is reduced and decreased facilitation (at 50–100 Hz) when the only change induced is glutamate accumulation in the synaptic cleft. We therefore measured field potentials in response to stimulation at various frequencies in brain slices during “epileptogenesis” (exposure to albumin in sACSF) compared with controls (sACSF alone). Comparison of the field potential amplitude and absolute integral during the first five stimuli revealed a significant reduction in both measures only under 100 Hz stimulation [amplitude: 1.13 ± 0.12 vs 0.46 ± 0.03 mV, 1.44 ± 0.36 vs 0.47 ± 0.09 mV; and area: 2.4 ± 0.2 vs 0.9 ± 0.02 V · s, 4.4 ± 1.1 vs 0.5 ± 0.7 V · s, first vs fifth stimulus, control (n = 5) and treated (n = 4), respectively, p < 0.05]. Comparing field potential duration (measured at 1/3 maximal amplitude) for the first versus the fifth stimulus among different frequencies did not reveal any changes in control slices. In contrast, in treated slices the field potential was significantly prolonged at 10 and 20 Hz (10 Hz: 7.5 ± 0.4 vs 9.4 ± 5.5 ms, 6.5 ± 0.7 vs 13.1 ± 2.6 ms, 20 Hz: 6.0 ± 0.9 vs 6.8 ± 0.8 ms, 6.6 ± 0.8 vs 12.9 ± 2.9 ms for first vs fifth stimulus in control and treated, respectively, p < 0.05) (Fig. 7c). Interestingly, in the “treated” group, stimulation-induced frequency-dependent, long-lasting epileptiform discharges occurred most reliably during 10 Hz stimulation (4 of 4 slices, n = 3 animals), and sometimes at 20 Hz (3 of 4 slices) and 5 Hz (2 of 4 slices), but never at higher frequencies (Fig. 7d). Epileptiform discharges were observed in one control slice without any apparent frequency dependence (5–50 Hz, 1 of 5 slices, n = 3 animals) (Fig. 7d).
Figure 7.

Recording in vitro shows frequency-dependent increased neuronal excitability and hypersynchronous network activity during albumin-mediated epileptogenesis. a, b, Neocortical field potential recordings of brain slices during stimulation trains of 50 pulses at 2, 10, and 100 Hz. Field responses were facilitated in the albumin-treated slices, observed as increased duration of the population spikes (see inset in a, b). c, Comparison of the average field potential duration (at 1/3 maximal amplitude) for the fifth to the first evoked response reveals maximal facilitation at 10 Hz. d, Percentage of slices showing prolonged, paroxysmal discharges. *p < 0.05.
Together, our experiments show that exposure to albumin in vitro induces changes in neuronal excitability and that evoked network activity facilitates, and often turns into, robust epileptiform discharges upon repetitive stimulation. We found that 10–20 Hz is the most reliable frequency, as was also predicted by our K+ recording data (Fig. 3d) and by our model in the case of reduction in [K+]o clearance with or without glutamate accumulation (Fig. 4g) (see Discussion).
Discussion
The primary goal of the present study was to study the role of astrocytes in epileptogenesis. With the BBB disruption and albumin-induced models of epileptogenesis, the following findings were obtained early during epileptogenesis: (1) Similar significant changes in astrocytic gene expression occurred following the two treatments. (2) Transcriptional data predicted disturbed homeostasis of extracellular potassium and glutamate. (3) Intracellular recordings confirmed reduced astrocytic glutamate uptake, which did not, however, seem to account for increased neuronal excitability. (4) Recordings with ISMEs confirmed activity-dependent impaired [K+]o buffering. (5) A NEURON-based model predicted that reduced [K+]o buffering leads to frequency- and NMDAR-dependent facilitation of EPSPs, with maximal facilitation ∼20 Hz, while reduced clearance of glutamate results in a modest increase in the amplitude and duration of a single EPSP with decreased facilitation during repetitive stimulation. (6) Finally, extracellular recordings in cortical slices confirmed the frequency-dependent facilitation of synaptic activity predicted by the model and showed epileptiform discharges at preferred stimulation frequencies of 10–20 Hz. Overall, the present study suggests a key role for reduced astrocytic-mediated clearance of [K+]o in activity-dependent facilitation of synaptic activity during epileptogenesis.
The working hypothesis for the present study is based on our previous experimental results, which demonstrated that BBB breakdown induces epileptogenesis (Seiffert et al., 2004; Ivens et al., 2007; Tomkins et al., 2007) and increases the expression of the astrocytic marker, GFAP, within hours following the epileptogenic treatment. This rapid astrocytic response was observed before the emergence of epileptiform activity, leading to the hypothesis that astrocytes play a role in the epileptogenic process [see also Tian et al. (2005) and Ding et al. (2007)]. Indeed, changes in the structure and function of astrocytes are found in a wide variety of brain insults, including epilepsy, in both animals and man (Schröder et al., 1999; Kivi et al., 2000; Bordey et al., 2001; Herman, 2002; Jauch et al., 2002). However, the role of astrocytic dysfunction in disease progression and neuronal dysfunction is not well understood [for review, see Heinemann et al. (1999), Seifert et al. (2006), and Schwarcz (2008)]. Since BBB breakdown causes albumin extravasation from brain vessels and its specific uptake by astrocytes (Ivens et al., 2007), we put forward the hypothesis that under BBB breakdown astrocytic gene expression is directly modulated by albumin. A role for serum albumin in epileptogenesis is also supported by experiments showing that albumin induced focal epileptiform activity in a dose-dependent manner (Seiffert et al., 2004). Importantly, albumin-induced epileptogenesis is observed only after a window period of several hours (in vitro) or days (in vivo), with no apparent effect on neuronal membrane characteristics, firing properties, or amplitude and duration of single EPSCs (Fig. 3) (Seiffert et al., 2004). Furthermore, the specific uptake of albumin by astrocytes, together with the rapid changes in the level of astrocyte-specific proteins (Ivens et al., 2007), led us to hypothesize that a concerted astrocytic transcriptional response may underlie epileptogenesis under these conditions. Using microarray technology, we confirmed that a large number of astrocytic genes do show significant changes in expression levels as early as 8 h following an epileptogenic event, several days before epileptic activity emerges (Seiffert et al., 2004; Ivens et al., 2007). However, it is plausible that under pathological conditions, different cell populations may express previously unexpressed transcripts, thus confounding our results. Nevertheless, the cell-specific mRNA changes in response to albumin exposure that were evident in our astrocytic- and neuronal-enriched cultures constitute further support for our conclusion that the observed changes do occur preferentially in astrocytes (Fig. 2). The cluster analysis and the strikingly high correlation between expression profiles of both BBB breakdown and albumin treatments at the various time points support the notion that the extravasation of the most abundant serum protein, albumin, through the injured vessels plays a role in the transcriptional modulation of astrocytic genes. An alternative hypothesis—that albumin itself is disruptive to the BBB, leading to the extravasation of some other blood-derived mediator—is less likely, since local application of albumin did not increase BBB permeability to large molecules, as previously measured using systemic injection of Evans-blue (Seiffert et al., 2004).
Since astrocytes are known to be key contributors to [K+]o buffering, we searched our microarrays for expression levels of astrocytic K+ channels (Barres et al., 1990). Indeed, Kir 4.1 (but not other K+ channels) (Fig. 2a), previously shown to be expressed in neocortical astrocytes (Higashi et al., 2001; Hibino et al., 2004), was downregulated, leading to activity-dependent accumulation of [K+]o in treated cortical slices (Fig. 3) (Ivens et al., 2007). Reduced [K+]o clearance has been reported in the injured brain (D'Ambrosio et al., 1999), and a loss of IKIR has been found in reactive astrocytes around freeze lesions (Bordey et al., 2001), after ischemic insults (Köller et al., 2000) and direct injuries (Schröder et al., 1999), and in epileptic Tsc1 knock-out mice (Jansen et al., 2005) as well as in human subjects with temporal lobe epilepsy (TLE) (Bordey and Sontheimer, 1998; Hinterkeuser et al., 2000; Kivi et al., 2000; Jauch et al., 2002). It is noteworthy that KIR 4.1 knock-out mice display seizure activity very early in life consistent with the idea that downregulation of KIR 4.1 channels may contribute to epileptogenesis (Djukic et al., 2007). The hypothesis that elevated [K+]o could lead to seizure initiation (Fertziger and Ranck, 1970) is not a new one; however, in this study we show that a selective downregulation of the KIR 4.1 channel occurs before the emergence of epileptic activity, thus highlighting the potential role of potassium accumulation in epileptogenesis. To what extent KIR channels contribute to the spatial buffering of [K+]o is not entirely known and may differ between brain regions. In hippocampal slices, low concentrations of Ba2+ augmented stimulus-induced K+ by 147% (to >9 mm) (Gabriel et al., 1998), while in the neocortex, Ba2+ slowed down the clearance of iontophoretically applied K+ by only 70% (Ivens et al., 2007). Notably, in both preparations, epileptogenesis was associated with a reduced effect of Ba2+ indicating reduced IKIR. The relatively high [K+]o found during stimulation in the neocortex in our study may reflect impairments in additional buffering mechanisms such as reduced expression of leak K+ channels [not supported by our microarray data, but see Päsler et al. (2007)], or the downregulation of gap junction proteins (Fig. 2). However, a recent study in connexin knock-out mice (which lack gap junctions in the hippocampus) showed an almost conserved capacity for potassium clearance (Wallraff et al., 2006). In addition, due to their rectification properties, the contribution of KIR channels to K+ clearance becomes critical when local [K+]o are high (Newman, 1993; Chen and Nicholson, 2000), thus augmenting their role in buffering potassium during high-frequency stimulation (Fig. 3d). Finally, reduced [K+]o clearance is expected to enhance potassium accumulation due to delayed neuronal repolarization and facilitated synaptic potentials (Fig. 4).
Due to the lack of pharmacological tools that specifically block astrocytic KIR channels, it is difficult to experimentally determine its role in controlling neuronal excitability. We therefore used computer simulations that predicted that synaptic accumulation of potassium will lead to frequency-dependent facilitation of EPSPs. Importantly, [K+]o accumulation under these conditions did not have an effect on a single EPSC, consistent with the observation of normal field potentials during epileptogenesis (Seiffert et al., 2004). In contrast, EPSCs were strongly facilitated at stimulation frequencies between 10 and 50 Hz (maximum around 20 Hz) due to membrane depolarization and increased NMDA conductance. Under these conditions, facilitation (>300%) was associated with synaptic [K+]o levels reaching ∼8 mm. This value may seem high considering that during normal neuronal activity (Heinemann et al., 1990) or that recorded in the present study (Fig. 3). Yet these levels were measured using ISMEs at a distance from the narrow synaptic cleft (<200 Å) (Egelman and Montague, 1999; Savtchenko et al., 2000). In fact, the model predicts that during neuronal activity, [K+]o levels in the extracellular space near the soma are three times lower (due to diffusion and astrocytic uptake) than levels in the synaptic cleft, in line with the results of Somjen et al. (2008).
In addition to the “potassium hypothesis,” our molecular experiments point to reduced expression of astrocytic glutamate transporters in both albumin- and DOC-treated rats. In the normal brain, passive diffusion and transport clears released glutamate into neurons (via members of the solute carrier protein family—SLC1A) and astrocytes (specifically SLC1A2, SLC1A3, and SLC1A4). The relative contribution of each of these mechanisms in the neocortex is unknown. Studies in the hippocampus indicated that astrocytes play a central role in the removal of synaptic glutamate (Bergles and Jahr, 1998), whereas neuronal transport plays a negligible role (Sarantis et al., 1993), leading to the hypothesis that downregulation of astrocytic glutamate transporters entails glutamate accumulation in the synaptic cleft and increased excitability. A reduction in glutamate uptake mechanisms has previously been reported in various neurodegenerative diseases frequently associated with BBB breakdown (Rothstein et al., 1992; Zlokovic, 2008). However, evidence for the role of glutamate uptake mechanisms in epilepsy remains inconclusive (Eid et al., 2008). Although Tanaka et al. (1997) demonstrated spontaneous seizures in SLC1A2 knock-out mice, studies of tissue from human TLE patients have failed to provide conclusive evidence for reduced glutamate uptake (Tessler et al., 1999; Proper et al., 2002). Moreover, the consequences of reduced glutamate uptake are still controversial, with considerable heterogeneity between various brain regions and preparations (Hestrin et al., 1990; Sarantis et al., 1993; Turecek and Trussell, 2000; Arnth-Jensen et al., 2002). To determine the functional consequences of the observed transcriptional response on glutamate homeostatic mechanisms, we recorded whole-cell glutamatergic currents before and after application of DHK, a selective inhibitor of the astrocytic glutamate transporter SLC1A2 (Arriza et al., 1994; Rothstein et al., 1994), in slices from control and treated rats. In agreement with previous studies, we found no effect for DHK on single EPSCs or EPSCs evoked by low-frequency stimulation (Hestrin et al., 1990; Diamond and Jahr, 2000; Takayasu et al., 2004). We did, however, find increased glutamatergic currents under DHK with stimulation frequencies of >20 Hz in control slices, similar to the effect of TBOA in the hippocampus (Arnth-Jensen et al., 2002). In contrast, in slices from albumin-treated animals, DHK had no effect on EPSCs, supporting the observed downregulation of SLC1A2. Yet, despite the reduced DHK effect, we did not observe an increase in EPSCs 24 h following the epileptogenic insult. This finding may be explained by activation of compensatory mechanisms (e.g., upregulation of neuronal glutamate uptake proteins) (Fig. 2a), excessive activation of presynaptic mGluR2 (Scanziani et al., 1997; Iserhot et al., 2004) or altered expression of NMDA receptor subunits.
Our computer model predicted that reduced glutamate uptake alone will result in reduced synaptic facilitation due to desensitization of AMPA receptors. These predictions are in agreement with previous studies in brainstem slices showing a frequency-dependent decrease in AMPA currents after incubation with the glutamate uptake blockers, THA and DHK (Turecek and Trussell, 2000), but stand in contrast to recordings in hippocampal neurons showing increased NMDA-mediated EPSPs under similar conditions (Arnth-Jensen et al., 2002). The difference may be because the latter study was conducted with holding voltages of +40 mV, resulting in a higher open probability of NMDAR operated ion channels. While our molecular and electrophysiological experiments do indicate a reduction in astrocytic glutamate uptake during the early stages of epileptogenesis, both the electrophysiological data and computer simulations suggest that only at high stimulation frequencies (>100 Hz), does sufficient transmitter accumulate to facilitate EPSCs (see below).
Changes in astrocytic membrane potential were not implemented in our model. Extracellular accumulation of potassium results in astrocytic depolarization, which inhibits glutamate uptake (or even causes a reversal of the uptake mechanism) (Szatkowski et al., 1990). Simulating a concurrent reduction in both uptake mechanisms, resulted in a reduction of the stimulation frequency at which maximal facilitation occurs (from ∼20 to 10 Hz). The simulations predicted that when both [K+]o and glutamate accumulate in the synapse, more NMDA receptors are activated at any given [K+]o (Fig. 6). In addition, we expect such depolarization to further reduce the cationic-coupled glutamatergic transport, thus enhancing voltage-dependent NMDA currents. The optimal frequency for synaptic facilitation recorded in brain slices exposed to albumin in sACSF was ∼20 Hz, which as predicted by the model, was mainly due to reduced clearance of [K+]o. This stimulation frequency—20 Hz—also proved to be the minimal stimulation frequency at which we measured [K+]o accumulation during epileptogenesis. It is striking that repetitive stimulation under these conditions often resulted in seconds-long, seizure-like activity, highlighting the potential role for BBB breakdown and brain exposure to serum albumin in neuronal hypersynchronicity, enhanced excitability, and the generation of seizures. A plausible hypothesis would be that the increased, repeated activation of NMDA receptors leads to nonspecific synaptic plasticity, thus strengthening excitatory synapses and causing persistent hyperexcitability (Li and Prince, 2002; Shao and Dudek, 2004). This premise also provides a satisfactory explanation for the efficacy of NMDAR antagonists in improving cortical function in animal studies of brain injury and stroke—conditions in which the BBB is frequently impaired (Hickenbottom and Grotta, 1998; Sonkusare et al., 2005) and seizures are often observed. While further studies are needed to confirm this hypothesis, we propose astrocytic reaction in the injured cortex, and specifically impaired buffering of extracellular K+, as novel targets for the prevention and treatment of injury-related neocortical epilepsies.
Footnotes
This study was supported by the Sonderforschungsbereich TR3 (A.F., U.H.), the German-Israel Foundation for Scientific Research and Development (A.F.), the Israel Science Foundation (566/07, A.F.), Minerva Stiftung (Y.D.), the United States-Israel Binational Science Foundation (A.F., D.K.), and the CURE Foundation (D.K., A.F.).
References
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Araque et al., 2001.Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen et al., 2002.Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Arriza et al., 1994.Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres et al., 1990.Barres BA, Chun LL, Corey DP. Ion channels in vertebrate glia. Annu Rev Neurosci. 1990;13:441–474. doi: 10.1146/annurev.ne.13.030190.002301. [DOI] [PubMed] [Google Scholar]
- Bergles and Jahr, 1998.Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral–commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey and Sontheimer, 1998.Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- Bordey et al., 2001.Bordey A, Lyons SA, Hablitz JJ, Sontheimer H. Electrophysiological characteristics of reactive astrocytes in experimental cortical dysplasia. J Neurophysiol. 2001;85:1719–1731. doi: 10.1152/jn.2001.85.4.1719. [DOI] [PubMed] [Google Scholar]
- Butt and Kalsi, 2006.Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacheaux et al., 2009.Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-β signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy et al., 2008.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry et al., 1995.Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Chen and Nicholson, 2000.Chen KC, Nicholson C. Spatial buffering of potassium ions in brain extracellular space. Biophys J. 2000;78:2776–2797. doi: 10.1016/S0006-3495(00)76822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani et al., 1978.Ciani S, Krasne S, Miyazaki S, Hagiwara S. A model for anomalous rectification: electrochemical-potential-dependent gating of membrane channels. J Membr Biol. 1978;44:103–134. doi: 10.1007/BF01976035. [DOI] [PubMed] [Google Scholar]
- Dai et al., 2005.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio et al., 1999.D'Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Impaired K+ homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci. 1999;19:8152–8162. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouiche and Frotscher, 1991.Derouiche A, Frotscher M. Astroglial processes around identified glutamatergic synapses contain glutamine synthetase: evidence for transmitter degradation. Brain Res. 1991;552:346–350. doi: 10.1016/0006-8993(91)90103-3. [DOI] [PubMed] [Google Scholar]
- Diamond, 2005.Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond and Jahr, 2000.Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- Ding et al., 2007.Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic et al., 2007.Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman DM, Montague PR. Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys J. 1999;76:1856–1867. doi: 10.1016/s0006-3495(99)77345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes—key players in human mesial temporal lobe epilepsy? Epilepsia. 2008;49(Suppl 2):42–52. doi: 10.1111/j.1528-1167.2008.01492.x. [DOI] [PubMed] [Google Scholar]
- Fertziger AP, Ranck JB., Jr Potassium accumulation in interstitial space during epileptiform seizures. Exp Neurol. 1970;26:571–585. doi: 10.1016/0014-4886(70)90150-0. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Eilers A, Kivi A, Kovacs R, Schulze K, Lehmann TN, Heinemann U. Effects of barium on stimulus induced changes in extracellular potassium concentration in area CA1 of hippocampal slices from normal and pilocarpine-treated epileptic rats. Neurosci Lett. 1998;242:9–12. doi: 10.1016/s0304-3940(98)00012-3. [DOI] [PubMed] [Google Scholar]
- Geursen A, Grigor MR. Serum albumin secretion in rat milk. J Physiol. 1987;391:419–427. doi: 10.1113/jphysiol.1987.sp016746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronau I, Moran S. Optimal implementations of UPGMA and other common clustering algorithms. Inf Process Lett. 2007;104:205–210. [Google Scholar]
- Heinemann U, Schaible HG, Schmidt RF. Changes in extracellular potassium concentration in cat spinal cord in response to innocuous and noxious stimulation of legs with healthy and inflamed knee joints. Exp Brain Res. 1990;79:283–292. doi: 10.1007/BF00608237. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Gabriel S, Schuchmann S, Eder C. Contribution of astrocytes to seizure activity. Adv Neurol. 1999;79:583–590. [PubMed] [Google Scholar]
- Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59:S21–S26. doi: 10.1212/wnl.59.9_suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Sah P, Nicoll RA. Mechanisms generating the time course of dual component excitatory synaptic currents recorded in hippocampal slices. Neuron. 1990;5:247–253. doi: 10.1016/0896-6273(90)90162-9. [DOI] [PubMed] [Google Scholar]
- Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. 2004;279:44065–44073. doi: 10.1074/jbc.M405985200. [DOI] [PubMed] [Google Scholar]
- Hickenbottom SL, Grotta J. Neuroprotective therapy. Semin Neurol. 1998;18:485–492. doi: 10.1055/s-2008-1040901. [DOI] [PubMed] [Google Scholar]
- Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- Hinterkeuser S, Schröder W, Hager G, Seifert G, Blümcke I, Elger CE, Schramm J, Steinhäuser C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–2096. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Iserhot C, Gebhardt C, Schmitz D, Heinemann U. Glutamate transporters and metabotropic receptors regulate excitatory neurotransmission in the medial entorhinal cortex of the rat. Brain Res. 2004;1027:151–160. doi: 10.1016/j.brainres.2004.08.052. [DOI] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia. 2005;46:1871–1880. doi: 10.1111/j.1528-1167.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Jauch R, Windmüller O, Lehmann TN, Heinemann U, Gabriel S. Effects of barium, furosemide, ouabaine and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res. 2002;925:18–27. doi: 10.1016/s0006-8993(01)03254-1. [DOI] [PubMed] [Google Scholar]
- Kager H, Wadman WJ, Somjen GG. Simulated seizures and spreading depression in a neuron model incorporating interstitial space and ion concentrations. J Neurophysiol. 2000;84:495–512. doi: 10.1152/jn.2000.84.1.495. [DOI] [PubMed] [Google Scholar]
- Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol. 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufer D, Ogle WO, Pincus ZS, Clark KL, Nicholas AC, Dinkel KM, Dumas TC, Ferguson D, Lee AL, Winters MA, Sapolsky RM. Restructuring the neuronal stress response with anti-glucocorticoid gene delivery. Nat Neurosci. 2004;7:947–953. doi: 10.1038/nn1296. [DOI] [PubMed] [Google Scholar]
- Kivi A, Lehmann TN, Kovács R, Eilers A, Jauch R, Meencke HJ, von Deimling A, Heinemann U, Gabriel S. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci. 2000;12:2039–2048. doi: 10.1046/j.1460-9568.2000.00103.x. [DOI] [PubMed] [Google Scholar]
- Köller H, Schroeter M, Jander S, Stoll G, Siebler M. Time course of inwardly rectifying K(+) current reduction in glial cells surrounding ischemic brain lesions. Brain Res. 2000;872:194–198. doi: 10.1016/s0006-8993(00)02434-3. [DOI] [PubMed] [Google Scholar]
- Kuffler SW, Potter DD. Glia in the leech central nervous system: Physiological properties and neuron-glia relationship. J Neurophysiol. 1964;27:290–320. doi: 10.1152/jn.1964.27.2.290. [DOI] [PubMed] [Google Scholar]
- Lester RAJ, Clements JD, Westbrook GL, Jahr CE. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 1990;346:565–567. doi: 10.1038/346565a0. [DOI] [PubMed] [Google Scholar]
- Li H, Prince DA. Synaptic activity in chronically injured, epileptogenic sensory-motor neocortex. J Neurophysiol. 2002;88:2–12. doi: 10.1152/jn.00507.2001. [DOI] [PubMed] [Google Scholar]
- Lux HD, Neher E. The equilibration time course of [K+]o in cat cortex. Exp Brain Res. 1973;17:190–205. doi: 10.1007/BF00235028. [DOI] [PubMed] [Google Scholar]
- Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I, Janigro D. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia. 2007;48:732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Myme CI, Sugino K, Turrigiano GG, Nelson SB. The NMDA-to-AMPA ratio at synapses onto layer 2/3 pyramidal neurons is conserved across prefrontal and visual cortices. J Neurophysiol. 2003;90:771–779. doi: 10.1152/jn.00070.2003. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131–140. doi: 10.1227/01.neu.0000097715.11966.8e. discussion 141–142. [DOI] [PubMed] [Google Scholar]
- Newman EA. Inward-rectifying potassium channels in retinal glial (Muller) cells. J Neurosci. 1993;13:3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Päsler D, Gabriel S, Heinemann U. Two-pore-domain potassium channels contribute to neuronal potassium release and glial potassium buffering in the rat hippocampus. Brain Res. 2007;1173:14–26. doi: 10.1016/j.brainres.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Pavlovsky L, Browne RO, Friedman A. Pyridostigmine enhances glutamatergic transmission in hippocampal CA1 neurons. Exp Neurol. 2003;179:181–187. doi: 10.1016/s0014-4886(02)00016-x. [DOI] [PubMed] [Google Scholar]
- Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MGA, Schrama LH, van Veelen CWM, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PNE. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2002;125:32–43. doi: 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: A novel functional genomics compendium with automated data mining and query reformulation support. Bioinformatics. 1998;14:656–664. doi: 10.1093/bioinformatics/14.8.656. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Saftenku EE. Modeling of slow glutamate diffusion and AMPA receptor activation in the cerebellar glomerulus. J Theor Biol. 2005;234:363–382. doi: 10.1016/j.jtbi.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J Physiol. 1984;347:641–657. doi: 10.1113/jphysiol.1984.sp015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarantis M, Ballerini L, Miller B, Silver RA, Edwards M, Attwell D. Glutamate uptake from the synaptic cleft does not shape the decay of the non-NMDA component of the synaptic current. Neuron. 1993;11:541–549. doi: 10.1016/0896-6273(93)90158-n. [DOI] [PubMed] [Google Scholar]
- Savtchenko LP, Antropov SN, Korogod SM. Effect of voltage drop within the synaptic cleft on the current and voltage generated at a single synapse. Biophys J. 2000;78:1119–1125. doi: 10.1016/S0006-3495(00)76670-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schröder W, Hager G, Kouprijanova E, Weber M, Schmitt AB, Seifert G, Steinhäuser C. Lesion-induced changes of electrophysiological properties in astrocytes of the rat dentate gyrus. Glia. 1999;28:166–174. [PubMed] [Google Scholar]
- Schröder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, Steinhäuser C. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia. 2000;41:S181–S184. doi: 10.1111/j.1528-1157.2000.tb01578.x. [DOI] [PubMed] [Google Scholar]
- Schwarcz R. Early glial dysfunction in epilepsy. Epilepsia. 2008;49(Suppl 2):1–2. doi: 10.1111/j.1528-1167.2008.01487.x. [DOI] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood–brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LR, Dudek FE. Increased excitatory synaptic activity and local connectivity of hippocampal CA1 pyramidal cells in rats with kainate-induced epilepsy. J Neurophysiol. 2004;92:1366–1373. doi: 10.1152/jn.00131.2004. [DOI] [PubMed] [Google Scholar]
- Somjen GG, Kager H, Wadman WJ. Computer simulations of neuron-glia interactions mediated by ion flux. J Comput Neurosci. 2008;25:349–365. doi: 10.1007/s10827-008-0083-9. [DOI] [PubMed] [Google Scholar]
- Sonkusare SK, Kaul CL, Ramarao P. Dementia of Alzheimer's disease and other neurodegenerative disorders—memantine, a new hope. Pharmacol Res. 2005;51:1–17. doi: 10.1016/j.phrs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Su ZZ, Leszczyniecka M, Kang DC, Sarkar D, Chao W, Volsky DJ, Fisher PB. Insights into glutamate transport regulation in human astrocytes: Cloning of the promoter for excitatory amino acid transporter 2 (EAAT2) Proc Natl Acad Sci U S A. 2003;100:1955–1960. doi: 10.1073/pnas.0136555100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–446. doi: 10.1038/348443a0. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Iino M, Ozawa S. Roles of glutamate transporters in shaping excitatory synaptic currents in cerebellar Purkinje cells. Eur J Neurosci. 2004;19:1285–1295. doi: 10.1111/j.1460-9568.2004.03224.x. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tessler S, Danbolt NC, Faull RL, Storm-Mathisen J, Emson PC. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience. 1999;88:1083–1091. doi: 10.1016/s0306-4522(98)00301-7. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O, Kaufer D, Korn A, Shelef I, Golan H, Reichenthal E, Soreq H, Friedman A. Frequent blood-brain barrier disruption in the human cerebral cortex. Cell Mol Neurobiol. 2001;21:675–691. doi: 10.1023/A:1015147920283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O, Friedman O, Ivens S, Reiffurth C, Major S, Dreier JP, Heinemann U, Friedman A. Blood-brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis. 2007;25:367–377. doi: 10.1016/j.nbd.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Traub RD, Buhl EH, Gloveli T, Whittington MA. Fast rhythmic bursting can be induced in layer 2/3 cortical neurons by enhancing persistent Na+ conductance or by blocking BK channels. J Neurophysiol. 2003;89:909–921. doi: 10.1152/jn.00573.2002. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Control of synaptic depression by glutamate transporters. J Neurosci. 2000;20:2054–2063. doi: 10.1523/JNEUROSCI.20-05-02054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet EA, da Costa Araújo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130:521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58:168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Fernald RD. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J Comput Biol. 2005;12:1047–1064. doi: 10.1089/cmb.2005.12.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]