

Abstract
Background:
Mutations in the Twinkle (PEO1) gene are a recognized cause of autosomal dominant progressive external ophthalmoplegia (adPEO), resulting in the accumulation of multiple mitochondrial DNA (mtDNA) deletions and cytochrome c oxidase (COX)-deficient fibers in skeletal muscle secondary to a disorder of mtDNA maintenance. Patients typically present with isolated extraocular muscle involvement, with little apparent evidence of the clinical heterogeneity documented in other mtDNA maintenance disorders, in particular POLG-related disease.
Methods:
We reviewed the clinical, histochemical, and molecular genetics analysis of 33 unreported patients from 26 families together with all previous cases described in the literature to define the clinical phenotype associated with PEO1 mutations.
Results:
Ptosis and ophthalmoparesis were almost universal clinical features among this cohort, with 52% (17/33) reporting fatigue and 33% (11/33) having mild proximal myopathy. Features consistent with CNS involvement were rarely described; however, in 24% (8/33) of the patients, cardiac abnormalities were reported. Mitochondrial histochemical changes observed in muscle showed remarkable variability, as did the secondary mtDNA deletions, which in some patients were only detected by PCR-based assays and not Southern blotting. Moreover, we report 7 novel PEO1 variants.
Conclusions:
Our data suggest a shared clinical phenotype with variable mild multiorgan involvement, and that the contribution of PEO1 mutations as a cause of adPEO may well be underestimated. Direct sequencing of the PEO1 gene should be considered in adPEO patients prior to muscle biopsy.
GLOSSARY
- adPEO
= autosomal dominant progressive external ophthalmoplegia;
- COX
= cytochrome c oxidase;
- IOSCA
= infantile-onset spinocerebellar ataxia;
- mtDNA
= mitochondrial DNA;
- PEO
= progressive external ophthalmoplegia;
- SANDO
= sensory ataxic neuropathy, dysarthria, and ophthalmoparesis;
- SDH
= succinate dehydrogenase.
Mutations in an increasing number of nuclear genes involved in the maintenance of mitochondrial DNA (mtDNA) are being described, associated with an extensive spectrum of clinical phenotypes ranging from severe encephalopathy in infancy and childhood to late-onset progressive external ophthalmoplegia (PEO), ataxia, and myopathy. Multiple mtDNA deletion disorders are genetically heterogeneous, and may be linked to recessive mutations in polymerase gamma, POLG,1 or TYMP,2 encoding thymidine phosphorylase, or dominant mutations in POLG, POLG2, encoding the accessory beta-subunit of polγ,3 SLC25A4, encoding adenine nucleotide translocator 1 (ANT-1),4 OPA1,5,6 RRM2B,7 or PEO1, encoding Twinkle,8 the hexameric, replicative mtDNA helicase which is a key protein of the mtDNA replisome.
Pathogenic mutations in the PEO1 gene have been associated with a number of different clinical presentations.8–13 Recessive PEO1 mutations cause severe, early-onset disorders of mtDNA maintenance, such as infantile-onset spinocerebellar ataxia (IOSCA)14 and a hepatocerebral mtDNA depletion disorder,15,16 although long-term follow up of IOSCA patients indicates that severe epilepsy, migraine, and psychiatric symptoms can manifest later.17 Conversely, the majority of adult-onset cases present with autosomal dominant PEO (adPEO) characterized by ptosis and ophthalmoparesis, with cytochrome c oxidase (COX)-deficient and ragged-red fibers and multiple mtDNA deletions in muscle. Patients with PEO1-linked adPEO may have other clinical features including proximal muscle weakness, ataxia, peripheral neuropathy, cardiomyopathy, cataracts, and depression8; the sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) phenotype has also been described.18
We present the clinical and laboratory findings in 33 patients with PEO1 mutations, highlighting the phenotypic spectrum of this disorder.
METHODS
Subjects.
We identified and studied 26 unrelated probands with PEO and a suspected diagnosis of mitochondrial disease who had been referred to established UK National Commissioning Group-funded Mitochondrial Diagnostic Centers at Oxford, Newcastle, and London, or diagnostic services in Munich and Bonn, Germany, for clinical assessment and molecular or biochemical analysis. For the majority of cases, PEO1 gene testing was considered appropriate in view of the clinical presentation of PEO and the presence of multiple mtDNA deletions in muscle. PEO1 analysis was initiated in 3 patients due to the presence of PEO and a family history consistent with an autosomal dominant inheritance pattern (patients 13, 24, and 25) and in another patient due to clinical phenotype, family history, and presence of COX-deficient and ragged-red fibers on muscle biopsy (patient 9), although the presence of secondary mtDNA deletions was not investigated. For patient 5-1, PEO1 gene analysis was undertaken in spite of normal mtDNA rearrangement screening in muscle and very mild histochemical changes due to the clinical phenotype. Patients 5-2 and 5-3 were subsequently screened after the mutation was identified in patient 5-1. The clinical features of all 26 probands and an additional 7 family members (33 patients in total) are summarized in table e-1 on the Neurology® Web site at www.neurology.org. PEO1 sequencing, together with SLC25A4 gene analysis, was performed when initial screening for POLG mutations did not reveal a molecular defect.
Muscle histology and histochemistry.
For patients who underwent open or needle muscle biopsy, mitochondrial changes were assessed in muscle using standard histologic and histochemical assessments. Cryostat sections (10 μm) were cut from transversely orientated muscle blocks and subjected to COX, succinate dehydrogenase (SDH), and sequential COX-SDH histochemical staining to identify COX-deficient fibers.19 The SDH section was used to determine the number of fibers exhibiting increased levels of enzyme activity in the subsarcolemmal region, so-called ragged blue fibers.
mtDNA studies.
Multiple mtDNA deletions were investigated in skeletal muscle DNA by Southern blotting or long range PCR protocols as described previously.20
PEO1 gene screening.
The 5 coding exons and adjacent intronic regions of the PEO1 gene were amplified and sequenced in the forward and reverse directions using the ABI Prism 3130 or 3730 Genetic Analyzer and Big Dye terminator kit 3.1 or 1.1 (Applied Biosystems, Foster City, CA). Primers and PCR cycling conditions were as previously described.21 All substitutions were compared to the cDNA sequence (GenBank Accession number AF292005) and confirmed by resequencing. The frequency of novel PEO1 substitutions in UK control alleles (n = 180 chromosomes) was determined by primer extension and matrix-associated laser desorption/ionization time of flight (MALDI-TOF; Sequenom, San Diego, CA) mass spectrometry.
Standard protocol approvals, registration, and patient consents.
This study was approved and performed under the ethical guidelines issued by each of our institutions for clinical studies, with written informed consent obtained from all subjects.
Statistical analysis.
Allele frequencies were compared using Fisher exact test. Exact 95% confidence intervals were determined by the method of Clopper and Pearson.22
RESULTS
Clinical phenotype.
The clinical features of 33 patients from 26 pedigrees are summarized in table e-1 and table 1. Mean age at onset was 42 years (range 8-65 years) with current age 57 years (range 19-78 years) (table 2). The most frequent clinical presentations of our patients were ptosis (32 patients) and PEO (31 patients). This was often accompanied by fatigue (17 patients) and mild proximal muscle weakness (11 patients) resulting in minimal to mild disability only. Other muscle-related symptoms such as muscle pain (4 patients) and dysphagia (4 patients) were less frequent. Cardiac findings occurred in 8 patients including ventricular enlargement (4 patients), nonfatal arrhythmias (3 patients, in 1 patient both), and nonspecific ECG changes (2 patients), suggesting that myocardial involvement and conduction defects may be part of the Twinkle phenotype. Other symptoms were relatively uncommon and included endocrinologic dysfunction (4 patients), migraine (3 patients), visual impairment (4 patients), lethargy (3 patients), gastrointestinal symptoms (3 patients), hearing loss (3 patients), cataracts (3 patients), and epilepsy (1 patient). The occurrence of these symptoms in our patients, albeit mild and minimally disabling, implies that adPEO may be accompanied by multiorgan involvement; however, this needs to be further elucidated.
Table 1 Summary of the clinical data of 33 patients (26 probands) with pathogenic mutations in the PEO1 gene
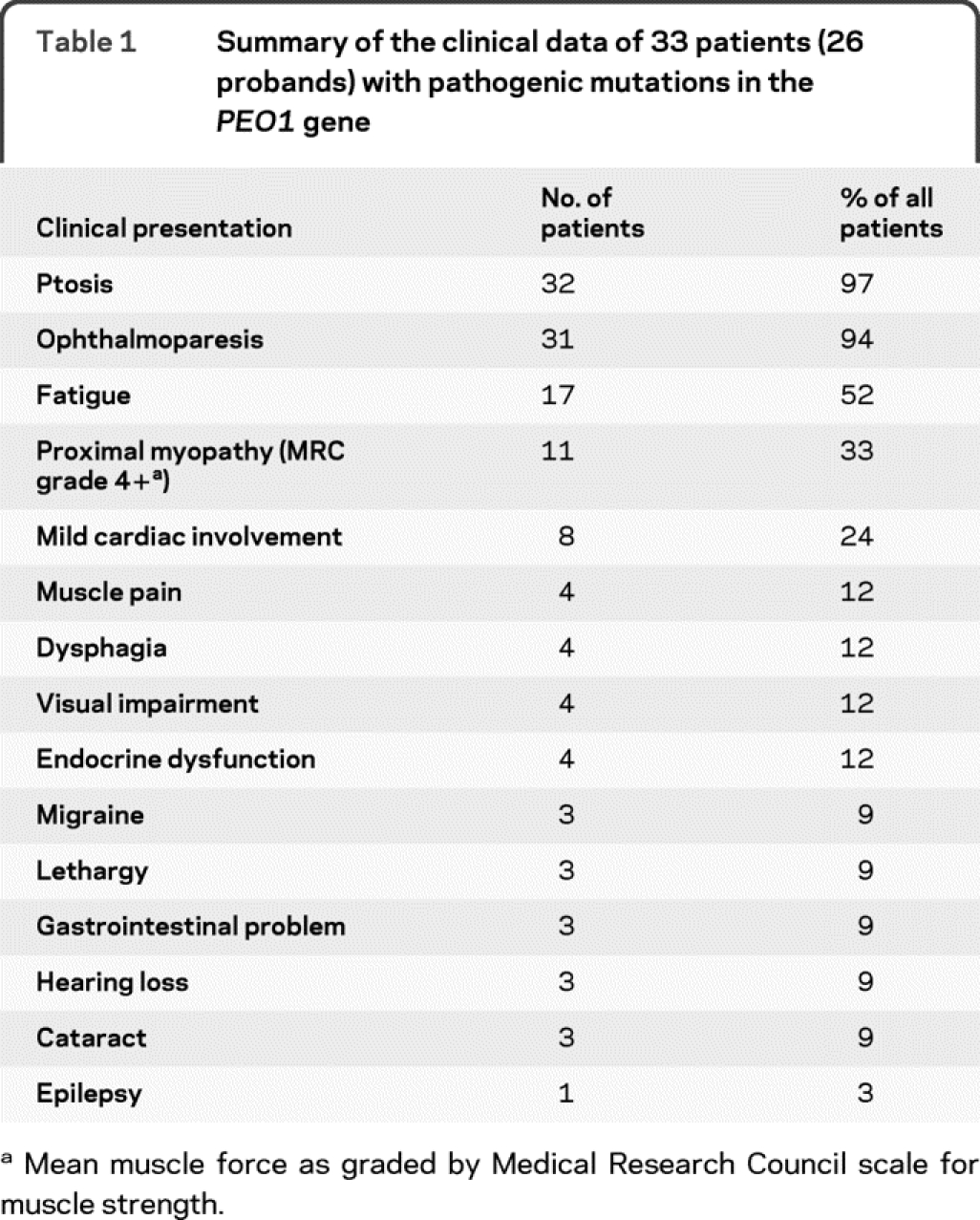
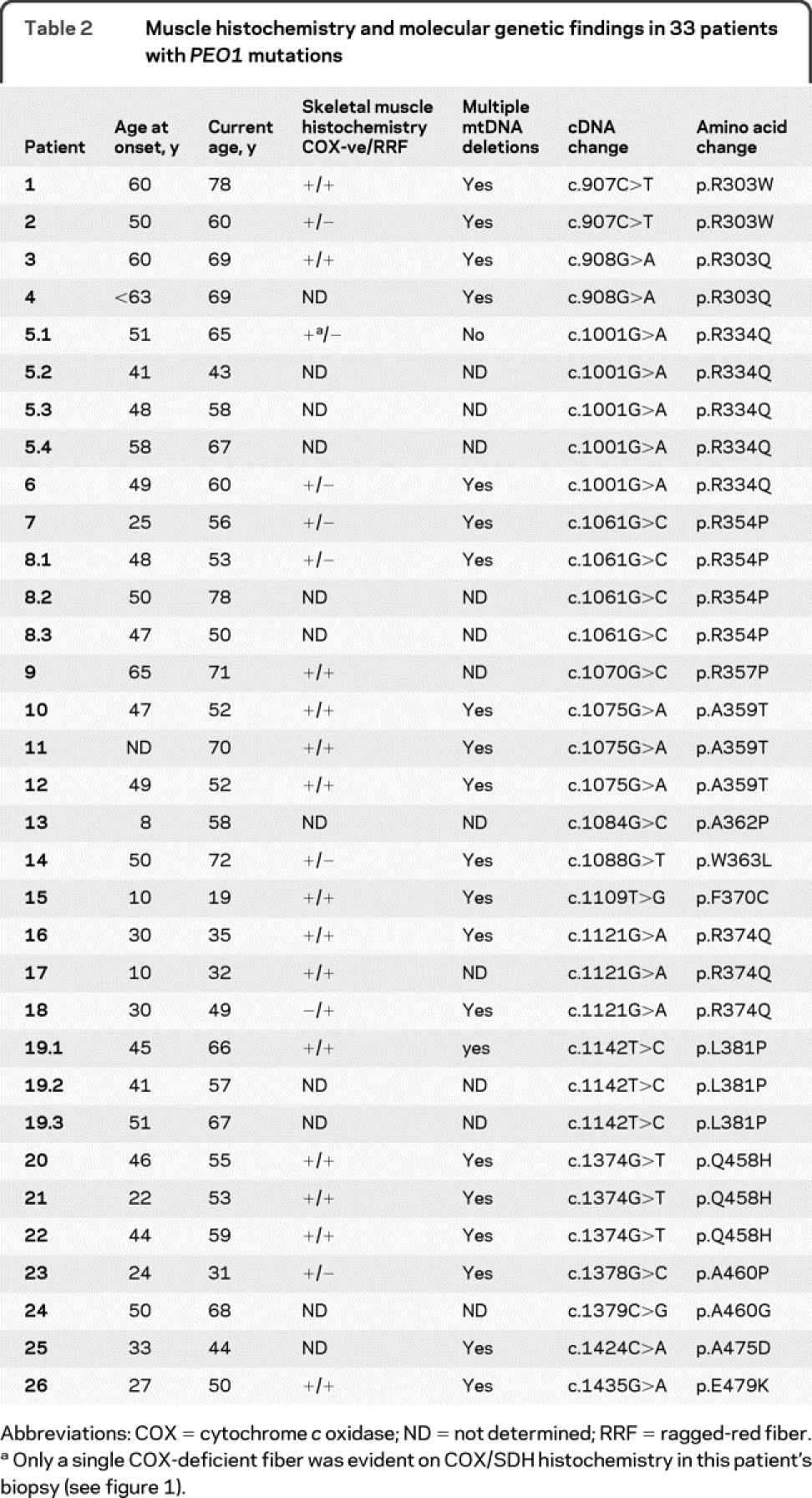
Table 2 Muscle histochemistry and molecular genetic findings in 33 patients with PEO1 mutations
Muscle histology and histochemistry.
Abnormal muscle biopsy findings, in particular the finding of COX-deficient fibers, are recognized as a pathologic hallmark of mtDNA maintenance disorders associated with multiple mtDNA deletions. Of the 33 patients we studied (26 probands), 23 underwent skeletal muscle biopsy for diagnostic purposes; 3 probands (patients 13, 24, and 25) were not biopsied, with mtDNA maintenance gene testing being undertaken on the grounds of a suggestive clinical presentation and dominant family history (table 2, table e-1); patient 4 was biopsied but was not subjected to histochemical analysis.
All 22 available muscle biopsies showed evidence of mitochondrial abnormalities, but with variable severity. The biopsy of patient 5-1 only revealed a single, COX-deficient (SDH positive) fiber, whereas that of patient 26 showed marked histocytochemical abnormalities (figure 1). For the majority of patients, a quantitative assessment of these changes was not possible but only a few COX-deficient fibers were described (patients 2, 3, 7, 8-1, 10, 12, 14, 15, 19.1, 20, 21, 22, and 23).

Figure 1 Skeletal muscle histochemistry and mtDNA analysis in patients with PEO1 mutations
(A, B) Dual COX-SDH histochemistry showing a mosaic distribution of COX-deficient muscle fibers (blue) among fibers with normal COX activity (brown). Panel A shows the mild biopsy findings in patient 5-1 (p.R334Q mutation), contrasting with the markedly abnormal pattern of COX deficiency demonstrated in patient 26 (p.E479K mutation) shown in B. (C) Long-range PCR of skeletal muscle mtDNA (extracted from a tissue homogenate) from these patients, confirming the absence of detectable mtDNA deletions in patient 5-1. Lane 1, 1kb ladder (Mr); lane 2, adult control muscle (C); lane 3, patient 5-1; lane 4, patient 26 showing multiple mtDNA deletions (highlighted by an asterisk).
Detection of multiple mtDNA deletions in muscle.
Muscle DNA samples were screened for the presence of multiple mtDNA deletions by long-range PCR in 22 of the probands. Variable mtDNA deletions were detected in all patients with the exception of 1 case, patient 5-1, whose biopsy had revealed minimal histochemical changes (figure 1). In some cases, marked abnormalities were detected using this assay (patient 26, figure 1), reflecting the severity of the histochemical defect. Southern blotting was performed as a diagnostic test in 15 patients; 11 patients had detectable mtDNA rearrangements on Southern blotting, but 4 patients (patients 5-1, 6, 11, and 18) only showed wild type, full-length mtDNA. A similar finding has been reported previously in patients with PEO1 mutation23 and other mtDNA maintenance gene (e.g., POLG) disorders.24,25
Identification of PEO1 gene mutations.
We identified 16 different mutations within the 26 unrelated probands; all changes were heterozygous missense mutations (figure 2; figure e-1). For clarity, we refer to the mutation on the basis of the predicted amino acid substitution although details of the cDNA changes are presented in table 2 and table e-1. Interestingly, all PEO1 mutations described in our collective and all those described previously in adPEO patients, with the exception of a 13-amino acid duplication,8 are missense changes; no nonsense, frameshift, or splice site mutations have been detected in adPEO patients to date.

Figure 2 Schematic highlighting the organization of the PEO1 gene (exons 1-5), and the location of the 16 different PEO1 mutations detected in our patient cohort
All mutations are found within exons 1 and 2; novel PEO1 variants are shown in pink shading. The primase-related domain is highlighted in yellow, the linker region in orange, and the helicase domain in red.
Seven PEO1 mutations (p.R303W, p.R303Q, p.R334Q, p.R354P, p.A359T, p.R374Q, p.Q458H) were identified in more than one family; the remaining 9 mutations were unique to individual families. Seven of the 16 mutations identified are previously unreported (p.A362P, p.W363L, p.F370C, p.Q458H, p.A460G, p.A460P, p.A475D). All pathogenic PEO1 mutations were excluded in a large collection of control chromosomes, with the allele frequencies of all reported PEO1 mutations shown in table e-1.
Affected relatives of patients 5, 8, and 19 were subjected to mutation testing (table 2 and table e-1), although formal segregation studies were not possible for the majority of families. For patient 15, analysis of parental samples indicated that p.F370C has arisen de novo in the proband, corroborated by the absence of any family history of PEO. For patient 25, p.A475D was shown to segregate with disease in father and son. Interestingly, we identified novel mutations (p.F370C and p.A475D) at 2 codons where different amino acid substitutions (p.F370L and p.A475P/p.A475T) have previously been reported as pathogenic,8–10 providing further evidence that these novel PEO1 mutations cause disease; the finding of the p.Q458H mutation in 3 families (patients 20, 21, and 22) likewise supports pathogenicity at this site.
As described in previous studies, the 16 pathogenic mutations were clustered within exons 1 and 2 of the PEO1 coding sequence, and found within different domains of the protein11 (table 2 and table e-1): 3 mutations were present in the primase-related domain, 7 in the linker domain, 1 at the N-terminal end of the helicase domain, and 5 in the helicase domain between the Walker A and B motifs. All 7 of the novel PEO1 mutations identified show a high degree of phylogenetic sequence conservation for the affected amino acid; 4 codons are conserved at least across all vertebrates (p.A362, p.F370, p.Q458, and p.A475), and the other 2 codons (p.W363 and p.A460) are conserved across all mammals except opossum (figure e-1). Mutations in POLG and SLC25A4 (ANT1) were excluded in these patients. In spite of its size, we were unable to identify any clear genotype-phenotype correlations in our patient cohort.
DISCUSSION
Familial PEO associated with multiple mtDNA deletions was first described in 198926 and since then PEO has been used as a clinical hallmark of multiple mtDNA deletion syndromes, leading to the discovery of several disease genes. Mutations in SLC25A4 (ANT1)4, PEO1,8 POLG,27 POLG2,3 and most recently RRM2B7 have been described in autosomal dominant PEO families. Routine genetic analysis of these genes in patients with PEO and multiple mtDNA deletions has led to the better characterization of these genetic defects. Within the frequently used PEO plus phenotype, recognizable clinical syndromes were characterized for the frequent autosomal dominant and recessive POLG mutations. However, there is less known about PEO1 defects. Mutations in POLG have been identified in a wide range of mitochondrial diseases, including severe childhood encephalopathy with epilepsy associated with liver failure (Alpers-Huttenlocher syndrome), adult onset spinocerebellar ataxia, and sensory nerve degeneration with or without epilepsy.1 In addition, Leigh syndrome28 and parkinsonism and premature ovarian failure causing early infertility have been described.29 Although the first pathogenic POLG mutations were detected in autosomal dominant PEO plus families, autosomal recessive POLG disease appears to be more frequent. The clinical presentation of dominant and recessive cases may be similar and multiple mutations can complicate the inheritance.
The clinical presentation of PEO1 mutations seems to be less variable, and there is a clear difference between the more frequent autosomal dominant and the rare recessive disease. We were able to find detailed clinical information on 70 individuals from 29 families reported in the literature to date (table e-2). 9–13,18,30–38 In most patients, PEO was the only clinical presentation; however, proximal muscle weakness,30,32,36 sensory-axonal neuropathy,10,17 ataxia,33 late-onset dementia,13 and parkinsonism35 were also reported in a small number of cases.
IOSCA and severe infantile hepatoencephalopathy, clinically resembling POLG deficiency, have been described in association with autosomal recessive PEO1 mutations and mtDNA depletion.14–16 Most of these cases are caused by a Finnish founder mutation (p.Y508C). Although the clinical presentations of autosomal recessive and autosomal dominant POLG and PEO1 mutations show similarities, their phenotypes may help to differentiate between these 2 genetic defects.
Ptosis and ophthalmoparesis are the predominant clinical characteristics of autosomal dominant PEO1 mutation in our cohort and previously published data36,39; however, isolated ptosis can occur (patient 8-3). Proximal myopathy, present in 11/33 patients in our cohort, has been reported previously in the literature, albeit to a lesser extent.11,12,17,30,32,34–36,39,40 Overt muscle wasting in one of the subjects has been noted previously, including wasting of shoulder and quadriceps muscles,32 facial weakness,30 and neck flexor weakness,30,35,36 as well as being reported in deceased family members suspected of harboring the genetic defect.39 Muscle pain and cramps have only been documented previously in one other cohort.39
Bulbar symptoms including dysphagia,11,32,34,40 dysphonia, and dysarthria11,17,34,39 are more common than might occur by chance. Presenile cataracts, present in 3 subjects in our cohort, have also been described previously.11,12,39 Epilepsy and migraine have only been noted in association with Twinkle mutation in isolated cases,11,17 while depression, dementia, and neuropsychiatric symptoms are more widespread.11,12,17,35,39
Cardiac sequelae as reported in 8/33 subjects are a common finding including sinus bradycardia and ischemic heart disease,39 right bundle branch block,30 palpitations,30,32 dilated cardiomyopathy, atrial arrhythmias,32 and hypertension.23 In addition, deceased family members suspected of harboring the mutation had documented cardiomyopathy.32
Respiratory muscle weakness and failure was occasionally described previously, which warrants further investigation30,34,35 and has also been reported in deceased family members with other cardinal features of the condition.32 Although no severe respiratory failure was detected in our patient collective, we cannot exclude that the frequently observed daytime fatigue is at least partially caused by hypoventilation. Therefore we suggest more thorough investigations of respiratory function in patients with mutations in PEO1. Endocrine dysfunction includes thyroid disease,11,39 diabetes,12 premature ovarian failure,11 and mild primary hypogonadism.39 Neuropathy18,35 and parkinsonian features9 are infrequently present.
We could not discern clear genotypic/phenotypic correlations within the patients studied, and earlier onset of symptoms was not associated with increased disease severity; however, we can suggest in patients with adPEO, a PEO1 defect is highly likely. We have only identified 5 adPEO families with dominant POLG mutations and 1 adPEO family with a dominant SLC25A4 mutation at our 3 centers. These clinical observations are in keeping with previously published results and provide important information about the disease prognosis, clinical management, and counseling of PEO1 mutation families.
We detected 16 different PEO1 mutations in 26 families; 9 of these have been reported previously (figure 2). However, we have identified 7 novel PEO1 mutations in this study (p.A362P, p.W363L, p.F370C, p.Q458H, p.A460G, p.A460P, and p.A475D). Evidence for pathogenicity of the novel changes is highly suggestive for the following reasons: 1) they affected conserved amino acids which are located close to previously described pathogenic mutations; 2) they were not present in a large number of ethnically matched control DNA samples; 3) they were associated with muscle histochemical abnormalities characteristic of mtDNA disease, and/or multiple mtDNA deletions (no muscle biopsy studies for p.A362P or p.A460G mutations); 4) in families where samples were available they cosegregated with the clinical phenotype, consistent with an autosomal dominant pattern of inheritance. Although segregation studies were not possible in all families, other evidence to support a pathogenic role for these changes included the mutation of amino acids already reported as linked to adPEO, and the finding of the same, novel PEO1 mutation in more than one family (table 2 and table e-1). Interestingly, all mutations found in our patients and in previous studies are either missense or in-frame duplications (1 case; reference 8). The absence of nonsense, frameshift, or splice-site PEO1 mutations suggests the possibility that they are either not compatible with life or do not cause a PEO plus phenotype. Furthermore, in patient 15, the p.F370C mutation was shown to have arisen de novo. To our knowledge, this is the fifth reported example of a de novo PEO1 gene mutation.10,11,21 De novo mutation at the identical amino acid site (c.1110C>A, p.F370L) was also previously described, raising the possibility that this region could be a mutation hot spot.10
Our study provides important information about the clinical phenotype and disease prognosis in adPEO due to PEO1 mutation, which can be used in the clinical management of individual patients and counseling of these families. These data suggest that neurologic sequelae of autosomal dominant PEO1 mutations are relatively benign; however, the possible involvement of other organs would advocate screening for predominantly cardiac and endocrine abnormalities. Sequencing of the coding region of the PEO1 gene is recommended in all patients with an adPEO phenotype, irrespective of whether the muscle biopsy findings and long-range PCR screen for mtDNA rearrangements are normal. We therefore suggest that in families with a predominant adPEO phenotype with or without mild additional symptoms, sequencing of the PEO1 gene should be performed prior to a muscle biopsy.
ACKNOWLEDGMENT
The authors thank the referring clinicians, the patients, and their families for contributing to this study. The mitochondrial diagnostic laboratories in Oxford, Newcastle, and London (UCLH/Institute of Neurology) are funded by the National Commissioning Group to provide the UK “Rare Mitochondrial Disease of Adults and Children” Service.
DISCLOSURE
Dr. Fratter reports no disclosures. Dr. Gorman receives research support from the UK NIHR Biomedical Research Centre for Aging and Age-related Disease awarded to the Newcastle upon Tyne Trust Foundation Hospitals NHS Trust. Dr. Stewart, Dr. Buddles, Dr. Smith, Dr. Evans, and Dr. Seller report no disclosures. Dr. Poulton receives research support from the Wellcome Trust, the MRC, and the Angus Memorial Mitochondrial Fund. Dr. Roberts reports no disclosures. Dr. Hanna serves as Deputy Editor of the Journal of Neurology, Neurosurgery, and Psychiatry and receives research support from the MRC Centre for Neuromuscular Diseases and the MRC Centre for Translational Research in Neuromuscular Disease Mitochondrial Disease Patient Cohort (UK). Dr. Rahman serves as Communicating Editor for the Journal of Inherited Metabolic Diseases and has received research support from the Medical Research Council, SPARKS, and Jeans 4 Genes. Dr. Omer has received funding for travel from Eisai Inc. and GlaxoSmithKline. Dr. Klopstock has received funding for travel and speaker honoraria from Dr. Willmar Schwabe GmbH & Co. KG and receives research support from Santhera Pharmaceuticals and the German Ministry of Education and Research (BMBF). Dr. Schoser reports no disclosures. Dr. Kornblum has received funding for travel and speaker honoraria from Genzyme Corporation and receives research support from the German Ministry of Education and Research (BMBF). Dr. Czermin, Dr. Lecky, Dr. Blakely, and Dr. Craig report no disclosures. Dr. Chinnery serves as an Associate Editor of Brain and receives research support from the Wellcome Trust, the MRC, and the UK Parkinson's Disease Society. Dr. Turnbull receives research support from Wellcome Trust, the MRC Centre for Neuromuscular Diseases, and the MRC Centre for Translational Research in Neuromuscular Disease Mitochondrial Disease Patient Cohort (UK). Dr. Horvath receives research support from the Deutsche Forschungsgemeinschaft, the Newcastle upon Tyne Hospitals NHS Charity, and the Academy of Medical Sciences (UK). Dr. Taylor receives research support from the Wellcome Trust and the MRC Centre for Translational Research in Neuromuscular Disease Mitochondrial Disease Patient Cohort (UK).
Supplementary Material
Address correspondence and reprint requests to Professor R.W. Taylor, Mitochondrial Research Group, Institute of Ageing and Health, The Medical School, Newcastle University, Framlington Place, Newcastle upon Tyne, NE2 4HH, UK r.w.taylor@ncl.ac.uk
Supplemental data at www.neurology.org
*These authors contributed equally to this work.
Disclosure: Author disclosures are provided at the end of the article.
Received September 29, 2009. Accepted in final form February 3, 2010.
REFERENCES
- 1.Horvath R, Hudson G, Ferrari G, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain 2006;129:1674–1684. [DOI] [PubMed] [Google Scholar]
- 2.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999;283:689–692. [DOI] [PubMed] [Google Scholar]
- 3.Longley MJ, Clark S, Yu Wai Man C, et al. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet 2006;78:1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaukonen J, Juselius JK, Tiranti V, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000;289:782–785. [DOI] [PubMed] [Google Scholar]
- 5.Hudson G, Amati-Bonneau P, Blakely EL, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 2008;131:329–337. [DOI] [PubMed] [Google Scholar]
- 6.Amati-Bonneau P, Valentino ML, Reynier P, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 2008;131:338–351. [DOI] [PubMed] [Google Scholar]
- 7.Tyynismaa H, Ylikallio E, Patel M, Molnar MJ, Haller RG, Suomalainen A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am J Hum Genet 2009;85:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spelbrink JN, Li FY, Tiranti V, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet 2001;28:223–231. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Ding Y, Du A, Zhang B, Zhao G, Ding M. A novel Twinkle (PEO1) gene mutation in a Chinese family with adPEO. Mol Vis 2008;14:1995–2001. [PMC free article] [PubMed] [Google Scholar]
- 10.Jeppesen TD, Schwartz M, Colding-Jørgensen E, Krag T, Hauerslev S, Vissing J. Phenotype and clinical course in a family with a new de novo Twinkle gene mutation. Neuromuscul Disord 2008;18:306–309. [DOI] [PubMed] [Google Scholar]
- 11.Virgilio R, Ronchi D, Hadjigeorgiou GM, et al. Novel Twinkle (PEO1) gene mutations in mendelian progressive external ophthalmoplegia. J Neurol 2008;255:1384–1391. [DOI] [PubMed] [Google Scholar]
- 12.Van Hove JL, Cunningham V, Rice C, et al. Finding twinkle in the eyes of a 71-year-old lady: a case report and review of the genotypic and phenotypic spectrum of Twinkle-related dominant disease. Am J Med Genet A 2009;149:861–867. [DOI] [PubMed] [Google Scholar]
- 13.Echaniz-Laguna A, Chanson JB, Wilhelm JM, et al. A novel variation in the Twinkle linker region causing late-onset dementia. Neurogenetics 2010;11:21–25. [DOI] [PubMed] [Google Scholar]
- 14.Nikali K, Suomalainen A, Saharinen J, et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet 2005;14:2981–2990. [DOI] [PubMed] [Google Scholar]
- 15.Sarzi E, Goffart S, Serre V, et al. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann Neurol 2007;62:579–587. [DOI] [PubMed] [Google Scholar]
- 16.Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lönnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain 2007;130:3032–3040. [DOI] [PubMed] [Google Scholar]
- 17.Lönnqvist T, Paetau A, Valanne L, Pihko H. Recessive twinkle mutations cause severe epileptic encephalopathy. Brain 2009;132:1553–1562. [DOI] [PubMed] [Google Scholar]
- 18.Hudson G, Deschauer M, Busse K, Zierz S, Chinnery PF. Sensory ataxic neuropathy due to a novel C10Orf2 mutation with probable germline mosaicism. Neurology 2005;64:371–373. [DOI] [PubMed] [Google Scholar]
- 19.Taylor RW, Schaefer AM, Barron MJ, McFarland R, Turnbull DM. The diagnosis of mitochondrial muscle disease. Neuromuscul Disord 2004;14:237–245. [DOI] [PubMed] [Google Scholar]
- 20.Murphy JL, Blakely EL, Schaefer AM, et al. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 2009;131:2832–2840. [DOI] [PubMed] [Google Scholar]
- 21.Hudson G, Deschauer M, Taylor RW, et al. POLG1, C10ORF2, and ANT1 mutations are uncommon in sporadic progressive external ophthalmoplegia with multiple mitochondrial DNA deletions Neurology 2006;66:1439–1441. [DOI] [PubMed] [Google Scholar]
- 22.Armitage P, Berry G, Matthews JNS. Statistical Methods in Medical Research. Oxford: Blackwell; 2002. [Google Scholar]
- 23.Deschauer M, Kiefer R, Blakely EL, et al. A novel Twinkle gene mutation in autosomal dominant progressive external ophthalmoplegia. Neuromuscul Disord 2003;13:568–572. [DOI] [PubMed] [Google Scholar]
- 24.Van Goethem G, Martin JJ, Van Broeckhoven C. Progressive external ophthalmoplegia characterized by multiple deletions of mitochondrial DNA: unraveling the pathogenesis of human mitochondrial DNA instability and the initiation of a genetic classification. Neuromolecular Med 2003;3:129–146. [DOI] [PubMed] [Google Scholar]
- 25.Winterthun S, Ferrari G, He L, et al. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology 2005;64:1204–1208. [DOI] [PubMed] [Google Scholar]
- 26.Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989;339:309–311. [DOI] [PubMed] [Google Scholar]
- 27.Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 2001;28:211–212. [DOI] [PubMed] [Google Scholar]
- 28.Taanman JW, Rahman S, Pagnamenta AT, et al. Analysis of mutant DNA polymerase gamma in patients with mitochondrial DNA depletion. Hum Mutat 2009;30:248–254. [DOI] [PubMed] [Google Scholar]
- 29.Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 2004;364:875–882. [DOI] [PubMed] [Google Scholar]
- 30.Lewis S, Hutchison W, Thyagarajan D, Dahl HH. Clinical and molecular features of adPEO due to mutations in the Twinkle gene. J Neurol Sci 2002;201:39–44. [DOI] [PubMed] [Google Scholar]
- 31.Agostino A, Valletta L, Chinnery PF, et al. Mutations of ANT1, Twinkle, and POLG1 in sporadic progressive external ophthalmoplegia (PEO). Neurology 2003;60:1354–1356. [DOI] [PubMed] [Google Scholar]
- 32.Kiechl S, Horváth R, Luoma P, et al. Two families with autosomal dominant progressive external ophthalmoplegia. J Neurol Neurosurg Psychiatry 2004;75:1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Houshmand M, Panahi MS, Hosseini BN, Dorraj GH, Tabassi AR. Investigation on mtDNA deletions and twinkle gene mutation (G1423C) in Iranian patients with chronic progressive external ophthalmoplegia. Neurol India 2006;54:182–185. [PubMed] [Google Scholar]
- 34.Naïmi M, Bannwarth S, Procaccio V, et al. Molecular analysis of ANT1, TWINKLE and POLG in patients with multiple deletions or depletion of mitochondrial DNA by a dHPLC-based assay. Eur J Hum Genet 2006;14:917–922. [DOI] [PubMed] [Google Scholar]
- 35.Baloh RH, Salavaggione E, Milbrandt J, Pestronk A. Familial parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch Neurol 2007;64:998–1000. [DOI] [PubMed] [Google Scholar]
- 36.Rivera H, Blázquez A, Carretero J, et al. Mild ocular myopathy associated with a novel mutation in mitochondrial twinkle helicase. Neuromuscul Disord 2007;17:677–680. [DOI] [PubMed] [Google Scholar]
- 37.Negro R, Zoccolella S, Dell'aglio R, et al. Molecular analysis in a family presenting with a mild form of late-onsetautosomal dominant chronic progressive external ophthalmoplegia. Neuromuscul Disord 2009;19:423–426. [DOI] [PubMed] [Google Scholar]
- 38.Vandenberghe W, Van Laere K, Debruvne F, Van Broeckhoven C, Van Goethem G. Neurodegenerative parkinsonism and progressive external ophthalmoplegia with a Twinkle mutation. Mov Disord 2009;24:308–309. [DOI] [PubMed] [Google Scholar]
- 39.Suomalainen A, Majander A, Wallin M, et al. Autosomal dominant progressive external ophthalmoplegia with multiple deletions of mtDNA: clinical, biochemical and molecular genetic features of the 10q-linked disease. Neurology 1997;48:1244–1253. [DOI] [PubMed] [Google Scholar]
- 40.Li FY, Tariq M, Croxen R, et al. Mapping of autosomal dominant progressive external ophthalmoplegia to a 7-cM critical region on 10q24. Neurology 1999;53:1265–1271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.